

Abstract
Background
Opitz G/BBB syndrome is a heterogeneous disorder characterised by variable expression of midline defects including cleft lip and palate, hypertelorism, laryngealtracheoesophageal anomalies, congenital heart defects, and hypospadias. The X-linked form of the condition has been associated with mutations in the MID1 gene on Xp22. The autosomal dominant form has been linked to chromosome 22q11.2, although the causative gene has yet to be elucidated.
Methods and results
In this study, we performed whole exome sequencing on DNA samples from a three-generation family with characteristics of Opitz G/BBB syndrome with negative MID1 sequencing. We identified a heterozygous missense mutation c.1189A>C (p.Thr397Pro) in SPECC1L, located at chromosome 22q11.23. Mutation screening of an additional 19 patients with features of autosomal dominant Opitz G/BBB syndrome identified a c.3247G>A ( p.Gly1083Ser) mutation segregating with the phenotype in another three-generation family.
Conclusions
Previously, SPECC1L was shown to be required for proper facial morphogenesis with disruptions identified in two patients with oblique facial clefts. Collectively, these data demonstrate that SPECC1L mutations can cause syndromic forms of facial clefting including some cases of autosomal dominant Opitz G/BBB syndrome and support the original linkage to chromosome 22q11.2.
INTRODUCTION
Opitz G/BBB syndrome is a genetically heterogeneous, multiple congenital anomalies syndrome diagnosed on the presence of characteristic clinical features. Opitz originally described two separate syndromes, the BBB syndrome and the G syndrome, which were characterised by hypertelorism, hypospadias and variable other midline defects. Due to the clinical overlap, these two syndromes were later combined into one entity, Opitz G/BBB syndrome or simply Opitz syndrome.1
Opitz syndrome is inherited in either an autosomal dominant or X linked pattern with multiple reported families showing male-to-male transmission.2–7 Linkage analysis of 10 families identified one locus on Xp22 and a second locus on 22q11.2.8 Five families were linked to D22S345 on chromosome 22q11.2 with a LOD score of 3.53 at zero recombination. Crossover events for markers D22S421 and D22S685 placed the Opitz syndrome gene within the 32 cM interval at chromosome 22q11.2, bordered distally by D22S685 and proximally by D22S421.8 The X linked form of Opitz is associated with mutations in the MID1 gene at chromosome Xp22.2 which encodes a microtubule-associated RING B-box coiled-coil domain protein.9
Opitz syndrome is a clinically heterogeneous disorder with variable expression in both the X linked and autosomal dominant families, and characterised by distinctive facial features including hypertelorism, a prominent forehead, broad nasal bridge and anteverted nares. Congenital anomalies include hypospadias, cleft lip/palate, laryngealtracheoesophageal abnormalities, imperforate anus and cardiac defects. Developmental delay and intellectual disability are variable. Hypospadias and anal anomalies were found more commonly in male patients with MID1 mutations than in those without.10,11
Using whole exome sequencing (WES), we identified a missense mutation in SPECC1L segregating with the phenotype of suspected autosomal dominant Opitz in a three-generation pedigree (see figure 1A). Subsequently, we sequenced the gene in an additional 19 probands and identified a second family with a novel missense mutation in SPECC1L and clinical features of Opitz.5 This second family also had a three-generational pedigree with the SPECC1L mutation segregating with the distinguishing phenotype (see figure 1B). This study provides further evidence that Opitz is a genetically heterogeneous syndrome and that SPECC1L mutations account for a subset of the autosomal dominant cases.
Figure 1.

(A) Pedigree of Family A. (B) Pedigree of Family B. (C) A schematic of SPECC1L protein showing that the T397P mutation lies in the same coiled-coil domain (CCD) as the previously reported Q415P mutation and that the G1083S mutation lies in C-terminal calponin homology domain (CHD). (D) DNA analysis. Trace from proband, AIII.2 and (E) trace from proband BIII.5.
PATIENTS AND METHODS
Patients
Family A
Family A presented to genetics at the Children’s Hospital of Philadelphia after the birth of their second child. The proband, individual III.2 (figure 2A), was the second boy born to a 24-year-old G2 mother (figure 2C) and was referred to genetics for multiple congenital anomalies including a congenital diaphragmatic hernia (CDH), bilateral cleft lip and palate, micrognathia, and dysmorphic facial features. Echocardiogram and brain MRI were normal, and he required monitoring for right grade two vesicoureteral reflux, and possible left sided hearing loss. At 12 months of age, his height was at the 15th centile, weight was at the 30th centile and head circumference was at the 85th centile. He was noted to have a prominent forehead, hypertelorism, broad nasal bridge, down-slanting palpebral fissures, extra ear crus bilaterally and micrognathia. Bilateral cleft lip had been repaired. He had truncal hypotonia with some delay of motor milestones, but his speech and cognition were felt to be age appropriate.
