

Abstract
The methylation of DNA bases plays an important role in numerous biological processes including development, gene expression, and DNA replication. Salmonella is an important foodborne pathogen, and methylation in Salmonella is implicated in virulence. Using single molecule real-time (SMRT) DNA-sequencing, we sequenced and assembled the complete genomes of eleven Salmonella enterica isolates from nine different serovars, and analysed the whole-genome methylation patterns of each genome. We describe 16 distinct N6-methyladenine (m6A) methylated motifs, one N4-methylcytosine (m4C) motif, and one combined m6A-m4C motif. Eight of these motifs are novel, i.e., they have not been previously described. We also identified the methyltransferases (MTases) associated with 13 of the motifs. Some motifs are conserved across all Salmonella serovars tested, while others were found only in a subset of serovars. Eight of the nine serovars contained a unique methylated motif that was not found in any other serovar (most of these motifs were part of Type I restriction modification systems), indicating the high diversity of methylation patterns present in Salmonella.
Introduction
The methylation of DNA is important in all kingdoms of life as a mechanism of epigenetic control [1–3]. Methylation is achieved through the action of methyltransferase enzymes (MTases), which covalently attach methyl groups to DNA bases. In eukaryotes, 5-methylcytosine (m5C) is the most common methylation. In contrast, N6-methyladenine (m6A) is the most frequent methylation in prokaryotes, although N4-methylcytosine (m4C) and m5C are also widespread.
Methylation in eukaryotes has been well studied and is known to mediate diverse processes including growth, development, and disease [4]. In prokaryotes, methylation is a key component of restriction-modification (RM) systems, which protect cells from foreign DNA. RM systems are composed of multiple proteins, including at least one MTase, which recognizes and methylates a base contained within a specific sequence motif, and one endonuclease, or REase, which cleaves foreign DNA with a methylation pattern different from that of the host DNA. RM systems are subdivided into four main classes that differ in subunit composition, motif characteristics, cofactor requirements, and location of DNA cleavage (for review, see [5]). In brief, Type 1 RM systems are composed of two restriction subunits (R), two methylation subunits (M) and one specificity subunit (S), which recognizes specific DNA sequences. Recognized motifs are asymmetric and bipartite. Type II systems include one R and one M subunit which can function independently, and recognized motifs are mostly symmetric. Type III systems are hetero-oligomers composed of a mod subunit (recognizes and modifies DNA) and a res subunit that is only active in a mod-res complex. The only RM systems that recognize methylated, instead of unmethylated sites, are Type IV. Methylation in bacteria also influences critical processes including gene regulation, cell cycle control, pathogenicity, and DNA repair [2].
Despite the important implications of bacterial methylation, its distribution, diversity, and functional consequences have not been extensively investigated. This paucity of data can, in part, be attributed to technological limitations. Methylation studies in eukaryotes have been facilitated by the development of detection methods for m5C, including bisulfite conversion, which allows for genome-wide modification analyses. Comparable methods have not been available for the detection of m6A and m4C until recent advances in sequencing technology. SMRT sequencing couples whole-genome sequencing with the simultaneous detection of base modifications using kinetic signals during DNA polymerization [6, 7]. This new technology has led to insights regarding the methylomes of several bacterial species [8–12]. However, methylation is widespread throughout the bacterial kingdom and is very diverse [13]. Thus, more studies are needed to gain a comprehensive understanding of the distribution and diversity of methylation motifs and their associated MTases, and ultimately to comprehend methylation functions and evolutionary history in these organisms.
Salmonella enterica is the leading cause of death and hospitalizations due to foodborne pathogens each year [14]. Previous studies have shown that the methylation of the Gm6ATC motif by the MTase Dam is an essential factor in the virulence of Salmonella, and that a lack of methylation leads to attenuation in animal models [15]. Subsequent studies have elucidated the mechanisms by which some virulence genes are regulated by Dam, including the plasmid-encoded fimbriae (pef) locus [16] and the std fimbrial operon [17]. In addition, Dam regulates both the phase variation of STM2209-STM2208 which alters lipopolysaccharide O-antigen side chain length [18], and the phase variation of the phage P22 glucosyltransferase (gtr) operon which controls O-antigen glucosylation [19]. Thus, it is possible that the methylation of other motifs in Salmonella also may have implications for virulence, pathogenicity, and other functions. Here, we sequenced and closed the genomes of six Salmonella enterica isolates from five serovars. We then analysed their methylomes, along with the methylomes of four additional serovars that we sequenced previously [11, 20–22], and employed a bioinformatics approach to identify methyltransferases and match them to observed methylated motifs in the genomes. We also examined how methylation patterns varied between Salmonella serovars.
