

Abstract
Alzheimer's disease (AD) is characterized by the presence of senile plaques and neurofibrillary tangles in the neocortex and hippocampus of AD patients. In addition, a marked decrease in synaptic contacts has been detected in these affected brain areas. Due to its prevalence in the aging population, this disease has been the focus of numerous studies. The data obtained from those studies suggest that the mechanisms leading to the formation of the hallmark lesions of AD might be linked. One of such mechanisms seems to be the dysregulation of calcium homeostasis that results in the abnormal activation of calpains. Calpains are a family of Ca2+-dependent cysteine proteases that play a key role in multiple cell functions including cell development, differentiation and proliferation, axonal guidance, growth cone motility, and cell death, among others. In this paper, we briefly reviewed data on the structure of these proteases and their regulation under normal conditions. We also summarized data underscoring the participation of calpains in the neurodegenerative mechanisms associated with AD.
1. Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the aging population. This disease develops over time and leads to significant cognitive deficits affecting memory, insight, judgment, abstraction, and language functions [1]. AD affects more than 5 million people in the United States and this number is projected to rise to 35 million by 2050 [2, 3]. This estimate underscores both the scope of this health care issue for the society as a whole and the need for the development of therapeutic options for these patients.
The diagnosis of this neurodegenerative disease relies on the presence of senile plaques and neurofibrillary tangles in affected brain areas at autopsy. These AD hallmark lesions are the results of the pathological deposition of proteins normally present throughout the brain. Senile plaques are composed of extracellular deposits of beta-amyloid (Aβ) derived by proteolytic cleavage from the amyloid precursor protein (APP) [4–10]. Neurofibrillary tangles, on the other hand, are intracellular bundles of self-assembled tau proteins [11–38]. The formation of both senile plaques and neurofibrillary tangles is associated with progressive and irreversible degeneration of neuronal processes and the loss of synaptic connections [39–50].
Initially, multiple studies focused on defining the characteristics of AD and on the analysis of the composition of senile plaques and neurofibrillary tangles. More recently, data have been obtained on the molecular mechanisms that link the formation of these lesions and underlie neurodegeneration and cell death in AD and related disorders. Calpains seem to play a key role in such mechanisms. Calpains are Ca2+-dependent proteases in which activity is dysregulated in AD and other neurodegenerative diseases [51–54]. A growing body of evidence suggests that the abnormal activation of calpains might modulate not only the formation of senile plaques and neurofibrillary tangles but also the development of synaptic pathology in AD. These data reviewed below position calpains at the crossroads of the mechanisms involved in the formation of the main pathological alterations associated with AD. Furthermore, these findings underscore the importance of calpains as potential targets to block the activation of the signaling cascades leading to degeneration in AD. In turn, this information could be applied to the design of therapeutic options and prevention strategies for this devastating disease.
In this review, we first summarized data on the properties of calpains and the mechanisms underlying their activation. Then, we reviewed findings on the dysregulation of calpain in the context of AD and its deleterious consequences for the morphology and function of affected brain areas. Finally, we examined the effects of experimental manipulations that could prevent such effects.
2. The Calpain Family
Calpains constitute a family of Ca2+-dependent cysteine proteases involved in multiple and very diverse cell functions including cell development, proliferation, and differentiation, cell motility, growth cone motility and guidance, apoptosis, learning, and memory, among others [51, 52]. Originally, two members of this family were identified: calpain 1 (μ-calpain) and calpain 2 (m-calpain), also known as conventional or classical calpains [51]. More recently, the other 14 members of the calpain family have been identified in mammals [51, 55]. In contrast to classical calpains that are ubiquitously distributed, the expression of some of these unconventional or nonclassical calpains is tissue specific. For example, calpain 3a and calpain 8 are mainly present in skeletal and smooth muscle cells, respectively. Calpain 11, on the other hand, is highly expressed in testes, while calpain 13 is concentrated in the lung and skin. Others, like calpains 5, 7, 9, and 10 have more widespread distributions [51]. None of these new members of the calpain family are highly expressed in the central nervous system (CNS) [51]. Thus, we will focus this review on the conventional calpains.
Both calpain 1 and calpain 2 are heterodimers composed of a large catalytic subunit (~80 kDa) and a small regulatory subunit (~30 kDa). The amino acid sequence of these subunits is highly conserved in mammals [51]. Based on this sequence, the catalytic subunit has been divided into four distinct domains (reviewed in [51]). Domain 1 corresponds to the N-terminal region of this subunit and undergoes autolysis upon Ca2+ binding. Domain 2 contains a catalytic unit formed by Cys, His, and Asn residues. This unit is characteristic of cysteine proteases like papain or cathepsin [56]. Domain 3 links the Ca2+ binding and the catalytic domains and regulates calpain activity. It might also bind phospholipids [57–59]. Domain 4 has partial homology with calmodulin and contains several EF-hand calcium-binding motifs [60]. The regulatory subunit can be divided into two domains, a calmodulin-like domain and a glycine-rich domain [61].
Calpain 1 and calpain 2 are highly expressed throughout the CNS. While both proteases are present in neurons and glial cells, their relative abundance differs. Calpain 1 is more abundant in neurons, and calpain 2 is prominent in glial cells [62]. Analysis of the subcellular localization of these enzymes demonstrated that calpain 1 is concentrated in the cell bodies and is also detected in the processes extended by central neurons, although at lower levels. In addition, calpain 1 is present at synaptic sites. Within the presynaptic element, calpain 1 is enriched between synaptic vesicles. Calpain 1 immunoreactivity has been also detected in postsynaptic densities as well as in dendritic spines [63]. Calpain 2 immunoreactivity is mainly present in white matter and in myelinated axons. Astrocytic processes are also enriched in calpain 2 [62, 63].
Calpains cleave proteins generating large fragments. Initially, it was thought that these proteases' cleavage sites have a Leu or a Val residue in the P2 position [64]. More recently, it has been shown that the cleavage site specificity is determined by conformation rather than amino acid sequence [65–68]. The specificities of the substrates for both calpains are very similar but not identical. In vitro studies have shown that more than 100 proteins could be cleaved by calpains. Calpain substrates include cytoskeletal proteins (cadherin, catenin, desmin, dystrophin, gelsolin, filamin, fodrin, microtubule-associated proteins MAP 1 and MAP 2, neurofilament proteins, spectrin, tau, talin, troponin, tubulin, vimentin, and vinculin), signal transduction proteins (calcium/calmodulin-dependent protein kinase, epidermal growth factor (EGF) kinase, pp60, protein kinase C, calcineurin, and caspases 3, 7, 8, 9, 12, and 14), synaptic proteins (dynamin 1, postsynaptic density (PSD) 95, N-methyl-D-aspartic acid (NMDA) glutamate receptors, and metabotropic glutamate receptor GluR1), and transcription factors (p53), among others [51, 53].
Calpain activation is tightly regulated to prevent massive proteolytic activity in the cell. The main regulator of this activity is Ca2+ [51]. The binding of Ca2+ to either calpain results in conformational changes that initiate their proteolytic activity. While calpain 1 is activated in the presence of micromolar concentrations of Ca2+, calpain 2 requires millimolar concentrations [51]. The Ca2+ requirement for either calpain is significantly higher than the concentration of this ion in living cells. This fact has prompted a series of studies on potential mechanisms that could lead to a decrease in the Ca2+ requirement for the activation of these proteases in living cells. In vitro studies have shown that the interaction of calpains with a series of activator molecules lowers the concentration of Ca2+ required to initiate the activation of these proteases. Among them, phospholipids (i.e., phosphatidylinositol) are the most studied. However, the phospholipid to calpain molar ratio required for lowering the Ca2+ requirement for either calpain activation is not likely to be achieved in the cellular context [69–71]. Nevertheless, it cannot be ruled out that higher concentrations of Ca2+ are achieved in limited cellular areas where Ca2+ is not easily detectable by available methods. In turn, these local micromolar or millimolar Ca2+ concentrations could trigger calpain activation in distinct subcellular domains.