Figure 2.
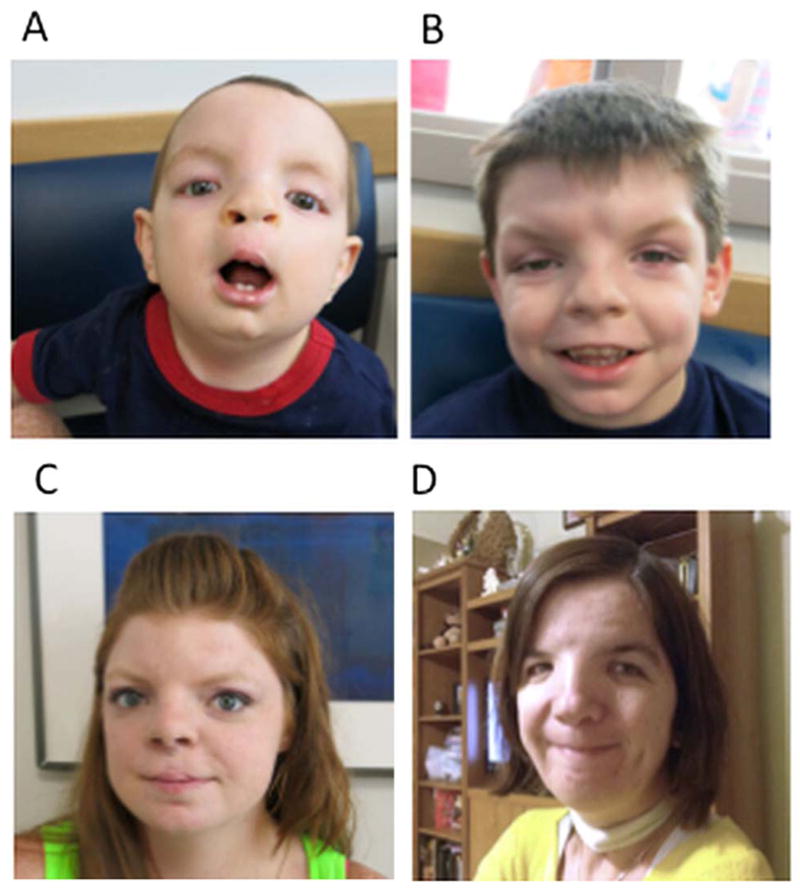
(A) Family A, III.3. (B) Family A, III.2. (C) Family A, II.2. (D) Family B, III.5.
The proband’s brother (figure 2B) had a history of tracheomalacia, inguinal and umbilical hernias, metopic craniosynostosis, critical aortic stenosis, and subsequent poststenotic dilation of the aortic root. Surgical repair of the metopic synostosis was first attempted at 12 months of age but was not completed due to tracheomalacia and complications with intubation. Repair was then pursued around 20 months of age due to persistent presentation of headaches.
Mild delays in speech acquisition were noted although cognition was appropriate for age. His facial features included hypertelorism, prominent forehead with prominent metopic suture shown in figure 2B and in a 3D head CT in online supplementary figure, broad nasal bridge, widow’s peak hairline, arched eyebrows, down-slanting palpebral fissures and a vertical groove in the nasal tip. There was no history of hypospadias in either boy. The mother (figure 2C) had a history of bilateral cleft lip and palate, congenital umbilical hernia and bicornuate uterus. In addition, she had hypertelorism, a prominent forehead, broad nasal root and repaired bilateral cleft lip. She reported that her mother, sister and the sister’s children all had similar facial features (see table 1).
Table 1.