Materials and Methods
We selected five serovars of Salmonella enterica subs enterica from our in-house strain collection at the FDA-CFSAN. These included Salmonella enterica subs enterica serovar (S. Bareilly), S. Abaetetuba, S. Abony, S. Anatum, S. Bredeney, S. Montevideo, and two isolates of S. Enteritidis. We also included data from four serovars we sequenced previously, S. Javiana, S. Typhimurium, S. Heidelberg, and S. Cubana [11, 20–22] (see Table 1 for strain names and accession numbers).
Table 1. Summary of Salmonella genomes sequenced in this study.
| Serovar | Chromosome size (bp) | Plasmid size (bp) | GenBank Accession (chromosome) | GenBank Accession (plasmid) | Phage | MTase on phage (specificity, if known) | MTase on plasmid (specificity, if known) |
|---|---|---|---|---|---|---|---|
| S. Bareilly CFSAN000189 | 4730612 | 78193 | CP006053.1 | CP006054.1 | Salmon_Fels_1_NC_010391 Gifsy_1_NC_010392 | _ | M.SbaUORF280P |
| S. Abony CFSAN001275 | 4737447 | NA | CP007534.1 | _ | Entero_ST64T_NC_004348 Gifsy_2_NC_010393 | _ | _ |
| S. Anatum CFSAN000665 | 4706101 | NA | CP007531.1 | _ | Salmon_Fels_1_NC_010391 Gifsy_1_NC_010392 | M.SenAnaORF14155P | _ |
| S. Cubana CFSAN002050 | 4977480 | 166,668 122,863 | CP006055.1 | CP006056.1 CP006057.1 | Gifsy_1_NC_010392 Salmon_vB_SemP_Emek_NC_018275 | _ | M.Sen2050ORF235P(GATC) M.Sen2050ORF245P M.Sen2050ORF400P M.Sen2050ORF480P(CAGCTG) |
| S. Heidelberg CFSAN002069 | 4783943 | 110,363 37,679 | CP005390.2 | CP005389.2 CP005391.2 | Entero_P22_NC_002371 Gifsy_2_NC_010393 | M.Sen2069ORF4005P (GATC) | M.Sen2069ORF23325P |
| S. Heidelberg CFSAN002064 | 4783867 | 37692 | CP005995.1 | CP005994.1 | Entero_P22_NC_002371 Gifsy_2_NC_010393 | M.Sen2069ORF21380P (GATC) | _ |
| S. Javiana CSFAN001992 | 4634161 | 24,012 17,094 | CP004027.1 | CP004026.1 CP004028.1 | Gifsy_2_NC_010393 Salmon_RE_2010_NC_019488 Entero_PsP3_NC_005340 | M.SenJORF19790P (GATC) | _ |
| S. Montevideo CFSAN000255 | 4694375 | NA | CP007530.1 | _ | Salmon_vB_SosS_Oslo_NC_018279 Entero_Fels_2_NC_010463 | M.Sen255II (ATGCAT) | _ |
| S. Enteritidis CFSAN000158 | 4679662 | 59369 | CP007528.1 | CP007529.1 | Salmon_RE_2010_NC_019488 Gifsy_2_NC_010393 | M.Sen158III (GATC) | _ |
| S. Enteritidis CFSAN000111 | 4679081 | 39599 | CP007598.1 | CP007599.1 | Gifsy_2_NC_010393 Salmon_RE_2010_NC_019488 | M.Sen1427ORF7910P (GATC) | _ |
| S. Typhimurium CFSAN001921 | 4859931 | 3,609 4,675 221,009 | CP006048.1 | CP006052.1 CP006051.1 CP006050.1 | Salmon_ST64B_NC_004313 Gifsy_1_NC_010392 Gifsy_2_NC_010393 Entero_ST104_NC_005841 | _ | M.SenTFORF23885P (CAGCTG) M.SenTFORF24805P (CCNGG) |
Each strain was plated onto Trypticase Soy Agar and incubated overnight at 37°C. Cells were then inoculated into Trypticase Soy Broth for DNA extraction. A 1 ml-aliquot was pelleted, and genomic DNA was extracted using the DNeasy Blood and Tissue kit from Qiagen (Qiagen, CA, USA). All samples were analyzed at the exponential stage of growth.
