

Background: Intrinsic protein disorder has been hypothesized to be advantageous for protein evolution.
Results: Parallel evolution of structured and disordered dihydrofolate reductases afforded comparable increases in activity without substantially changing the dynamic properties of the starting scaffolds.
Conclusion: Ordered and disordered enzymes can be similarly evolvable.
Significance: Understanding how structural (dis)order influences evolutionary pathways may aid efforts to engineer existing proteins for new tasks.
Keywords: Directed Evolution, Enzyme, Enzyme Catalysis, Intrinsically Disordered Protein, Protein Dynamic
Abstract
Intrinsically disordered proteins are ubiquitous in nature. To assess potential evolutionary advantages and disadvantages of structural disorder under controlled laboratory conditions, we directly compared the evolvability of weakly active ordered and disordered variants of dihydrofolate reductase by genetic selection. The circularly permuted Escherichia coli enzyme, which exists as a molten globule in the absence of ligands, and a well folded deletion mutant of the Bacillus stearothermophilus enzyme served as starting points. Both scaffolds evolved at similar rates and to similar extents, reaching near-native activity after three rounds of mutagenesis and selection. Surprisingly, however, the starting structural properties of the two scaffolds changed only marginally during optimization. Although the ordered and disordered proteins accumulated distinct sets of mutations, the changes introduced likely improved catalytic efficiency indirectly in both cases by bolstering the network of dynamic conformational fluctuations that productively couple into the reaction coordinate.
Introduction
Intrinsically disordered proteins, found in all domains of life, are essential to key cellular processes such as signaling and regulation (1–3). Rather than adopt rigid tertiary structures, disordered proteins exist as dynamic ensembles of many roughly isoenergetic conformational states (4). Such plasticity confers fast binding kinetics and high specificity, despite modest affinity, and can be advantageous for molecular recognition and protein modification (5, 6). Substantial structural disorder has even been shown to be compatible with enzyme catalysis (7).
Although intrinsic disorder contravenes the traditional view that proteins should possess a stable, well defined tertiary fold to be fully functional, intrinsically disordered proteins are evolutionarily persistent (8, 9). Indeed, such biopolymers often possess properties linked to high evolvability, including low sequence complexity, few intramolecular contacts, and a large fraction of solvent-exposed amino acid residues (10, 11). Statistical surveys of protein families have suggested that disordered sequences generally undergo more rapid evolution than their ordered counterparts (12). Disordered regions in transcription factors, for example, have been found to be particularly susceptible to positive Darwinian selection, facilitating functional adaptation (9). Because residues introduced into a flexible structure are free to sample multiple structural conformations, divergence and innovation are potentially easier than in a rigid scaffold.
Although disordered sequences may increase adaptive potential, other studies have emphasized the importance of structure and stability for accessing new or improved functions (13–15). Mutations often cause deleterious losses in thermodynamic and kinetic stability (16), so an ordered scaffold may compensate for destabilization more easily than a disordered one. In fact, high inter-residue contact density has been shown to correlate with increased rates of sequence evolution in yeast proteins (17); a large number of buried residues imparts robustness, enhancing the evolvability of solvent-exposed residues. Predictions that extra stability allows a protein to accept a wider range of mutations while maintaining its native structure have been borne out in selection experiments with enzymes like cytochrome P450 (18) and chorismate mutase (19). Because stable proteins from thermophilic organisms tolerate a greater number of destabilizing mutations than their mesophilic counterparts, they are often preferred as starting points for enzyme design projects.
Directed evolution (20, 21) provides a means of directly comparing the evolvability of intrinsically disordered and homologous structured proteins under controlled laboratory conditions. The small (∼18-kDa), well studied enzyme dihydrofolate reductase (DHFR)3 is an attractive model system for this purpose. DHFR catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate by stereospecific hydride transfer from the cofactor NADPH (Fig. 1A), an activity essential for de novo purine biosynthesis and hence cell growth and proliferation (22).
FIGURE 1.
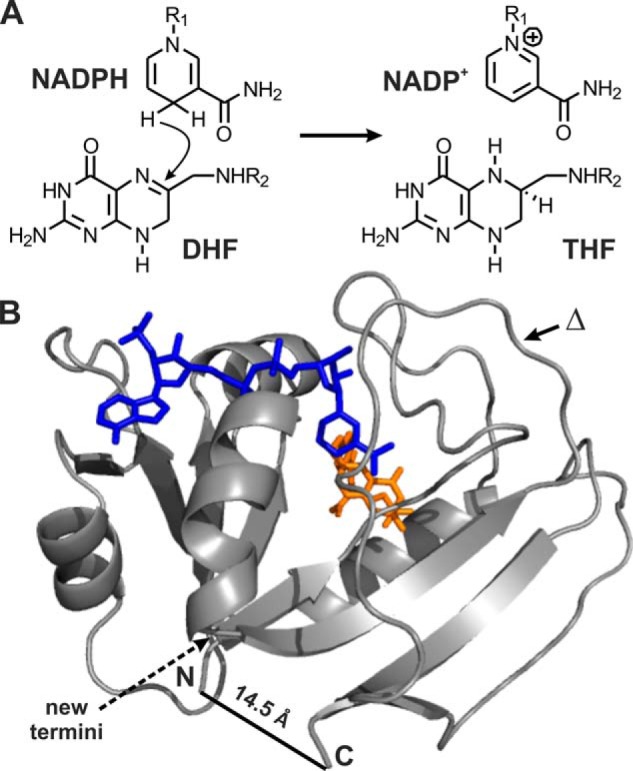
Dihydrofolate reductase. A, DHFR catalyzes the reduction of DHF to 5,6,7,8-tetrahydrofolate (THF) by stereospecific hydride transfer from NADPH. B, the enzyme has a mixed α/β structure and binds the nicotinamide (blue) and folate (orange) in a cleft between two domains (Protein Data Bank 1RX2). The circularly permuted variant cpDHFR was constructed by linking the original termini with a GSG peptide linker and generating new termini by cleavage between residues 107 and 108 (dashed arrow). The deletion variant was generated by removing residue 121 from BsDHFR (indicated by Δ).
Despite low sequence homology, DHFRs typically fold into two-domain structures, consisting of a central eight-stranded β-sheet and four flanking α-helices (Fig. 1B). Circular permutation of the well studied Escherichia coli enzyme (EcDHFR) yields intrinsically disordered yet weakly active catalysts (23, 24). Biochemical and spectroscopic characterization showed that such permutants behave like molten globules in the absence of ligands but become more structured upon addition of NADP+ and the folate antagonist methotrexate (MTX). Although detailed kinetic analyses were never performed, the catalytic activity of circularly permuted DHFR variants in the presence of substrates was reported to be 10–100 times lower than for EcDHFR (23, 24).
In this study, we evolved a circularly permuted variant of the E. coli enzyme (cpDHFR) in parallel with a more structured homolog from Bacillus stearothermophilus (BsDHFR), exploiting in vivo selection for DHFR activity to increase catalytic efficiency. Analysis of the optimized catalysts and their respective evolutionary trajectories sheds light on how (dis)order shapes protein evolution.
EXPERIMENTAL PROCEDURES
Bacterial Strains
E. coli XL1-Blue (Stratagene) was used for general cloning and amplification purposes (25). The DHFR-deficient E. coli strain MH829, created by replacement of folA by a kanamycin resistance cassette, was used for selection experiments (26). The tetracycline-resistant DHFR-deficient E. coli strain MH829tet was used for protein expression. It was constructed by horizontal gene transfer using XL1-Blue cells as the tet(M) donor. Tetracycline-resistant cells were subsequently selected for tetracycline and kanamycin resistance.
DNA Manipulation
Molecular cloning was performed according to standard procedures (27). Oligonucleotide synthesis and DNA sequencing were carried out by Microsynth (Balgach, Switzerland).
