

Background: K-Ras4B modulates downstream signaling at different lipid microdomains.
Results: K-Ras4B farnesyl group spontaneously inserts into the disordered lipid microdomains, but phosphorylation prohibits the farnesyl membrane insertion.
Conclusion: The farnesyl may determine K-Ras4B function in different membrane microdomain environments.
Significance: Figuring out K-Ras4B localization at different membrane microdomains is important for a more complete understanding Ras-effector interactions mediating signaling pathways.
Keywords: Cooperativity, Phospholipid, Phosphorylation, Post-translational Modification (PTM), Protein Isoprenylation, HVR, Membrane Microdomains
Abstract
K-Ras4B belongs to a family of small GTPases that regulates cell growth, differentiation and survival. K-ras is frequently mutated in cancer. K-Ras4B association with the plasma membrane through its farnesylated and positively charged C-terminal hypervariable region (HVR) is critical to its oncogenic function. However, the structural mechanisms of membrane association are not fully understood. Here, using confocal microscopy, surface plasmon resonance, and molecular dynamics simulations, we observed that K-Ras4B can be distributed in rigid and loosely packed membrane domains. Its membrane binding domain interaction with phospholipids is driven by membrane fluidity. The farnesyl group spontaneously inserts into the disordered lipid microdomains, whereas the rigid microdomains restrict the farnesyl group penetration. We speculate that the resulting farnesyl protrusion toward the cell interior allows oligomerization of the K-Ras4B membrane binding domain in rigid microdomains. Unlike other Ras isoforms, K-Ras4B HVR contains a single farnesyl modification and positively charged polylysine sequence. The high positive charge not only modulates specific HVR binding to anionic phospholipids but farnesyl membrane orientation. Phosphorylation of Ser-181 prohibits spontaneous farnesyl membrane insertion. The mechanism illuminates the roles of HVR modifications in K-Ras4B targeting microdomains of the plasma membrane and suggests an additional function for HVR in regulation of Ras signaling.
Introduction
Ras GTPases are small 21-kDa proteins that regulate essential cellular functions including cell survival, proliferation, motility, and cytoskeletal organization (1, 2). The three major Ras isoforms, H-Ras, N-Ras, and K-Ras, have a highly conserved N-terminal catalytic domain (G-domain) and a flexible C-terminal 22–25-amino acid-long, hypervariable region (HVR).4 K-Ras has two splice variants, K-Ras4A and K-Ras4B. K-Ras4B is expressed at higher levels and plays a vital role in growth and development (3–5). Activation varies across isoforms, resulting from distinct interaction patterns with different membrane microdomains that serve as signaling platforms (6–9). Ras proteins primarily bind the membrane through their HVRs (10), although the N-terminal catalytic domains may also contribute (11). H-Ras, N-Ras, and K-Ras4A are modified with farnesyl and palmitoyl lipid groups on their HVRs. Lipidation helps anchor the proteins to the plasma membrane. K-Ras4B lacks palmitoyl groups in its HVR, containing a single farnesyl prenylation. Instead, it has a unique polybasic region, with half of the residues being positively charged Lys. These together with the farnesyl group help localize K-Ras4B to membrane microdomains (12, 13) and increase the specificity to its binding partner proteins.
HVR farnesylation plays an important role in Ras signaling (14), but the structural mechanisms and consequences of farnesyl-induced membrane binding are poorly understood. Recent studies have uncovered the mechanism of K-Ras4B enrichment in the plasma membrane that involves continuous sequestration from endomembranes by cytosolic PDEδ (15). However, the basis for Ras localization in certain parts of plasma membrane is unknown. H-Ras is predominantly found on lipid rafts (16–18). In contrast, K-Ras is localized predominantly in the disordered parts (19–21). Constitutively activated K-Ras (G12V) interacts with non-raft regions (22), suggesting that K-Ras4B is preferentially localized in liquid-disordered lipid domains (23). K-Ras localization on lipid rafts is a subject of ongoing debate. It was argued that both H-Ras and K-Ras are in cholesterol-rich lipid rafts/caveolae fractions (24, 25). Ras clustering and intramembrane localization are believed to play an important role in the signaling (11, 19–21, 23, 26). We hypothesized that membrane composition may play an important role in HVR interaction with the membrane. The studies show that K-Ras4B can be differentially distributed in the membrane depending on membrane fluidity. The data suggest that K-Ras4B signaling is impacted by association with different microdomains of the membrane.
Our studies show that K-Ras4B is distributed between rigid lipid raft and disordered non-raft regions of the plasma membrane. The farnesyl group binds cooperatively to membrane phospholipids depending on membrane fluidity. We observe that the farnesyl group of K-Ras4B inserts into loosely packed bilayers containing unsaturated phospholipids. Conversely, saturated phospholipids that pack tightly into rigid lipid domains are less permissive of farnesyl insertion. Studies of the modeled HVR peptides verify that the farnesyl group spontaneously inserts into the lipid bilayer containing unsaturated phospholipids, but the insertion is restricted by phosphorylation. Our data further point to the farnesyl group being responsible for oligomerization of the protein. We speculate that if the farnesyl group is not inserted into the membrane, it may serve in cooperative binding to other K-Ras4B molecules, thus leading to dimerization. In this case, the protein binds the membrane through electrostatic interactions between its positively charged HVR and the negatively charged membrane phospholipids. In the case of disordered phospholipid domains, the farnesyl group burial in the phospholipids tends to hinder its interactions.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
The murine alveolar type II lung epithelial E10 cell line was provided as a kind gift by Dr. A. Maciag (NCI-Frederick). Cells were cultured in Connaught Medical Research Laboratories1065 basal medium supplemented with 10% tetracycline-free FCS (Sigma), 1% penicillin/streptomycin and 2 mm l-glutamine (Invitrogen). Cells were transduced with hemagglutinin (HA)-tagged K-Ras4B-G12V cDNA in a tetracycline regulated retroviral expression vector (obtained from Dr. Geoffrey J. Clark, University of Louisville). Individual blasticidin-resistant colonies were isolated by ring cloning. HA-KRas4B-G12V expression was optimized and induced with a final concentration of 2 μg/ml doxycycline (Fisher). For immunohistochemistry, cells were grown in the presence of 200 μg/ml GM1 (Avanti Polar Lipids Inc., Alabaster, AL) for 1 h before fixing.
Immunocytochemistry
E10 cells grown on glass coverslips were fixed in 4% paraformaldehyde for 30 min at 4 °C. Cells were blocked in PBS containing 5% bovine serum albumin and 0.1% Triton X-100 (Sigma) for 1 h at 4 °C. GM1 was labeled using the Vybrant Alexa Fluor 594 Lipid Raft Labeling kit (Invitrogen). Samples were subsequently probed with Alexa Fluor 488-conjugated anti-HA (Invitrogen) primary antibody overnight at 4 °C for K-Ras4B-G12V labeling. Slides were mounted with ProLong Gold antifade reagent (Invitrogen) and analyzed using Zeiss LSM 700 confocal microscope. Staining controls were performed with normal rabbit or mouse IgG.
Nanodiscs
The stabilized lipid bilayers called nanodiscs were assembled according to published protocols (27, 28). First, membrane scaffold protein (MSP1D1) was expressed as a His6 fusion in BL21DE3-Star cell and purified to homogeneity using Ni2+-His-bind resin (Novagen). Pure fractions of MSP1D1 were pooled and dialyzed against storage buffer (20 mm Tris-HCl, pH 7.4, 0.1 m NaCl, 0.5 mm EDTA, and 0.1% NaN3). The protein concentration was adjusted to ∼0.4 mm using an Amicon ultracentrifugal filter device. The following phospholipids were utilized for making nanodiscs: dipalmitoylphosphatidylcholine (DPPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS). We also used phosphatidylethanolamine-based lipids for the purpose of immobilization: 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE) and dipalmitoylphosphatidylethanolamine (DPPE). DPPC was dissolved in chloroform and mixed with DPPE phospholipids (Avanti Lipids) in a 95:5 molar ratio. The mixture was subsequently dried at room temperature using an Eppendorf Vacufuge and resuspended in storage buffer containing a 2-fold molar excess of sodium cholate. MSP1D1 was added to the phospholipid mixture such that the molar ratio of MSP1D1:DPPE:DPPC was 1:5:95. The protein and phospholipid mixture was stored at 37 °C for 1 h. The concentration of sodium cholate in solution was reduced by a 5-fold dilution in storage buffer. The sample was dialyzed extensively against the storage buffer to remove all traces of sodium cholate. DOPC and DOPS nanodiscs were prepared in a similar manner to DPPC nanodiscs.