In addition to Ca2+, calpastatin has a key role in the regulation of calpain. Calpastatin, a heat-stable protein ranging from ~70 to ~140 kDa of apparent molecular weight depending on the cell type, is considered a specific endogenous inhibitor of calpains [72]. It is ubiquitously expressed and has a widespread cytosolic distribution [73]. Immunocytochemical analysis showed calpastatin immunoreactivity throughout the dendritic tree in pyramidal neurons and Purkinje cells. On the other hand, weak calpastatin labeling was detected in glial cells [74]. The calpastatin molecule contains four inhibitory units [75–77]. Each of these units binds to one calpain molecule [75–77]. Therefore, the ratio calpain/calpastatin plays a key role in the regulation of calpain activity [78–80]. The inhibitory effect of calpastatin requires Ca2+-dependent high-affinity binding to three sites of calpain [51].
Phosphorylation might be also involved in the regulation of calpain activity. Both calpains can be phosphorylated in multiple sites of the catalytic subunit [51]. The kinases responsible for the phosphorylation of some of these sites have already been identified. Thus, two of those sites seem to be phosphorylated by protein kinase C (PKC), two by protein kinase A (PKA), two by calmodulin kinase II, one site by casein kinase I, and another by protein kinase G [51]. Although the role of the phosphorylation of each site in calpain activation has not been completely elucidated, data suggest that phosphorylation mediated by mitogen-activated kinases (ERK/MAP kinases) activates these proteases [81]. On the other hand, phosphorylation by PKA may decrease their activity [82, 83].
3. Dysregulation of Calpain Activity in AD and Related Disorders
As briefly reviewed above, calpain activation is a tightly regulated process to prevent deleterious consequences of massive proteolytic activity. These regulatory mechanisms seem to decline with aging resulting in an increased calpain activity [84–87]. In addition, these proteases seem to be abnormally activated under pathological conditions. Thus, several studies have reported significantly increased activation of calpains in AD brains [88–94]. The hyperactivation of calpain in the context of AD is the result of several factors including enhanced intracellular Ca2+ concentration and decreased calpastatin levels. Experiments performed using an AD culture model system showed that oligomeric Aβ induced a significant (5-fold) and instantaneous rise in Ca2+ in hippocampal neurons [95–97]. These studies also addressed the source of the Ca2+ influx leading to calpain activation in the context of AD taking advantage of specific blockers of Ca2+ release from the endoplasmic reticulum, the major source of intracellular Ca2+, and BAPTA, a chelator of extracellular Ca2+ [98]. The data obtained showed that Aβ induces calpain activation by enhancing extracellular Ca2+ influx [95–97]. The mechanisms underlying the regulation of the enhanced Ca2+ influx and calpain activation in AD have also been studied. Both NMDA receptors and voltage-gated calcium channels (VGCC) have been implicated in such regulation in central neurons [99–101]. Specific NMDA receptor inhibitors, MK801 [102], and the FDA approved NMDA receptor antagonist memantine [103] significantly attenuated the initial Aβ-induced increase of Ca2+ and blocked Aβ-induced calpain activation in cultured hippocampal neurons [95–97]. In contrast, nimodipine, an L-type VSCCs blocker [104], did not decrease the Aβ-induced activation of calpain [95–97]. These data suggest that NMDA receptors play an important role in mediating the sustained Ca2+ influx and enhanced calpain activity induced by aggregated Aβ. Furthermore, these studies suggested that factors that affect NMDA receptor-mediated Ca2+ influx could modulate calpain activation and its deleterious effects in central neurons. Cholesterol seems to play such a role. Recently, it has been shown that cholesterol regulates calpain activity in the context of AD [105]. Cholesterol is a risk factor for AD. A population-based study demonstrated that not only high cholesterol levels but also moderately elevated cholesterol levels in midlife represent a significant risk factor for AD [106, 107]. Interestingly, cholesterol has been implicated in the susceptibility of cells to Ca2+ influx [108–111]. Thus, elevated membrane cholesterol actually increased the susceptibility of cells to Aβ-induced elevation in Ca2+ influx leading to cell death via a calpain-dependent mechanism [111, 112]. By regulating the NMDA receptor content and changing their localization in membrane microdomains at the synaptic sites, cholesterol modulates the ability of Aβ to induce Ca2+ influx, leading to calpain activation in hippocampal neurons [111, 112].
Calpastatin also seems to play an important role in the regulation of calpain activation in AD. Thus, it has been shown that calpastatin is markedly depleted in the cortex of AD brains at late stages of the disease as compared to age-matched controls. Focal areas of calpastatin depletion have been also detected along dystrophic neurites at early stages of AD [74]. On the other hand, no changes in calpastatin levels were detected in neurons less susceptible to neurodegeneration in AD like Purkinje cells [113]. This decrease in calpastatin levels is the result of the proteolytic activity of caspases and calpain [74]. In turn, the decrease in the ratio calpastatin to calpain causes calpain hyperactivation perpetuating this cellular deleterious effect [79, 113–115].
4. The Role of Calpain in the Generation of Pathological Manifestations of AD
The data reviewed above provide insights into the abnormal calpain activation in AD brains. These findings have been complemented with immunocytochemical studies on the distribution of calpain in AD. These studies have shown intense calpain immunoreactivity in dystrophic neurites associated with neurofibrillary tangles, neuritic plaques, and neuropil threads in the hippocampal region and entorhinal cortex of subjects suffering from AD and other tauopathies [116–118]. Based on this information, it was hypothesized that the enhanced calpain activity observed in AD brains plays an important role in the formation of senile plaques, neurofibrillary tangles, and synaptic dysfunction in the context of AD (Figure 1). Senile plaques are mainly composed of Aβ 1–40 and Aβ 1–42 peptides [4]. These peptides are the result of the sequential proteolytic cleavage of APP by two proteases. First, β-secretase (β-site APP-cleaving enzyme (BACE 1)) cuts APP at the N-terminus of the Aβ domain leaving a 99-amino-acid-long C-terminal fragment. Then, the γ-secretase complex (presenilin 1, presenilin 2, nicastrin, APH1, and PEN2) cuts the C-terminus to generate Aβ peptides of 38 to 43 amino acids in length [5, 119–121]. Of these peptides, Aβ 1–42 is regarded as the main pathogenic species [122–130]. Therefore, increased levels of BACE 1 could lead to enhanced production of Aβ, its aggregation, and consequently the activation of signaling pathways associated with the neurodegenerative process. This seems to be the case in AD patients. Thus, a 2-fold increase in BACE1 levels has been detected in AD brains [127–129]. Recently, it has been shown that the calpain/calpastatin system can modulate the pathological deposition of Aβ. Calpain activation increases the levels of BACE1 in a transgenic mouse model of AD [130]. In addition, the bidirectional regulation of the activation of the calpain/calpastatin system and Aβ metabolism has been established [131]. Using transgenic mice overexpressing APP, these authors showed that calpastatin deficiency enhanced calpain activation, Aβ production, and increased mortality. In turn, the increased levels of Aβ lead to enhanced Ca2+ influx and increased calpain activation [131].
Figure 1.
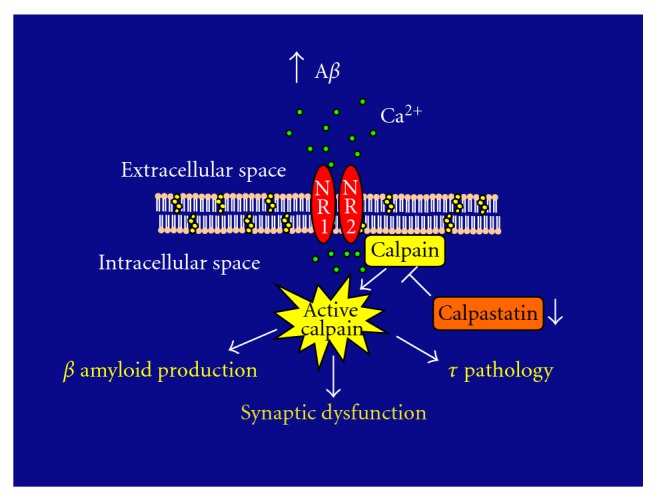
In the context of Alzheimer's disease, increased levels of beta-amyloid (Aβ) induce calcium (Ca2+) influx through NMDA receptors (NR1 and NR2) in hippocampal neurons. This Ca2+ influx and a decrease in calpastatin levels result in the dysregulation of calpain activity leading to the cleavage of a series of proteins involved in the formation of senile plaques and neurofibrillary tangles as well as in synaptic dysfunction.