Plasmid Construction
Plasmids pKTCTET-0 and pKTNTET-0 were obtained from Dr. Kathrin Roderer (28). Plasmid His-pKTNTET-0 (4011 bp) was constructed by ligating the NdeI/NheI cleavage fragment from pKTNTET-0 with a synthetic DNA fragment (5′-TA ATG CAT CAT CAT CAT CAT CAT TCG TCG GGC CAT ATG) encoding the His tag and linker sequence. In this way, the original NdeI restriction site of pKTNTET-0 was eliminated and a new NdeI restriction site introduced.
Gene Construction
The E. coli folA gene was isolated from E. coli strain K12 (29) using the primer EcDHFR_NdeI_fwd (5′-AC TAT CAT ATG ATT AGC CTG ATT GCG GCG CT) and EcDHFR_SpeI_rev (5′-C CTA CAC TAG TTA TTA GCG GCG TTC CAG AAT TTC AAA GCA A) and ligated into NdeI/SpeI-cleaved pKTCTET-0 to give pKTCTET-ec.
The gene encoding cpDHFR was derived from the gene encoding EcDHFR. It was constructed by overlap extension PCR of two fragments. The first fragment was obtained with primer A (5′-AC TAT CAT ATG CAG AAA CTG TAT CTG ACC CAT AT), which has an NdeI restriction site, and primer B (5′-CAG GCT AAT CAT GCC GCT GCC GCG GCG TTC CAG AAT TTC AAA GC), which includes a sequence that encodes a GSG linker and is partially complementary to the second fragment. The second fragment was amplified using primer C (5′-CTG GAA CGC GCG GGC AGC GGC ATG ATT AGC CTG ATT GCA GCA CT), which contains the GSG linker sequence, and primer D (5′-C CTA CAC TAG TTA TTA CGC TTT CGG CAG AAA CTG TTC AT), which contains two stop codons and an SpeI restriction site. The combined fragments were ligated into NdeI/SpeI-cleaved pKTCTET-0 to give pKTCTET-cp.
The gene for BsDHFR was a generous gift from Prof. Judith Klinman. It was amplified by primers bsDHFR_fwd (5′-AC TAT CAT ATG ATT TCG CAC ATT GTG GCA AT), which introduced an NdeI restriction site, and bsDHFR_rev (5′-CCT ACA CTA GTT ATT ATT TCG CCT TTT CCG CTC), which introduced two stop codons and an SpeI cleavage site. Ligation into NdeI/SpeI-cleaved pKTCTET-0 afforded pKTCTET-bs.
The gene encoding the Δ121BsDHFR variant was constructed by a QuikChange reaction, using the primers Δ121bs_fwd (5′-GAC AAA AAT TTT TGC TTC CCC CGG CGA TAC GT) and Δ121bs_rev (5′-GGA AGC AAA AAT TTT TGT C). Ligation into NdeI/SpeI-cleaved His-pKTNTET-0 gave His-pKTNTET-Δbs.
Library Construction
The genes encoding cpDHFR and Δ121BsDHFR were mutagenized by error-prone PCR (GeneMorph II Random Mutagenesis Kit, Stratagene) using the T7_fwd and T7_rev primers, which hybridize upstream and downstream of the gene, respectively. The PCR products were purified via a 2% agarose gel and the NucleoSpin gel extraction kit (Macherey-Nagel). The NdeI/SpeI-digested fragments were ligated into NdeI/SpeI-cleaved pKTCTET-0. About 100 ng of the ligation product was transformed into 50 μl of electrocompetent XL1-Blue cells for library quality control. After 1 h of incubation in SOC medium at 37 °C, the cells were plated on LB agar plates containing 50 μg/ml ampicillin. Six colonies were picked and sequenced to estimate the average number of newly introduced mutations. About 1 μg of ligation product was transformed into 50 μl of electrocompetent MH829 cells. The cells were transferred to 1 ml of SOC medium containing 50 μg/ml thymidine and incubated for 1 h at 30 °C. Afterward, 100 μl of the cell suspension was plated on LB agar plates containing 50 μg/ml thymidine and 50 μg/ml ampicillin to determine the transformation efficiency. The remaining cells were incubated overnight in 10 ml of LB liquid medium, supplemented with 50 μg/ml thymidine and 50 μg/ml ampicillin to ensure optimal growth conditions. The resulting cultures were used for library selection experiments.
Library Selection Experiments
The precultured cells were washed three times in M9 liquid medium (6 mg/ml Na2HPO4, 3 mg/ml KH2PO4, 1 mg/ml NH4Cl, 0.5 mg/ml NaCl, 0.2% (w/v) d(+)-glucose, 5 μg/ml thiamine-HCl, 1 mm MgSO4, 0.1 mm CaCl2, 50 μg/ml ampicillin) and subsequently incubated in M9 liquid medium for 1 h at 30 °C. The cultures were diluted to 103–105 cells (assuming that an A600 of 1 corresponds to 108 cells/ml), and 5 × 102 to 103 cells were plated on nonselective and selective plates containing increasing concentrations of tetracycline. Selection experiments were carried out on M9 agar plates, pH 7.0, containing M9 liquid medium plus 15 g/liter agar and 0–100 ng/ml tetracycline. The plates were incubated at 30 °C for 1–5 days. Full complementation was achieved when the number of cells growing on the selective plates equaled the number of cells growing on nonselective plates.
Complementation Experiments
For liquid growth assays, single colonies were used to inoculate 5-ml precultures in LB-AT medium (25 g/liter LB medium, 50 μg/ml ampicillin, 50 μg/ml thymidine). After overnight incubation, 1 ml of precultured cells was grown in 5 ml of fresh LB-AT medium for 4 h at 30 °C, 230 rpm. For growth tests, 1 ml of cells was washed three times in 500 μl of M9 medium and subsequently incubated in the same medium for 1 h at 30 °C. For growth curves, 4 ml of liquid LB-AT medium or 4 ml of liquid M9 medium was inoculated to a starting A600 of 0.05. The cultures were incubated at 30 °C and 230 rpm, and the A600 was recorded for 1–5 days. The experiments were conducted in triplicate in the presence and absence of 5 ng/ml tetracycline.
For growth assays on M9 agar plates, single colonies were used to inoculate 5 ml of LB-AT precultures. The precultured cells were washed three times in liquid M9 medium and subsequently incubated in M9 medium for 1 h at 30 °C. The cultures were used for three-dimensional streak outs on M9 agar plates (pH 7.0) containing 0–100 ng/ml tetracycline. The plates were incubated at 30 °C for 1 to 5 days.
EcDHFR Production and Purification
Electrocompetent MH829tet cells were co-transformed with pKTCTET-ec, containing the gene for EcDHFR, and pT7POLTS, which encodes the T7 polymerase (28). 500 ml of LB medium, containing 50 μg/ml ampicillin, 30 μg/ml chloramphenicol, and 50 μg/ml thymidine (LB-ACT), were inoculated with an overnight preculture to a starting A600 of 0.05 and incubated at 37 °C until an A600 of 0.8–1 was reached. Gene expression was induced with 2 μg/ml tetracycline. Cells were harvested by centrifugation after 4–16 h of growth at 37 °C and washed with 50 ml of 0.9% NaCl. Cell pellets were stored at −20 °C.
Cells were solubilized in 50 mm Tris-HCl, 1 mm EDTA, pH 7.0, and lysed by sonication in the presence of 1 mg/ml lysozyme. After DNase (5 μg/ml) and RNase (10 μg/ml) treatment, the solution was centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatant was stirred with 45% (v/v) NH4(SO4)2 for 30 min at 4 °C. After centrifugation at 14,000 rpm for 20 min at 4 °C, the supernatant was dialyzed against 10 mm potassium phosphate buffer, pH 7.0, 50 mm KCl, 0.1 mm EDTA at 4 °C. The protein was purified by ion exchange chromatography on a diethylaminoethyl (DEAE)-Sepharose column pre-equilibrated with 10 mm potassium phosphate buffer, pH 7.0, 50 mm KCl, 0.1 mm EDTA. Protein was eluted with a gradient of 0–1 m KCl. Enzyme-containing fractions were concentrated with a 3-kDa Amicon Ultra-15 Centrifugal Filter Unit (Millipore) and injected onto a Superdex 75 size exclusion column pre-equilibrated with 50 mm HEPES, pH 6.8, 150 mm NaCl, and 3 mm 2-mercaptoethanol. The correct mass of the purified protein was verified by electrospray mass spectrometry (ESI-MS).