Surface Plasmon Resonance Experiments
Before immobilization on surface plasmon resonance (SPR) sensor chips the phospholipid nanodiscs were dialyzed against 20 mm HEPES, pH 7.6, and 0.1 m NaCl. The immobilization was carried out on one flowcell of the CM5 sensor chip (Biacore) using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) to obtain a maximum 6000 response units using the immobilization wizard on the Biacore T100 control software. Ethanolamine was immobilized on the control flowcell of the same sensor chip for referencing purposes. To study the binding of farnesylated and non-farnesylated hypervariable region peptides to the nanodiscs, the peptides were adjusted to the required concentrations in 50 mm Tris-citrate, pH 6.5, 50 mm NaCl, 5 mm MgCl2, 10 mm β-mercaptoethanol. Before each run the running buffer was applied to the sensor chip for equilibration. The affinity wizard was used for binding and dissociation tests. All experiments were performed at 25 °C with a flow rate of 10 μl/min and a contact time of 240 s. The regeneration time and dissociation time were set to 30 s and 240 s, respectively. A solution of 2 m NaCl was used to regenerate the sensor chips. After successive rounds of association and dissociation the response was adjusted to baseline using the regeneration procedure.
Modification of the HVR Domain
The production of farnesylated HVR domain was done according to our published paper (29). We attached farnesyl-cysteine methyl ester to HVR peptide using succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), a bifunctional cross-linker.
Preparing the HVR Peptide of K-Ras4B for Molecular Dynamics Simulations
We generated a peptide containing the sequence of HVR from K-Ras4B, KEKMSKDGKKKKKKSKTKCVIM, using the CHARMM program (30). The HVR sequence contains positively charged Lys residues. The initially created 22-residue-long peptide was very stiff but gradually relaxed after solvation and molecular dynamics simulations. Ensembles of conformations in aqueous environment were obtained after a 50-ns production run, and these water-relaxed conformations were used as initial configurations in the lipid bilayer simulations. To simulate the HVR peptide on the lipid bilayer, we modified the C-terminal tail of the peptide via farnesylation at Cys-185 and phosphorylation at Ser-181. The first modification was a replacement of the lipidated side chain, the farnesyl group with the Cys-185 side chain, and the second involved the addition of phosphate (PO43−) group to the Ser-181 side chain. Although the standard CHARMM (30) program provides parameters for phosphorylated amino acids, no parameters for the farnesylated Cys residue are currently available. Thus, we first created the molecular topology for the farnesyl group using the Avogadro software (31). Then we calculated partial charges, bond lengths, angles, and torsional angles for the atoms in the farnesyl group using the Gaussian09 program (32) on a Biowulf cluster at the NIH. The calculated parameters can be directly adopted in the CHARMM (30) program.
Construction of Lipid Bilayer Systems with the Peptide
The HVR peptides, one with the farnesyl at Cys-185, KEKMSKDGKKKKKKSKCF, and the other with farnesyl at Cys-185 and phosphoryl at Ser-181, KEKMSKDGKKKKKKSPKTKCF, were initially placed on the lipid bilayer surface. In the peptide sequence, CF denotes the farnesylated Cys-185, and Sp represents the phosphorylated Ser-181. Two types of zwitterionic lipid bilayers, containing DPPC and DOPC, and an anionic lipid bilayer containing DOPC/DOPS (mole ratio 4:1) were used in the simulations. Similar anionic lipid bilayers have been extensively simulated (33–42). For each combination of two HVR peptides with three types of lipid bilayers, six independent simulations labeled HP1 to HP6 were performed. The farnesylated S181D phosphomimetic peptide, KEKMSKDGKKKKKKDKTKCF, was also tested.
The lipid bilayers were generated using the bilayer building protocol as described in our previous publications (91, 92). At both bilayer leaflets, pseudo spheres interacting through the van der Waals (vdW) force field were generated (43, 44). 10-ps dynamics was performed on the spheres, ensuring that the spheres were evenly distributed on the bilayer surfaces. Each sphere representing the phosphate atom of lipid was then converted into a lipid molecule. In the conversion, fully hydrated lipids were randomly selected from a library of preequilibrated gel state for DPPC and liquid crystalline state for DOPC and DOPS. Three different lipid bilayers with the peptide held rigid were minimized in order to remove overlaps of the alkane chains and gradually relax the system. For DPPC, the cross-sectional area per lipid and the headgroup distance across the bilayer are 48.6 Å2 and 47.1 Å at 25 °C, respectively (45). For DOPC and DOPS, they are 72.4 Å2 and 36.7 Å and 65.3 Å2 and 38.4 Å at 30 °C, respectively (46, 47). At these temperatures, DPPC is in the gel phase, and both DOPC and DOPS are in the liquid phase. We used the updated CHARMM (30) all-atom additive force field for lipids (C36) (48) and the modified TIP3P water model (49) in the bilayer construction and the production runs. The system contains Na+ and Cl− to satisfy a total ion concentration near 100 mm.
Production Runs
To obtain starting points, we generated at least 10 different initial configurations for each HVR peptide for the relaxation process and determined the best initial configuration for each system. In each starting point, the HVR peptide was located at the surface of the lipid bilayers; the peptide backbone and the farnesyl or phosphoryl groups were not initially inserted into the lipid bilayer. In the pre-equilibrium stages, all systems were subjected to a series of minimizations for the solvents around the harmonically restrained peptides. After minimizations, 2-ns dynamics was performed on each system with the restrained peptide until the solvent reached 303 K. At the final pre-equilibrium stage, the peptide was gradually relaxed by removing the harmonic restraints through dynamic cycles and adapted to the surrounding heat bath. In the dynamics, long range electrostatic interactions were calculated by using the particle mesh Ewald method. The Langevin temperature control was used to maintain the constant temperature at 303 K, and Nosé-Hoover Langevin piston pressure control was used to sustain the pressure at 1 atm. In the production runs to 30 ns, our simulation employed the NPAT (constant number of atoms, pressure, surface area, and temperature) ensemble with a constant normal pressure applied in the direction perpendicular to the membrane. In the production runs from 30 ns to 100 ns, the simulations employed the NPT ensemble. The NAMD (50) parallel computing code was employed in the production runs on a Biowulf cluster at the NIH. We analyzed the simulation trajectories after 30 ns, discarding initial transients, with the CHARMM programming package (30).
RESULTS
K-Ras4B Is Distributed between Lipid Raft and Non-raft Membrane Domains
K-Ras4B was reported to localize either in lipid raft (LR) or in non-raft (NR) regions. To address this discrepancy, we used immunocytochemistry to study K-Ras4B co-localization with an LR marker sialylated ganglioside GM1 in E10 mouse type II alveolar epithelial cells stably transduced with HA-tagged human K-Ras4B-G12V (Fig. 1). K-Ras4B expression was induced with doxycycline. Expression of HA-KRas4B-G12V was optimally induced using a final doxycycline concentration of 2 μg/ml.
FIGURE 1.
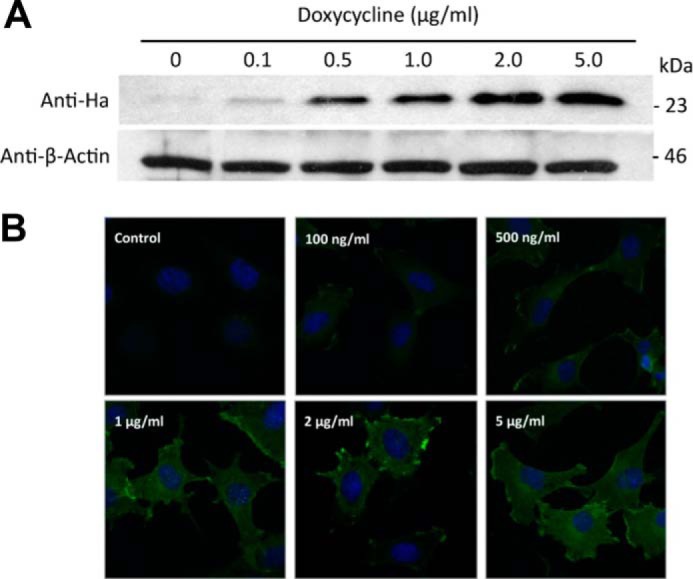
Induction of HA-tagged K-Ras4B-G12V expression in mouse type II alveolar epithelial cells. A, immunoblot analysis of whole cell lysates of E10 cells induced to express HA-tagged K-Ras4B-G12V using increasing concentrations of doxycycline was performed using HRP-conjugated anti-HA antibody (GenScript), with anti-β-Actin (Santa Cruz) used as a loading control. Cells were lysed in ice-cold radioimmune precipitation assay buffer before sonication. Confocal microscopy analysis of HA-KRas4B expression was performed as described, without GM1 staining.
GM1 is commonly used to identify proteins that partition into LRs (51). GM1 was recognized by fluorescent cholera toxin B and detected by confocal microscopy (52). In the K-Ras4B/GM1 co-localization experiment, we observed significant areas in the plasma membrane where K-Ras4B co-localizes with GM1 marked LRs (Fig. 2). We find that at 37 °C, K-Ras4B is distributed between both GM1-marked LRs and NR regions (Manders M1 and M2 correlation coefficients are 0.79 and 0.84, respectively). At 18 °C, with presumably less membrane fluidity, LR staining is “condensed” and perhaps more prevalent. Under these conditions, K-Ras4B still localizes with LRs (M1 and M2 are 0.76 and 0.89, respectively). Conversely, at 42 °C, GM1 staining is more distributed throughout the plasma membrane, likely due to increased membrane fluidity. Ras localization at the plasma membrane still occurs with M1 and M2 of 0.89 and 0.94, respectively. Thus, K-Ras4B localization in LRs occurred at high and low temperatures.