Prevalence of phenotypic features in patients with MID1 mutations compared with phenotypic features of Family A and Family B
| Prevalence of features in Opitz G/BBB syndrome* | Family A | Family B | ||||
|---|---|---|---|---|---|---|
| AIII.2 | AIII.1 | A11.2 | BIII.5 | BII.6 | ||
| Sex | M | M | F | F | M | |
| Index | Brother | Mother | Index | Father | ||
| Facial features | ||||||
| Ocular hypertelorism | +++ | + | + | + | + | + |
| Prominent forehead | ++ | + | + | + | + | + |
| Widow’s peak | ++ | − | + | − | + | + |
| Broad nasal bridge | ++ | + | + | + | + | + |
| Anteverted nares | ++ | − | − | + | ||
| Micrognathia | + | + | + | + | + | |
| Down-slanted palpebral fissures | + | + | + | + | + | + |
| Central groove in nasal tip | − | + | − | + | ||
| Posteriorly rotated ears | − | − | + | + | ||
| LTE anomalies | +++ | − | + | − | + | |
| Hypospadias | +++ | − | − | NA | NA | − |
| Cleft lip and/or cleft palate | ++ | + | − | + | + | − |
| Congenital heart defect | ++ | − | AS | − | ||
| Imperforate or ectopic anus | ++ | − | − | − | − | |
| Midline brain defects | ++ | − | ||||
| Diaphragmatic hernia | + | + | − | − | − | |
| Umbilical/inguinal hernia | + | + | + | + | + | |
| Craniosynostosis | − | Metopic | − | Sagittal | ||
| Hearing loss | ? | + | ||||
| Developmental delay | ++ | Mild | Mild | − | Mild | |
| Intellectual disability | ++ | − | − | − | − | − |
| Other | Aortic dilation | Bicornuate uterus | Cardiomegaly | Congenital GI obstruction | ||
| Family history | AI.2: hypertelorism, bicornuate uterus | BII.8: hypertelorism, CL/CP | ||||
| AII.3 and AIII.3: hypertelorism AIII.4: hypertelorism, undescended testicles, hip dysplasia |
BI.2, BII.1, BII.3, BII.5, BIII.4: hypertelorism | |||||
+++ major feature; ++ minor features; + previously reported/present in patient.
AS, aortic stenosis; GI, gastrointestinal; LTE, laryngealtracheoesophageal; NA, not applicable.
Genetic testing to date had been uninformative and included a normal male karyotype, normal SNP genome-wide microarray, and MID1 sequencing and deletion/duplication testing negative for Opitz syndrome in both brothers. Sequencing of EFNB1 for craniofrontonasal dysplasia was also normal in the proband. Sequencing of GPC3 for Simpson–Golabi–Behmel syndrome and TGFBR1 and TGFBR2 sequencing for Loeys–Dietz syndrome were normal in the brother.
Family B
Family B was previously described by Judith Allanson in 1988 and is reported as Family 1 in the Robin et al 1995 article establishing the linkage of the autosomal dominant form of Opitz to chromosome 22q11.2.5,8,11 The proband, individual III.5 (figure 2D; table 1), was an affected girl with swallowing difficulties, stridor, micrognathia, cleft palate, bilateral hearing loss, mild ventricular dilatation, sagittal craniosynostosis (without history of surgery), ureteral reflux, umbilical hernia and cardiomegaly, thought to be secondary to chronic hypoxia. Her facial features included a broad prominent nasal root and bridge, mild central groove in the tip of the nose, prominence of the metopic suture and both parietal areas, marked widow’s peak, downslanting palpebral fissures, hypertelorism, posteriorly rotated ears, and a wide and poorly defined philtrum. The proband’s father and his four siblings were also felt to be affected based on the findings of megalencephaly, hypertelorism, down-slanting palpebral fissure, high broad nasal bridge, wide nasal base with a hooked tip, and long columella. The father was reported to have a congenital upper gastrointestinal obstruction. One paternal uncle had a unilateral cleft lip and palate. Hypospadias was not present in the father or two affected uncles. The proband’s sister had hypertelorism and high broad nasal bridge.