Senile plaques are often surrounded by dystrophic neurons that contain tau aggregates known as neurofibrillary tangles. Many studies have focused on the composition and the mechanisms underlying the formation of tau aggregates. Those studies have identified two types of tau filaments in the neurofibrillary tangles: straight and paired helical filaments. Hyperphosphorylated forms of tau form both types of filaments [132–142]. Tau hyperphosphorylation has been attributed to the increased activity of several kinases, including cyclin-dependent kinase 5 (CDK5), glycogen synthase kinase β (GSKβ), and the mitogen-activated protein kinase (MAPK) in hippocampal neurons [138, 143–145]. Tau phosphorylation can be modulated by calpain. Thus, it has been shown that calpain cleaves the inhibitory domain of GSK3 generating two fragments of 40 and 30 kDa. This cleavage enhanced activity of the kinase [146]. Calpains also modulate the activity of CDK5. Physiologically, CDK 5 is activated by p35 and its cleaved product p25. The latter has a longer half life than p35 and therefore it is a more potent activator of CDK5. The cleavage of p35 to p25 is mediated by calpain [147–149]. In addition, calpains activate ERK/MAP kinases [150]. Experiments using calpeptin, a cell permeable calpain inhibitor, blocked both ERK 1 and ERK 2 activation. Conditions that blocked the activation of these kinases induced a decreased in tau phosphorylation and resulted in enhanced neuronal survival in the presence of Aβ [136].
Besides tau phosphorylation, calpain activation might play a role in tau-mediated neurodegeneration by inducing tau cleavage. In vitro studies have shown that both fetal and adult tau isoforms are rapidly proteolyzed by calpains [151–153]. On the other hand, tau present in paired-helical filaments is considerably more resistant to proteolysis by these proteases [153–156]. These findings suggest that phosphorylation might regulate the susceptibility of tau to calpain-mediated cleavage [153]. Support for this hypothesis was obtained using cultured hippocampal neurons treated with okadaic acid. Tau obtained from cultured neurons incubated in the presence of this phosphatase inhibitor was more resistant to calpain cleavage than tau extracted from nontreated control neurons [90]. Nevertheless, the effect of phosphorylation on calpain-mediated tau cleavage seems to be complex. This effect might depend on the site phosphorylated and/or the extent of phosphorylation under pathological conditions since highly phosphorylated fetal isoforms are readily cleaved by calpain [153].
Calpain-mediated tau cleavage seems to play an important role under neurodegenerative conditions [157–164]. It has been shown that calpain activation results in the generation of several N-terminal tau fragments. One of such tau fragments, of ~20 kDa of apparent molecular weight, has been detected in mitochondria present in synaptosomal fractions obtained from AD brains [158]. The levels of this tau fragment are partially reduced when cultured neurons are treated with a calpain inhibitor [158]. The presence of this fragment could impair mitochondria function. It has been shown that tau fragments containing the 26–44 tau amino acids affect mitochondria oxidative phosphorylation acting at the level of the adenine nucleotides translocator contributing to synapse dysfunction [159].
A smaller tau fragment generated by calpain cleavage has been detected in mature hippocampal neurons treated with aggregated Aβ oligomers [160, 161]. Aβ-induced tau cleavage mediated by calpain 1 leads to the generation of a neurotoxic 17 kDa tau fragment (tau 45–230) in cultured hippocampal neurons [160, 161]. This fragment has also been detected in the neocortex of AD brains as well as in brain areas affected by other tauopathies [90]. Furthermore, when expressed in neuronal and nonneuronal cell types or in an in vivo Drosophila model system, the 17 kDa tau fragment produced cell death in the absence of Aβ oligomers [160, 161, 163]. These data provided strong evidence for an important role of calpain 1 and the generation of the 17 kDa tau fragment in the progression of Aβ-mediated neurodegeneration. It is worth noting that calpain 2 activation cleaves tau generating a smaller tau fragment that lacks neurotoxic effects in central neurons [164].
In addition to the presence of senile plaques and neurofibrillary tangles, AD brains are characterized by a decrease in synaptic contacts. This synaptic loss seems to be the best morphological correlate of the functional deficits observed in the mid to late stages of AD [39, 40]. Although no significant decline in synapse number has been detected in the earliest stages of the disease, a stage of synaptic dysfunction seems to precede frank synapse loss [41, 46, 165]. By cleaving proteins in the presynaptic terminals and/or the postsynaptic elements, calpain could induce profound functional changes in affected hippocampal neurons [166].
Changes in proteins involved in synaptic vesicle biogenesis and/or recycling at synaptic terminals are responsible, at least in part, for the synaptic dysfunction detected in AD [43]. Recently, data obtained using culture and animal models of AD showed that Aβ induced a significant reduction in dynamin 1 levels that preceded synapse loss. Dynamin 1 is a neuron-specific mechanochemical GTPase highly enriched in presynaptic terminals. Dynamin 1 pinches off synaptic vesicles, freeing them from the membrane and allowing them to reenter the synaptic vesicle pool to be refilled for future release [167, 168]. Decreased levels of this protein lead to the depletion of synaptic vesicles and the accumulations of invaginated pits at presynaptic membranes adjacent to the synaptic clefts [95–97, 169, 170]. The Aβ-induced decrease in dynamin 1 is a result, at least partially, of calpain-mediated proteolysis [95–97, 171].
Calpain dysregulation could also affect proteins in the postsynaptic element. Thus, it has been shown that calpain activation results in the breakdown of several proteins leading to changes in the ultrastructure of the postsynaptic density (PSD). These changes in the PSD are accompanied by the rapid cleavage of PSD-95 and the NMDA receptor subunits NR1 and NR2A and 2B [172–175]. This cleavage is regulated by the tyrosine kinase Fyn phosphorylation of the NR2B subunit [176]. Calpain has been shown to truncate also mGluR1 exacerbating NMDA-mediated neurotoxicity [176–178].
An additional mechanism by which calpain activation can result in synaptic dysfunction involved the cleavage of PKA leading to a decrease in both the regulatory and the catalytic subunits of this kinase. The decrease in PKA activity attenuates CREB activation impairing memory [179, 180].
5. Future Directions and Concluding Remarks
Taken together, the data reviewed above strongly suggest that calpains play an important role in the neurodegeneration mechanisms underlying AD in the aging population. Based on these data, it has been tempting to speculate that calpain inhibition could be a useful tool to prevent neurodegeneration in this disease. To obtain further insights into the beneficial effects of blocking calpain activation, several studies have been conducted using experimental approaches to either suppress the expression of these proteases or prevent their abnormal activation. Early studies using gene deletion of the calpain regulatory subunit by homologous recombinant techniques showed that the depletion of these proteases is embryonic lethal [181]. Later, studies have been performed using specific antisense oligonucleotides. The data obtained showed that indeed the suppression of calpain 1 expression by means of these specific probes prevented oxidative stress-induced cell injury in human hepatic cancer cell lines [182]. Other studies have assessed the effects of calpain inhibitors on cell death in culture model systems of AD. Leupeptin and E64 calpain inhibitors had protective effects in those cultures [160, 183–186]. The idea that calpain could be a potential therapeutic target in neurodegenerative diseases has been reinforced by studies performed in vitro and in culture using the calpain inhibitor MDL 28170. Those studies suggested a protective effect of calpain inhibitors against excitotoxicity [178, 187, 188]. Unfortunately, the potential use of known calpain inhibitors as therapeutic tools in AD is limited due to their low cellular penetration, poor selectivity, and kinetics. Recently, A-705053, a novel calpain inhibitor with improved pharmacokinetics, has been characterized [189]. This benzoylalanine-derived ketoamide is capable of inhibiting calpain in nanomolar concentrations and has improved oral bioavailability, water solubility, and metabolic stability [189]. This calpain inhibitor is highly effective in preventing calpain-mediated cleavage of dynamin 1 and tau in cultured hippocampal neurons. This inhibitor is effective not only when added prior to the Aβ treatment but also when added simultaneously with Aβ and even when added after Aβ has triggered the neurodegeneration process [171]. The calpain inhibitor A-705253 also prevents Aβ oligomer-induced neurodegeneration of the nucleus basalis magnocellularis [190]. These experiments raised the possibility that more potent calpain inhibitors could have beneficial effects even at late stages of AD. Moreover, initial studies using calpain inhibitors in mouse and rat models of AD showed an encouraging recovery of cognitive function in these animals when they were treated with calpain inhibitors at an early age [186, 190]. Together, these data underscore the potential importance of calpain inhibitors as promising therapeutic tools in AD and related neurodegenerative diseases.