FIGURE 2.

Confocal microscopy images of HA-K-Ras4B co-localization with the GM1 LR marker in E10 cells incubated in culture media at 18 °C (upper panels), 37 °C (middle panels), and 42 °C (lower panels) for 15 min before fixing. Shown are slides stained with DAPI (blue), anti-HA antibody conjugated with Alexa-Fluor 488 (green), and cholera toxin subunit B labeled with Vybrant Alexa Fluor 594 (red) for GM1 recognition. The merged images show co-localization of K-Ras4B and GM1 at 18 °C (Mander's coefficient = 0.764, 0.887), 37 °C (Mander's coefficient = 0.794, 0.839), and 42 °C (Mander's coefficient = 0.891, 0.940). The images were generated using Zeiss LSM 700 confocal microscope, and the data were analyzed using LSM Browser Software (Zeiss).
The Farnesylated HVR Peptide Shows Cooperative Binding toward Membrane Phospholipids
LRs contain saturated lipids that pack tightly to form rigid microdomains (53). The NR regions are known to accumulate unsaturated lipids with kinks in the acyl chains. These lipids pack loosely against each other and create fluid membrane areas. We hypothesized that membrane fluidity affects K-Ras4B distribution in the plasma membrane. To mimic rigid and loosely packed membrane microdomains, we prepared SPR sensor chips with cross-linked stabilized lipid bilayers, known as nanodiscs, containing saturated and unsaturated phospholipids. To study K-Ras4B binding to rigid microdomains, we assembled nanodiscs using a phospholipid mixture consisting of DPPC:DPPE (95:5 molar ratio). DPPC is often used to mimic rigid membrane microdomains (54). To mimic disordered lipid domains, we used mixtures of DOPC:DOPE and DOPS:DOPE (both in 95:5 molar ratio) lipids. These unsaturated phospholipids are also commonly used to study loosely packed domains (54). To understand the effect of farnesylation on membrane binding, SPR experiments were performed with increasing concentrations of the farnesylated or non-farnesylated HVR peptide, which was injected onto DPPC, DOPC, and DOPS immobilized in nanodiscs on the CM5 chip. The baseline-corrected, normalized response units at equilibrium for various injection points were plotted versus concentration, and the Hill equation was used to fit the curves (Fig. 3). We calculated the equilibrium dissociation constant (KD) for the non-farnesylated and farnesylated HVR peptides, and the Hill coefficient (HC) in each case for DPPC, DOPC, and DOPS phospholipids (Table 1). The KD values for the non-farnesylated HVR peptide for DPPC, DOPC, and DOPS phospholipids were in the low micromolar range. The non-farnesylated peptide binds all phospholipid nanodiscs with increasing KD values: 1.59 ± 0.01, 2.10 ± 0.01, and 5.06 ± 0.04 μm for DPPC, DOPC, and DOPS nanodiscs, respectively. Binding may involve electrostatic interactions between positively charged Lys residues in the HVR peptide and the phosphate in the phospholipid headgroups. This is in agreement with recent observations (55). Post-translational modifications significantly alter the binding mode between the HVR and the nanodiscs. Compared with the non-farnesylated peptide, the farnesylated peptide slightly increases the binding affinities with KD values of 1.08 ± 0.01, 1.06 ± 0.02, and 1.21 ± 0.00 μm for DPPC, DOPC, and DOPS nanodiscs, respectively. The binding event exhibits pronounced cooperativity with the farnesylated peptide. HCs that measure the degree of cooperativity differ significantly. It is not possible to compare between the KD values for the non-farnesylated and farnesylated HVR peptides due to this cooperative behavior. However, in all cases, the farnesylated peptide reaches saturation at ∼10-fold lower concentration than the non-farnesylated peptide. Binding of the farnesylated peptide to DPPC nanodiscs is highly cooperative with HC of 6.17 ± 0.28. The peptide binds the more fluid nanodiscs, DOPC and DOPS, with less cooperativity (HCs are 2.91 ± 0.11 and 2.55 ± 0.01, respectively). The mode of interaction of K-Ras4B with membranes can differ depending on the level of saturation of the acyl chains. On the other hand, the HC of non-farnesylated peptide is ∼1 on all phospholipids. Among the tested lipids, DOPS forms the most fluid bilayers. In DOPS phospholipids, an HC value of ∼2 points to dimer formation. However, a cooperativity of 6, as in the case of gel phase DPPC bilayers, would indicate binding of 6 ligands on one binding site; DOPC bilayers of intermediate fluidity allow cooperative binding of 3 peptides. DPPC phospholipids exist in gel phase at temperatures below 41 °C as opposed to DOPC and DOPS, which have subzero transition temperatures. This means that DOPC and DOPS nanodiscs do not display temperature-dependent fluidity at temperatures above zero. DPPC, on the other hand, does display this temperature-dependent fluidity.
FIGURE 3.
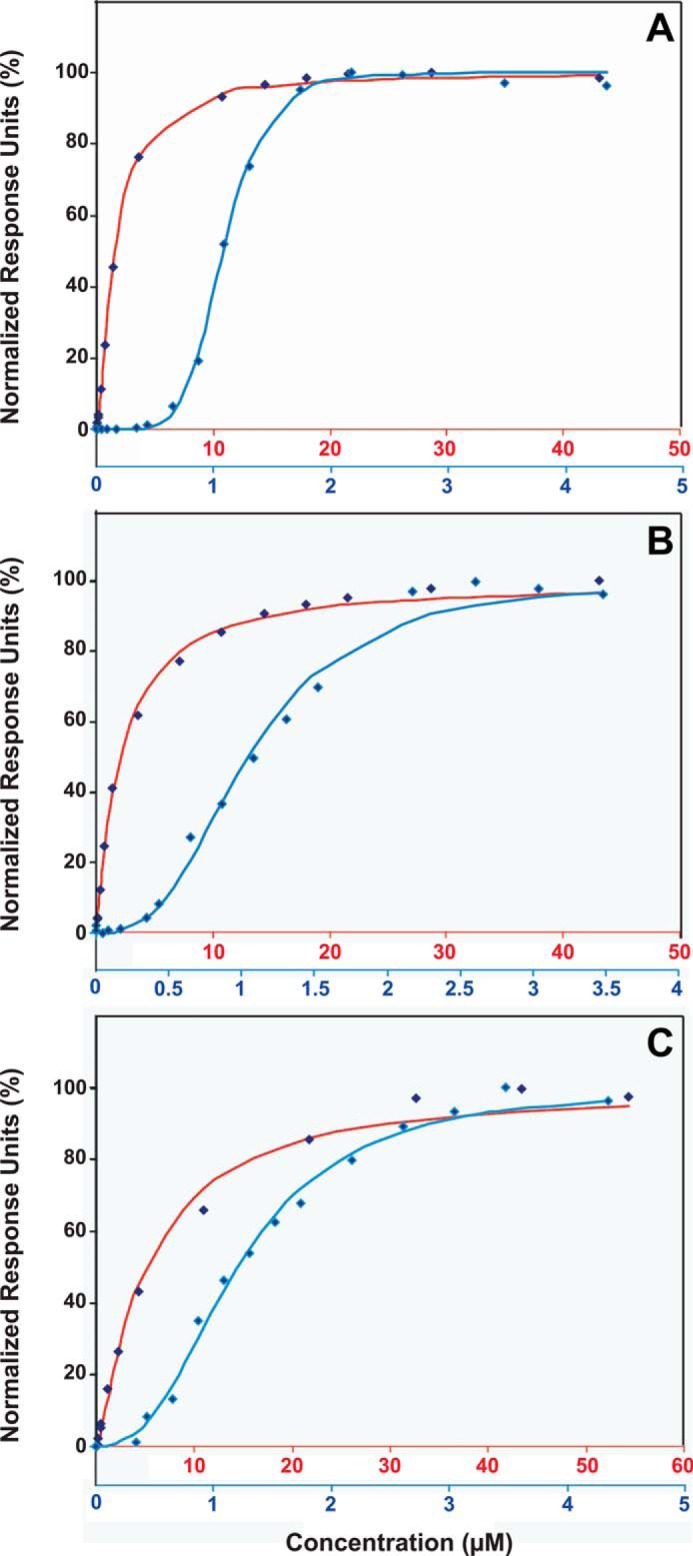
SPR titration of DPPC (A), DOPC (B), and DOPS (C) nanodiscs with non-farnesylated (red) and farnesylated HVR (blue) of K-Ras4B. The normalized response units at equilibrium for various injection points were plotted versus concentration, and the Hill equation was used to fit the curves. Red and blue horizontal labels of concentration correspond to non-farnesylated and farnesylated HVRs, respectively.