Exome sequencing and bioinformatics variant
After written informed consent was obtained, genomic DNA was extracted from the peripheral-blood lymphocytes of the proband and both parents, and from the saliva of the affected brother and maternal grandmother in Family A. Exome capture was carried out for the proband and both parents using NimbleGen SeqCap EZ Human Exon Library V.3.0 (Roche NimbleGen, Madison, Wisconsin, USA), guided by the manufacturer’s protocols. In brief, the qualified genomic DNA was isolated from peripheral-blood samples and randomly sheared to 200–300 bp fragments, followed by end-repair, a-tailing and pair-end index adapter ligation. The libraries were subsequently clustered on the cBOT instrument, multiplexing four samples per flowcell lane and sequenced using pair-end reads for 101 cycles with a paired-end mode on the Illumina HiSeq2000 following the manufacturer’s instructions (Illumina, San Diego, California, USA). Base calling was performed by the Illumina CASAVA software (V.1.8.2) with default parameters. All the raw reads were aligned to the reference human genome (UCSC hg19) using the Burrows–Wheeler aligner and single nucleotide variants (SNVs) and small insertions/deletions (indels) were identified using the Genome Analysis Tool Kit (GATKv2.6).12,13 The kinship coefficient was calculated between every two samples via KING to confirm reported relationships.14 Annovar11 and SnpEff were used to functionally annotate the variants and to categorise them into missense, nonsense, splice-altering variants and coding frameshift indels, which are likely to be deleterious compared with synonymous and non-coding variants.15,16 Under autosomal dominant mode of inheritance, we excluded variants that: (1) were synonymous variants; (2) present in unaffected father; (3) had a minor allele frequency of >0.005 in 1000 Genomes Project, 6503 exomes from the NHLBI Exome Sequencing Project (ESP6500SI; http://evs.gs.washington.edu/EVS/) or our inhouse database of >1500 sequencing exomes; (4) had a conservation score GERP++ < 2; and (5) were predicted by SIFT/PolyPhen2 scores to be benign variants.17–19
Sanger sequencing
Validation of the mutation candidate was performed by Sanger sequencing in all the available family members using the standard techniques of PCR amplicons with primer set 5′ CTACCAGCCCCTCACATCG 3′ and 5′ AGTTCCTGGGTAATGTGCTGT 3′.
This study was approved by the Institutional Review Boards at The Children’s Hospital of Philadelphia and the National Human Genome Research Institute, the National Institutes of Health.
Sequencing of additional patients with Opitz G/BBB
After written informed consent was obtained, genomic DNA was extracted from the peripheral-blood lymphocytes of 25 additional families with clinically diagnosed Opitz syndrome. Six probands were found to have MID1 mutations and all probands had normal chromosomal microarrays. The remaining 19 probands without MID1 mutations were sequenced for SPECC1L mutations by Sanger sequencing for exons 1–17 with standard techniques of PCR (see online supplementary table S1 for primer sets).
Mutagenesis
The p.Thr397Pro and p.Gly1083Ser mutations were created in full-length in a murine SPECC1L-GFP expression construct, described previously,20 using the Q5 site-directed mutagenesis kit (NEB, Ipswich, Massachusetts, USA) according to manufacturer’s protocol. The p.Gln415Pro and C-terminal calponin homology domain truncated (ΔCHD) constructs were created previously.20
Cell culture
SPECC1L-GFP expression constructs containing either wildtype or Thr397Pro, Gly1083Ser, Gln415Pro and ΔCHD mutations were transfected into U2OS osteosarcoma cells (ATCC, Manassas, Virginia, USA) using Viafect (Promega, Madison, Wisconsin, USA) or TransIT (Mirus Bio, Madison, Wisconsin, USA) according to manufacturer’s protocol. U2OS cells were grown in standard DMEM supplemented with 10% fetal bovine serum (FBS). Transfections and immunostaining were carried out on coverslips in 24-well plates. Cells were fixed in 4% paraformaldehyde (PFA) for 10 min, and blocked in phosphate buffered saline (PBS) with 1% goat serum and 0.1% Tween. Acetylated α-tubulin antibody (Sigma, St Louis, Missouri, USA) was used at 1:1000 dilution. After staining, coverslips were mounted in VectaShield containing DAPI (Vector Labs, Burlingame, California, USA).
RESULTS
Identification of SPECC1L mutation in Family A by WES
The WES generated a total of 50 443 SNVs and 4799 indels in the proband, 50 533SNVs and 4808 indels in the mother and 50 592 SNVs and 4811 indels in the father. We applied the filtering strategy as described in the Patients and methods section, filtering variants to exclude those who had an minor allele frequency (MAF) >0.5% or predictive of benign variant. Of these, 27 variants (see online supplementary table S2) were shared between the two affected patients (proband and mother), but not in the healthy father (figure 1A). Those variants included a missense variant c.1189A>C (p.Thr397Pro) in SPECC1L (mendelian inheritance in man (MIM) 614140; NM_015330.3), which is required for proper facial morphogenesis.20 The mutation was absent from 1000 Genomes Project, ESP6500SI, or additional exome sequencing data of over 1500 WES samples that we had previously sequenced in our inhouse database. Sanger sequencing of five family members, consisting of one unaffected and four affected individuals, confirmed its presence in proband, affected brother, mother and maternal grandmother (figure 1D).