Production and Purification of cpDHFR and Variants
Electrocompetent MH829tet cells were co-transformed with pT7POLTS and the pKTCTET-cp plasmid encoding the cpDHFR variant of interest. The proteins were produced as described for EcDHFR. After cell lysis and centrifugation, the pellet was resuspended by stirring for 40 min in 25 ml of 1.5 m NaCl, 60 mm EDTA, 2% Triton X-100 at 4 °C. The solution was centrifuged at 14,000 rpm for 20 min at 4 °C, and the pellet washed three times (100 mm Tris-HCl, pH 7.0, 20 mm EDTA). After solubilization in 100 mm potassium phosphate buffer, pH 7.0, containing 6 m urea and 3 mm 2-mercaptoethanol at room temperature, the solution was centrifuged at 14,000 rpm for 20 min at 4 °C. The protein was dialyzed against refolding buffer (10 μm protein, 25 mm Tris-HCl, pH 8.0, 0.5 mm EDTA, 3 mm 2-mercaptoethanol) at 4 °C. Precipitated proteins were separated by centrifugation, and the supernatant was dialyzed against 10 mm potassium phosphate buffer, pH 7.0, 50 mm KCl, 0.1 mm EDTA at 4 °C. The protein was subsequently purified as described for EcDHFR. The correct mass of all variants was verified by ESI-MS.
BsDHFR Production and Purification
Electrocompetent MH829tet cells were co-transformed with pT7POLTS and pKTCTET-bs. The protein was produced as described for EcDHFR and purified in analogy to the procedure described previously (30). Briefly, after cell lysis, the supernatant was heat-precipitated at 55 °C for 20 min. The re-centrifuged protein solution was concentrated with a 3-kDa Amicon Ultra-15 Centrifugal Filter Unit (Millipore) and injected into a Superdex 75 size exclusion column in 50 mm HEPES, pH 6.8, 150 mm NaCl. The correct mass of the purified protein was verified by ESI-MS.
Production and Purification of Δ121BsDHFR and Variants
Electrocompetent MH829tet cells were co-transformed with pT7POLTS and the His-pKTNTET-bs plasmid encoding the Δ121BsDHFR variant of interest. Gene expression was induced with 0.5 μg/ml tetracycline, and the cells were harvested by centrifugation after 14–16 h at 18 °C. Cell pellets were stored at −20 °C. After cell lysis, the soluble fraction was loaded onto a nickel-nitrilotriacetic acid-agarose column (Qiagen) in 50 mm potassium phosphate buffer, pH 8.0, 300 mm NaCl, 10 mm imidazole and washed with the same buffer containing 30 mm imidazole. The protein was eluted in the same buffer with 250 mm imidazole. The elution fraction was concentrated with a 3-kDa Amicon Ultra-15 Centrifugal Filter Unit (Millipore) and injected onto a Superdex 75 size exclusion column pre-equilibrated with 50 mm HEPES, pH 6.8, 150 mm NaCl. Proteins were eluted from the column with the same buffer. The mass of all variants was verified by ESI-MS.
Protein Concentration Determination
Protein concentrations were determined with the Coomassie PlusTM kit (Pierce) and by absorbance spectroscopy using the calculated extinction coefficient at 280 nm determined by the Bioinformatics Resource Portal ExPASy. Extinction coefficients for Δ121BsDHFR and its variants were determined as described previously (31).
Enzyme Kinetics
Kinetic measurements were performed at 25 °C using final enzyme concentrations of 2–200 nm in PBS, pH 7.4, containing 5 mm DTT. The concentrations of substrate stock solutions were determined for NADPH at 340 nm (ϵ 6,220 m−1 cm−1). For DHF, an extinction coefficient of 28,000 m−1 cm−1 at 282 nm was used. Initial rates were determined by monitoring the decrease in signal at 340 or 365 nm at low and high substrate concentrations, respectively. The extinction coefficient of 11,800 m−1 s−1 at 340 nm was reported previously (32), and the extinction coefficient of 5,740 m−1 s−1 at 365 nm was determined from the difference in absorbance at 365 nm before and after the reaction in a two-chamber cuvette. Initial velocities were plotted as a function of NADPH or DHF concentration, and the steady-state parameters kcat and Km were determined using the Michaelis-Menten equation.
Circular Dichroism (CD) Spectroscopy
Far-UV CD experiments were performed on a CD spectropolarimeter (model 202) from AVIV Instruments Inc. Spectra were recorded at 25 °C in 10 mm potassium phosphate buffer, pH 7.4, in the presence or absence of 25 μm MTX with 5–7 μm protein. Spectra were obtained by averaging five wavelength scans taken in 0.5- nm steps, with a signal averaging time of 2 s and a bandwidth of 1 nm.
Fluorescence Spectroscopy
For ANS binding studies, the fluorescence spectra were recorded at 25 °C in 100 mm potassium phosphate buffer, 50 mm KCl, 0.1 mm EDTA, pH 7.4, with a fluorescence spectrometer (Photon Technology International). Spectra were obtained in 1-cm quartz cuvettes by averaging three wavelength scans with a signal averaging time of 1 s, in 1-nm steps. Excitation and emission slits were 10 nm. For ANS binding studies, 10 μm ANS solution was excited at 370 nm, and fluorescence was monitored at 400–600 nm in the presence and absence of 10 μm protein.
Chemical Denaturation
GdnHCl-induced transition curves were obtained by fluorescence spectroscopy on a 96-well plate reader (Thermo Scientific) at 25 °C using 0.15 mg/ml protein in 100 mm potassium phosphate buffer, 50 mm KCl, 0.1 mm EDTA, pH 7.4, containing 0 to 7.4 m GdnHCl. Samples were prepared starting from native and unfolded protein to check for reversibility. Fluorescence spectra were recorded from 300 to 450 nm. The bandwidth was 1 nm for excitation at 280 nm and 10 nm for emission. The shift of the wavelength of the emission maximum was determined as a function of the GdnHCl concentration and evaluated by nonlinear regression according to Ref. 33 with the modification reported in Ref. 34. Spectra were recorded in the presence and absence of 25 μm MTX.
Temperature-induced Unfolding
Temperature-induced denaturation was monitored on a CD spectropolarimeter (model 202) from AVIV Instruments Inc. at 25 °C in 10 mm potassium phosphate buffer, pH 7.4, in the presence or absence of 25 μm MTX with 5–7 μm protein. Denaturation was examined by measuring the CD signal at 222 nm while heating from 10 to 90 °C in 0.5 °C steps. Equilibration time at each time point was 60 s.
1H NMR Spectroscopy
NMR spectra were recorded in 100 mm potassium phosphate buffer, pH 7.4, and 10% D2O at 25 °C on a 600 MHz Bruker Avance II 600 spectrometer. Protein concentration was 50 μm; the concentration of MTX was 75 μm.
Tryptic Digests
DHFR variants (0.5 mg/ml) were incubated with 0.05 mg/ml bovine pancreatic trypsin (Fluka) at 15 and 25 °C. At different time points, 30-μl aliquots were removed from the reaction mixture and quenched with 10 μl phenylmethanesulfonyl fluoride (50 mm in 2-propanol). The samples were separated via SDS-PAGE, and band intensities were quantified with the program ImageJ (National Institutes of Health, Bethesda). Intensities were normalized to the band intensity of an undigested sample.