TABLE 1.
Summary of fitting the Hill equation to binding curves of non-farnesylated and farnesylated HVR peptides to DPPC, DOPC, and DOPS phospholipids
| Phospholipid | Non-farnesylated HVR peptide |
Farnesylated HVR peptide |
||
|---|---|---|---|---|
| KD | Hill coefficient | KD | Hill coefficient | |
| μm | μm | |||
| DPPC | 1.59 ± 0.01 | 1.24 ± 0.01 | 1.08 ± 0.01 | 6.17 ± 0.28 |
| DOPC | 2.10 ± 0.01 | 1.12 ± 0.00 | 1.06 ± 0.02 | 2.91 ± 0.11 |
| DOPS | 5.06 ± 0.04 | 1.24 ± 0.01 | 1.21 ± 0.00 | 2.55 ± 0.01 |
To test the relationship between packing of phospholipids in the bilayer and the degree of cooperativity, the binding of farnesylated HVR peptide to DPPC phospholipids was studied at three different temperatures, 25 °C, 35 °C, and 45 °C (Fig. 4). At 25 °C, the farnesylated peptide binds to DPPC phospholipids with high cooperativity (HC 6.17 ± 0.28). When the temperature was increased to 35 °C, the cooperativity decreased drastically with a corresponding HC of 2.69 ± 0.04. An increase to 45 °C results in further decrease of HC to only 2.39 ± 0.02. In summary, increasing the temperature decreased binding cooperativity of farnesylated HVR peptides in DPPC phospholipids. This is consistent with the transition of DPPC phospholipids from a gel phase to more fluid-like disordered packing as the temperature approaches the transition temperature of 41 °C (56). A more disordered packing would allow for insertion of the farnesyl groups into lipid bilayers, thereby decreasing cooperativity. This finding indicates that K-Ras4B oligomerization on lipid bilayers may be dependent on bilayer fluidity.
FIGURE 4.
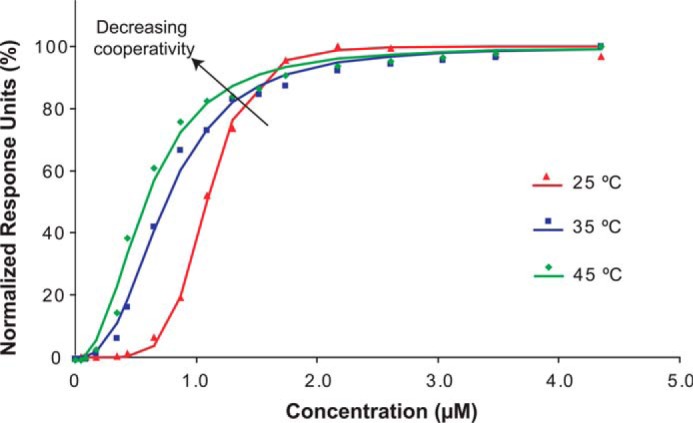
Binding cooperativity of farnesylated HVR of K-Ras4B to DPPC phospholipids decreases with an increase in temperature. At 25 °C, the peptide binds DPPC nanodiscs with high cooperativity (HC = 6.17 ± 0.28). When the temperature is increased to 35 °C, the cooperativity decreases (HC = 2.69 ± 0.04). At the temperature of 45 °C, the cooperativity is further reduced (HC = 2.39 ± 0.02).
The Farnesyl Group Inserts into Loosely Packed Phospholipid Bilayers
The idea that K-Ras4B oligomerization is related to bilayer fluidity leads us to propose that in loosely packed membrane domains the farnesyl moiety inserts more readily into the lipid bilayer. To test this hypothesis, we used an anti-farnesyl antibody to detect farnesyl group insertion and exposure to the solvent in rigid and loosely packed phospholipid bilayers in SPR experiments with the farnesylated HVR peptide. A solution of 5 μm farnesylated HVR peptide was passed over DPPC and DOPC nanodiscs on a sensor chip. The peptide was probed with 0.2 mg/ml anti-farnesyl antibody. On DPPC nanodiscs the antibody induced increased response units, suggesting its accumulation on the tested phospholipids (data not shown). The antibody recognized the farnesyl epitope because it was exposed to the solvent. With DOPC-based nanodiscs, no significant change in response units was observed, indicating that the farnesyl epitope was not available for antibody recognition (data not shown). The farnesyl group does not insert into the DPPC bilayer. Instead, it is exposed and available to bind the anti-farnesyl antibody. Control experiments, injecting buffer instead of the antibody for DPPC and DOPC nanodiscs, respectively, indicate that there is no artifact induced by the buffer (data not shown).
Spontaneous Membrane Insertion of the Farnesyl Group Depends on Lipid Fluidity and Is Inhibited by Phosphorylation
To probe the molecular mechanisms of farnesyl insertion into phospholipid bilayers, we performed all-atoms molecular dynamics simulations on the HVR peptide of K-Ras4B. The Cys-185 side chain at the C terminus of HVR peptide is modified with the farnesyl group. The HVR peptide was initially placed on the surface of the lipid bilayers with three different phospholipid compositions: the zwitterionic DPPC bilayer in the gel phase, the zwitterionic DOPC bilayer in the liquid phase, and the anionic DOPC:DOPS (mole ratio 4:1) bilayer in the liquid phase. The initial configuration for each bilayer system ensures that the peptide backbone and the farnesyl group are not inserted into the lipid bilayers before the start of the simulations. During the simulation, however, we observed that peptide-lipid interaction and spontaneous membrane insertion of the farnesyl group are lipid-dependent. In the gel-phased DPPC bilayer, the HVR peptide dismissed the lipid bilayer, whereas the peptide clung rather strongly to lipids in both the liquid-phased zwitterionic DOPC and anionic DOPC:DOPS (4:1) bilayers (Fig. 5). Our single peptide simulations are not able to reproduce the experimentally observed cooperativity of binding of the HVR peptides, as the simulations represent an extremely low peptide concentration. Under this condition, the HVR peptide can be “lifted“ from the rigid DPPC bilayer due to the unfavorable repelling force exerted on the farnesyl by the lipid headgroups. Otherwise the HVR peptide cooperatively binds the phospholipids through oligomerization of the farnesyl groups as the peptide concentration and, thus, cooperativity is increased. In both fluidic bilayers, we observed that the farnesyl group inserted spontaneously into the liquid hydrophobic core. Of particular interest is insertion kinetics. In the anionic bilayer, spontaneous farnesyl insertion occurred in less than 1 ns, suggesting that the HVR peptide preferentially binds the anionic bilayer.
FIGURE 5.

Snapshots of farnesylated HVR peptides of K-Ras4B, HP1, HP2, and HP3, at a simulation time of t = 100 ns, representing the peptide interaction with the zwitterionic DPPC bilayer in the gel phase (A), the zwitterionic DOPC bilayer in the liquid phase (B), and the anionic DOPC:DOPS (mole ratio 4:1) bilayer in the liquid phase (C). In the peptides, hydrophobic residues are shown in white, polar and Gly residues are shown in green, negatively charged residues are shown in red, and positively charged residues are in blue. The thick yellow stick represents the farnesyl group. Shown is the time series of the deviation, Δz, from the upper bilayer leaflet for the center of mass of the HVR peptide and selected carbon atoms in the farnesyl group for the HP1 (D), HP2 (E), and HP3 peptides (F). The light gray area denotes the interior of the lipid bilayers.
In addition to farnesylation at Cys-185, we phosphorylated Ser-181. K-Ras4B HVR phosphorylation alters the mechanism of peptide-lipid interaction (9). We observed that in the zwitterionic DPPC and DOPC bilayers, the HVR peptide lost contact with the lipids (Fig. 6). In the anionic bilayer, although the N-terminal portion of the HVR peptide interacted with lipids, the C-terminal farnesyl flipped away from the surface of the lipid bilayer. No spontaneous farnesyl insertion was observed during the simulations, suggesting that the phosphorylation at Ser-181 prohibits the farnesyl insertion into the lipid bilayer. We have repeated the molecular dynamics simulations of the S181D phosphomimetic peptide on the same lipid bilayers. The S181D point mutation in the HVR peptide readily mimicked the phosphorylation effect at Ser-181 (data not shown). No spontaneous farnesyl insertion was observed during the simulations as observed for the phosphorylated HVR peptide, suggesting that the S181D mutant also removes the farnesyl from the lipid bilayer.
FIGURE 6.

Snapshots of farnesylated and phosphorylated HVR peptides of K-Ras4B, HP4, HP5, and HP6, at a simulation time of t = 100 ns, representing the peptide interaction with the zwitterionic DPPC bilayer in the gel phase (A), the zwitterionic DOPC bilayer in the liquid phase (B), and the anionic DOPC:DOPS (mole ratio 4:1) bilayer in the liquid phase (C). In the peptides, hydrophobic residues are shown in white, polar and Gly residues are shown in green, negatively charged residues are in red, and positively charged residues are in blue. Thick yellow and red sticks represent the farnesyl and phosphoryl groups, respectively. Shown is the time series of the deviation, Δz, from the upper bilayer leaflet for the center of mass of HVR peptide and selected carbon atoms in the farnesyl group for the HP4 (D), HP5 (E), and HP6 peptides (F). The light gray area denotes the interior of the lipid bilayers.