Screening of additional patients
To further establish the association between SPECC1L and Opitz syndrome, we sequenced its coding region in an additional 19 patients with features of Opitz syndrome. The sequencing analysis identified a heterozygous missense mutation, c.3247G>A (p.Gly1083Ser), in one proband (Family B); Family B has been previously linked to 22q11.2 by Robin et al.8 We tested one other family linked to 22q11.2 besides Family B and did not find a mutation in SPECC1L. Mutation screening of additional family members confirmed segregation of the mutation with the phenotype. The p.Gly1083Ser mutation occurs in the CHD of SPECC1L (figure 1C) and predicted to be damaging with high probability according to the pathogenic score algorithms SIFT, Polyphen2, LRT and MutationTaster, and is not found in the 1000 Genomes Projects ESP6500SI or our inhouse database.
Functional analysis
Expression of SPECC1L-GFP results in stabilisation of a subset of microtubules (figure 3A) that appear in a lattice-like pattern and colocalise with acetylated α-tubulin (figure 3B,C), as previously described.20 We used this property of SPECC1L as a functional assay to assess the effect of SPECC1L Thr397Pro and Gly1083Ser mutations. Both Thr397Pro-GFP (figure 3D–F) and Gly1083Ser-GFP (figure 3G–I) mutant proteins show a drastic reduction in their ability to stabilise microtubules, consistent with these variants being pathological. The altered expression pattern of both Thr397Pro-GFP and Gly1083Ser-GFP mutant proteins is similar to that of the previously described Gln415Pro mutation (figure 3J–L). All three mutant SPECC1L-GFP proteins show a largely punctate expression pattern with poor, non-contiguous association with stabilised microtubules, in contrast to a robust, elaborate network of stabilised microtubules seen with wildtype SPECC1L-GFP. Interestingly, while Gln415Pro and Thr397Pro lay in the second coiled-coil domain, the Gly1083Ser mutation is located in the C-terminal CHD which was also previously shown to be required for SPECC1L stabilisation of microtubules (figure 3M–O).20 Synthetic mutations outside these domains do not strongly affect microtubule stabilisation (data not shown).20 Thus, mutations affecting the second coiled-coil domain or the CHD are likely to affect the same aspect of SPECC1L function and result in similar phenotypes.
Figure 3.

SPECC1L-T397P and SPECC1L-G1083S mutant proteins are defective in stabilising microtubules. U2OS cells were transfected with wildtype or mutant green fluorescent protein (GFP)-tagged Specc1l expression constructs. Wildtype SPECC1L-GFP expression (A; green) stabilises a subset of microtubules that colocalise with acetylated-α-tubulin (B, C; red). Compared with wildtype protein (A–C), both SPECC1L-T397P (D–F) and SPECC1L-G1083S (G–I) mutant proteins fail to stabilise microtubules efficiently and show a punctuate expression pattern, similar to SPECC1L-Q415P mutant protein ( J–L). SPECC1L-ΔCHD shows a diffuse expression pattern with near complete absence of microtubule stabilisation (M–O). Nuclei are stained with DAPI (blue).
DISCUSSION
Here we report mutations in SPECC1L as a cause of autosomal dominant Opitz syndrome in a subset of patients. WES analysis identified a novel mutation in SPECC1L on chromosome 22q11.23 to be the most probable disease causing mutation in a family with features suggestive of autosomal dominant Opitz syndrome. Sanger sequencing confirmed this result and the mutation was found to segregate with the clinical findings in three generations. Gene sequencing in 19 additional patients with suspected autosomal dominant Opitz syndrome revealed one additional novel mutation which segregated with the phenotype in a three-generation pedigree. Importantly, the SPECC1L gene, located at chromosome 22q11.23, is within one Mb of the D22S345 marker, which was previously linked to autosomal dominant Opitz syndrome.8 From Robin et al, we tested two families that were linked to 22q11.2; the other family did not have a mutation in SPECC1L, which may be due to locus heterogeneity associated with autosomal dominant Opitz syndrome or due to a mutation in a non-coding area of the SPECC1L not captured by our analysis such as a promoter region or intron. Interestingly, there have been case reports of patients with the Opitz syndrome phenotype and 22q11.2 deletions;21–23 however, SPECC1L is not contained within the 2.54 Mb typically deleted region associated with 22q11.2 deletion syndrome. Further study is needed to inquire whether there are other Opitz syndrome causing genes on chromosome 22.