DNA was sheared to approximately 10 kb using a Covaris g-TUBE (Covaris, Inc.; Woburn, MA). SMRTbell 10 kb template libraries were prepared using DNA Template Prep Kit 2.0 and the Low-Input 10 kb Library Protocol (Pacific Biosciences; Menlo Park, CA, USA). In brief, DNA was concentrated, repaired, ligated to hairpin adapters, and purified. Incompletely formed SMRTbell templates were digested with a combination of Exonucleases III and VII. Adapters were annealed, and SMRT sequencing was carried out on the PacBioRS II (Pacific Biosciences; Menlo Park, CA, USA) using standard protocols.
Analysis of sequence reads was implemented using SMRT Analysis 1.10 and the SMRT Portal 2.0 platform (Pacific Biosciences). De novo assembly was performed using the Hierarchical Genome Assembly Process (HGAP) with default parameters [23]. HGAP consists of three steps to ensure high accuracy. First, Basic Local Alignment with Successive Refinement (BLASR) is used to align all reads to the longest seed reads and a consensus is generated to create pre-assembled reads. Preassembled reads are then assembled using the Celera assembler. Finally, all reads are mapped to the de novo assembly and final consensus and accuracy scores are determined using the Quiver consensus algorithm. HGAP outputs assemblies with overlapping regions at the ends. Coordinates of this region were identified using dot plots in Gepard [24], and trimmed from one end to circularize the genome. Genomes were checked manually for even sequencing coverage. Genomes were annotated using the NCBI (National Center for Biotechnology Information) Prokaryotic Genomes Automatic Annotation Pipeline [25] (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). Prophages were detected using PHAST [26]. Only prophages scored as intact are reported here. We excluded putative intact prophages that did not show significant sequence similarity to known phages using the Basic Local Alignment Search Tool (BLAST) sequence alignment tool with default parameters.
Motif Detection and Analysis was also carried out using SMRT Analysis 1.1 and the RS_Modification_and_Motif_Analysis.1 protocol as described at http://www.pacb.com/pdf/TN_Detecting_DNA_Base_Modifications.pdf. Interpulse durations (IPDs) were measured based on the kinetic signals [7] and processed as described previously [6]. At each position in the genome, the observed IPD was compared to the IPD of an in-silico control using a two-sample t-test, and a QV score was calculated as QV = -10 log (p-value). Bases were accepted as modified based on a minimum QV threshold value. QV 30 was used as a threshold for preliminary analyses. A plot of QV versus coverage was then constructed using publicly available R scripts found at: https://github.com/PacificBiosciences/motif-finding. The observed bimodal distribution of kinetic data, resulting from modified and unmodified positions, was then used to determine a more stringent QV threshold (S1 Fig). Only sites with a minimum of 25x coverage were included. Motifs were identified using the algorithm MotifMaker. m6A and m4C motifs can be reliably detected with 25x coverage across all positions in the genome, but m5C requires either significantly higher coverage (~100x) or Tet-methylation for confident detection. In this study we report only m6A and m4C methylations. To identify MTases, assembled genomes were scanned for homologs of RM system genes using in-house software (e value > 1e-11) to identify putative MTases as previously described [10]. Predicted specificities were assigned to candidate MTases based on specificities of the known MTases. The presence of functional motifs and information regarding the placement of the gene within the genome were also used to support or reject those assignments, as were known characteristics of different MTase types. For example, Type III MTases and most Type IIG systems only methylate one strand of their recognition sequence, whereas Type I systems have bipartite recognition sequences. MTase candidates with predicted specificities were matched where possible with observed motifs found in our motif analyses. If a single candidate MTase existed for an observed motif, then that gene was assumed to be responsible for that particular specificity. If multiple candidates existed for a single motif, no MTase was assigned. When making assignments of new motifs to specific MTases, we always cross-checked the matched gene against other similar genes in REBASE and against the unassigned motifs from the more than 700 other genomes for which we have PacBio data. In many cases, the same motif occurred in a different genome with an essentially identical methyltransferase or specificity subunit protein sequence, adding weight to the strength of the assignment. Raw processed PacBio data files were deposited in the Sequence Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/sra) (S2 Table) and MTase information and sequences were deposited in REBASE (http://rebase.neb.com/rebase/rebase.html).