RESULTS
Characterization of the Intrinsically Disordered Starting Point
EcDHFR was circularly permuted as described by Gudkov and co-workers (23) by linking the N and C termini with a Gly-Ser-Gly tripeptide and cleaving the chain between residues 107 and 108 (Fig. 1B). Our construct, cpDHFR, differs from the literature variant in retaining the starting methionine at the linkage site and the original glutamine at position 108 (Fig. 2). It nevertheless exhibits all the hallmarks of a molten globule (Fig. 3). Far-UV CD spectra showed that cpDHFR is pre-dominantly α-helical, whereas EcDHFR has higher β-strand content (Fig. 3A). Temperature denaturation (Fig. 3B) and GdnHCl-induced unfolding (Fig. 3C) are less cooperative for cpDHFR than for EcDHFR, indicating multiple unfolding conformations. As is typical for disordered proteins, the circularly permuted protein also binds the hydrophobic dye ANS, giving rise to enhanced and blue-shifted fluorescence (Fig. 3D). Finally, broad peaks and poor dispersion in the 1H NMR spectrum of cpDHFR (Fig. 3E) attest to an ensemble of rapidly interconverting species.
FIGURE 2.

Sequence alignment of EcDHFR, cpDHFR, and Δ121BsDHFR. EcDHFR is represented in duplicate for better comparison with cpDHFR. The first sequence is highlighted in green, the second sequence in red. The linker sequence inserted for cpDHFR construction is highlighted in yellow. Δ121BsDHFR is numbered according to BsDHFR; the deleted residue is indicated by –.
FIGURE 3.
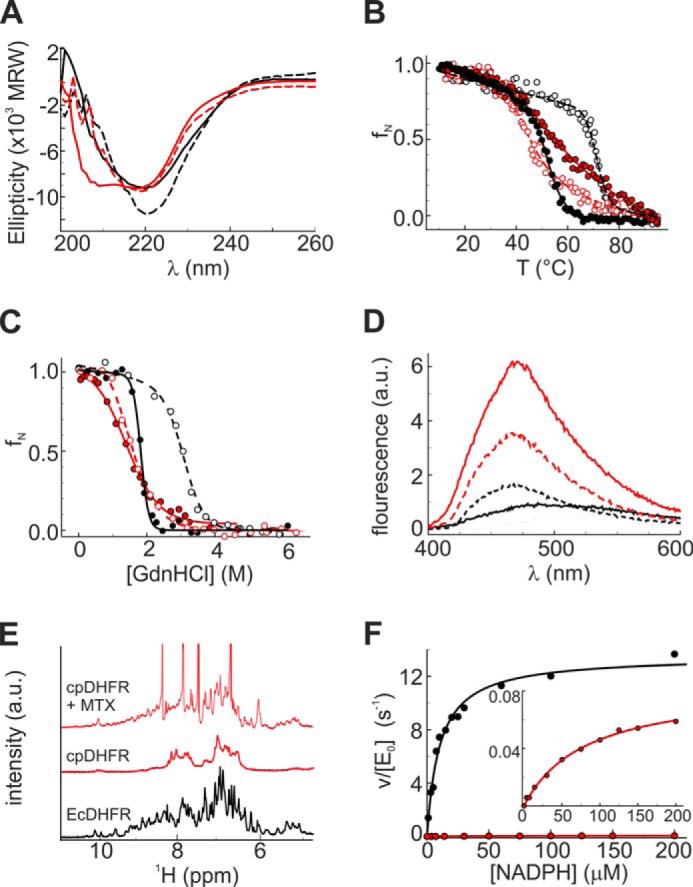
Biophysical and kinetic characterization of cpDHFR (red) and EcDHFR (black). A, far-UV CD spectra. B, temperature-induced unfolding. C, GdnHCl-induced unfolding; D, ANS binding were measured in 100 mm potassium phosphate buffer, pH 7.4. Measurements in the presence of 25 μm MTX are shown as dashed lines and open symbols. D, free ANS is shown as a black dotted line. E, NMR spectra were recorded in 100 mm potassium phosphate buffer, pH 7.4, including 10% D2O. F, initial rate constants for the catalyzed reaction were measured as a function of NADPH in PBS buffer, pH 7.4, at 25 °C. The inset shows a blowup of the Michaelis-Menten plot for cpDHFR.
As seen previously for other DHFR permutants (23, 24), the competitive inhibitor MTX induces conformational changes in cpDHFR (Fig. 3). In contrast to EcDHFR, which showed only a small increase in the CD signal at 218 nm, MTX binding to cpDHFR causes a dramatic shift in the CD spectrum, indicating a transition from a predominantly α-helical to a mixed α/β-structure similar to apoEcDHFR (Fig. 3A). Substantial ordering of the cpDHFR structure by MTX is further indicated by increased cooperativity of temperature-induced denaturation (Fig. 3B), weaker ANS binding (Fig. 3D), and greater peak dispersion in the 1H NMR spectrum (Fig. 3E). Regardless, cpDHFR still appears to be substantially less structured than EcDHFR even when complexed with ligand.
Although not natively folded, cpDHFR is catalytically active (Fig. 3F) but roughly 20,000-fold less efficient than EcDHFR (Table 1). The permuted protein has an approximately 100-fold lower kcat value than EcDHFR and Km values for DHF and NADPH that are 15- and 8-fold higher, respectively.
TABLE 1.
Steady-state kinetic and biophysical parameters for the DHFR variants
DHFR activity was determined in PBS buffer, pH 7.4, at 25 °C by monitoring the change in absorbance at 340 nm at low substrate concentrations and 365 nm at high substrate concentrations, respectively. Biophysical parameters were determined in 100 mm potassium/phosphate buffer, pH 7.4, at 25 °C.
| Parameter | EcDHFR | cpDHFR | cpDHFR.4 | BsDHFR | Δ121BsDHFR | Δ121BsDHFR.3 |
|---|---|---|---|---|---|---|
| kcat (s−1) | 13 ± 1 | 0.09 ± 0.03 | 1.9 ± 0.6 | 14 ± 2 | 0.30 ± 0.03 | 8.5 ± 0.5 |
| KmDHF (μm) | 4.7 ± 0.6 | 73 ± 7 | 21 ± 6 | 4.5 ± 0.8 | 78 ± 19 | 9.9 ± 0.1 |
| KmNADPH(μm) | 10 ± 1 | 83 ± 20 | 18 ± 3 | 14 ± 4 | 10 ± 2 | 9 ± 2 |
| kcat/( KmDHF· KmNADPH) (m−2 s−1) | 2.8 × 1011 | 1.5 × 107 | 5.0 × 109 | 2.2 × 1011 | 3.8 × 108 | 9.5 × 1010 |
| Tm (°C) | 53 ± 1 | 50 ± 7 | 57 ± 3 | 63 ± 1 | 43 ± 1 | 40 ± 2 |
| Tm (°C) + MTX | 77 ± 2 | 45 ± 1 | 57 ± 1 | 88 ± 2 | 58 ± 1 | 59 ± 2 |
| ΔG0 (kcal mol−1) | 10 ± 1 | 1.8 ± 0.1 | 1.9 ± 0.1 | 7.4 ± 1 | 2.0 ± 0.1 | 2.0 ± 0.1 |
| ΔG0 (kcal mol−1) + MTX | 15 ± 1 | 3.1 ± 0.2 | 3.1 ± 0.2 | 18 ± 1 | 7.9 ± 0.7 | 7.9 ± 0.9 |
Creation and Characterization of a Well Structured Counterpart
For the purpose of comparison, a well structured enzyme with activity similar to that of cpDHFR was engineered starting from a thermostable DHFR from B. stearothermophilus (BsDHFR) (30). To reduce the efficiency of this highly active catalyst (Table 1), two circularly permuted and three deletion variants were constructed. The permuted proteins were designed as described for cpDHFR, using the same linker to connect the original termini and introducing new termini between either Val-109/Asp-110 and Asp-110/Arg-111 (BsDHFR numbering). The deletion variants were obtained by deleting Arg-52 and Phe-121, which are located in mutationally sensitive loop regions of the protein (35), alone and in combination. The resulting variants were analyzed by their ability to complement an E. coli DHFR knock-out strain that requires an extra-chromosomal DHFR for growth (26). Most of the transformants grew at wild-type levels; only cells producing Δ121BsDHFR showed reduced growth.