The SPECC1L gene was first identified by Saadi et al20 to be disrupted by a balanced translocation, t(1;22)(21.3;q11.23) in a female patient with bilateral oromedial-canthal (Tessier IV) clefts, cleft palate, bilateral ocular hypoplasia and unilateral calcaneovarus foot deformity. The coding regions of SPECC1L were subsequently sequenced in 23 patients with oblique facial clefts and a de novo missense mutation, c.1244A>C (p.Gln415Pro), was identified in one individual.20 This mutation is located within the second coiled-coil domain of the SPECC1L protein, similar to the c.1189A>C (p.Thr397Pro) identified in Family A of this report.
SPECC1L gene encodes a ‘cross-linking’ protein that functionally interacts with both microtubules and the actin cytoskeleton and is necessary for cell adhesion and migration.20 Morpholino-based knockdown of a SPECC1L ortholog in zebrafish (specc1lb) results in loss of mandible and bilateral clefts between median and lateral elements of the ethmoid plate, which are structures analogous to the frontonasal process (FNP) and the paired maxillary processes (MxP), respectively.24 Lineage tracing analysis revealed that cranial neural crest cells contributing to the FNP fail to integrate with the MxP populations, while cells contributing to lower jaw structures were able to migrate to their destined pharyngeal segment but failed to converge to form mandibular elements.24 The function of SPECC1L in migration and adhesion of FNP and MxP is consistent with our patient phenotypes of hypertelorism and orofacial clefting. It is unclear why mutations in SPECC1L, and even the same domain, result in the two distinct phenotypes of Opitz syndrome and oblique facial clefts. Although facial malformations are major portions of the phenotype in both conditions, a larger cohort of patients with SPECC1L mutations will be needed to examine genotype–phenotype correlation.
Saadi et al20 demonstrated that the c.1244A>C ( p.Gln415Pro) mutation identified in the patient with oblique facial cleft significantly interfered with the ability of SPECC1L to bind to and stabilise microtubules in an in vitro cell assay. Similarly, the Thr397Pro-GFP mutant protein (Family A) and Gly1083Ser-GFP (Family B) mutant proteins show a drastic reduction in their ability to stabilise microtubules. It is important to note that the p.Thr397Pro and the p.Gln415Pro mutations are both located in the second coiled-coil domain and manifest a similar inability in microtubule stabilisation. In contrast, the p.Gly1083Ser mutation is located in the CHD, located at the C-terminus, which has been shown to facilitate actin binding.20 However, Saadi et al20 demonstrated that expression of a truncated form of SPECC1L protein lacking the CHD (SPECC1L-ΔCHD) completely abolished the formation of stabilised microtubules as well. Thus, the second coiled-coil domain and the CHD interaction with actin cytoskeleton are required for microtubule stabilisation function of SPECC1L. It is therefore consistent that the two mutations we report here, in the second coiled-coil domain and the CHD respectively, manifest similar patient phenotypes.
The combination of hypertelorism, facial cleft and CDH has previously been reported in Opitz G/BBB with MID1 mutation as well as in other craniofacial disorders with midline defects including craniofrontonasal dysplasia with EFNB1 mutation and Simpson–Golabi with GPC3 mutation.25–27 Many of the other genes associated with CDH, including EFNB1, GPC3 and SLT3, have been found to be critical to cell migration.27 Given SPECC1L’s involvement in cell adhesion and migration, it is therefore reasonable to speculate that the SPECC1L mutation might also explain the CDH seen in the proband of Family A.
MID1 and SPECC1L mutations result in Opitz syndrome and both of their gene products are involved in microtubule stability. Using the green fluorescent protein (GFP) as a tag, MID1 has been shown to be a microtubule-associated protein that affects microtubule dynamics in MID1 overexpressing cells.28 The MID1 protein consists of a triparatite motif at the N-terminal which includes a RING finger, two B-boxes, and a coiled-coil domain. The C-terminus has a fibronectin Type III domain, a cells (simian CV-1) in origin, and carrying the Simian virus 40 (SV40) (COS) domain and a B30.2 domain.29 Most mutations in X linked Opitz syndrome are found in the C-terminus domains and when these mutations are introduced into in vitro cellular assays, they abolish microtubule association of these proteins.28 It is clear that mutations in MID1 and SPECC1L have a similar cellular and clinical phenotype.