The Conformation of the HVR Peptides Determines the Membrane Interaction
During the simulations, the HVR peptides generally remained unfolded, although transient 310 and α-helical structures in HP1, HP3, and HP5 peptides can be observed. Asp-173 and Gly-174 are commonly involved in these helical structures. The helical structures facilitate formation of salt bridges between Asp-173 and nearby Lys residues (Fig. 7, A and B). The HP1, HP4, and HP5 peptides exhibit water-relaxed conformations, whereas the HP2, HP3, and HP6 peptides present membrane-mediated conformations. Without phosphorylation, the HVR peptide yields a slightly collapsed conformation. The radius of gyration is decreased by ∼2 Å from the initial values. However, upon phosphorylation, the peptide collapses significantly with the radius of gyration decreasing by ∼4 Å from the initial values. The bend or kinked backbone chain can be observed near phosphorylated Ser-181 in which the negatively charged phosphate (PO43−) group is encompassed by nearby Lys side chains (Fig. 7, B–D).
FIGURE 7.

Snapshots depicted at t = 100 ns for the HP3 peptide on the anionic DOPC:DOPS (mole ratio 4:1) bilayer in the liquid phase (A), at t = 60 ns for the HP5 peptide on the zwitterionic DOPC bilayer in the liquid phase (B), at t = 100 ns for the HP4 peptide on the zwitterionic DPPC bilayer in the gel phase (C), and at t = 80 ns for the HP6 peptide on the anionic DOPC:DOPS (mole ratio 4:1) bilayer in the liquid phase (D). In the peptide ribbon, hydrophobic residues are shown in white, polar and Gly residues are shown in green, negatively charged residues in red, and positively charged residues are in blue. Thick yellow and red sticks represent the farnesyl and phosphoryl groups, respectively.
To understand the effects of post-translational modifications in the HVR-membrane interaction, we calculated the interaction energies of both farnesyl and phosphoryl groups with their surrounding environments. The farnesyl tends to interact with the solvent, including lipids and water (Fig. 8A). For the HP2 and HP3 peptides in the membrane-bound state, the farnesyl interacts strongly with lipids, whereas the farnesyl interacts intensively with water in the membrane-unbound state. The interaction of the farnesyl with its own backbone residues is relatively weak due to the lack of the hydrophobic residues in the HVR. This suggests that K-Ras4B HVR has functionally evolved into abundant positive charges to prevent the hydrophobic farnesyl group folding back onto its backbone. Unlike other Ras isoforms, the K-Ras4B HVR contains a sole prenylation, and thus the HVR should release the farnesyl group from its backbone to effectively anchor in the membrane. However, the farnesyl is not able to bind the membrane in the presence of phosphorylation. The interaction of the phosphoryl with its own backbone residues is formidable, almost 10 times stronger than the farnesyl-peptide interaction (Fig. 8B). The strong electrostatic interaction between the phosphoryl group at Ser-181 and the surrounding Lys residues and the consequent bending at the C-terminal portion of the peptide, cages the farnesyl with the backbone residues. Thus, the phosphorylation removes the farnesyl-lipid interaction but increases both the farnesyl-water and farnesyl-peptide interactions (Fig. 8C).
FIGURE 8.
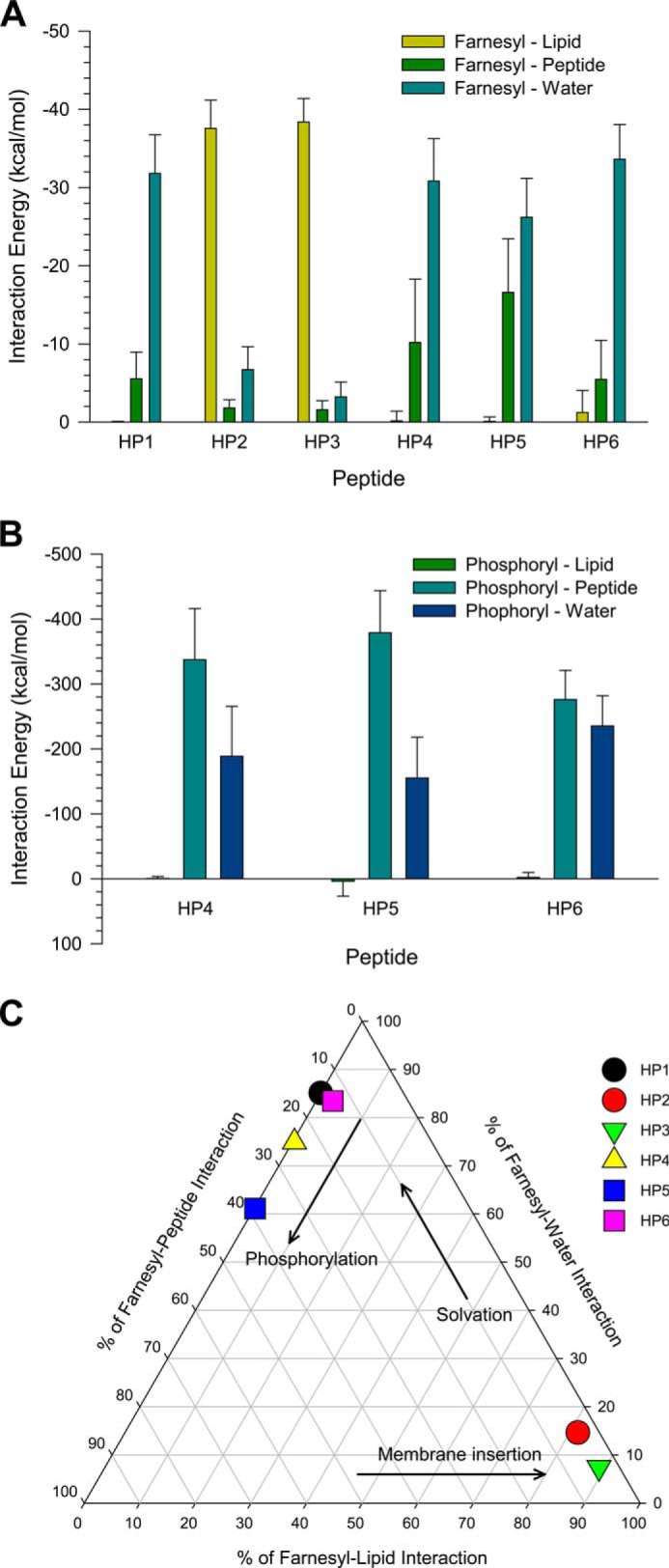
Shown are the averaged total interaction energies of the farnesyl (A) and phosphoryl groups (B) in the HVR peptides of K-Ras4B with the surrounding environments including lipid, its own peptide, and water. C, interaction map representing the percentage of farnesyl interactions with surrounding environments including lipid, its own peptide, and water. The interaction energies of each farnesyl with lipid, peptide, and water are calculated and then averaged over the time.
DISCUSSION
Prenylation is a relatively common modification affecting as many as 0.5% of all cellular proteins (57), including central players in signal transduction (58), cellular trafficking (59), cytoskeletal function (60), regulation of cell growth and polarity (61), viral replication (62), and protein folding (63). Farnesylated proteins are frequently involved in pathogenesis of common human diseases. Cancer (64), cardiac dysfunction (65), developmental disorders, and premature aging are some of the clinical conditions in which farnesylated proteins play a significant role. Inhibitors of protein farnesylation are widely used (66). Although farnesyltransferase inhibitors are largely unsuccessful in cancer therapy due to off-target effects and alternative prenylation mechanisms (1), their use for treatment of African sleeping sickness (67), malaria (68), and pediatric Hutchinson-Gilford Progeria Syndrome (69) has been proposed. Development of inhibitors of farnesyl-directed membrane binding is ongoing, and some of these are entering clinical trials (70, 71); however, toxicity is an on-going concern.
Studies of farnesyl interaction with phospholipids generally assume its insertion into the lipid bilayer (72, 73). Meister et al. (74) showed that the farnesyl moiety inserts into the lipid bilayer when sufficient external pressure is applied. It has also been postulated that the association rate of K-Ras with all cell membranes must be equal (15). However, the detailed mechanism of farnesyl-induced membrane binding is unknown. Even the role of farnesylation in protein binding to the plasma membrane has been questioned. A farnesyl-deficient C186S mutant of human Ras p21 GTPase was used to establish the role of farnesylation in membrane binding (75). Notwithstanding, later studies showed that this mutant never exits the endoplasmic reticulum and, therefore, has never had a chance to interact with the membrane (76). It has also been pointed out that fully processed K-Ras4B can be eluted from the membrane with high ionic strength solutions (77). This suggests that K-Ras4B binding to the plasma membrane is strongly influenced by electrostatic interactions between the HVR polylysine region and the membrane anionic lipids.