In vitro studies confirmed that Δ121BsDHFR is less efficient than WT BsDHFR (Table 1 and Fig. 4). Deletion of Phe-121 reduced kcat values 48-fold and increased the Km value for DHF 17-fold; the Km value for NADPH remained unchanged. As a result, the catalytic efficiency of Δ121BsDHFR is nearly 1000-fold lower than that of the WT enzyme and only an order of magnitude higher than that of cpDHFR (Table 1).
FIGURE 4.
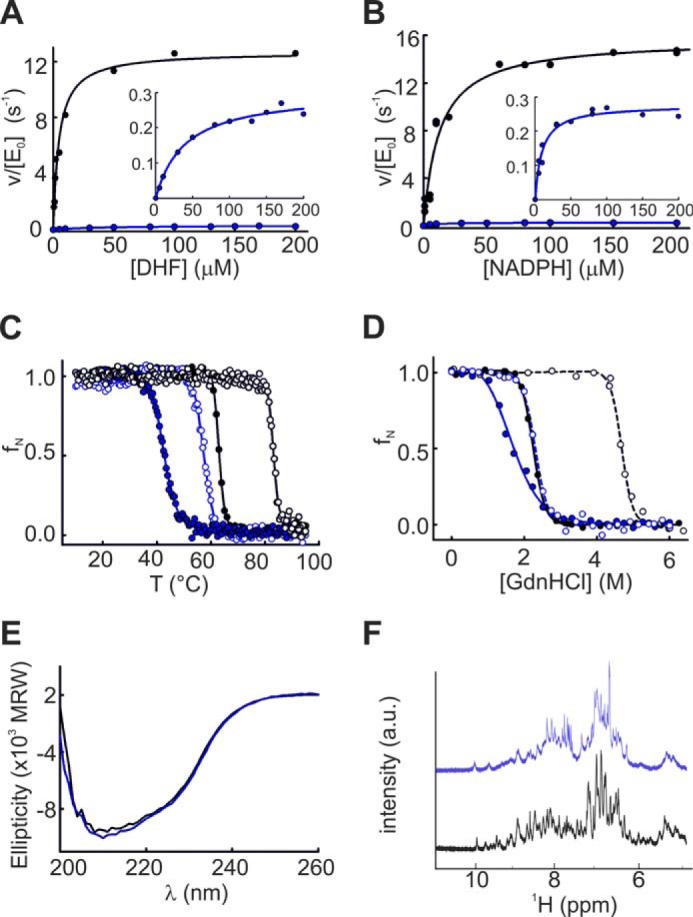
Kinetic and biophysical characterization of BsDHFR (black) and Δ121BsDHFR (blue). Initial rate constants as a function of DHF (A) and NADPH (B) concentration were measured in PBS buffer, pH 7.4, at 25 °C. The data for Δ121BsDHFR are rescaled in the insets. Temperature-induced unfolding (C), GdnHCl-induced unfolding (D), and far-UV CD spectra (E) were measured in 100 mm potassium phosphate buffer, pH 7.4, at 25 °C. a.u., arbitrary units. Measurements in the presence of MTX are shown in dashed lines (C and D). NMR spectra (F) were recorded in 100 mm potassium phosphate buffer, pH 7.4, including 10% D2O.
Although WT BsDHFR is thermostable, removal of Phe-121 also decreased the Tm of the enzyme by 20 °C (Fig. 4C) and the ΔG0 for chemical denaturation by 5 kcal/mol (Fig. 4D and Table 1). Nonetheless, the unfolding behavior of Δ121BsDHFR is still highly cooperative (Fig. 4, C and D). Moreover, its far-UV CD spectrum is indistinguishable from that of BsDHFR (Fig. 4E), and its 1H NMR spectrum shows good peak dispersion (Fig. 4F), consistent with a well structured protein.
Laboratory Evolution under Selection Pressure
The disordered and structured scaffolds were subjected to three rounds of random mutagenesis and selection. Gene libraries, constructed by error-prone PCR, were placed under the control of the tunable tetracycline-dependent Ptet promoter. Active catalysts were identified by their ability to complement the DHFR deficiency of the E. coli knock-out strain under auxotrophic conditions (26). In this system, selection pressure can be steadily increased simply by reducing the tetracycline concentration and thus lowering the intracellular concentration of the DHFR variants (Fig. 5) (36).
FIGURE 5.
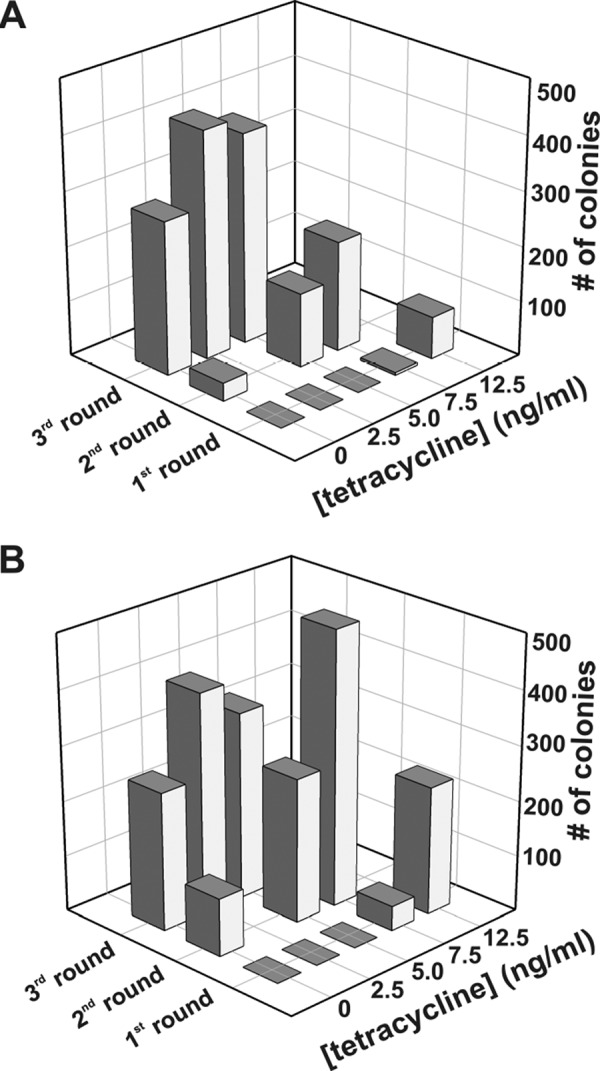
Evolutionary optimization. Complementation of MH829 cells producing variants of cpDHFR (A) or Δ121BsDHFR (B) as a function of tetracycline concentration over three rounds of laboratory evolution.
In the first selection round, cells grew only at high tetracycline concentrations because of the low activity of the starting variants. A slight advantage was observed for the Δ121BsDHFR library, most likely due to the higher starting activity of the deletion variant. After the second round of mutagenesis, 7% of the colonies producing cpDHFR variants and 21% of the colonies producing Δ121BsDHFR variants grew in the absence of inducer. After three rounds of mutagenesis, the number of complementing clones growing under the most stringent selection conditions increased to roughly 50% for both libraries (Fig. 5).