Results and Discussion
Genome Assemblies
All genomes were assembled into a single, circular chromosomal contig and up to three plasmids. Consensus accuracy scores were at least 99.9999% for all assemblies. Sizes of Salmonella chromosomes ranged from 4,547,600 – 4,977,480 bp, plasmid sizes ranged from 3,609–221,009 bp (Table 1). Sequences were deposited in GenBank. Putative prophages and BLAST alignment data are reported in Table 1.
Methylation Patterns
This is the first comparative report of genome-wide methylation patterns in the pathogenic bacteria Salmonella enterica. We analyzed the methylomes of five Salmonella enterica subsp. enterica serovars, including two isolates of S. Enteritidis. We also sequenced and released their closed genomes. We present those results, along with data from four additional Salmonella serovars, S. Javiana, S. Typhimurium, S. Heidelberg, and S. Cubana, which we analyzed previously [11, 20–22]. In total, we observed 18 motifs among the nine Salmonella serovars, 16 m6A motifs, one m4C motif, m4CCWWGG, and one Type I MTase which encodes both m6A and m4C activities, Gm6ATGN5G4m GC (Fig 1; an underscore represents the base which is methylated in the opposite DNA strand; W = A or T). Eight of the motifs were novel, i.e., they have not been previously observed in any bacterial species. We were able to match 13 of the Salmonella motifs to their respective MTase enzymes in most of the serovars tested (S1 Table).
Fig 1. The methylomes of eleven Salmonella serovars.
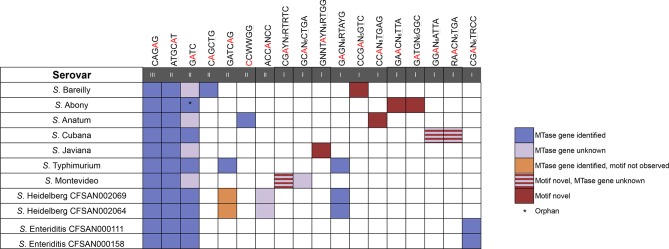
Violet = MTase identified, light violet = MTase unknown, red = novel motif, orange = MTase present in genome, motif not observed. Light violet/red stripe = novel motif, MTase unknown. Roman numerals indicate MTase type (I – III). Note the majority of novel MTases are Type I systems.
Several motifs were common among multiple serovars, while other motifs were unique to specific serovars. All Salmonella serovars examined contained the methylated motifs ATGCm6AT, CAGm6AG, and Gm6ATC. In all serovars, we identified a Type III MTase responsible for the methylation of CAGm6AG, and an extremely common Type II MTase was found to methylate the ATGCm6AT motif (see Table 2 for a list of enzyme names specific to each strain). The methylation of ATGCm6AT was never complete (38–78.5%). This MTase is usually active in Salmonella, although rarely active in E. coli, and is not thought to be an essential gene [27]. Confident assignment of an MTase to the Gm6ATC motifs could only be performed in eight of the eleven isolates: two were orphan MTases, and the remaining were common Type II enzymes. In multiple serovars, we identified candidate enzymes that have the potential to methylate this motif (Table 3).
Table 2. Summary of motifs and methyltransferases found in each Salmonella genome.