In summary, the data briefly reviewed above provide strong support for the role of calpains in AD. They also highlight the tantalizing possibility that these proteases could serve as targets for the development of therapeutic interventions for this disease, one of the main challenges for decades to come in AD research.
Acknowledgments
The author gratefully acknowledges the assistant of Ashlee E. Lang and Alexandra Nicholson in the preparation of this paper and Figure 1, respectively. Research from this laboratory cited here was supported by NIH Grant RO1 NS39080 and Alzheimer's Association Grant 57869 to A. Ferreira.
References
- 1.Chiu M. J., Chen T. F., Yip P. K., Hua M. S., Tang L. Y. Behavioral and psychologic symptoms in different types of dementia. Journal of the Formosan Medical Association. 2006;105(7):556–562. doi: 10.1016/S0929-6646(09)60150-9. [DOI] [PubMed] [Google Scholar]
- 2.Hebert L. E., Scherr P. A., Bienias J. L., Bennett D. A., Evans D. A. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Archives of Neurology. 2003;60(8):1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 3.Mebane-Sims I. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2009;5:234–270. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Glenner G. G., Wong C. W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 5.Haass C., Selkoe D. J. Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide. Cell. 1993;75(6):1039–1042. doi: 10.1016/0092-8674(93)90312-E. [DOI] [PubMed] [Google Scholar]
- 6.Yankner B. A., Mesulam M. M. β-amyloid and the pathogenesis of Alzheimer's disease. The New England Journal of Medicine. 1991;325(26):1849–1857. doi: 10.1056/NEJM199112263252605. [DOI] [PubMed] [Google Scholar]
- 7.Tam J. H. K., Pasternak S. H. Amyloid and Alzheimer’s disease: inside and out. Canadian Journal of Neurological Sciences. 2012;39:286–289. doi: 10.1017/s0317167100013408. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe D. J. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Annals of the New York Academy of Sciences. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 9.Golde T. E. The Aβ hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathology. 2005;15(1):84–87. doi: 10.1111/j.1750-3639.2005.tb00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinha S., Lieberburg I. Cellular mechanisms of β-amyloid production and secretion. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wisniewski H. M., Narang H. K., Terry R. D. Neurofibrillary tangles of paired helical filaments. Journal of the Neurological Sciences. 1976;27(2):173–181. doi: 10.1016/0022-510X(76)90059-9. [DOI] [PubMed] [Google Scholar]
- 12.Goedert M., Wischik C. M., Crowther R. A., Walker J. E., Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(11):4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3(4):519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 14.Goedert M., Jakes R., Crowther R. A., et al. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(11):5066–5070. doi: 10.1073/pnas.90.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.del C. Alonso A., Grundke-Iqbal I., Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nature Medicine. 1996;2(7):783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 16.Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y.-C., Zaidi M. S., Wisniewski H. M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. The Journal of Biological Chemistry. 1986;261(13):6084–6089. [PubMed] [Google Scholar]
- 17.Grundke-Iqbal I., Vorbrodt A. W., Iqbal K., Tung Y. C., Wang G. P., Wisniewski H. M. Microtubule-associated polypeptides tau are altered in Alzheimer paired helical filaments. Molecular Brain Research. 1988;4(1):43–52. doi: 10.1016/0169-328x(88)90017-4. [DOI] [PubMed] [Google Scholar]
- 18.Grundke-Iqbal I., Iqbal K., Tung Y.-C., Quinlan M., Wisniewski H. M., Binder L. I. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(13):44913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillozet A. L., Weintraub S., Mash D. C., Mesulam M. M. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Archives of Neurology. 2003;60(5):729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 20.Iqbal K., del C. Alonso A., Chen S., et al. Tau pathology in Alzheimer disease and other tauopathies. Biochimica et Biophysica Acta. 2005;1739(2):198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Kosik K. S., Joachim C. L., Selkoe D. J. Microtubule-associated protein tau (τ) is a major antigenic component of paired helical filaments in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(11):4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosik K. S., Greemberg S. M. Tau protein and Alzheimer’s disease. In: Terry R., Katzman R., Bick K. L., editors. Alzheimer’s Disease. New York, NY, USA: Raven Press Ltd; 1994. pp. 335–344. [Google Scholar]
- 23.Braak H., Braak E., Mandelkow E. M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathologica. 1994;87(6):554–567. doi: 10.1007/s004010050124. [DOI] [PubMed] [Google Scholar]
- 24.Lee V. M.-Y., Balin B. J., Otvos L., Trojanowski J. Q. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251(4994):675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 25.Lee V. M.-Y., Goedert M., Trojanowski J. Q. Neurodegenerative tauopathies. Annual Review of Neuroscience. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 26.Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42(3):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 27.Rapoport M., Dawson H. N., Binder L. I., Vitek M. P., Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(9):6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberson E. D., Scearce-Levie K., Palop J. J., et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 29.Spillantini M. G., Goedert M. Tau protein pathology in neurodegenerative diseases. Trends in Neurosciences. 1998;21(10):428–433. doi: 10.1016/S0166-2236(98)01337-X. [DOI] [PubMed] [Google Scholar]
- 30.Wood J. G., Mirra S. S., Pollock N. J., Binder L. I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (τ) Proceedings of the National Academy of Sciences of the United States of America. 1986;83(11):4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 32.Hyman B. T., Augustinack J. C., Ingelsson M. Transcriptional and conformational changes of the tau molecule in Alzheimer's disease. Biochimica et Biophysica Acta. 2005;1739(2):150–157. doi: 10.1016/j.bbadis.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 33.Geschwind D. H. Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron. 2003;40(3):457–460. doi: 10.1016/S0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- 34.Ballatore C., Lee V. M. Y., Trojanowski J. Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nature Reviews Neuroscience. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 35.Morris M., Maeda S., Vossel K., Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Novak P., Prcina M., Kontsekova E. Tauons and prions: infamous cousins? Alzheimer's Disease. 2011;26:413–430. doi: 10.3233/JAD-2011-110194. [DOI] [PubMed] [Google Scholar]
- 37.Calignon A., Polydoro M., Suarez-Calvet M., et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L., Drouet V., Wu J. W., et al. Trans-synaptic spread of tau pathology in vivo. PlosOne. 2012;7 doi: 10.1371/journal.pone.0031302.e31303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terry R. D., Masliah E., Salmon D. P., et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 40.DeKosky S. T., Scheff S. W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Annals of Neurology. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 41.Tiraboschi P., Hansen L. A., Alford M., Masliah E., Thal L. J., Corey-Bloom J. The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease. Neurology. 2000;55(9):1278–1283. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 42.Selkoe D. J. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 43.Yao P. J. Synaptic frailty and clathrin-mediated synaptic vesicle trafficking in Alzheimer's disease. Trends in Neurosciences. 2004;27(1):24–29. doi: 10.1016/j.tins.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Hamos J. E., DeGennaro L. J., Drachman D. A. Synaptic loss in Alzheimer's disease and other dementias. Neurology. 1989;39(3):355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 45.Marcello E., Epis R., Saraceno C., Di Luca M. Synaptic dysfunction in Alzheimer’s disease. In: Kreutz M. R., Sala C., editors. Synaptic Plasticity. Weinheim, Germany: Springer; 2012. pp. 573–601. [DOI] [PubMed] [Google Scholar]
