

Abstract
Just as Koch’s postulates formed the foundation of early infectious disease study, Stanley Falkow’s molecular Koch’s postulates define best practice in determining whether a specific gene contributes to virulence of a pathogen. Fundamentally, these molecular postulates state that if a gene is involved in virulence, its removal will compromise virulence. Likewise, its reintroduction should restore virulence to the mutant. These approaches are widely employed in Cryptococcus neoformans, where gene deletion via biolistic transformation is a well-established technique. However, the complementation of these mutants is less straightforward. Currently, one of three approaches will be taken: the gene is reintroduced at the original locus, the gene is reintroduced into a random site in the genome, or the mutant is not complemented at all. Depending on which approach is utilized, the mutant may be complemented but other genes are potentially disrupted in the process. To counter the drawbacks of the current approaches to complementation we have created a new tool to assist in this key step in the study of a gene’s role in virulence. We have identified and characterized a small gene-free region in the C. neoformans genome dubbed the “safe haven”, and constructed a plasmid vector that targets DNA constructs to this preselected site. The plasmid vector integrates with high frequency, effectively complementing a mutant strain without disrupting adjacent genes. qRT-PCR of the flanking genes on either side of the safe haven site following integration of the targeting vector revealed no changes in their expression, and no secondary phenotypes were observed in a range of phenotypic assays including an intranasal murine infection model. Combined, these data confirm that we have successfully created a much-needed molecular resource for the Cryptococcus community, enabling the reliable fulfillment of the molecular Koch’s postulates.
Introduction
In 1884, Robert Koch proposed a set of four criteria designed to establish a causative relationship between a microbe and a disease that defined infectious disease research in the 19th and 20th centuries [1]. Koch’s postulates stated that the causative agent of a disease must be found in abundance in all organisms suffering from the condition, but should not be found in association with healthy individuals. Furthermore, the etiological agents of the disease must be able to be isolated from an infected individual, and the isolated microbe should subsequently cause disease when introduced into a healthy individual [1]. Finally, the etiological agent must be able to be reisolated from the inoculated host, and be identified as identical to the original causative agent.
When the era of molecular genetics began nearly a century later, the need arose to redefine Koch’s postulates. As molecular studies of disease-causing microbes advanced, Stanley Falkow modified Koch’s postulates in order to apply them to the molecular genetics of pathogenicity [2]. Following equivalent criteria to his predecessor, Falkow extended the postulates to require a virulence phenotype under investigation to be associated with a pathogenic species, and the deletion of the genes required for this phenotype to result in a reduction or loss of virulence. Additionally, and perhaps most importantly, the subsequent reintroduction of the wild-type gene should restore pathogenicity, proving that the reduction of virulence observed in the mutant is indeed due to the loss of the gene of interest [2]. These molecular Koch’s postulates now serve as the foundation of molecular genetic studies in pathogenic species.
The experimental methodology employed in creating targeted gene deletion strains differs between species. Common methods include biolistic, chemical and protoplast-mediated transformation, all of which rely on homologous recombination to replace the gene of interest with a selectable marker. The degree of success via these approaches is species-dependent. In Saccharomyces cerevisiae, gene deletion through homologous recombination occurs readily, with a 30–60% chance of the desired mutant being created in a single transformation attempt and the entire process taking less than a week [3,4]. In contrast, in species such as Mycobacterium tuberculosis, gene deletion mutants are still extremely difficult to create [5].
Importantly, successful molecular genetic analysis of the role of a gene in virulence does not end with creating a single mutant strain. To fulfill Falkow’s molecular Koch’s postulates, and to prove any reduction in virulence is due to the deletion of the gene of interest rather than unanticipated consequences of the gene deletion process, the mutant strain must be complemented. Complementation of a gene deletion strain involves reintroducing a wild-type copy of the deleted gene back into the genome. As with the deletion process, the methodology of complementation also varies between species. In Escherichia coli, the reintroduction of the wild-type gene is usually straightforward, achieved via introduction of a plasmid that carries the wild-type gene of interest which is then maintained extrachromosomally [6]. In S. cerevisiae, one of three approaches is usually employed: the gene of interest is reintroduced in a high copy 2μ plasmid [7], in a low copy centromeric plasmid [8], or by integrating it into a predetermined location in the genome [9]. In Candida albicans, constructs can be targeted to integrate at the well characterized and highly expressed RPS10 locus, disrupting this allele in the process [10,11]. These types of well-utilized approaches enable molecular Koch’s postulates to be easily fulfilled in the majority of organisms in which targeted gene deletions can be made.
While molecular genetic studies were originally restricted to model systems, many other organisms of interest, including pathogens, can now also be easily manipulated. One such pathogen is Cryptococcus neoformans, a basidiomycete yeast that primarily infects immunocompromised individuals, disseminating via the lungs to cause life-threatening meningoencephalitis. In developed countries, the mortality rate of cryptococcosis is up to 20% [12,13]. Limited treatment availability in developing countries results in a much poorer patient prognosis; without treatment the mortality rate can be as high as 100% [12,13].
In the pursuit of a deeper understanding of this pathogenic species, manipulation of the C. neoformans genome has become common, although it is not without challenges. Gene deletion via biolistic transformation in C. neoformans typically results in homologous recombination frequencies of 1–10% [14–16], far lower than in S. cerevisiae. In addition, C. neoformans is also unable to stably maintain plasmids, making complementation of mutants more difficult.