The two families presented in this report show many of the classic features associated with Opitz syndrome, but also show some unusual features. The brother of the proband in Family A had surgical repair of metopic craniosynostosis and the original proband in Family B was reported to have a prominent metopic ridge and sagittal craniosynostosis, which were not surgically repaired. A detailed history of the sagittal craniosynostosis documented in Allanson’s 1988 paper5 in the proband in Family B and why it was not surgically repaired was difficult to ascertain, making further natural history studies important to further clarify the significance of this finding. Synostosis of the cranial sutures is not a common feature of Opitz and the presence of this finding may help distinguish cases caused by SPECC1L mutations, especially hypertelorism associated with metopic synostosis as this type of synostosis usually results in hypotelorism (see online supplementary figure). Hypospadias is a common finding in both autosomal dominant and X linked Opitz G/BBB syndrome 10 but was not seen in any of the affected male patients in either family in this report. Therefore, absence of hyospadias in a family with suspected autosomal dominant Opitz syndrome should also lead a clinician to consider the possibility of SPECC1L mutation. Further studies are indicated to determine the prevalence of SPECC1L mutations in patients with syndromic clefting including those with autosomal dominant Opitz syndrome.
Supplementary Material
Acknowledgments
We thank the patients and families involved in the research. We would like to thank Diana Acevedo and Abby Stork (University of Kansas Medical Center, Kansas City, Kansas, USA) for assistance with experiments.
Funding
This project was supported in part by the National Institutes of Health Kansas IDeA Network for Biomedical Research Excellence grant (National Institute of General Medical Sciences P20 GM103418, I.S.), Kansas Intellectual and Developmental Disabilities Research Center grant (P30 Eunice Kennedy Shriver National Institute of Child Health and Human Development, HD 002528, I.S.), and Center of Biomedical Research Excellence grant (National Institute of General Medical Sciences P20 GM104936, I.S.), the Division of Intramural Research of the National Human Genome Research Institute, the National Institutes of Health.
Footnotes
Contributors
Manuscript writers: PK, MHH, DL, EHZ and MM. Study design: MM, EHZ, IS, NRW and PK. Family A sequencing and data analysis: DL, ML, MJF and HH. Family B sequencing and data analysis: AFM, RAH and PK. Figure construction: MHH, DL, EMM, IS and NRW. Microtubule studies: IS and NRW. Patient evaluation and phenotyping Family A: MHH, DS, EMM, RMC, ML, DMM, MAD, MJF, JPJ, CH, HH and EHZ. Patient evaluation and phenotyping Family B and 24 non-specc1l carriers: PK, MM, EHZ and JEA. Opitz GBBB study coordinators Children’s Hospital of Philadelphia: MHH, EMM, RMC, DMM and JPJ. Opitz GBBB study coordinators NIH: PK, RAH and AFM.
Competing interests
None.
Patient consent
Obtained.
Ethics approval
Institutional Review Boards at The Children’s Hospital of Philadelphia and the National Human Genome Research Institute, NIH.
Provenance and peer review
Not commissioned; externally peer reviewed.
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/jmedgenet-2014-102677).
References
- 1.Cappa M, Borrelli P, Marini R, Neri G. The Opitz syndrome: a new designation for the clinically indistinguishable BBB and G syndromes. Am J Med Genet. 1987;28:303–9. doi: 10.1002/ajmg.1320280207. [DOI] [PubMed] [Google Scholar]
- 2.Funderburk SJ, Stewart R. The G and BBB syndromes: case presentations, genetics, and nosology. Am J Med Genet. 1978;2:131–44. doi: 10.1002/ajmg.1320020204. [DOI] [PubMed] [Google Scholar]
- 3.Farndon PA, Donnai D. Male to male transmission of the G syndrome. Clin Genet. 1983;24:446–8. doi: 10.1111/j.1399-0004.1983.tb00101.x. [DOI] [PubMed] [Google Scholar]
- 4.Stoll C, Geraudel A, Berland H, Roth M, Dott B. Male-to-male transmission of the hypertelorism-hypospadias (BBB) syndrome. Am J Med Genet. 1985;20:221–5. doi: 10.1002/ajmg.1320200203. [DOI] [PubMed] [Google Scholar]