- 46.Bertoni-Freddari C., Fattoretti P., Pieroni M., Meier-Ruge W., Ulrich J. Enlargement of synaptic size as a compensative reaction in aging and dementia. Pathology Research and Practice. 1992;188(4-5):612–615. doi: 10.1016/S0344-0338(11)80066-X. [DOI] [PubMed] [Google Scholar]
- 47.Gastard M. C., Troncoso J. C., Koliatsos V. E. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Annals of Neurology. 2003;54(3):393–398. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- 48.Honer W. G. Pathology of presynaptic proteins in Alzheimer's disease: more than simple loss of terminals. Neurobiology of Aging. 2003;24(8):1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Hsiao K., Chapman P., Nilsen S., et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 50.King D. L., Arendash G. W. Maintained synaptophysin immunoreactivity in Tg2576 transgenic mice during aging: correlations with cognitive impairment. Brain Research. 2002;926(1-2):58–68. doi: 10.1016/S0006-8993(01)03294-2. [DOI] [PubMed] [Google Scholar]
- 51.Goll D. E., Thompson V. F., Li H., Wei W., Cong J. The calpain system. Physiological Reviews. 2003;83(3):731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 52.Ono Y., Sorimachi H. Calpains- an elaborate proteolytic system. Biochimica et Biophysica Acta. 2012;1824:224–236. doi: 10.1016/j.bbapap.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Vosler P. S., Brennan C. S., Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Molecular Neurobiology. 2008;38(1):78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nixon R. A. The calpains in aging and aging-related diseases. Ageing Research Reviews. 2003;2(4):407–418. doi: 10.1016/S1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki K., Sorimachi H. A novel aspect of calpain activation. FEBS Letters. 1998;433(1-2):1–4. doi: 10.1016/S0014-5793(98)00856-4. [DOI] [PubMed] [Google Scholar]
- 56.Moldoveanu T., Hosfield C. M., Lim D., Elce J. S., Jia Z., Davies P. L. A Ca2+ switch aligns the active site of calpain. Cell. 2002;108(5):649–660. doi: 10.1016/S0092-8674(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 57.Hosfield C. M., Elce J. S., Davies P. L., Jia Z. Crystal structure of calpain reveals the structural basis for Ca2+-dependent protease activity and a novel mode of enzyme activation. EMBO Journal. 1999;18(24):6880–6889. doi: 10.1093/emboj/18.24.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strobl S., Fernandez-Catalan C., Braun M., et al. The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(2):588–592. doi: 10.1073/pnas.97.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tompa P., Emori Y., Sorimachi H., Suzuki K., Friedrich P. Domain III of calpain is a Ca2+-regulated phospholipid-binding domain. Biochemical and Biophysical Research Communications. 2001;280(5):1333–1339. doi: 10.1006/bbrc.2001.4279. [DOI] [PubMed] [Google Scholar]
- 60.Sorimachi H., Suzuki K. The structure of calpain. Journal of Biochemistry. 2001;129(5):653–664. doi: 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- 61.Carafoli E., Molinari M. Calpain: a protease in search of a function? Biochemical and Biophysical Research Communications. 1998;247(2):193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- 62.Hamakubo T., Kannagi R., Murachi T., Matus A. Distribution of calpains I and II in rat brain. Journal of Neuroscience. 1986;6(11):3103–3111. doi: 10.1523/JNEUROSCI.06-11-03103.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perlmutter L. S., Siman R., Gall C., Seubert P., Baudry M., Lynch G. The ultrastructural localization of calcium-activated protease 'calpain' in rat brain. Synapse. 1988;2(1):79–88. doi: 10.1002/syn.890020111. [DOI] [PubMed] [Google Scholar]
- 64.Murachi T., Hatanaka M., Hamakubo T. Calpains and neuropeptide metabolism. In: Chichester T. E., editor. Neuropeptides and Their Peptidases. Chichester, UK: Ellis Horwood; 1987. pp. 202–228,. [Google Scholar]
- 65.Croall D. E., Slaughter C. A., Wortham H. S., Skelly C. M., DeOgny L., Moomaw C. R. Polyclonal antisera specific for the proenzyme form of each calpain. Biochimica et Biophysica Acta. 1992;1121(1-2):47–53. doi: 10.1016/0167-4838(92)90335-B. [DOI] [PubMed] [Google Scholar]
- 66.Fischer S., Vandekerckhove J., Ampe C., Traub P., Weber K. Protein-chemical identification of the major cleavage sites of the Ca2+ proteinase on murine vimentin, the mesenchymal intermediate filament protein. Biological Chemistry Hoppe-Seyler. 1986;367(11):1147–1152. doi: 10.1515/bchm3.1986.367.2.1147. [DOI] [PubMed] [Google Scholar]
- 67.Harris A. S., Croall D. E., Morrow J. S. The calmodulin-binding site in α-fodrin is near the calcium-dependent protease-I cleavage site. The Journal of Biological Chemistry. 1988;263(30):15754–15761. [PubMed] [Google Scholar]
- 68.Stabach P. R., Cianci C. D., Glantz S. B., Zhang Z., Morrow J. S. Site-directed mutagenesis of αII spectrin at codon 1175 modulates its μ-calpain susceptibility. Biochemistry. 1997;36(1):57–65. doi: 10.1021/bi962034i. [DOI] [PubMed] [Google Scholar]
- 69.Coolican S. A., Hathaway D. R. Effect of L-α-phosphatidylinositol on a vascular smooth muscle Ca2+-dependent protease. Reduction of the Ca2+ requirement for autolysis. The Journal of Biological Chemistry. 1984;259(19):11627–11630. [PubMed] [Google Scholar]
- 70.Saido T. C., Mizuno K., Suzuki K. Proteolysis of protein kinase C by calpain: effect of acidic phospholipids. Biomedica Biochimica Acta. 1991;50(4–6):485–489. [PubMed] [Google Scholar]
- 71.Saido T. C., Shibata M., Takenawa T., Murofushi H., Suzuki K. Positive regulation of μ-calpain action by polyphosphoinositides. The Journal of Biological Chemistry. 1992;267(34):24585–24590. [PubMed] [Google Scholar]
- 72.Kiss R., Kovács D., Tompa P., Perczel A. Local structural preferences of calpastatin, the intrinsically unstructured protein inhibitor of calpain. Biochemistry. 2008;47(26):6936–6945. doi: 10.1021/bi800201a. [DOI] [PubMed] [Google Scholar]
- 73.Thompson V. F., Goll D. E. Purification of m-calpain, m-calpain, and calpastatin from animal tissues. In: Elce J. S., Totowa N. J., editors. Methods in Molecular Biology. Calpain Methods and Protocols. Vol. 144. Humana Press; 2000. pp. 3–6. [PubMed] [Google Scholar]
- 74.Rao M. V., Mohan P. S., Peterhoff C. M., et al. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. Journal of Neuroscience. 2008;28(47):12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Emori Y., Kawasaki H., Imajoh S., Imahori K., Suzuki K. Endogenous inhibitor for calcium-dependent cysteine protease contains four internal repeats that could be responsible for its multiple reactive sites. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(11):3590–3594. doi: 10.1073/pnas.84.11.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maki M., Takano E., Mori H., Kannagi R., Murachi T., Hatanaka M. Repetitive region of calpastatin is a functional unit of the proteinase inhibitor. Biochemical and Biophysical Research Communications. 1987;143(1):300–308. doi: 10.1016/0006-291x(87)90665-6. [DOI] [PubMed] [Google Scholar]
- 77.Maki M., Takano E., Mori H., Sato A., Murachi T., Hatanaka M. All four internally repetitive domains of pig calpastatin possess inhibitory activities against calpains I and II. FEBS Letters. 1987;223(1):174–180. doi: 10.1016/0014-5793(87)80531-8. [DOI] [PubMed] [Google Scholar]
- 78.Barnoy S., Maki M., Kosower N. S. Overexpression of calpastatin inhibits L8 myoblast fusion. Biochemical and Biophysical Research Communications. 2005;332(3):697–701. doi: 10.1016/j.bbrc.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 79.Higuchi M., Tomioka M., Takano J., et al. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. The Journal of Biological Chemistry. 2005;280(15):15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- 80.Maekawa A., Lee J. K., Nagaya T., et al. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. Journal of Molecular and Cellular Cardiology. 2003;35(10):1277–1284. doi: 10.1016/S0022-2828(03)00238-4. [DOI] [PubMed] [Google Scholar]
- 81.Glading A., Chang P., Lauffenburger D. A., Wells A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. The Journal of Biological Chemistry. 2000;275(4):2390–2398. doi: 10.1074/jbc.275.4.2390. [DOI] [PubMed] [Google Scholar]