There are currently three widely used approaches for complementing a gene deletion in C. neoformans. The simplest of these is to not complement at all, with a surprising number of publications passing peer review without including this important control. Sometimes this approach is accompanied with data from a sexual cross to show the mutant phenotype is linked to the inserted selectable marker; linkage does not, however, prove that any observed phenotypes are not caused by polar effects of the selectable marker on genes adjacent to the insertion site. When complementation is performed, the most common method is to integrate a wild-type copy of the gene randomly into the genome [17,18]. However, protein-coding genes account for 85% of the C. neoformans genome, and when taking into consideration the large number of miscRNAs also transcribed [19], it is highly likely that this random integration will disrupt other genes and produce confounding phenotypes. It is possible to address this problem via the creation of multiple, independent complemented strains to find a common phenotype. Ethically, only one of these strains can be employed in a murine virulence model, making it impossible to determine whether virulence has been affected by interruption of another non-target virulence-associated gene until after infection of the animals. An alternative complementation approach is to reintroduce the wild-type gene back into its native location [20]. However, this may influence the expression of adjacent genes due to the small intergenic regions and frequently overlapping transcripts of the C. neoformans genome [19].
In this study, we further analyze the two current methodologies for complementation in C. neoformans and provide evidence of their shortcomings. To counter the drawbacks of these approaches, we have developed a new molecular tool for complementation that obviates these problems. Following an analysis of gene distribution in the genome, we have characterised a small gene-free region termed the “safe haven” and developed a vector to facilitate integration of genetic constructs at this site. We have confirmed that integration of the vector at the safe haven has no effect on the transcription of flanking genes, on a range of virulence-associated phenotypes, or on virulence itself. Beyond providing a much-needed resource to facilitate the satisfaction of Falkow’s molecular Koch’s postulates in C. neoformans, our targeted safe haven approach also provides an effective avenue for the introduction of other useful genetic constructs such as fluorescent proteins and reporter constructs.
Methods
Bioinformatics
The genome sequence of the C. neoformans var. grubii type strain H99 was obtained from the Broad Institute “Cryptococcus neoformans var. grubii H99 Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org/)” Sequence analysis to determine the numbers of convergent, divergent and tandemly arrayed gene pairs, and the size of their intergenic regions, was performed in Excel using the relevant H99 gene file from the Broad Institute.
Strains and growth conditions
C. neoformans was cultured in YPD (2% bacto-peptone, 2% agar, 1% yeast extract and 2% glucose) media at 30°C unless stated otherwise. Biolistic transformants were selected on YPD medium containing 100 μg/mL nourseothricin (Werner Bioagents, Germany), G418 (Sigma, USA) or hygromycin B (Life Technologies, USA). All strains (S1 Table) were stored in 15% glycerol at -80°C until use, at which point they were maintained on YPD at 4°C for a maximum of two weeks. E. coli Mach1 cells (Invitrogen, USA) served as the host strain for transformation and propagation of all plasmids using lysogeny broth supplemented with 100 μg/ml ampicillin (Sigma, USA) [21].
Creation of a targeted integration vector
Plasmids constructed in this study are listed in S2 Table and primers in S3 Table. PCR for construct generation was performed using Phusion High Fidelity DNA Polymerase (New England Biolabs, USA). The intergenic region between H99 ORFs CNAG_00777 and CNAG_00778 was PCR amplified from genomic DNA as two fragments: chromosome 1 coordinates 2,045,692–2,046,441 using primers UQ2920 and UQ3107, and 2,046,485–2,047,234 using UQ2916 and UQ2917. A third fragment was amplified using UQ2918 and UQ2919 from a synthetic gBlock (IDT, USA) consisting of chromosome 1 nucleotides 2,045,692–2,045,827 followed by the restriction site-containing linker sequence GGCGCGCCACGATACTTGTGTTAATTAA and then chromosome 1 nucleotides 2,047,035–2,047,234. The three fragments were joined via overlap PCR using primers UQ2916 and UQ3107, purified, then the 1.5 kb product included in a second overlap PCR reaction using primers UQ3106 and UQ3107 to join them to the nourseothricin resistance marker NAT (amplified from pCH233 using primers UQ2915 and UQ3106) to give a 3.2 kb product.
pBluescript II SK- [22] was subsequently amplified with primers UQ3108 and UQ3109; both the 3.2 kb overlap and pBluescript II SK- products were then cut with NcoI and BglII, and ligated together prior to transformation into E. coli. The final product, pSDMA25, contained the safe haven, linker and nourseothricin resistance marker sequences in place of the pBluescript II SK- f1 origin of replication, maintaining the intact multicloning site and blue/white screening capability of the parent plasmid. Equivalent plasmids bearing G418 resistance (amplified from pJAF1 [23] using primers UQ2915 and UQ3106 to give pSDMA57) or hygromycin resistance (amplified from pJAF15 using primers UQ2915 and UQ3106 to give pSDMA58) markers were generated using the same strategy.
Construction of mutant strains
A deletion construct for the ADE2 gene was generated using overlap PCR, joining the ADE2 5’ region (primers UQ1439 and UQ1440), the G418 resistance marker NEO (UQ234 and UQ235) and ADE2 3’ region (UQ1441 and UQ1442); H99 genomic DNA was used as the template for ADE2 and pJAF1 for NEO [23]. C. neoformans transformation was carried out via biolistic particle delivery as previously described onto media containing G418 [24]. The ade2Δ strain SA26 was identified via its pink coloration and adenine auxotrophy, with correct integration confirmed by Southern blot.