The farnesyl group in the K-Ras4B HVR interacts differently with different phospholipid bilayers (Fig. 9). It can insert into loosely packed bilayers containing unsaturated phospholipids, whereas tightly packed saturated phospholipids in rigid lipid domains do not permit farnesyl insertion. Although farnesylation is required for membrane insertion (15), K-Ras4B protein attachment to membrane microdomains may be modulated through electrostatic interactions of the positively charged polybasic region of the HVR with anionic phospholipid headgroups (78). A similar result is reported for Rnd3, a K-Ras like GTP binding protein, that binds the membrane through a positively charged C-terminal HVR (79). Solvent-exposed farnesyl group engages in favorable farnesyl-farnesyl or farnesyl-protein interactions manifested by increased cooperativity when the farnesylated HVR binds to membrane phospholipids. The increasing cooperativity in HVR binding to the rigid DPPC bilayer suggests that K-Ras4B may oligomerize in lipid raft regions of the membrane and form clusters. The clusters may include Ras effectors and their partners, forming a multivalent network with gel-like properties of dynamic proteins and lipids that vary in time and space (80–82). Signaling in the clusters can be thought of as transient, allostery, or cooperativity-driven cluster re-forming interactions. Evolution appears to have exploited membrane composition in two oligomerization modes of K-Ras4B, farnesyl- and catalytic domain-mediated, which may favor distinct effectors under certain environments. Fig. 9 presents a schematic overview of our results.
FIGURE 9.
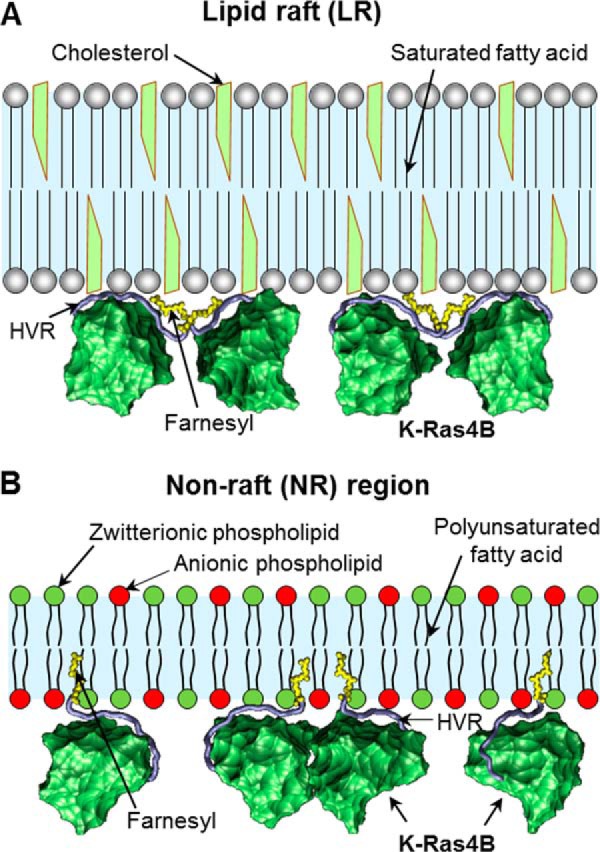
Model for membrane association of K-Ras4B at different plasma membrane microdomains. A, LR is rich in cholesterol and contains saturated lipids such as sphingolipids. It is highly ordered and tightly packed into an ordered gel phase. The K-Ras4B farnesyls are not able to insert into the LR microdomain. Instead, they bind together via farnesyl-farnesyl interactions leading to high cooperativity. B, NR microdomain primarily contains phosphatidylserine and phosphatidic acid with acidic headgroup. These lipids are polyunsaturated and pack into a less ordered fluid-like phase. The farnesyl groups can insert spontaneously into the NR microdomain.
Phosphorylation dramatically affects the spontaneous membrane insertion of the farnesyl group. Our studies demonstrated that attachment of the phosphoryl group at Ser-181 in the HVR removes the farnesyl from the fluidic lipid bilayers even in the presence of the anionic phospholipids. Phosphomimetic mutant S181D, a mimetic of phosphorylation, enhances tumor growth (83). It was also reported that K-Ras with a phosphomimetic residue at position 181 induces apoptosis (84). These works point to the biological significance of K-Ras4B phosphorylation (83). Protein kinase C, a ligand of 14-3-3, mediates the phosphorylation at Ser-181 in the K-Ras4B HVR, which is believed to dissociate K-Ras4B from the plasma membrane through electrostatic repulsion with the negatively charged phospholipid headgroups (85–87) and Ras trafficking in the cytoplasm to an internal membrane (9). Alternatively, phosphomimetic oncogenic K-Ras4B may stay associated with the membrane (88), albeit with altered distribution in the plasma membrane, from low density to high density microdomains. This could also take place after ejection from the membrane.
To summarize, the farnesyl moiety preferentially interacts with the loosely packed phospholipid bilayers and spontaneously inserts into the hydrophobic core of the lipid bilayers, an action facilitated by the anionic phospholipids. However, the farnesyl is not able to insert into rigid domains of the phospholipid bilayers, such as lipid rafts or caveolae. In such a membrane-unbound state, the farnesyls may oligomerize through their hydrophobic interactions resulting in a cooperative binding mode of K-Ras4B. Ras isoforms preferentially localize in different membrane microdomains, which play a role in preferred effector interactions. Which effector interacts with Ras at any specific time is a key cell decision, and how higher membrane fluidity in the K-Ras4B environment plays a role is still not entirely clear. Facilitated access of Raf could be one important factor (23, 89). Our work affirms the importance of microdomain properties in affecting K-Ras4B behavior at the membrane, and provides a mechanistic account of farnesylated and phosphorylated K-Ras4B HVR under different environments. Therapeutically, interference with K-Ras4B at any stage is important for inhibiting K-Ras4B signaling in cancer (90). Even though it is still unclear how to therapeutically interrupt K-Ras4B behavior at the membrane while avoiding a high level of toxicity, it is vital to fully understand the biological mechanisms of the highly oncogenic K-Ras4B isoform.
Acknowledgments
We thank Dr. A. Maciag for providing murine alveolar type II lung epithelial E10 cell line. We also thank David Corcoran for help with the confocal microscopy experiments. All simulations had been performed using the high performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, Maryland (biowulf.nih.gov).
Note Added in Proof
Parts of the description of the molecular dynamics calculations under “Experimental Procedures” were previously published in Jang et al. (Jang, H., Arce, F. T., Ramachandran, S., Kagan, B. L., Lal, R., and Nussinov, R. (2013) Familial Alzheimer's disease Osaka mutant (ΔE22) β-barrels suggest an explanation for the different Aβ1–40/42 preferred conformational states observed by experiment. J. Phys. Chem. B 117, 11518–11529) and Jang et al. (Jang, H., Crozier, P. S., Stevens, M. J., and Woolf, T. B. (2004) How environment supports a state: molecular dynamics simulations of two states in bacteriorhodopsin suggest lipid and water compensation. Biophys. J. 87, 129–145) without citation. This error has been corrected.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA135341 (to V. G.), Frederick National Laboratory for Cancer Research Contract HHSN261200800001E, and by the Intramural Research Program, Frederick National Laboratory Center for Cancer Research. This work was also supported by American Cancer Society Grant RGS-09-057-01-GMC (to V. G.).
- HVR
- hypervariable region
- DPPC
- dipalmitoylphosphatidylcholine
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phosphoserine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine
- DPPE
- dipalmitoylphosphatidylethanolamine
- SPR
- surface plasmon resonance
- LR
- lipid raft
- NR
- non-raft
- HC
- Hill coefficient
- GM1
- monosialotetrahexosylganglioside.