- 82.Shiraha H., Glading A., Chou J., Jia Z., Wells A. Activation of m-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-calpain. Molecular and Cellular Biology. 2002;22(8):2716–2727. doi: 10.1128/MCB.22.8.2716-2727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith S. D., Jia Z., Huynh K. K., Wells A., Elce J. S. Glutamate substitutions at a PKA consensus site are consistent with inactivation of calpain by phosphorylation. FEBS Letters. 2003;542(1–3):115–118. doi: 10.1016/S0014-5793(03)00361-2. [DOI] [PubMed] [Google Scholar]
- 84.Averna M., De Tullio R., Salamino F., Minafra R., Pontremoli S., Melloni E. Age-dependent degradation of calpastatin in kidney of hypertensive rats. The Journal of Biological Chemistry. 2001;276(42):38426–38432. doi: 10.1074/jbc.M101936200. [DOI] [PubMed] [Google Scholar]
- 85.Benuck M., Banay-Schwartz M., DeGuzman T., Lajtha A. Changes in brain protease activity in aging. Journal of Neurochemistry. 1996;67(5):2019–2029. doi: 10.1046/j.1471-4159.1996.67052019.x. [DOI] [PubMed] [Google Scholar]
- 86.Manya H., Inomata M., Fujimori T., et al. Klotho protein deficiency leads to overactivation of μ-calpain. The Journal of Biological Chemistry. 2002;277(38):35503–35508. doi: 10.1074/jbc.M206033200. [DOI] [PubMed] [Google Scholar]
- 87.Sloane J. A., Hinman J. D., Lubonia M., Hollander W., Abraham C. R. Age-dependent myelin degeneration and proteolysis of oligodendrocyte proteins is associated with the activation of calpain-1 in the rhesus monkey. Journal of Neurochemistry. 2003;84(1):157–168. doi: 10.1046/j.1471-4159.2003.01541.x. [DOI] [PubMed] [Google Scholar]
- 88.Saito K. I., Elce J. S., Hamos J. E., Nixon R. A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(7):2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saitoh T., Masliah E., Jin L. W., Cole G. M., Wieloch T., Shapiro I. P. Protein kinases and phosphorylation in neurologic disorders and cell death. Laboratory Investigation. 1991;64(5):596–616. [PubMed] [Google Scholar]
- 90.Ferreira A., Bigio E. H. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Molecular Medicine. 2011;17(7-8):676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peterson C., Goldman J. E. Alterations in calcium content and biochemical processes in cultured skin fibroblasts from aged and Alzheimer donors. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(8):2758–2762. doi: 10.1073/pnas.83.8.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karlsson J. O., Blennow K., Holmberg B., et al. Increased proteolytic activity in erythrocytes from patients with Alzheimer's disease. Dementia. 1992;3(4):200–204. [Google Scholar]
- 93.Patrick G. N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 94.Tsuji T., Shimohama S., Kimura J., Shimizu K. M-calpain (calcium-activated neutral proteinase) in alzheimer's disease brains. Neuroscience Letters. 1998;248(2):109–112. doi: 10.1016/S0304-3940(98)00348-6. [DOI] [PubMed] [Google Scholar]
- 95.Kelly B. L., Vassar R., Ferreira A. β-amyloid-induced dynamin 1 depletion in hippocampal neurons: a potential mechanism for early cognitive decline in Alzheimer disease. The Journal of Biological Chemistry. 2005;280(36):31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kelly B. L., Ferreira A. β-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. The Journal of Biological Chemistry. 2006;281(38):28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 97.Kelly B. L., Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;147(1):60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abdel-Hamid K. M., Baimbridge K. G. The effects of artificial calcium buffers on calcium responses and glutamate-mediated excitotoxicity in cultured hippocampal neurons. Neuroscience. 1997;81(3):673–687. doi: 10.1016/S0306-4522(97)00162-0. [DOI] [PubMed] [Google Scholar]
- 99.Dodart J. C., Bales K. R., Gannon K. S., et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nature Neuroscience. 2002;5(5):452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 100.Rovira C., Arbez N., Mariani J. Aβ(25–35) and Aβ(1–40) act on different calcium channels in CA1 hippocampal neurons. Biochemical and Biophysical Research Communications. 2002;296(5):1317–1321. doi: 10.1016/S0006-291X(02)02072-7. [DOI] [PubMed] [Google Scholar]
- 101.Qiu Z., Gruol D. L. Interleukin-6, β-amyloid peptide and NMDA interactions in rat cortical neurons. Journal of Neuroimmunology. 2003;139(1-2):51–57. doi: 10.1016/S0165-5728(03)00158-9. [DOI] [PubMed] [Google Scholar]
- 102.Huettner J. E., Bean B. P. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(4):1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bullock R. Efficacy and safety of memantine in moderate-to-severe Alzheimer disease: the evidence to date. Alzheimer Disease and Associated Disorders. 2006;20(1):23–29. doi: 10.1097/01.wad.0000201847.29836.a5. [DOI] [PubMed] [Google Scholar]
- 104.Fanelli R. J., McCarthy R. T., Chisholm J. Neuropharmacology of nimodipine: from single channels to behavior. Annals of the New York Academy of Sciences. 1994;747:336–350. doi: 10.1111/j.1749-6632.1994.tb44421.x. [DOI] [PubMed] [Google Scholar]
- 105.Nicholson A. M., Ferreira A. Cholesterol and neuronal susceptibility to beta-amyloid toxicity. Trends in Cognitive Sciences. 2011;5:35–56. [PMC free article] [PubMed] [Google Scholar]
- 106.Kivipelto M., Helkala E. L., Hänninen T., et al. Midlife vascular risk factors and late-life mild cognitive impairment: a population-based study. Neurology. 2001;56(12):1683–1689. doi: 10.1212/wnl.56.12.1683. [DOI] [PubMed] [Google Scholar]
- 107.Solomon A., Kivipelto M., Wolozin B., Zhou J., Whitmer R. A. Midlife serum cholesterol and increased risk of Alzheimer's and vascular dementia three decades later. Dementia and Geriatric Cognitive Disorders. 2009;28(1):75–80. doi: 10.1159/000231980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bastiaanse E. M. L., Atsma L. D. E., Kuijpers M. M. C., Van der Laarse A. The effect of sarcolemmal cholesterol content on intracellular calcium ion concentration in cultured cardiomyocytes. Archives of Biochemistry and Biophysics. 1994;313(1):58–63. doi: 10.1006/abbi.1994.1358. [DOI] [PubMed] [Google Scholar]
- 109.Hartmann H., Eckert A., Muller W. E. Apolipoprotein E and cholesterol affect neuronal calcium signalling: the possible relationship to β-amyloid neurotoxicity. Biochemical and Biophysical Research Communications. 1994;200(3):1185–1192. doi: 10.1006/bbrc.1994.1576. [DOI] [PubMed] [Google Scholar]
- 110.Kawahara M., Kuroda Y. Intracellular calcium changes in neuronal cells induced by Alzheimer's β-amyloid protein are blocked by estradiol and cholesterol. Cellular and Molecular Neurobiology. 2001;21(1):1–13. doi: 10.1023/A:1007168910582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nicholson A. M., Ferreira A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to β-Amyloid-induced calpain activation and tau toxicity. Journal of Neuroscience. 2009;29(14):4640–4651. doi: 10.1523/JNEUROSCI.0862-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nicholson A. M., Methner D. N. R., Ferreira A. Membrane cholesterol modulates β-amyloid-dependent Tau cleavage by inducing changes in the membrane content and localization of N-methyl-D-aspartic acid receptors. The Journal of Biological Chemistry. 2011;286(2):976–986. doi: 10.1074/jbc.M110.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vaisid T., Kosower N. S., Katzav A., Chapman J., Barnoy S. Calpastatin levels affect calpain activation and calpain proteolytic activity in APP transgenic mouse model of Alzheimer's disease. Neurochemistry International. 2007;51(6-7):391–397. doi: 10.1016/j.neuint.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 114.Blomgren K., Hallin U., Andersson A. L., et al. Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. The Journal of Biological Chemistry. 1999;274(20):14046–14052. doi: 10.1074/jbc.274.20.14046. [DOI] [PubMed] [Google Scholar]
- 115.Mellgren R. L., Mericle M. T., Lane R. D. Proteolysis of the calcium-dependent protease inhibitor by myocardial calcium-dependent protease. Archives of Biochemistry and Biophysics. 1986;246(1):233–239. doi: 10.1016/0003-9861(86)90468-6. [DOI] [PubMed] [Google Scholar]