To create wild-type strains carrying the empty safe haven vector, pSDMA25 was linearized with either AscI, BaeI or PacI as indicated and biolistically transformed into the recipient strain; to create an ade2Δ strain carrying the empty vector, BaeI was used. To complement the ade2Δ mutant using random integration, ADE2 was PCR amplified (UQ1439 and UQ1442) and cloned as a HindIII/EcoRI fragment into HindIII/EcoRI cut pBluescript II SK- to give pSDMA42. ADE2 was subsequently subcloned as an XbaI/XhoI fragment into XbaI/XhoI-cut NAT resistance vector pCH233 to create pSDMA55. pSDMA55 was then linearized with PvuII and biolistically transformed into the ade2Δ strain SA26. To complement the ade2Δ mutant by reintroducing the gene at the wild-type locus, ADE2 (UQ1439 and UQ3342), NAT (UQ3343 and UQ3344) and the ADE2 3’ region (UQ3345 and UQ3243) were amplified then joined via overlap PCR, with the product transformed into the ade2Δ strain SA26. To complement the ade2Δ mutant using targeted safe haven integration, ADE2 was subcloned from pSDMA42 as a SacII/XhoI fragment into SacII/XhoI-cut pSDMA25 to create pSDMA54. pSDMA54 was subsequently linearized using AscI and biolistically transformed into the ade2Δ strain SA26. All transformants were selected on YPD supplemented with nourseothricin, and confirmed by Southern blot analysis.
Multiplex colony PCR
Multiplex PCR was employed to confirm correct integration of the safe haven-targeting constructs. 12 μL of sterile water and a small amount of Cryptococcus cells on the end of a pipette tip were added to a PCR tube for each reaction and incubated for 10 minutes at 94°C. 2.5 μL of each of the 10 mM primer stocks (UQ1768, UQ2962, UQ2963 and UQ3348), 2.5 μL Taq buffer, 0.5 μL 10 mM dNTPs and 0.1 μL of Taq polymerase (New England Biolabs, USA) were then added and the reaction returned to the PCR machine. The cycling parameters were 35 cycles of 94°C for 20 seconds, 55°C for 20 seconds and 72°C for 90 seconds. Products were visualized using electrophoresis with a 1% TAE agarose gel.
Quantitative real-time PCR
C. neoformans strains were grown in YNB with shaking at 30°C for 16 hours. Cultures were harvested, cell pellets frozen and lyophilized, and total RNA isolated using TRIzol reagent (Life Technologies, USA). cDNA was generated using the Superscript III First-Strand Synthesis System (Invitrogen, USA). Primers for ADE2 (CNAG_02294), the genes flanking ADE2 (CNAG_02293 and CNAG_02295) and the genes flanking the safe haven insertion site (CNAG_00777 and CNAG_00778) were designed to span exon–exon boundaries. Quantitative real-time PCR (qRT-PCR) was performed using SYBR Green Supermix (Applied Biosystems) and an Applied Biosystems 7900HT Fast Real Time PCR System with the following cycling conditions: denaturation at 95°C for 10 minutes, followed by amplification and quantification in 45 cycles of 95°C for 15 seconds and 60°C for 1 minute, with melting-curve profiling at 95°C for 2 minutes, 60°C for 15 seconds, and 95°C for 15 seconds. Relative gene expression was quantified using SDS 1.3.1 (Applied Biosystems) based on the 2-ΔΔCT method [25]. The housekeeping actin-encoding gene ACT1 was used as a control for normalization. One-way analysis of variance was performed using the unpaired, two-tailed t-test in GraphPad Prism Version 6.0c (GraphPad Software, USA). P-values of <0.05 were considered statistically significant.
Murine inhalation model of cryptococcosis
For murine infection assays, 6-week-old female BALB/c mice were infected via nasal inhalation [26]. For each strain, 10 mice were inoculated with a 50 μL drop containing 5 × 105 cryptococcal cells. Mice were weighed daily, and once their body weight had decreased to 80% of pre-infection weight (or on the final day of the experiment), were euthanized using CO2 inhalation. For survival assays, Kaplan-Meier survival curves were plotted using GraphPad Prism 6.0 (GraphPad Software, USA). Significance was tested by using Students t-test on the logged data. P-values of <0.05 were considered significant.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes by the National Health and Medical Research Council. The protocol was approved by the Molecular Biosciences Animal Ethics Committee (AEC) of The University of Queensland (AEC approval no. SCMB/439/13/UQ/NHMRC). Infection was performed under methoxyflurane anesthesia, and all efforts were made to minimize suffering through adherence to the Guidelines to Promote the Wellbeing of Animals Used for Scientific Purposes as put forward by the National Health and Medical Research Council (Australia).
Results
Identifying locations in the Cryptococcus genome suitable for construct integration
Given it is an essential part of molecular genetic studies in C. neoformans, the creation of complemented derivatives of mutant strains has long proven a surprisingly problematic process. The sequencing of two C. neoformans var. neoformans strains revealed the genome of this species to be very gene-rich [19]. Consequently, reintroduction of a wild-type copy of the gene of interest through random integration is essentially a round of insertional mutagenesis which will more than likely disrupt another gene in the process, explaining the frequent phenotypic defects identified in complemented C. neoformans strains. Reinsertion at the original locus is no better, potentially leaving the new strain indistinguishable from a wild-type contaminant, and abolishing polar effects (or introducing new ones) on adjacent genes that may have contributed to the original mutant phenotype.
To provide a controlled, consistent complementation process we searched the genome for potential locations to which we could target an integrating vector without influencing other genetic factors. Our first consideration regarding suitability of a defined insertion site was gene distribution. Ideally, we wanted to find a gene-poor region of the genome to target our construct. One option was to target one of the large transposon fragment-rich centromeres, which are devoid of genes [19,27]. As the transcriptional fate of a construct targeted to such a region is unknown and highly likely different to wild-type, and as the repeat-rich nature of the region would make targeting difficult, we chose to instead identify a location elsewhere in the genome. Unfortunately, our previous analysis of gene distribution revealed that C. neoformans has a consistent distribution of genes across the genome, unlike species such as S. cerevisiae that exhibit reduced gene density in subtelomeric regions [28]. As no obvious gene-poor regions exist, it became necessary to systematically analyze all intergenic regions in the genome to identify the best candidate location.