| Serovar | Enzyme Assignment | Gene Locus_Tag (GenBank) | Type | Sub- Type | Motif Observed | Motif Uni-que* | % Methyl-ated 5'-3'/3'-5' | Number Methyl- ated Motifs (5'-3' strand/ 3'-5' strand) | Number Motifs in Genome (5'-3' strand/ 3'-5' strand) |
|---|---|---|---|---|---|---|---|---|---|
| S. Bareilly CFSAN000189 | M.SbaUI | SEEB0189_17520 | III | beta | CAGm6AG | no | 97.7 | 5652 | 5787 |
| M.SbaUII | SEEB0189_19945 | II | beta | Cm6AGCTG | no | 88.2 | 1466 | 1662 | |
| M.SbaUIII | SEEB0189_19740 | I | gamma | CCGm6ANNNNNGTC | yes | 98.6/ 98.6 | 482/ 482 | 489/ 489 | |
| M.SbaUIV | SEEB0189_02925 | II | beta | ATGCm6AT | no | 78.5 | 1093 | 1392 | |
| M.SbaUDam | SEEB0189_02450 | Orphan | _ | G6mATC | no | 98.6/ 98.6 | 37148 | 37688 | |
| S. Abony CFSAN001275 | M.SenAboI | SEEA0014_11325 | III | beta | CAGm6AG | no | 96.8 | 5391 | 5569 |
| M.SenAboII | SEEA0014_03225 | II | beta | ATGCm6AT | no | 38 | 283 | 744 | |
| M.SenAboIV | SEEA0014_08865 | I | gamma | GAm6ACNNNNNNNTTA | yes | 94.9/ 93.5 | 410/ 404 | 432/ 432 | |
| M.SenAboDam | SEEA0014_03700 | Orphan | alpha | G6mATC | no | 95.1/ 95.1 | 35607 | 37436 | |
| M1.SenAboIII | SEEA0014_08700 | I | gamma | G6mATGNNNNNG4mGC/ G4mCCNNNNNCATC | yes | 96.1/ 31.0 | 1260/ 406 | 1311/ 1311 | |
| M2.SenAboIII | SEEA0014_08705 | I | gamma | G6mATGNNNNNG4mGC/ G4mCCNNNNNCATC | yes | 96.1/ 31.0 | 1260/ 406 | 1311/ 1311 | |
| S. Anatum CFSAN000665 | M.SenAnaI | SEEA1592_11695 | I | gamma | CCm6ANNNNNNNNTGAG | yes | 99.7/ 99.4 | 354/ 353 | 355/ 355 |
| M.SenAnaII | SEEA1592_09525 | III | beta | CAGm6AG | no | 100.0 | 5509 | 5511 | |
| M.SenAnaIII | SEEA1592_17520 | II | beta | ATGCm6AT | no | 66.7/ 66.7 | 674 | 1010 | |
| M.SenAnaIV | SEEA1592_11855 | II | beta | 4mCCWWGG | no | 83.3/ 83.3 | 1423 | 1708 | |
| M.SenAnaDam | SEEA1592_01330 | Orphan | _ | G6mATC | no | 99.8/ 99.8 | 37140 | 37224 | |
| S. Cubana CFSAN002050 | M.Sen2050I | CFSAN002050_08375 | III | beta | CAGm6AG | no | 95.1 | 6235 | 6558 |
| M.Sen2050II | CFSAN002050_23900 | II | beta | ATGCm6AT | no | 45.1/ 45.1 | 510 | 1131 | |
| _ | _ | I | _ | GGm6ANNNNNNATTA | yes | 92.7/ 92.3 | 459/ 457 | 495/ 495 | |
| _ | _ | I | _ | TCm6ANNNNNGTTY | yes | 95.5/ 92.3 | 1248/ 1338 | 1352/ 1352 | |
| S. Heidelberg CFSAN002064 | M.Sen2064I | CFSAN002064_15765 | I | gamma | Gm6AGNNNNNNRTAYG | no | 97.9/ 97.5 | 231/ 230 | 236/ 236 |
| M.Sen2064II | CFSAN002064_18310 | III | beta | CAGm6AG | no | 98.2 | 5587 | 5691 | |
| M.Sen2064III | CFSAN002064_10125 | II | beta | ATGCm6AT | no | 42.4 | 319 | 752 | |
| _ | _ | II | _ | ACCm6ANCC | no | 99.4 | 2703 | 2719 | |
| S. Heidelberg CFSAN002069 | M.Sen2069I | CFSAN002069_07060 | III | beta | CAGm6AG | no | 97.9 | 5816 | 5939 |
| M.Sen2069II | CFSAN002069_09575 | I | gamma | Gm6AGNNNNNNRTAYG | no | 97.5/ 97.9 | 238/ 239 | 244/ 244 | |
| M.Sen2069III | CFSAN002069_15235 | II | beta | ATGCm6AT | no | 42.2/ 42.2 | 217 | 514 | |