- 116.Adamec E., Mohan P., Vonsattel J. P., Nixon R. A. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathologica. 2002;104(1):92–104. doi: 10.1007/s00401-002-0528-6. [DOI] [PubMed] [Google Scholar]
- 117.Grynspan F., Griffin W. R., Cataldo A., Katayama S., Nixon R. A. Active site-directed antibodies identify calpain II as an early- appearing and pervasive component of neurofibrillary pathology in Alzheimer's disease. Brain Research. 1997;763(2):145–158. doi: 10.1016/S0006-8993(97)00384-3. [DOI] [PubMed] [Google Scholar]
- 118.Shimohama S., Suenaga T., Araki W., Yamaoaka Y., Shimizu K., Kimura J. Presence of calpain II immunoreactivity in senile plaques in Alzheimer's disease. Brain Research. 1991;558(1):105–108. doi: 10.1016/0006-8993(91)90722-8. [DOI] [PubMed] [Google Scholar]
- 119.Sisodia S. S., Kim S. H., Thinakaran G. Function and dysfunction of the presenilins. American Journal of Human Genetics. 1999;65(1):7–12. doi: 10.1086/302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vassar R., Bennett B. D., Babu-Khan S., et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 121.Edbauer D., Winkler E., Regula J. T., Pesold B., Steiner H., Haass C. Reconstitution of γ-secretase activity. Nature Cell Biology. 2003;5(5):486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 122.Jarrett J. T., Berger E. P., Lansbury P. T. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 123.Citron M., Oltersdorf T., Haass C., et al. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature. 1992;360(6405):672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 124.Citron M., Westaway D., Xia W., et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature Medicine. 1997;3(1):67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 125.Cai X. D., Golde T. E., Younkin S. G. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259(5094):514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki N., Cheung T. T., Cai X. D., et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 127.Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Archives of Neurology. 2002;59(9):1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 128.Yang L. B., Lindholm K., Yan R., et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Medicine. 2003;9(1):3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 129.Li Q., Südhof T. C. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. The Journal of Biological Chemistry. 2004;279(11):10542–10550. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- 130.Liang B., Duan B.-Y., Zhou X.-P., Gong J.-X., Luo Z.-G. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of alzheimer disease. The Journal of Biological Chemistry. 2010;285(36):27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Higuchi M., Iwata N., Matsuba Y., et al. Mechanistic involvement of the calpain-calpastatin system in Alzheimer neuropathology. The FASEB Journal. 2012;26:1204–1217. doi: 10.1096/fj.11-187740. [DOI] [PubMed] [Google Scholar]
- 132.Biernat J., Gustke N., Drewes G., Mandelkow E. M., Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11(1):153–163. doi: 10.1016/0896-6273(93)90279-Z. [DOI] [PubMed] [Google Scholar]
- 133.Biernat J., Mandelkow E.-M., Schroter C., et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO Journal. 1992;11(4):1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bramblett G. T., Goedert M., Jakes R., Merrick S. E., Trojanowski J. Q., Lee V. M.-Y. Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10(6):1089–1099. doi: 10.1016/0896-6273(93)90057-X. [DOI] [PubMed] [Google Scholar]
- 135.Brion J. P., Smith C., Couck A. M., Gallo J. M., Anderton B. H. Developmental changes in τ phosphorylation: fetal τ is transiently phosphorylated in a manner similar to paired helical filament-τ characteristic of Alzheimer's disease. Journal of Neurochemistry. 1993;61(6):2071–2080. doi: 10.1111/j.1471-4159.1993.tb07444.x. [DOI] [PubMed] [Google Scholar]
- 136.Rapoport M., Ferreira A. PD98059 prevents neurite degeneration induced by fibrillar β-amyloid in mature hippocampal neurons. Journal of Neurochemistry. 2000;74(1):125–133. doi: 10.1046/j.1471-4159.2000.0740125.x. [DOI] [PubMed] [Google Scholar]
- 137.Busciglio J., Lorenzo A., Yeh J., Yankner B. A. β-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14(4):879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 138.Ferreira A., Lu Q., Orecchio L., Kosik K. S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar Aβ . Molecular and Cellular Neurosciences. 1997;9(3):220–234. doi: 10.1006/mcne.1997.0615. [DOI] [PubMed] [Google Scholar]
- 139.Shea T. B., Deargay A. N., Ekinci F. J. Beta-amyloid induced hyperphosphorylation of tau in human neuroblastoma cells involves MAP kinase. Neuroscience Research Communications. 1998;22:45–49. [Google Scholar]
- 140.Pérez M., Hernández F., Gómez-Ramos A., Smith M., Perry G., Avila J. Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. European Journal of Biochemistry. 2002;269(5):1484–1489. doi: 10.1046/j.1432-1033.2002.02794.x. [DOI] [PubMed] [Google Scholar]
- 141.Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D. J. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Martin L., Latypova X., Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochemistry International. 2011;58(4):458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 143.Takashima A., Noguchi K., Sato K., Hoshino T., Imahori K. Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(16):7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ekinci F. J., Malik K. U., Shea T. B. Activation of the L voltage-sensitive calcium channel by mitogen- activated protein (MAP) kinase following exposure of neuronal cells to β- amyloid. MAP kinase mediates β-amyloid-induced neurodegeneration. The Journal of Biological Chemistry. 1999;274(42):30322–30327. doi: 10.1074/jbc.274.42.30322. [DOI] [PubMed] [Google Scholar]
- 145.Alvarez A., Toro R., Cáceres A., Maccioni R. B. Inhibition of tau phosphorylating protein kinase cdk5 prevents β-amyloid-induced neuronal death. FEBS Letters. 1999;459(3):421–426. doi: 10.1016/S0014-5793(99)01279-X. [DOI] [PubMed] [Google Scholar]
- 146.Goñi-Oliver P., Lucas J. J., Avila J., Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. The Journal of Biological Chemistry. 2007;282(31):22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- 147.Kusakawa G. I., Saito T., Onuki R., Ishiguro K., Kishimoto T., Hisanaga S. I. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. The Journal of Biological Chemistry. 2000;275(22):17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 148.Lee M.-S., Kwon Y. T., Li M., Peng J., Friedlander R. M., Tsai L.-H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405(6784):360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 149.Nath R., Davis M., Probert A. W., et al. Processing of cdk5 activator p35 to its truncated form (p25) by Calpain in acutely injured neuronal cells. Biochemical and Biophysical Research Communications. 2000;274:16–21. doi: 10.1006/bbrc.2000.3070. [DOI] [PubMed] [Google Scholar]
- 150.Veeranna, Kaji T., Boland B., et al. Calpain mediates calcium-induced activation of the Erk 1,2 MARK pathway and cytoskeletal phosphorylation in neurons: relevance to alzheimer's disease. American Journal of Pathology. 2004;165(3):795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guillozet-Bongaarts A. L., Garcia-Sierra F., Reynolds M. R., et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiology of Aging. 2005;26(7):1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 152.Johnson G. V. W., Jope R. S., Binder L. I. Proteolysis of tau by calpain. Biochemical and Biophysical Research Communications. 1989;163(3):1505–1511. doi: 10.1016/0006-291x(89)91150-9. [DOI] [PubMed] [Google Scholar]
- 153.Mercken M., Grynspan F., Nixon R. A. Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Letters. 1995;368(1):10–14. doi: 10.1016/0014-5793(95)00590-6. [DOI] [PubMed] [Google Scholar]
- 154.Yang L.-S., Ksiezak-Reding H. Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. European Journal of Biochemistry. 1995;233(1):9–17. doi: 10.1111/j.1432-1033.1995.009_1.x. [DOI] [PubMed] [Google Scholar]
- 155.Yang L. S., Gordon-Krajcer W., Ksiezak-Reding H. Tau released from paired helical filaments with formic acid or guanidine is susceptible to calpain-mediated proteolysis. Journal of Neurochemistry. 1997;69(4):1548–1558. doi: 10.1046/j.1471-4159.1997.69041548.x. [DOI] [PubMed] [Google Scholar]