Selecting a safe haven site in the C. neoformans genome
Our criteria for identifying a potential safe haven site for insertion of genetic constructs were twofold. First, the safe haven must be flanked by convergently transcribed genes; as no promoters in C. neoformans have been exhaustively characterized, we could not risk putting our constructs in a potential promoter region and disrupting transcription of a downstream gene. Second, the safe haven must be one of the larger intergenic regions in the genome to minimize the chance of inserted constructs influencing adjacent genes.
Of the almost 7,000 predicted protein-coding genes identified via the recently completed H99 genome project [17], roughly one third of gene pairs are tandemly transcribed, one third divergently transcribed, and one third convergently transcribed (Fig 1). Consistent with the compact nature of most characterized fungal promoters, tandemly and divergently transcribed genes in C. neoformans tend not to overlap. Furthermore, the inter-transcript distance is usually short, with 68% of tandem and 71% of divergent inter-transcript distances less than 500 bp. Inserting a construct within one of these small inter-transcript regions is likely to influence a promoter, making them unsuitable targets. In contrast, almost 73% of the 2,231 convergently transcribed gene pairs have overlapping transcripts (Fig 1); while these do not contain promoters, they still cannot be used as a potential insertion site without disrupting a known transcript.
Fig 1. Gene distribution and spacing in the Cryptococcal genome.
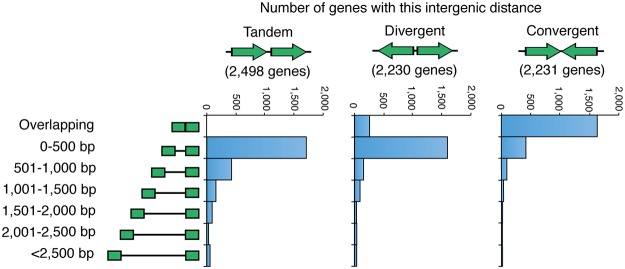
Distribution of intergenic distances in C. neoformans with the frequency of neighboring genes.
Of the 587 intergenic regions left between convergently transcribed genes with non-overlapping transcripts, the majority were deemed unsuitable as candidate insertion sites due to their small size; 418 had an inter-transcript distance of less than 500 bp. To minimize the probability of our inserted constructs affecting adjacent genes we decided to choose a site that was larger. However, as the H99 genome has on average one gene every 2,636 bp (when the centromeres are excluded), we also wanted to limit the upper size of the region to reduce the possibility of it containing an uncharacterized gene or transcript. We therefore chose to focus on the intergenic regions between convergently transcribed genes that had an inter-transcript distance between 1,500–2,000 bp in length. Only 18 intergenic regions met these criteria, and comparison of these regions to the extensive transcriptome data generated by Christine Cuomo, Guilhem Janbon and colleagues revealed that 12 contain miscRNAs of unknown function, making them unsuitable as a safe haven site [19]. Of the remaining 6 intergenic regions, we chose the smallest, reasoning that it should be less likely to contain an unidentified gene. This 1,544 bp intergenic region is found on chromosome 1 between the convergently transcribed CNAG_00777 and CNAG_00778 genes.
Creating an integration vector that targets the safe haven site
After selecting a safe haven site, we next designed a series of targeting vectors to facilitate integration of constructs of interest at this location. The three vectors created each contain the safe haven site sequence in the backbone of pBluescript II SK- to enable integration into the C. neoformans genome via homologous recombination, in addition to a linker sequence containing multiple rare-cutting restriction enzyme cut sites to facilitate linearization prior to biolistic transformation. Each vector also includes either the NAT (pSDMA25), NEO (pSDMA57) or HYG (pSDMA58) dominant selectable marker (Fig 2A). Importantly, as the safe haven and marker fragments were inserted into the pBluescript II SK- backbone in place of the f1 origin of replication, each plasmid still retains the original multicloning site and blue/white screening capabilities (Fig 2A).
Fig 2. Targeted integration vector.

A. The vector created has several key features. These include an intact multicloning site and blue-white screening for cloning the genetic construct of interest, a dominant selectable marker (here NAT, but also constructed in NEO and HYG versions), bla, a bacterial selection marker, and 5’ and 3’ flanking regions that are homologous to the safe haven site in the genome. The polylinker sequence contains three rare cutting restriction enzyme recognition sites that are used to linearize the vector so that the 5’ and 3’ regions are subsequently flanking the construct. B. Representation of the selected safe haven site, and how the linearized vector inserts via homologous recombination.
Linearization of the safe haven constructs is made possible by the inclusion of a linker containing cut sites for three rare cutting restriction enzymes—BaeI, AscI and PacI (Fig 2A). To use the vectors, the gene of interest is subcloned into the standard pBluescript II SK- multicloning site and sequenced to ensure it is error free. Subsequently, the vector is cut using either BaeI, AscI or PacI, a choice influenced by the sequence of the fragment that has been cloned into the multicloning site. Following linearization of the plasmid, the safe haven homology regions flanking the gene of interest facilitate homologous recombination at the safe haven site following biolistic transformation (Fig 2B).
Multiplex PCR reveals small non-homologous overlaps slightly inhibit integration rates
To simplify preliminary identification of transformants carrying the targeting vector at the desired safe haven location, we designed a diagnostic multiplex PCR (Fig 3A). The two outer primers yield a single band (2,177 bp) when a colony does not carry the safe haven vector at the correct location (Fig 3B). However, when the construct successfully integrates at the safe haven site, the additional primer binding sites introduced by the construct permit the amplification of two products (1,514 bp and 1,203 bp). In the event of tandem integration of the construct at the safe haven site, a 2,157 bp band would also be evident along with the 1,514 bp and 1,203 bp fragments, however we never observed this event.
Fig 3. Multiplex PCR for screening successful genomic integration events.