After three rounds of evolution, representative members from each library were isolated and sequenced (Fig. 6). The successful variants harbored four to six substitutions (Tables 2 and 3). Eight different sites in cpDHFR, distributed across the entire scaffold, were mutated. These include four consensus mutations (H18Y, P24S, M57R, and I117V), plus four additional changes that appeared in various combinations. In contrast, 14 completely different sites were targeted in Δ121BsDHFR, all of which are surface-exposed. Three consensus mutations (A49S, F118S, and G143W) emerged, but only A49S is present in every sequenced variant; eight of the remaining mutations are unique to a single clone.
FIGURE 6.

Sequence alignment of the starting points and six evolved variants of the first three rounds of directed evolution of cpDHFR (A) and Δ121BsDHFR (B). A, arrow indicates the original N terminus of EcDHFR. The amino acid residues are numbered as depicted in Fig. 2. The residues identified as DHFR sector residues are highlighted in green (A and B). The variants that were characterized in detail are highlighted in pink (A) and cyan (B), respectively.
TABLE 2.
cpDHFR variants obtained after three rounds of directed evolution
Residue numbering corresponds to the cpDHFR sequence shown in Fig. 2. The variant depicted in boldface type was biochemically characterized.
| Variant | Mutations |
|---|---|
| 1 | H18Y, S32R, M57R, I117V |
| 2 | I9V, H18Y, P24S, M57R, I117V |
| 3 | H18Y, P24S, M57R, I117V, A139T |
| 4 | I9V, H18Y, P24S, S32R, M57R, I117V |
| 5 | I9V, H18Y, P24S, M57R, M76L, I117V |
| 6 | H18Y, P24S, S32R, M57R, I117V, A139T |
TABLE 3.
Δ121BsDHFR variants obtained after three rounds of directed evolution
Residue numbering corresponds to the Δ121BsDHFR sequence shown in Figure 2. The variant depicted in boldface type was biochemically characterized.
| Variant | Mutations |
|---|---|
| 1 | A49S, V75M, F118S, G144W |
| 2 | A49S, A87T, F118S, G144W |
| 3 | A49S, A87T, F118S, G144W |
| 4 | I2T, K32R, A49S, F118S, G144W, P150R |
| 5 | D9G, A49S, R66H, A87T, P122S, K145N |
| 6 | K32R, A49S, P70A, F118S, E136G, G144W |
Characterization of Improved Variants
For a better understanding of the evolutionary changes at the molecular level, the variant from each library containing the most frequently occurring mutations (cpDHFR.4 and Δ121BsDHFR.3; see Tables 2 and 3) was characterized in detail. Following recloning, stringent growth tests with the DHFR-deficient strain transformed with the evolved enzymes confirmed full complementation.
Steady-state kinetic studies pinpointed increased catalytic activity as the reason for improved growth in both cases (Table 1 and Fig. 7). For the evolved cpDHFR variant, which has six mutations, catalytic efficiency rose 330-fold. The evolved Δ121BsDHFR variant has four mutations and showed a 290-fold increase. These improvements reflect 20–30-fold increases in the kcat parameter and smaller decreases in the Km values for the two substrates. Although evolution of Δ121BsDHFR recovered nearly full wild-type activity, the evolved cpDHFR variant is 17-fold less efficient, reflecting the lower activity of the disordered starting point.
FIGURE 7.
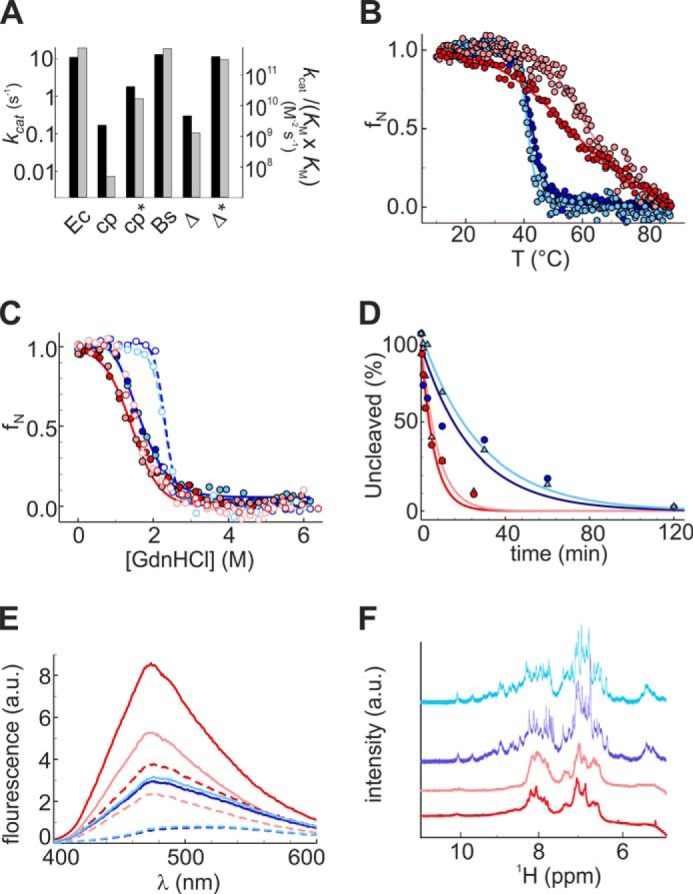
Characterization of the evolved variants cpDHFR.4 and Δ121BsDHFR.3. A, steady-state kinetic parameters for EcDHFR (Ec), cpDHFR (cp), cpDHFR.4 (cp*), BsDHFR (Bs), Δ121BsDHFR (Δ), and Δ121BsDHFR.3 (Δ*) were determined in PBS buffer, pH 7.4, at 25 °C. The turnover number kcat is given by the black bars, whereas the catalytic efficiency, kcat/(KmDHF·KmNADPH), is indicated by the gray bars. B, temperature-induced unfolding. C, GdnHCl-induced unfolding. D, tryptic digestion. E, ANS binding. F, NMR spectra were determined in 100 mm potassium phosphate buffer, pH 7.4, cpDHFR (red), cpDHFR.4 (pink), Δ121BsDHFR (blue), and Δ121BsDHFR.3 (cyan). a.u., arbitrary units. Measurements in the presence of MTX (C and E) are indicated as dashed lines.
The second-site mutations had relatively minor effects on protein structure and stability (Table 1). Evolved cpDHFR.4 showed a Tm increase relative to cpDHFR (Fig. 7B) but no differences in GdnHCl-induced unfolding (Fig. 7C) or proteolytic degradation (Fig. 7D). Moreover, the broad thermal transition (Fig. 7B), considerable albeit diminished ANS binding (Fig. 7E), and poor NMR signal dispersion (Fig. 7F) indicated that the variant retains a significant degree of intrinsic disorder. In contrast, temperature-induced unfolding, chemical denaturation studies, proteolytic degradation, ANS binding, and NMR spectra were indistinguishable for Δ121BsDHFR and its evolved variant (Fig. 7, B–F).
DISCUSSION
The active site of DHFR is located in a cleft between two protein subdomains (Fig. 1B). Structural and dynamic changes in these domains, induced by binding and dissociation of substrates, cofactors, and products (37), are critical to the function of the enzyme (22). NMR studies and molecular dynamics simulations of EcDHFR, supported by the results of site-directed mutagenesis, have produced maps of correlated conformational motions that productively channel bound intermediates along the reaction coordinate and facilitate hydride transfer (38–42). Disrupting these interaction networks can cause significant losses in stability and catalytic efficiency as seen for the deletion variant Δ121BsDHFR. In more extreme cases, such as the circularly permuted cpDHFR, structural integrity is lost as well, giving rise to intrinsic disorder.
Laboratory evolution provides a practical means to recover lost activity through second-site mutations. Three rounds of mutagenesis and genetic selection increased the efficiency of both Δ121BsDHFR and cpDHFR about 300-fold, affording variants that fully complement the DHFR deficiency of an E. coli auxotroph under stringent selection conditions. At least in this specific case, structural plasticity appears to have had a relatively modest impact on evolvability. Contrary to suggestions that disordered proteins evolve significantly faster than ordered proteins in nature (12), the compact and molten globular DHFR variants evolved at similar rates in this system. An even less structured protein than the circularly permuted DHFR variant might be needed to recapitulate such effects.