- 156.Liu M. C., Kobeissy F., Zheng W., Zhang Z., Hayes R. L., Wang K. K. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro. 2011;3(1, article e00051) doi: 10.1042/AN20100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reifert J., Hartung-Cranston D., Feinstein S. C. Amyloid β-mediated cell death of cultured hippocampal neurons reveals extensive tau fragmentation without increased full-length tau phosphorylation. The Journal of Biological Chemistry. 2011;286(23):20797–20811. doi: 10.1074/jbc.M111.234674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Amadoro G., Corsetti V., Stringaro A., et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. Journal of Alzheimer's Disease. 2010;21(2):445–470. doi: 10.3233/JAD-2010-100120. [DOI] [PubMed] [Google Scholar]
- 159.Atlante A., Amadoro G., Bobba A., et al. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochimica et Biophysica Acta. 2008;1777(10):1289–1300. doi: 10.1016/j.bbabio.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 160.Park S.-Y., Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. Journal of Neuroscience. 2005;25(22):5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Park S.-Y., Tournell C. E., Sinjoanu R. C., Ferreira A. Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience. 2007;144(1):119–127. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Amadoro G., Ciotti M. T., Costanzi M., Cestari V., Calissano P., Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2892–2897. doi: 10.1073/pnas.0511065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reinecke J. B., DeVos S. L., McGrath J. P., et al. Implicating calpain in tau-mediated toxicity in vivo. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023865.e23865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Garg S., Timm T., Mandelkow E. M., Mandelkow E., Wang Y. Cleavage of tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment. Neurobiology of Aging. 2011;32(1):1–14. doi: 10.1016/j.neurobiolaging.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 165.Yao P. J., Zhu M., Pyun E. I., et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiology of Disease. 2003;12(2):97–109. doi: 10.1016/S0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 166.Liu J., Liu M. C., Wang K. K. Calpain in the CNS: from synaptic function to neurotoxicity. Science Signaling. 2008;1(14, article re1) doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- 167.Clark S. G., Shurland D. L., Meyerowitz E. M., Bargmann C. I., van der Bliek A. M. A dynamin GTPase mutation causes a rapid and reversible temperature-inducible locomotion defect in C. elegans . Proceedings of the National Academy of Sciences of the United States of America. 1997;94(19):10438–10443. doi: 10.1073/pnas.94.19.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Damke H., Baba T., Warnock D. E., Schmid S. L. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. Journal of Cell Biology. 1994;127(4):915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Koenig J. H., Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. Journal of Neuroscience. 1989;9(11):3844–3860. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.van der Bliek A. M., Meyerowitz E. M. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351(6325):411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- 171.Sinjoanu R. C., Kleinschmidt S., Bitner R. S., Brioni J. D., Moeller A., Ferreira A. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochemistry International. 2008;53(3-4):79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bi R., Bi X., Baudry M. Phosphorylation regulates calpain-mediated truncation of glutamate ionotropic receptors. Brain Research. 1998;797(1):154–158. doi: 10.1016/S0006-8993(98)00433-8. [DOI] [PubMed] [Google Scholar]
- 173.Bi X., Rong Y., Chen J., Dang S., Wang Z., Baudry M. Calpain-mediated regulation of NMDA receptor structure and function. Brain Research. 1998;790(1-2):245–253. doi: 10.1016/S0006-8993(98)00067-5. [DOI] [PubMed] [Google Scholar]
- 174.Lu X., Rong Y., Baudry M. Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neuroscience Letters. 2000;286(2):149–153. doi: 10.1016/S0304-3940(00)01101-0. [DOI] [PubMed] [Google Scholar]
- 175.Vinade L., Petersen J. D., Do K., Dosemeci A., Reese T. S. Activation of calpain may alter the postsynaptic density structure and modulate anchoring of NMDA receptors. Synapse. 2001;40(4):302–309. doi: 10.1002/syn.1053. [DOI] [PubMed] [Google Scholar]
- 176.Wu H. Y., Hsu F. C., Gleichman A. J., Baconguis I., Coulter D. A., Lynch D. R. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. The Journal of Biological Chemistry. 2007;282(28):20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xu W., Wong T. P., Chery N., Gaertner T., Wang Y. T., Baudry M. Calpain-mediated mGluR1α truncation: a key step in excitotoxicity. Neuron. 2007;53(3):399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 178.Calò L., Bruno V., Spinsanti P., et al. Interactions between ephrin-B and metabotropic glutamate 1 receptors in brain tissue and cultured neurons. Journal of Neuroscience. 2005;25(9):2245–2254. doi: 10.1523/JNEUROSCI.4956-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Barco A., Alarcon J. M., Kandel E. R. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108(5):689–703. doi: 10.1016/S0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 180.Liang Z., Liu F., Grundke-Iqbal I., Iqbal K., Gong C. X. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. Journal of Neurochemistry. 2007;103(6):2462–2470. doi: 10.1111/j.1471-4159.2007.04942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Arthur J. S. C., Elce J. S., Hegadorn C., Williams K., Greer P. A. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Molecular and Cellular Biology. 2000;20(12):4474–4481. doi: 10.1128/MCB.20.12.4474-4481.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Taniguchi K., Umeshita K., Sakon M., et al. Suppression of oxidative stress-induced hepatocyte injury by calpain antisense. Journal of Surgical Research. 2003;111(1):23–27. doi: 10.1016/S0022-4804(03)00047-7. [DOI] [PubMed] [Google Scholar]
- 183.Di Rosa G., Odrljin T., Nixon R. A., Arancio O. Calpain inhibitors: a treatment for alzheimer's disease. Journal of Molecular Neuroscience. 2002;19(1-2):135–141. doi: 10.1007/s12031-002-0024-4. [DOI] [PubMed] [Google Scholar]
- 184.Brorson J. R., Marcuccilli C. J., Miller R. J. Delayed antagonism of calpain reduces excitotoxicity in cultured neurons. Stroke. 1995;26(7):1259–1267. doi: 10.1161/01.str.26.7.1259. [DOI] [PubMed] [Google Scholar]
- 185.McCollum A. T., Jafarifar F., Lynn B. C., et al. Inhibition of calpain-mediated cell death by a novel peptide inhibitor. Experimental Neurology. 2006;202(2):506–513. doi: 10.1016/j.expneurol.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 186.Battaglia F., Trinchese F., Liu S., Walter S., Nixon R. A., Arancio O. Calpain inhibitors, a treatment for Alzheimer's disease: position paper. Journal of Molecular Neuroscience. 2003;20(3):357–362. doi: 10.1385/JMN:20:3:357. [DOI] [PubMed] [Google Scholar]
- 187.Kunz S., Niederberger E., Ehnert C., et al. The calpain inhibitor MDL 28170 prevents inflammation-induced neurofilament light chain breakdown in the spinal cord and reduces thermal hyperalgesia. Pain. 2004;110(1-2):409–418. doi: 10.1016/j.pain.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 188.Li P. A., Howlett W., He Q. P., Miyashita H., Siddiqui M., Shuaib A. Postischemic treatment with calpain inhibitor MDL 28170 ameliorates brain damage in a gerbil model of global ischemia. Neuroscience Letters. 1998;247(1):17–20. doi: 10.1016/S0304-3940(98)00266-3. [DOI] [PubMed] [Google Scholar]
- 189.Lubisch W., Beckenbach E., Bopp S., et al. Benzoylalanine-derived ketoamides carrying vinylbenzyl amino residues: discovery of potent water-soluble calpain inhibitors with oral bioavailability. Journal of Medicinal Chemistry. 2003;46(12):2404–2412. doi: 10.1021/jm0210717. [DOI] [PubMed] [Google Scholar]
- 190.Granic I., Nyakas C., Luiten P. G. M., et al. Calpain inhibition prevents amyloid-β-induced neurodegeneration and associated behavioral dysfunction in rats. Neuropharmacology. 2010;59(4-5):334–342. doi: 10.1016/j.neuropharm.2010.07.013. [DOI] [PubMed] [Google Scholar]