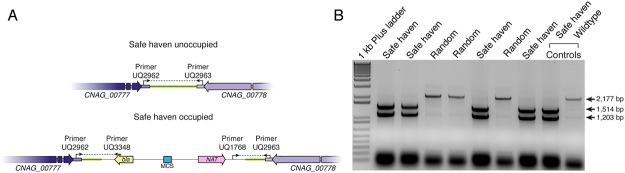
A. Depiction of the genome with the primers used in the multiplex PCR. B. Representative multiplex colony PCR results from transformants of H99 with the empty vector cut with BaeI. Transformants yielding two bands indicate the construct has integrated correctly, while incorrect transformants only have one band.
While a simple PCR analysis is a convenient method to rapidly screen transformants, it does not discriminate between strains that may carry additional, ectopic copies of the vector. To ensure that our transformants carry only a single copy of our vector, and that copy was at the safe haven site, we always perform Southern blot analysis prior to the strain being used for further work. Of 76 positive PCR colonies tested in this way so far, only 13 were incorrect as identified by Southern blot.
Due to the necessary inclusion of a linker sequence in the targeting vector, a small stretch of this sequence remains on the ends of the product following linearization—sequence that differs from the safe haven flanking sequence on chromosome 1. Cutting with PacI or AscI leaves up to 26 bp of non-homologous sequence, while BaeI digestion leaves only 3 bp of overhang, of which 2 bp are identical to the genome sequence. To determine whether these small non-homologous ends inhibit correct integration of a construct we performed a series of transformations of wild-type strain H99, integrating the empty pSDMA25 safe haven construct linearized with each of the linker-cutting enzymes. PacI- and AscI-digested vector incorporated at the correct position in 16% and 10% of resistant transformants, respectively. These frequencies are towards the upper end of published C. neoformans rates of homologous integration [14–16]. In contrast, BaeI-digested vector incorporated at the correct location in over 58% of resistant transformants, much higher than is typically observed (Fig 4). Together, these data indicate that while AscI and PacI digestion yield excellent homologous integration rates of the vector by current standards in C. neoformans, BaeI is the restriction enzyme of choice when no BaeI site exists in the genetic construct cloned into the safe haven vector.
Fig 4. Non-homologous ends inhibit successful homologous integration.
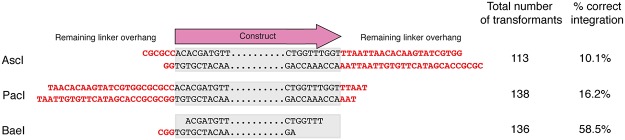
Illustration of the polylinker sequence after being cut with each of three rare cutting restriction enzymes. The construct depiction indicates where the marker and gene as well as the two flanking regions would be located after linearization. The red sequence shows residual polylinker sequence after digestion. Percent integration indicates the proportion of antibiotic resistant colonies in which the targeting vector was correctly integrated at the safe haven site as determined by multiplex PCR after biolistic transformation.
Integration of the targeted vector does not affect the expression of genes flanking the safe haven site, or alter virulence factor-related phenotypes
To confirm that insertion of the targeting vector did not affect expression of the two genes flanking the safe haven site (CNAG_00777 and CNAG_00778), qRT-PCR was performed using total RNA extracted from both strain H99 and an H99 transformant carrying the correctly integrated safe haven vector. Under standard growth conditions, there was no significant difference in the abundance of either CNAG_00777 or CNAG_00778 transcripts when comparing wild-type H99 to the H99 transformant carrying the empty safe haven vector (Fig 5). Furthermore, we found no observable change in the production of the key virulence factors urease, phospholipase, protease and melanin in the strain carrying the safe haven vector, nor was there a change in growth rate at either 30°C or 37°C (data not shown). These results further validate the suitability of the chosen site for construct targeting.
Fig 5. Integration of the vector into the safe haven site does not affect expression of flanking genes.

Transcript abundance of CNAG_00777 and CNAG_00778 relative to ACT1 with (H99 + empty vector) and without (H99) integration of vector at the safe haven site. Values show mean, error bars show S.E.M.
Employing an ade2Δ mutant to test the various approaches to complementation
The purpose of creating the targeting vectors was to ensure we could efficiently and reproducibly complement mutant strains without introducing unwanted secondary mutations. To verify that the targeting vector functioned as desired, we first deleted the well-characterized phosphoribosylaminoimidazole carboxylase-encoding ADE2 gene [29]. Loss of ADE2 results in the easily observable phenotypes of mutant colonies turning pink due to accumulation of the purine biosynthetic intermediate P-ribosylaminoimidazole, as well as adenine auxotrophy [30].
To compare our new targeting method to established complementation protocols we complemented the ade2Δ mutant using three different approaches. First, we used random integration by transforming the ade2Δ mutant with a linearized plasmid carrying wild-type ADE2 and the NAT selectable marker, but no targeting sequence. As independent random integrants will each have the wild-type ADE2 gene/nourseothricin-resistance cassette inserted at a different site in the genome, we selected four transformants for initial analysis. After observing that all four strains complemented the ade2Δ strain’s mutant phenotypes, two were randomly selected for further analysis: strains “Random #1” and “Random #2”. Second, we reinserted the wild-type gene at the original locus by targeting integration at the ade2Δ allele with a construct consisting of ADE2 plus the NAT marker at the 3’ end; this strain was named “Genomic Location”. Finally, the ade2Δ mutant was also complemented using targeted integration at the safe haven site to produce the strain “Safe Haven”. All three strategies complemented the ade2Δ mutant’s adenine auxotrophy and pink pigmentation (Fig 6). Furthermore, qRT-PCR confirmed that all three methods of complementation returned ADE2 expression levels to wild-type (Fig 7).
Fig 6. An ade2Δ mutant cannot grow on YNB media lacking adenine.