Although protein folding and stability might have been expected to co-evolve with function, optimization of Δ121BsDHFR and cpDHFR resulted in only subtle structural changes to the starting scaffolds. Thus, the Δ121BsDHFR.3 variant did not recover the thermostability of the parent scaffold but remained well packed after three rounds of evolution. The evolved circular permutant cpDHFR.4, however, retained significant disorder in the absence of ligands. In the latter case, reduced ANS binding and slightly more cooperative thermal denaturation provide evidence for some decrease in intrinsic disorder, but poor NMR signal dispersion indicates that the optimized cpDHFR variant is not native-like. Nevertheless, efficient complementation of the DHFR deficiency by cpDHFR shows that the dramatic disorder-to-order transition that accompanies ligand binding and subsequent catalytic events occurs on a time scale that is fully compatible with biologically relevant turnover times for folate metabolism (43).
Intriguingly, optimization of the compact and intrinsically disordered proteins followed distinct evolutionary trajectories. Indeed, the selected variants have no mutations in common. These distinct outcomes may reflect the low sequence identity (39%) between the starting scaffolds and/or the idiosyncratic perturbations to the parent enzymes, i.e. deletion as opposed to circular permutation. However, consistent with the greater tolerance of more ordered scaffolds to substitution (13–15), the structurally compact Δ121BsDHFR harbors a larger number of unique changes and fewer consensus substitutions than the disordered cpDHFR protein. Strikingly, all the Δ121BsDHFR mutations are at surface sites, which generally evolve more rapidly than buried residues (11, 44). Surface substitutions also predominate in the cpDHFR clones, although two buried residues, Ile-9 and Ile-117, were also conservatively mutated to valine.
Because the mutations in both proteins are generally distant from the active site, their influence on catalytic efficiency must necessarily be indirect. More detailed spectroscopic studies will be needed to elucidate the precise mechanistic consequences of these changes, but it is likely that mutation favorably alters the population distribution of ligand-bound conformations in both proteins, facilitating progression of the bound intermediates along the reaction coordinate (37). Both theoretical (45, 46) and experimental (47–49) studies have shown that a dynamic network of coupled motions influences DHFR catalysis, and evidence suggests that this network has been maintained over billions of years of evolution (50, 51).
Notable in this context, all the targeted sites in cpDHFR.4 are in contact with the sparse collection of physically contiguous and co-evolving amino acids identified by statistical coupling analysis of the DHFR protein family (Fig. 8) (52, 53). This network, called a sector (54), functionally links the active site to distantly positioned surface sites and is strongly correlated with the network of residues in DHFR undergoing millisecond conformational fluctuations. Although these sites need not be directly relevant to catalysis (48), the mutations fixed during directed evolution conceivably increased protein fitness by bolstering or extending this network, restoring critical dynamic motions that were disrupted by circular permutation. In contrast, in Δ121BsDHFR.3, which contains a less structurally disruptive deletion, the sector residue Ala-49 was mutated to serine. None of the other targeted residues is physically contiguous with the sector, suggesting that mutation at these sites either established new connectivities or enhanced activity by an alternative mechanism.
FIGURE 8.
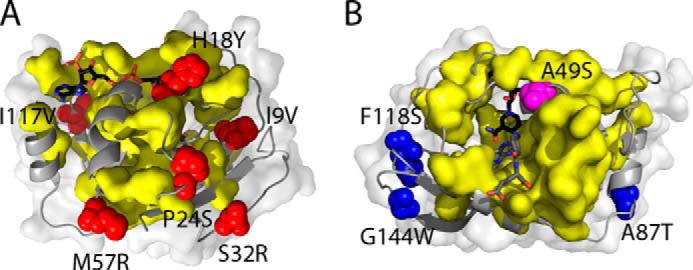
Location of mutations in the optimized DHFR variants relative to the sector of co-evolving residues in the DHFR family. A, in cpDHFR.4, all six mutations (red), mapped onto the crystal structure of EcDHFR (Protein Data Bank 1RX2), are physically contiguous with sector residues (yellow surface). B, mutations in Δ121BsDHFR.3 are mapped onto the crystal structure of BsDHFR (Protein Data Bank 1ZDR); they include one sector residue (A49S, magenta) and three other mutations (blue) that are not in contact with sector sites. Note that different perspectives were chosen for A and B to make all of the mutated sites visible. NADP+ and folate are shown with black and gray carbons, respectively.
These comparative selection experiments show that, with respect to a weak pre-existing enzymatic activity, ordered and disordered scaffolds can be similarly evolvable. In both scaffolds, relatively few, albeit distinct, mutations sufficed to counteract the deleterious effects of amino acid deletion or circular permutation and thereby improve binding and catalysis of DHF reduction. It is notable that optimization of the circularly permuted protein did not require sacrificing dynamic character to achieve high catalytic efficiency. Although further evolution of the disordered enzyme may afford more structured and stable catalysts, as seen during the optimization of an engineered molten globular chorismate mutase (55, 56), additional studies will be needed to assess whether such plasticity ultimately enhances the adaptive potential of this scaffold for completely new tasks.
Acknowledgments
We are grateful to Dr. Marc-Olivier Ebert for NMR measurements; Manuel Drefahl for cloning the circularly permuted BsDHFR variants; and Dr. Hajo Kries, Prof. Brian G. Miller, and Prof. Rama Ranganathan for helpful discussions.
This work was supported by the Swiss National Science Foundation and the ETH Zurich.
This article was selected as a Paper of the Week.
- DHFR
- dihydrofolate reductase
- ANS
- 1-anilinonaphthalene 8-sulfonate
- BsDHFR
- B. stearothermophilus DHFR
- cpDHFR
- circularly permuted variant of E. coli DHFR
- EcDHFR
- E. coli DHFR
- GdnHCl
- guanidine hydrochloride
- MTX
- methothrexate
- Δ121BsDHFR
- deletion variant of B. stearothermophilus DHFR
- DHF
- 7,8-dihydrofolate.