REFERENCES
- 1. Friday B. B., Adjei A. A. (2005) K-ras as a target for cancer therapy. Biochim. Biophys. Acta 1756, 127–144 [DOI] [PubMed] [Google Scholar]
- 2. Ellis C. A., Clark G. (2000) The importance of being K-Ras. Cell. Signal. 12, 425–434 [DOI] [PubMed] [Google Scholar]
- 3. Esteban L. M., Vicario-Abejón C., Fernández-Salguero P., Fernández-Medarde A., Swaminathan N., Yienger K., Lopez E., Malumbres M., McKay R., Ward J. M., Pellicer A., Santos E. (2001) Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol. 21, 1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koera K., Nakamura K., Nakao K., Miyoshi J., Toyoshima K., Hatta T., Otani H., Aiba A., Katsuki M. (1997) K-ras is essential for the development of the mouse embryo. Oncogene 15, 1151–1159 [DOI] [PubMed] [Google Scholar]
- 5. Plowman S. J., Williamson D. J., O'Sullivan M. J., Doig J., Ritchie A. M., Harrison D. J., Melton D. W., Arends M. J., Hooper M. L., Patek C. E. (2003) While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol. Cell. Biol. 23, 9245–9250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simons K., Vaz W. L. (2004) Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33, 269–295 [DOI] [PubMed] [Google Scholar]
- 7. Fujiwara T., Ritchie K., Murakoshi H., Jacobson K., Kusumi A. (2002) Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157, 1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murase K., Fujiwara T., Umemura Y., Suzuki K., Iino R., Yamashita H., Saito M., Murakoshi H., Ritchie K., Kusumi A. (2004) Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys. J. 86, 4075–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahearn I. M., Haigis K., Bar-Sagi D., Philips M. R. (2012) Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eisenberg S., Henis Y. I. (2008) Interactions of Ras proteins with the plasma membrane and their roles in signaling. Cell. Signal. 20, 31–39 [DOI] [PubMed] [Google Scholar]
- 11. Abankwa D., Hanzal-Bayer M., Ariotti N., Plowman S. J., Gorfe A. A., Parton R. G., McCammon J. A., Hancock J. F. (2008) A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 27, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welman A., Burger M. M., Hagmann J. (2000) Structure and function of the C-terminal hypervariable region of K-Ras4B in plasma membrane targeting and transformation. Oncogene 19, 4582–4591 [DOI] [PubMed] [Google Scholar]
- 13. Abraham S. J., Muhamed I., Nolet R., Yeung F., Gaponenko V. (2010) Expression, purification, and characterization of soluble K-Ras4B for structural analysis. Protein Expr. Purif. 73, 125–131 [DOI] [PubMed] [Google Scholar]
- 14. Lopez-Alcalá C., Alvarez-Moya B., Villalonga P., Calvo M., Bachs O., Agell N. (2008) Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 283, 10621–10631 [DOI] [PubMed] [Google Scholar]
- 15. Schmick M., Vartak N., Papke B., Kovacevic M., Truxius D. C., Rossmannek L., Bastiaens P. I. (2014) KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 157, 459–471 [DOI] [PubMed] [Google Scholar]
- 16. Rizzo M. A., Kraft C. A., Watkins S. C., Levitan E. S., Romero G. (2001) Agonist-dependent traffic of raft-associated Ras and Raf-1 is required for activation of the mitogen-activated protein kinase cascade. J. Biol. Chem. 276, 34928–34933 [DOI] [PubMed] [Google Scholar]
- 17. Roy S., Luetterforst R., Harding A., Apolloni A., Etheridge M., Stang E., Rolls B., Hancock J. F., Parton R. G. (1999) Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat. Cell Biol. 1, 98–105 [DOI] [PubMed] [Google Scholar]
- 18. Mineo C., James G. L., Smart E. J., Anderson R. G. (1996) Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J. Biol. Chem. 271, 11930–11935 [DOI] [PubMed] [Google Scholar]
- 19. Prior I. A., Hancock J. F. (2001) Compartmentalization of Ras proteins. J. Cell Sci. 114, 1603–1608 [DOI] [PubMed] [Google Scholar]
- 20. Abankwa D., Gorfe A. A., Inder K., Hancock J. F. (2010) Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 1130–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Janosi L., Li Z., Hancock J. F., Gorfe A. A. (2012) Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. U.S.A. 109, 8097–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niv H., Gutman O., Kloog Y., Henis Y. I. (2002) Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J. Cell Biol. 157, 865–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weise K., Kapoor S., Denter C., Nikolaus J., Opitz N., Koch S., Triola G., Herrmann A., Waldmann H., Winter R. (2011) Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. J. Am. Chem. Soc. 133, 880–887 [DOI] [PubMed] [Google Scholar]
- 24. Furuchi T., Anderson R. G. (1998) Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J. Biol. Chem. 273, 21099–21104 [DOI] [PubMed] [Google Scholar]
- 25. White M. A., Anderson R. G. (2001) Which Ras rides the raft? Nat. Cell Biol. 3, E172. [DOI] [PubMed] [Google Scholar]
- 26. Mor A., Philips M. R. (2006) Compartmentalized Ras/MAPK signaling. Annu. Rev. Immunol. 24, 771–800 [DOI] [PubMed] [Google Scholar]
- 27. Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 [DOI] [PubMed] [Google Scholar]
- 28. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., Sligar S. G. (2009) Chapter 11: Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chavan T. S., Abraham S., Gaponenko V. (2013) Application of reductive 13C-methylation of lysines to enhance the sensitivity of conventional NMR methods. Molecules 18, 7103–7119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 [Google Scholar]
- 31. Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeersch T., Zurek E., Hutchison G. R. (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J., Brothers E. N., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam N. J., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J. (2009) Gaussian 09, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 33. Gillman A. L., Jang H., Lee J., Ramachandran S., Kagan B. L., Nussinov R., Teran Arce F. (2014) Activity and architecture of pyroglutamate-modified amyloid-β (AβpE3–42) pores. J. Phys. Chem. B. 118, 7335–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gupta K., Jang H., Harlen K., Puri A., Nussinov R., Schneider J. P., Blumenthal R. (2013) Mechanism of membrane permeation induced by synthetic β-hairpin peptides. Biophys. J. 105, 2093–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Connelly L., Jang H., Arce F. T., Ramachandran S., Kagan B. L., Nussinov R., Lal R. (2012) Effects of point substitutions on the structure of toxic Alzheimer's β-amyloid channels: atomic force microscopy and molecular dynamics simulations. Biochemistry 51, 3031–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Connelly L., Jang H., Arce F. T., Capone R., Kotler S. A., Ramachandran S., Kagan B. L., Nussinov R., Lal R. (2012) Atomic force microscopy and MD simulations reveal pore-like structures of all-d-enantiomer of Alzheimer's β-amyloid peptide: relevance to the ion channel mechanism of AD pathology. J. Phys. Chem. B. 116, 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Capone R., Jang H., Kotler S. A., Kagan B. L., Nussinov R., Lal R. (2012) Probing structural features of Alzheimer's amyloid-β pores in bilayers using site-specific amino acid substitutions. Biochemistry 51, 776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Capone R., Jang H., Kotler S. A., Connelly L., Teran Arce F., Ramachandran S., Kagan B. L., Nussinov R., Lal R. (2012) All-d-enantiomer of β-amyloid peptide forms ion channels in lipid bilayers. J. Chem. Theory Comput. 8, 1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Capone R., Mustata M., Jang H., Arce F. T., Nussinov R., Lal R. (2010) Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 98, 2644–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jang H., Ma B., Lal R., Nussinov R. (2008) Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic Alzheimer β-amyloid ion channels. Biophys. J. 95, 4631–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jang H., Michaud-Agrawal N., Johnston J. M., Woolf T. B. (2008) How to lose a kink and gain a helix: pH independent conformational changes of the fusion domains from influenza hemagglutinin in heterogeneous lipid bilayers. Proteins 72, 299–312 [DOI] [PubMed] [Google Scholar]
- 42. Jang H., Arce F. T., Ramachandran S., Kagan B. L., Lal R., Nussinov R. (2014) Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs. Chem. Soc. Rev. 43, 6750–6764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woolf T. B., Roux B. (1994) Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 91, 11631–11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woolf T. B., Roux B. (1996) Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins 24, 92–114 [DOI] [PubMed] [Google Scholar]
- 45. Rand R. P., Parsegian V. A. (1989) Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta 988, 351–376 [Google Scholar]
- 46. Kucerka N., Tristram-Nagle S., Nagle J. F. (2005) Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208, 193–202 [DOI] [PubMed] [Google Scholar]
- 47. Petrache H. I., Tristram-Nagle S., Gawrisch K., Harries D., Parsegian V. A., Nagle J. F. (2004) Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 86, 1574–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durell S. R., Brooks B. R., Bennaim A. (1994) Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 98, 2198–2202 [Google Scholar]