All complemented strains created in this study have wild-type levels of growth on YNB media and are of normal color.
Fig 7. Expression of ADE2 and flanking genes in an ade2Δ strain complemented with targeted or random integration.
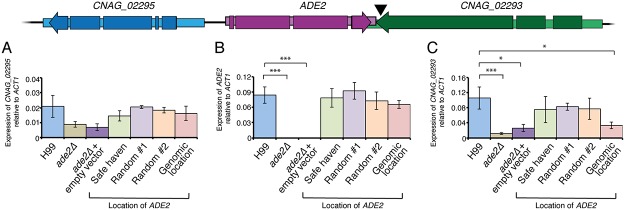
Expression of (A) CNAG_02295, (B) ADE2 and (C) CNAG_02293 relative to ACT1 in various strains. The black arrow indicates where the NAT selectable marker was inserted to complement in the strain “Genomic Location”. Values show mean, error bars show S.E.M. * = P<0.05; *** = P<0.01.
Complementation of ade2Δ using the safe haven strategy fully restores virulence, but random integration or targeting to the genomic locus does not
To compare the robustness of the three different approaches to complementation, we conducted virulence assays in a murine inhalation model using all four ade2Δ complemented strains—the two random integrants, the genomic location strain, and the safe haven strain. We tested these alongside wild-type and ade2Δ controls, as well as derivatives of these controls that carried the safe haven vector (Fig 8).
Fig 8. Virulence in a mouse model.
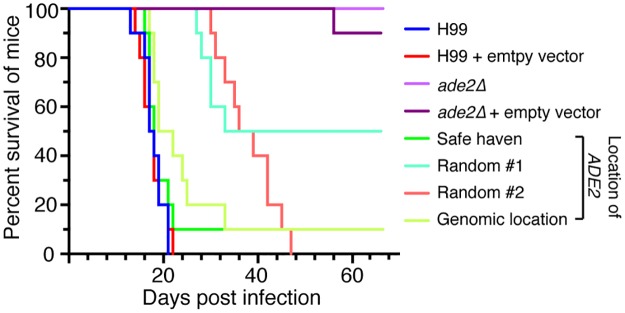
A. No significant difference was found between H99, H99 carrying the empty targeting plasmid, and the ade2Δ strain complemented with ADE2 using the safe haven targeting plasmid. While the ade2Δ mutant and the ade2Δ strain carrying the empty safe haven showed no significant difference from each other, a significant difference in survival was seen between each and H99 (P<0.0001). Significant differences were also observed between H99 and complemented strains Random #1, Random #2 and Genomic Location (P<0.0001, P<0.0001 & P<0.05 respectively).
As previously reported [29], the ade2Δ mutant was severely impaired for virulence compared to the wild-type strain. A crucial observation for this study was that the wild-type and ade2Δ controls carrying the empty safe haven vector exhibited equivalent virulence to their parental strains; that is, inserting the targeting vector at the safe haven site did not influence virulence in either the virulent wild-type strain, or the attenuated ade2Δ mutant.
Interestingly, the four complementation strains all exhibited differing survival times in the mouse model, despite our qRT-PCR results showing that each expressed ADE2 at wild-type levels under our tested condition. The ADE2 random integrants both partially restored virulence, but poorly. The strain in which the ADE2-NAT cassette had been targeted to the original ADE2 genomic location exhibited virulence more closely resembling wild-type, but was still statistically different. In contrast, the complemented strain carrying ADE2 at the safe haven site showed no statistical difference to wild-type, indicating a complete restoration of virulence. Of the three strategies employed for complementation, the safe haven approach was the only one that restored virulence to a level indistinguishable from wild-type, confirming that our safe haven approach was the superior complementation strategy.
The hazards of complementation via random integration or targeting the original genomic locus
We wanted to determine why the non-safe haven complementation strains failed to restore virulence to a wild-type level. Beginning with random integration strains Random #1 and Random #2, we employed an inverse PCR approach to identify the insertion sites of the ADE2/NAT vector in the genome of these strains. Sequencing of PCR products obtained revealed that, as could be expected, each disrupted another gene. In strain Random #1 the ADE2/NAT random integration vector had inserted into exon 4 of CNAG_06080 on chromosome 12, a gene whose product exhibits 48% similarity to the S. cerevisiae phosphatidylinositol phosphate phosphatase Sac1. Unsurprisingly, like the S. cerevisiae sac1Δ mutant, the Random #1 strain is an inositol auxotroph, and this likely explains the compromised virulence of this strain. In strain Random #2 the ADE2/NAT random integration vector had inserted 9 bp upstream of the start point of transcription of CNAG_05028 on chromosome 4, a gene whose product exhibits 34% similarity to the S. cerevisiae cysteine synthase Cys4. Again, just as the S. cerevisiae cys4Δ mutant is a cysteine auxotroph, so is strain Random #2, and this auxotrophy likely explains the compromised virulence. Importantly, neither of these auxotrophies are observable on either YPD or YNB, the two standard media that are used almost exclusively for routine cultivation of C. neoformans in the laboratory setting and which both contain inositol and cysteine.
Analysis of the Genomic Location complementation strain revealed a different type of problem. Our qRT-PCR results revealed that the ade2Δ mutant has a broad effect on the expression of all of the genes we investigated with this technique (S1 Fig). While this observation has not been reported before, we found it unsurprising; as Ade2 is required for de novo ATP and GTP synthesis, it is necessary for the production of two of the four nucleotides required for RNA synthesis. Complementation of the ade2Δ mutation using the safe haven approach or random integration resolves this phenotype. However, the Genomic Location strain does not return to normal expression levels for CNAG_02293. To ensure the entire wild-type ADE2 transcript was produced, the complementation construct we generated to create the Genomic Location strain included the NAT selectable marker slighty after the ADE2 transcriptional endpoint. The CNAG_02293 gene downstream of ADE2 is convergently transcribed, and like the majority of convergently transcribed genes in this species, the CNAG_02293 and ADE2 transcripts overlap. The consequence of this overlap is that our insertion of the NAT marker actually occurs in the 3’ end of the CNAG_02293 coding region, truncating the predicted 837 aa product by 85 residues. Furthermore, our qRT-PCR data shows that in this strain, CNAG_02293 transcript is significantly less abundant than in wild-type. As this gene is not a clear homolog of any well-characterised gene in another species, we cannot predict the biochemical reason underpinning why the Genomic Location strain exhibits a slightly, but still statistically significant, reduction in virulence.