REFERENCES
- 1. Dunker A. K., Silman I., Uversky V. N., Sussman J. L. (2008) Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764 [DOI] [PubMed] [Google Scholar]
- 2. Uversky V. N. (2011) Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103 [DOI] [PubMed] [Google Scholar]
- 3. Tompa P. (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 37, 509–516 [DOI] [PubMed] [Google Scholar]
- 4. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 5. Shoemaker B. A., Portman J. J., Wolynes P. G. (2000) Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 97, 8868–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vuzman D., Levy Y. (2012) Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 8, 47–57 [DOI] [PubMed] [Google Scholar]
- 7. Schulenburg C., Hilvert D. (2013) Protein conformational disorder and enzyme catalysis. Top. Curr. Chem. 337, 41–67 [DOI] [PubMed] [Google Scholar]
- 8. Brown C. J., Johnson A. K., Dunker A. K., Daughdrill G. W. (2011) Evolution and disorder. Curr. Opin. Struct. Biol. 21, 441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilsson J., Grahn M., Wright A. P. (2011) Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins. Genome Biol. 12, R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 11. Lin Y. S., Hsu W. L., Hwang J. K., Li W. H. (2007) Proportion of solvent-exposed amino acids in a protein and rate of protein evolution. Mol. Biol. Evol. 24, 1005–1011 [DOI] [PubMed] [Google Scholar]
- 12. Brown C. J., Takayama S., Campen A. M., Vise P., Marshall T. W., Oldfield C. J., Williams C. J., Dunker A. K. (2002) Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 55, 104–110 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Minasov G., Shoichet B. K. (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 [DOI] [PubMed] [Google Scholar]
- 14. Bloom J. D., Wilke C. O., Arnold F. H., Adami C. (2004) Stability and the evolvability of function in a model protein. Biophys. J. 86, 2758–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bloom J. D., Labthavikul S. T., Otey C. R., Arnold F. H. (2006) Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U.S.A. 103, 5869–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tokuriki N., Tawfik D. S. (2009) Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604 [DOI] [PubMed] [Google Scholar]
- 17. Bloom J. D., Drummond D. A., Arnold F. H., Wilke C. O. (2006) Structural determinants of the rate of protein evolution in yeast. Mol. Biol. Evol. 23, 1751–1761 [DOI] [PubMed] [Google Scholar]
- 18. Bloom J. D., Lu Z., Chen D., Raval A., Venturelli O. S., Arnold F. H. (2007) Evolution favors protein mutational robustness in sufficiently large populations. BMC Biol. 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Besenmatter W., Kast P., Hilvert D. (2007) Relative tolerance of mesostable and thermostable protein homologs to extensive mutation. Proteins 66, 500–506 [DOI] [PubMed] [Google Scholar]
- 20. Romero P. A., Arnold F. H. (2009) Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jäckel C., Kast P., Hilvert D. (2008) Protein design by directed evolution. Annu. Rev. Biophys. 37, 153–173 [DOI] [PubMed] [Google Scholar]
- 22. Schnell J. R., Dyson H. J., Wright P. E. (2004) Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 33, 119–140 [DOI] [PubMed] [Google Scholar]
- 23. Protasova NYu, Kireeva M. L., Murzina N. V., Murzin A. G., Uversky V. N., Gryaznova O. I., Gudkov A. T. (1994) Circularly permuted dihydrofolate reductase of E. coli has functional activity and a destabilized tertiary structure. Protein Eng. 7, 1373–1377 [DOI] [PubMed] [Google Scholar]
- 24. Uversky V. N., Kutyshenko V. P., Protasova NYu, Rogov V. V., Vassilenko K. S., Gudkov A. T. (1996) Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 5, 1844–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bullock W. O., Fernandez J. M., Short J. M. (1987) XL1-Blue: a high efficiency plasmid transformig recA Escherichia coli strain with β-galactosidase selection. BioTechniques 5, 376–379 [Google Scholar]
- 26. Herrington M. B., Chirwa N. T. (1999) Growth properties of a folA null mutant of Escherichia coli K12. Can. J. Microbiol. 45, 191–200 [PubMed] [Google Scholar]
- 27. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Roderer K. (2012) Structure, Function, and Directed Evolution of Shikimate Pathway Enzymes from Mycobacterium tuberculosis. Ph.D. thesis, ETH Zurich, Switzerland [Google Scholar]
- 29. Kast P., Asif-Ullah M., Jiang N., Hilvert D. (1996) Exploring the active site of chorismate mutase by combinatorial mutagenesis and selection: the importance of electrostatic catalysis. Proc. Natl. Acad. Sci. U.S.A. 93, 5043–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H. S., Damo S. M., Lee S. Y., Wemmer D., Klinman J. P. (2005) Structure and hydride transfer mechanism of a moderate thermophilic dihydrofolate reductase from Bacillus stearothermophilus and comparison to its mesophilic and hyperthermophilic homologues. Biochemistry 44, 11428–11439 [DOI] [PubMed] [Google Scholar]
- 31. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 32. Stone S. R., Morrison J. F. (1982) Kinetic mechanism of the reaction catalyzed by dihydrofolate reductase from Escherichia coli. Biochemistry 21, 3757–3765 [DOI] [PubMed] [Google Scholar]
- 33. Santoro M. M., Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 34. Clarke J., Fersht A. R. (1993) Engineered disulfide bonds as probes of the folding pathway of barnase: increasing the stability of proteins against the rate of denaturation. Biochemistry 32, 4322–4329 [DOI] [PubMed] [Google Scholar]
- 35. Horiuchi Y., Ohmae E., Tate S., Gekko K. (2010) Coupling effects of distal loops on structural stability and enzymatic activity of Escherichia coli dihydrofolate reductase revealed by deletion mutants. Biochim. Biophys. Acta 1804, 846–855 [DOI] [PubMed] [Google Scholar]
- 36. Neuenschwander M., Kleeb A. C., Kast P., Hilvert D. (2011) in Protein Engineering Handbook (Lutz S., Bornscheuer U. T., eds) pp. 537–561, Wiley-VCH, Weinheim [Google Scholar]
- 37. Fierke C. A., Johnson K. A., Benkovic S. J. (1987) Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26, 4085–4092 [DOI] [PubMed] [Google Scholar]
- 38. Boehr D. D., McElheny D., Dyson H. J., Wright P. E. (2006) The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 [DOI] [PubMed] [Google Scholar]
- 39. Boehr D. D., McElheny D., Dyson H. J., Wright P. E. (2010) Millisecond time scale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 1373–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boehr D. D., Schnell J. R., McElheny D., Bae S. H., Duggan B. M., Benkovic S. J., Dyson H. J., Wright P. E. (2013) A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry 52, 4605–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luk L. Y., Javier Ruiz-Pernía J., Dawson W. M., Roca M., Loveridge E. J., Glowacki D. R., Harvey J. N., Mulholland A. J., Tuñón I., Moliner V., Allemann R. K. (2013) Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. U.S.A. 110, 16344–16349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Z., Singh P., Czekster C. M., Kohen A., Schramm V. L. (2014) Protein mass-modulated effects in the catalytic mechanism of dihydrofolate reductase: Beyond promoting vibrations. J. Am. Chem. Soc. 136, 8333–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vendruscolo M. (2010) Enzymatic activity in disordered states of proteins. Curr. Opin. Chem. Biol. 14, 671–675 [DOI] [PubMed] [Google Scholar]
- 44. Franzosa E. A., Xia Y. (2009) Structural determinants of protein evolution are context-sensitive at the residue level. Mol. Biol. Evol. 26, 2387–2395 [DOI] [PubMed] [Google Scholar]
- 45. Rod T. H., Radkiewicz J. L., Brooks C. L., 3rd (2003) Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. U.S.A. 100, 6980–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong K. F., Selzer T., Benkovic S. J., Hammes-Schiffer S. (2005) Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc. Natl. Acad. Sci. U.S.A. 102, 6807–6812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rajagopalan P. T., Lutz S., Benkovic S. J. (2002) Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry 41, 12618–12628 [DOI] [PubMed] [Google Scholar]
- 48. Singh P., Sen A., Francis K., Kohen A. (2014) Extension and limits of the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. J. Am. Chem. Soc. 136, 2575–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L., Goodey N. M., Benkovic S. J., Kohen A. (2006) Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc. Natl. Acad. Sci. U.S.A. 103, 15753–15758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu C. T., Hanoian P., French J. B., Pringle T. H., Hammes-Schiffer S., Benkovic S. J. (2013) Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. U.S.A. 110, 10159–10164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Francis K., Stojkovic V., Kohen A. (2013) Preservation of protein dynamics in dihydrofolate reductase evolution. J. Biol. Chem. 288, 35961–35968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reynolds K. A., McLaughlin R. N., Ranganathan R. (2011) Hot spots for allosteric regulation on protein surfaces. Cell 147, 1564–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee J., Natarajan M., Nashine V. C., Socolich M., Vo T., Russ W. P., Benkovic S. J., Ranganathan R. (2008) Surface sites for engineering allosteric control in proteins. Science 322, 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Halabi N., Rivoire O., Leibler S., Ranganathan R. (2009) Protein sectors: Evolutionary units of three-dimensional structure. Cell 138, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pervushin K., Vamvaca K., Vögeli B., Hilvert D. (2007) Structure and dynamics of a molten globular enzyme. Nat. Struct. Mol. Biol. 14, 1202–1206 [DOI] [PubMed] [Google Scholar]
- 56. Butz M., Neuenschwander M., Kast P., Hilvert D. (2011) An N-terminal protein degradation tag enables robust selection of highly active enzymes. Biochemistry 50, 8594–8602 [DOI] [PubMed] [Google Scholar]