- 50. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dietrich C., Volovyk Z. N., Levi M., Thompson N. L., Jacobson K. (2001) Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. U.S.A. 98, 10642–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rouquette-Jazdanian A. K., Foussat A., Lamy L., Pelassy C., Lagadec P., Breittmayer J. P., Aussel C. (2005) Cholera toxin B-subunit prevents activation and proliferation of human CD4+ T cells by activation of a neutral sphingomyelinase in lipid rafts. J. Immunol. 175, 5637–5648 [DOI] [PubMed] [Google Scholar]
- 53. Pike L. J. (2003) Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 54. Dietrich C., Bagatolli L. A., Volovyk Z. N., Thompson N. L., Levi M., Jacobson K., Gratton E. (2001) Lipid rafts reconstituted in model membranes. Biophys. J. 80, 1417–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Calvert R. J., Ramakrishna G., Tepper S., Diwan B. A., Anderson L. M., Kritchevsky D. (2002) Alterations in membrane-bound and cytoplasmic K-ras protein levels in mouse lung induced by treatment with lovastatin, cholestyramine, or niacin: effects are highly mouse strain dependent. Biochem. Pharmacol. 64, 41–48 [DOI] [PubMed] [Google Scholar]
- 56. Leonenko Z. V., Finot E., Ma H., Dahms T. E., Cramb D. T. (2004) Investigation of temperature-induced phase transitions in DOPC and DPPC phospholipid bilayers using temperature-controlled scanning force microscopy. Biophys. J. 86, 3783–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cox A. D., Der C. J. (1992) Protein prenylation: more than just glue? Curr. Opin. Cell Biol. 4, 1008–1016 [DOI] [PubMed] [Google Scholar]
- 58. Sorek N., Bloch D., Yalovsky S. (2009) Protein lipid modifications in signaling and subcellular targeting. Curr. Opin. Plant Biol. 12, 714–720 [DOI] [PubMed] [Google Scholar]
- 59. Resh M. D. (2006) Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2, 584–590 [DOI] [PubMed] [Google Scholar]
- 60. Bifulco M. (2005) Role of the isoprenoid pathway in ras transforming activity, cytoskeleton organization, cell proliferation, and apoptosis. Life Sci. 77, 1740–1749 [DOI] [PubMed] [Google Scholar]
- 61. Bialek-Wyrzykowska U., Bauer B. E., Wagner W., Kohlwein S. D., Schweyen R. J., Ragnini A. (2000) Low levels of Ypt protein prenylation cause vesicle polarization defects and thermosensitive growth that can be suppressed by genes involved in cell wall maintenance. Mol. Microbiol. 35, 1295–1311 [DOI] [PubMed] [Google Scholar]
- 62. Einav S., Glenn J. S. (2003) Prenylation inhibitors: a novel class of antiviral agents. J. Antimicrob. Chemother. 52, 883–886 [DOI] [PubMed] [Google Scholar]
- 63. Pylypenko O., Schönichen A., Ludwig D., Ungermann C., Goody R. S., Rak A., Geyer M. (2008) Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. J. Mol. Biol. 377, 1334–1345 [DOI] [PubMed] [Google Scholar]
- 64. Fritz G. (2009) Targeting the mevalonate pathway for improved anticancer therapy. Curr. Cancer Drug Targets 9, 626–638 [DOI] [PubMed] [Google Scholar]
- 65. Nakagami H., Jensen K. S., Liao J. K. (2003) A novel pleiotropic effect of statins: prevention of cardiac hypertrophy by cholesterol-independent mechanisms. Ann. Med. 35, 398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wright L. P., Philips M. R. (2006) Thematic review series: lipid posttranslational modifications: CAAX modification and membrane targeting of Ras. J. Lipid Res. 47, 883–891 [DOI] [PubMed] [Google Scholar]
- 67. Buckner F. S., Eastman R. T., Yokoyama K., Gelb M. H., Van Voorhis W. C. (2005) Protein farnesyl transferase inhibitors for the treatment of malaria and African trypanosomiasis. Curr. Opin. Investig. Drugs 6, 791–797 [PubMed] [Google Scholar]
- 68. Liñares G. E., Rodriguez J. B. (2007) Current status and progresses made in malaria chemotherapy. Curr. Med. Chem. 14, 289–314 [DOI] [PubMed] [Google Scholar]
- 69. Gonzalez J. M., Pla D., Perez-Sala D., Andres V. (2011) A-type lamins and Hutchinson-Gilford progeria syndrome: pathogenesis and therapy. Front. Biosci. S3, 1133–1146 [DOI] [PubMed] [Google Scholar]
- 70. Bustinza-Linares E., Kurzrock R., Tsimberidou A. M. (2010) Salirasib in the treatment of pancreatic cancer. Future Oncol. 6, 885–891 [DOI] [PubMed] [Google Scholar]
- 71. Blum R., Cox A. D., Kloog Y. (2008) Inhibitors of chronically active ras: potential for treatment of human malignancies. Recent Pat. Anticancer Drug Discov. 3, 31–47 [DOI] [PubMed] [Google Scholar]
- 72. Pechlivanis M., Kuhlmann J. (2006) Hydrophobic modifications of Ras proteins by isoprenoid groups and fatty acids: more than just membrane anchoring. Biochim. Biophys. Acta 1764, 1914–1931 [DOI] [PubMed] [Google Scholar]
- 73. Prakash P., Gorfe A. A. (2013) Lessons from computer simulations of Ras proteins in solution and in membrane. Biochim. Biophys. Acta 1830, 5211–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Meister A., Nicolini C., Waldmann H., Kuhlmann J., Kerth A., Winter R., Blume A. (2006) Insertion of lipidated Ras proteins into lipid monolayers studied by infrared reflection absorption spectroscopy (IRRAS). Biophys. J. 91, 1388–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Willumsen B. M., Norris K., Papageorge A. G., Hubbert N. L., Lowy D. R. (1984) Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J. 3, 2581–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Choy E., Chiu V. K., Silletti J., Feoktistov M., Morimoto T., Michaelson D., Ivanov I. E., Philips M. R. (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98, 69–80 [DOI] [PubMed] [Google Scholar]
- 77. Quatela S. E., Sung P. J., Ahearn I. M., Bivona T. G., Philips M. R. (2008) Analysis of K-Ras phosphorylation, translocation, and induction of apoptosis. Methods Enzymol. 439, 87–102 [DOI] [PubMed] [Google Scholar]
- 78. Gomez G. A., Daniotti J. L. (2007) Electrical properties of plasma membrane modulate subcellular distribution of K-Ras. FEBS J. 274, 2210–2228 [DOI] [PubMed] [Google Scholar]
- 79. Madigan J. P., Bodemann B. O., Brady D. C., Dewar B. J., Keller P. J., Leitges M., Philips M. R., Ridley A. J., Der C. J., Cox A. D. (2009) Regulation of Rnd3 localization and function by protein kinase Cα-mediated phosphorylation. Biochem. J. 424, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nussinov R., Jang H. (2014) Dynamic multiprotein assemblies shape the spatial structure of cell signaling. Prog. Biophys. Mol. Biol. 116, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nussinov R., Jang H., Tsai C. J. (2014) Oligomerization and nanocluster organization render specificity. Biol. Rev. Camb. Philos. Soc. 10.1111/brv.12124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nussinov R. (2013) The spatial structure of cell signaling systems. Phys. Biol. 10, 045004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barceló C., Paco N., Morell M., Alvarez-Moya B., Bota-Rabassedas N., Jaumot M., Vilardell F., Capella G., Agell N. (2014) Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 74, 1190–1199 [DOI] [PubMed] [Google Scholar]
- 84. Bivona T. G., Quatela S. E., Bodemann B. O., Ahearn I. M., Soskis M. J., Mor A., Miura J., Wiener H. H., Wright L., Saba S. G., Yim D., Fein A., Pérez de Castro I., Li C., Thompson C. B., Cox A. D., Philips M. R. (2006) PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 21, 481–493 [DOI] [PubMed] [Google Scholar]
- 85. Ghomashchi F., Zhang X., Liu L., Gelb M. H. (1995) Binding of prenylated and polybasic peptides to membranes: affinities and intervesicle exchange. Biochemistry 34, 11910–11918 [DOI] [PubMed] [Google Scholar]
- 86. Ashery U., Yizhar O., Rotblat B., Kloog Y. (2006) Nonconventional trafficking of Ras associated with Ras signal organization. Traffic 7, 119–126 [DOI] [PubMed] [Google Scholar]
- 87. Plowman S. J., Ariotti N., Goodall A., Parton R. G., Hancock J. F. (2008) Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol. Cell. Biol. 28, 4377–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Barceló C., Paco N., Beckett A. J., Alvarez-Moya B., Garrido E., Gelabert M., Tebar F., Jaumot M., Prior I., Agell N. (2013) Oncogenic K-ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J. Cell Sci. 126, 4553–4559 [DOI] [PubMed] [Google Scholar]
- 89. Cho K. J., Kasai R. S., Park J. H., Chigurupati S., Heidorn S. J., van der Hoeven D., Plowman S. J., Kusumi A., Marais R., Hancock J. F. (2012) Raf inhibitors target ras spatiotemporal dynamics. Curr. Biol. 22, 945–955 [DOI] [PubMed] [Google Scholar]
- 90. Zimmermann G., Papke B., Ismail S., Vartak N., Chandra A., Hoffmann M., Hahn S. A., Triola G., Wittinghofer A., Bastiaens P. I., Waldmann H. (2013) Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 497, 638–642 [DOI] [PubMed] [Google Scholar]
- 91. Jang H., Arce F. T., Ramachandran S., Kagan B. L., Lal R., Nussinov R. (2013) Familial Alzheimer's disease Osaka mutant (ΔE22) β-barrels suggest an explanation for the different Aβ1–40/42 preferred conformational states observed by experiment. J. Phys. Chem. B 117, 11518–11529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jang H., Crozier P. S., Stevens M. J., Woolf T. B. (2004) How environment supports a state: molecular dynamics simulations of two states in bacteriorhodopsin suggest lipid and water compensation. Biophys. J. 87, 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]