Discussion
The current molecular tools and methodologies available for the study of C. neoformans are amongst the best in fungal species, making it not only a readily tractable pathogen but also a valid model system for the study of fundamental biological processes. However while gene deletions are relatively simple to create via techniques used widely within the C. neoformans research community, the approach to the equally important step of complementation is less well prescribed.
In this study we have analyzed the two complementation methods currently accepted by the field and explored their shortcomings. Results confirm that of the two methods, complementing via random integration shows the greatest potential for problems because it will likely interrupt other genes. As the vast majority of the C. neoformans genome is transcribed, the chance of the complementation construct disrupting a gene or promoter region is high. We had reasoned that because ADE2 expression in our random integration strains was equivalent to wild-type, that they should behave as wild-type in a murine inhalation model. This assumption was incorrect; both strains carried conditional lethal mutations that were not obvious using standard laboratory growth conditions, but were apparent upon infection of mice. To our knowledge, this study is the first to characterize insertion sites of complementation constructs in C. neoformans, despite the approach of random integration being the basis of insertional mutagenesis performed by others in this same species [31]. Our observations show that if a random insertion approach is employed, then care should be taken to identify the exact location of insertion of the complementation construct prior to animal experiments for ethical reasons to reduce the probability that secondary virulence defects are observed, resulting in unpublishable infection data.
The more conservative complementation approach employed by the community is reintroducing the wild-type gene along with a dominant selectable marker at the original genomic location. We also encountered problems with this strategy, with the highly compressed genome, short intergenic regions and overlapping transcripts of the organism resulting in our example of this strategy compromising the downstream gene. Retrospectively, we now know that the problems we encountered could have been avoided with the aid of the recently released transcriptome information by incorporating appropriate duplicated sequence in our insertion construct. While this information was not available when the strain was originally generated, this example does highlight the complexities that must be considered when retargeting the gene of interest to its original genomic location.
The major factor we needed to consider in this study was identifying an appropriate site in the Cryptococcus genome to target genetic constructs. In S. cerevisiae auxotrophy markers are widely utilized, and loci such as LEU2, URA3 and HIS3 serve as excellent locations for the insertion of genetic constructs. However in species where pathogenicity is being studied these same makers can be problematic due to the nutritional environment encountered in the infected animal. When auxotrophic and prototrophic strain pairs were assessed in a murine model in C. albicans, it was found that mutants auxotrophic for uracil, adenine and heme each showed a lower level of pathogenicity relative to control strains [32]. While these shortcomings can be overcome by targeting constructs to the highly expressed RPS10 locus [11,33], this example highlights the inherent risks associated with the use of auxotrophic markers in the study of pathogens. It is therefore unsurprising that while auxotrophic markers have been developed in C. neoformans, they are not widely used [34].
Here we characterized a safe haven site to be used for targeted integration of complementation constructs in C. neoformans and propose a strategy that has distinct advantages over current approaches. The safe haven strategy we employed is much simpler and more straightforward than existing methods of complementation. Our method allows for easy cloning, transformation and identification of positive inserts of a construct. It overcomes the current problems of influencing the expression of neighbouring genes, or disrupting them outright. We have successfully employed it to complement multiple mutants beyond those described in this manuscript, as well as to introduce tagged proteins and genes from other species. In all cases, the transformants have behaved exactly as we would expect. Based on this success, the use of the safe haven approach for introduction of genetic constructs has not only become the default approach in our own laboratory, but also in the laboratories of a number of our colleagues in the C. neoformans community. In short, we have designed a new methodology that we believe is the best option for complementing deletion strains and introducing genetic constructs into the genome of C. neoformans.
Supporting Information
Expression of CNAG_02295, CNAG_02293, CNAG00077 and CNAG00078 relative to ACT1 in various strains. Values show mean, error bars show S.E.M. * = P<0.05; *** = P<0.01.
(TIF)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We would like to thank Joe Heitman for coining the term “Safe Haven”.
Data Availability
All relevant data are within the paper and its supporting information files.