- 5.Allanson J. G syndrome: an unusual family. Am J Med Genet. 1988;31:637–42. doi: 10.1002/ajmg.1320310319. [DOI] [PubMed] [Google Scholar]
- 6.Wilson GN, Oliver WJ. Further delineation of the G syndrome: a manageable genetic cause of infantile dysphagia. J Med Genet. 1988;25:157–63. doi: 10.1136/jmg.25.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guion-Almeida ML, Richieri-Costa A. CNS midline anomalies in the Opitz G/BBB syndrome: report on 12 Brazilian patients. Am J Med Genet. 1992;43:918–28. doi: 10.1002/ajmg.1320430603. [DOI] [PubMed] [Google Scholar]
- 8.Robin NH, Feldman GJ, Aronson AL, Mitchell HF, Weksberg B, Leonard CO, Burton BK, Josephson KD, Laxova R, Aleck AA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11. 2. Nat Genet. 1995;11:459–61. doi: 10.1038/ng1295-459. [DOI] [PubMed] [Google Scholar]
- 9.Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli E, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997;17:285–91. doi: 10.1038/ng1197-285. [DOI] [PubMed] [Google Scholar]
- 10.So J, Suckow V, Kijas Z, Kalscheuer V, Moser B, Winter J, Baars M, Firth H, Lunt P, Hamel B, Meinecke P, Moraine C, Odent S, Schinzel A, van der Smaqt JJ, Devriendt K, Albrecht B, Gillessen-Kaesback G, van der Burgt I, Petrij F, Faivre L, McGaughran J, McKenzie F, Opitz JM, Cox T, Schweiger S. Mild phenotypes in a series of patients with Opits GBBB syndrome with MID1 mutations. Am J Med Genet. 2005;132A:1–7. doi: 10.1002/ajmg.a.30407. [DOI] [PubMed] [Google Scholar]
- 11.Robin NH, Opitz JM, Muenke M. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am J Med Genet. 1996;62:305–17. doi: 10.1002/(SICI)1096-8628(19960329)62:3<305::AID-AJMG20>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manichaikul A, Mychaleckyi JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cinqolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–9. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saadi I, Alkuraya FS, Gisselbrecht SS, Goessling W, Cavallesco R, Turbe-Doan A, Petrin AL, Harris J, Siddiqui U, Grix A, Hove HD, Leboulch P, Glover TW, Morton CC, Richieri-Costa A, Murray JC, Erickson RP, Maas RL. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am J Hum Genet. 2011;84:44–55. doi: 10.1016/j.ajhg.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11. 2 deletion. Am J Med Genet. 1995;59:103–13. doi: 10.1002/ajmg.1320590122. [DOI] [PubMed] [Google Scholar]
- 22.Fryburg JS, Lin KY, Golden WL. Chromosome 22q11. 2 deletion in a boy with Opitz (G/BBB) syndrome. Am J Med Genet. 1996;62:274–5. doi: 10.1002/(SICI)1096-8628(19960329)62:3<274::AID-AJMG13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Lacassie Y, Arriaza MI. Opitz GBBB syndrome and the 22q11. 2 deletion. Am J Med Genet. 1996;62:318. doi: 10.1002/(SICI)1096-8628(19960329)62:3<318::AID-AJMG21>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 24.Gfrerer L, Shubinets V, Hoyos T, Kong Y, Nguyen C, Pietschmann P, Morton CC, Maas RL, Liao EC. Functional analysis of SPECC1L in craniofacial development and oblique facial cleft pathogenesis. Plast Reconstr Surg. 2014;134:748–59. doi: 10.1097/PRS.0000000000000517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor J, Aftimos S. Congenital diaphragmatic hernia is a feature of Opitz G/BBB syndrome. Clin Dysmorphol. 2010;19:225–6. doi: 10.1097/MCD.0b013e32833b2bd3. [DOI] [PubMed] [Google Scholar]
- 26.Brooks AS, van Dooren M, Hoogeboom J, Gischler S, Willems PJ, Tibboel D. Congenital diaphragmatic hernia in a female patient with craniofrontonasal syndrome. Clin Dysmorphol. 2002;11:151–3. doi: 10.1097/00019605-200204000-00019. [DOI] [PubMed] [Google Scholar]
- 27.Slavotinek AM. Single gene disorders associated with congenital diaphragmatic hernia. 2007;145C:172–83. doi: 10.1002/ajmg.c.30125. [DOI] [PubMed] [Google Scholar]
- 28.Schweiger S, Foerster J, Lehmann T, Suckow V, Muller YA, Walter G, Davies T, Porter H, van Bokhoven H, Lunt PW, Traub P, Ropers HH. The Opitz syndrome gene product, MID1, associates with microtubules. Cell Biol. 1999;96:2794–9. doi: 10.1073/pnas.96.6.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem. 2006;281:8970–80. doi: 10.1074/jbc.M512755200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.