Funding Statement
This research was supported by the National Health and Medical Research Council, http://www.nhmrc.gov.au/, APP1049716 to JAF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Koch. 2 Die Aetiologie der Tuberkulose. 1884; 1–88 p. [Google Scholar]
- 2. Falkow S. Molecular Koch's postulates applied to microbial pathogenicity. Rev Infect Dis. 1988; 10 Suppl 2: S274–276. [DOI] [PubMed] [Google Scholar]
- 3. Hegemann JH, Heick SB. Delete and repeat: a comprehensive toolkit for sequential gene knockout in the budding yeast Saccharomyces cerevisiae . Methods Mol Biol. 2011; 765: 189–206. 10.1007/978-1-61779-197-0_12 [DOI] [PubMed] [Google Scholar]
- 4. Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae . Yeast. 1998; 14: 953–961. [DOI] [PubMed] [Google Scholar]
- 5. Parish T, Stoker NG. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology-Uk. 2000; 146: 1969–1975. [DOI] [PubMed] [Google Scholar]
- 6. Groisman EA. Principles of Bacterial Pathogenesis. 1st ed Academic Press; 2001. [Google Scholar]
- 7. Murray JAH. Bending the Rules—the 2-Mu Plasmid of Yeast. Molecular Microbiology. 1987; 1: 1–4. [DOI] [PubMed] [Google Scholar]
- 8. Clarke L, Carbon J. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 1980: 287: 504–509. [DOI] [PubMed] [Google Scholar]
- 9. Hill JE, Myers AM, Koerner TJ, Tzagoloff A. Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast. 1986; 2: 163–167. [DOI] [PubMed] [Google Scholar]
- 10. Goshorn AK, Grindle SM, Scherer S. Gene isolation by complementation in Candida albicans and applications to physical and genetic mapping. Infect Immun. 1992; 60: 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. CIp10, an efficient and convenient integrating vector for Candida albicans . Yeast. 2000; 16: 325–327. [DOI] [PubMed] [Google Scholar]
- 12. Chen SC. Cryptococcosis in Australasia and the treatment of cryptococcal and other fungal infections with liposomal amphotericin B.J Antimicrob Chemother. 2002; 49 Suppl 1: 57–61. [DOI] [PubMed] [Google Scholar]
- 13. Perfect JR. Cryptococcosis: a model for the understanding of infectious diseases. J Clin Invest. 2014; 124: 1893–1895. 10.1172/JCI75241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goins CL, Gerik KJ, Lodge JK. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet Biol. 2006; 43: 531–544. [DOI] [PubMed] [Google Scholar]
- 15. Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. Gene-transfer in Cryptococcus-neoformans by use of biolistic delivery of DNA. Journal of Bacteriology. 1993; 175: 1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davidson RC, Cruz MC, Sia RAL, Allen B, Alspaugh JA, Heitman J. Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans . Fungal Genetics and Biology. 2000; 29: 38–48. [DOI] [PubMed] [Google Scholar]
- 17. Ormerod KL, Morrow CA, Chow EWL, Lee IR, Arras SDM, Schirra HJ, et al. Comparative Genomics of Serial Isolates of Cryptococcus neoformans Reveals Gene Associated With Carbon Utilization and Virulence. G3-Genes Genomes Genetics. 2013; 3: 675–686. 10.1534/g3.113.008896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cramer KL, Gerrald QD, Nichols CB, Price MS, Alspaugh JA. Transcription factor Nrg1 mediates capsule formation, stress response, and pathogenesis in Cryptococcus neoformans . Eukaryotic Cell. 2006; 5: 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janbon G, Ormerod KL, Paulet D, Byrnes EJ 3rd, Yadav V, Chatterjee G, et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 2014; 10: e1004261 10.1371/journal.pgen.1004261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wills EA, Roberts IS, Del Poeta M, Rivera J, Casadevall A, Cox GM, et al. Identification and characterization of the Cryptococcus neoformans phosphomannose isomerase-encoding gene, MAN1, and its impact on pathogenicity.Molecular Microbiology. 2001; 40: 610–620. [DOI] [PubMed] [Google Scholar]
- 21. Sambrook J, Fritsch EF & Maniatis T. Molecular cloning: A laboratory manual. 1989. Cold Spring Harbor, New York: Cold spring harbor laboratory press, 2001. [Google Scholar]
- 22. Altingmees MA, Short JM. Pbluescript-Ii—Gene-Mapping Vectors.Nucleic Acids Research. 1989. 17: 9494–9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fraser JA, Subaran RL, Nichols CB, Heitman J. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell. 2003; 2: 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidson RC, Cruz MC, Sia RA, Allen B, Alspaugh JA, Heitman J. Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans . Fungal Genet Biol. 2000; 29: 38–48. [DOI] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 26. Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. Ur;ease as a virulence factor in experimental cryptococcosis. Infect Immun. 2000. 68: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans.Science. 2005. 307: 1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chow EWL, Morrow CA, Djordjevic JT, Wood IA, Fraser JA. Microevolution of Cryptococcus neoformans Driven by Massive Tandem Gene Amplification. Molecular Biology and Evolution. 2012; 29: 1987–2000. 10.1093/molbev/mss066 [DOI] [PubMed] [Google Scholar]
- 29. Perfect JR, Toffaletti DL, Rude TH. The gene encoding phosphoribosylaminoimidazole carboxylase (ADE2) is essential for growth of Cryptococcus neoformans in cerebrospinal fluid.Infect Immun. 1993; 61: 4446–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Donovan M, Schumuke JJ, Fonzi WA, Bonar SL, Gheesling-Mullis K, Jacob GS, et al. Virulence of a phosphoribosylaminoimidazole carboxylase-deficient Candida albicans strain in an immunosuppressed murine model of systemic candidiasis. Infection and Immunity. 2001; 69: 2542–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Idnurm A, Walton FJ, Floyd A, Reedy JL, Heitman J. Identification of ENA1 as a virulence gene of the human pathogenic fungus Cryptococcus neoformans through Signature-Tagged Insertional Mutagenesis.Eukaryotic Cell. 2009; 8: 315–326. 10.1128/EC.00375-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirsch DR, Whitney RR. Pathogenicity of Candida albicans auxotrophic mutants in experimental infections. Infect Immun. 1991; 59: 3297–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brand A, MacCallum DM, Brown AJ, Gow NA, Odds FC. Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RPS10 locus. Eukaryot Cell. 2004; 3: 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sudarshan S, Davidson RC, Heitman J, Alspaugh JA. Molecular analysis of the Cryptococcus neoformans ADE2 gene, a selectable marker for transformation and gene disruption.Fungal Genet Biol. 1999; 27: 36–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of CNAG_02295, CNAG_02293, CNAG00077 and CNAG00078 relative to ACT1 in various strains. Values show mean, error bars show S.E.M. * = P<0.05; *** = P<0.01.
(TIF)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its supporting information files.