| _ | _ | ACCm6ANCC | 99 | 2747 | 2774 | ||||
| S. Javiana CFSAN001992 | M.SenJI | CFSAN001992_09405 | III | beta | CAGm6AG | no | 97.8 | 5410 | 5523 |
| M.SenJII | CFSAN001992_11490 | I | gamma | CCm6AYNNNNNRTANNC | yes | 98.1/ 97.7 | 474/ 472 | 483/ 483 | |
| M.SenJIII | CFSAN001992_16620 | II | beta | ATGCm6AT | no | 58.8/ 58.8 | 803 | 1364 | |
| _ | _ | G6mATC | no | 98.9/ 98.9 | 36330 | 36738 | |||
| S. Montevideo CFSAN000255 | M.Sen255I | Y007_00590 | III | beta | CAGm6AG | no | 99.4 | 5504 | 5535 |
| M.Sen255II | Y007_12075 | II | beta | ATGCm6AT | no | 50.0/ 50.0 | 387 | 774 | |
| _ | _ | I | _ | CG6mAYNNNNNNNRTRTC | yes | 99.1/ 98.9 | 439/ 438 | 443/ 443 | |
| _ | _ | II | alpha | G6mATC | no | 99.1/ 99.1 | 36866 | 37204 | |
| _ | _ | I | _ | GCm6ANNNNNNCTGA | no | 98.6/ 99.5 | 554/ 559 | 562/ 562 | |
| S. Enteriditis CSFAN000111 | M.Sen1427II | SEEE1427_7355 | I | gamma | CGm6ANNNNNNTRCC | no | 98.4/ 97.9 | 1721/ 1712 | 1749/ 1749 |
| _ | _ | II | G6mATC | no | 98.8/ 98.8 | 36824 | 37256 | ||
| M.Sen1427I | SEEE1427_9465 | CAGm6AG | no | 99.2 | 5505 | 5549 | |||
| M.Sen1427III | ATGC6mAT | no | 43 | 315 | 732 | ||||
| S. Enteriditis CFSAN000158 | M.Sen158I | SEEE0968_18850 | III | beta | CAGm6AG | no | 98.1 | 5490 | 5599 |
| M.Sen158II | SEEE0968_20955 | I | gamma | CGm6ANNNNNNTRCC | no | 99.0/ 98.0 | 1739/ 1722 | 1757/ 1757 | |
| M.Sen158III | SEEE0968_03950 | II | beta | ATGC6mAT | no | 41.3/ 41.3 | 302 | 732 | |
| S. Typhimurium CFSAN001921 | M.SenTFI | CFSAN001921_15255 | III | beta | CAGm6AG | no | 89.3 | 5635 | 6308 |
| M.SenTFII | CFSAN001921_17800 | I | gamma | CRTm6AYNNNNNNCTC | no | 90.7/ 89.1 | 233/ 229 | 257/ 257 | |
| M.SenTFIII | CFSAN001921_00055 | II | beta | ATGCm6AT | no | 60.9 | 630 | 1035 | |
| SenTFIV | CFSAN001921_17955 | II | alpha | GATC6mAG | no | 94.3 | 2841 | 3011 |
*A unique motif refers to one that has not been previously observed in any bacterial species.
Table 3. Methyltransferases identified in the Salmonella serovars, but not assigned to a motif.
| Serovar | Enzyme Assignment | Type | SubType | Motif (if known) |
|---|---|---|---|---|
| S. Bareilly CFSAN000189 | M.SbaUORF19730P | I | gamma | - |
| M.SbaUORF280P | II | beta | - | |
| S. Abony CFSAN001275 | M.SenAboORF8720P | I | gamma | - |
| S. Anatum CFSAN000665 | M.SenAnaDamP | Orphan | alpha | G6mATC |
| M.SenAnaORF14155P | II | alpha | G6mATC | |
| S. Cubana CFSAN002050 | M.Sen2050DamP | Orphan | alpha | G6mATC |
| M.Sen2050ORF235P | II | alpha | G6mATC | |
| M.Sen2050ORF245P | II | gamma | - | |
| M.Sen2050ORF400P | II | gamma | - | |
| M.Sen2050ORF480P | II | beta | - | |
| M.Sen2050ORF4940P | I | gamma | - | |
| M.Sen2050ORF5885P | I | gamma | - | |
| S. Heidelberg CFSAN002064 | M.Sen2064DamP | Orphan | alpha | G6mATC |
| M.Sen2064ORF21380P | II | alpha | G6mATC | |
| Sen2064ORF15615P | II | G,S | GATC6mAG | |
| S. Heidelberg CFSAN002069 | M.Sen2069DamP | Orphan | alpha | G6mATC |
| M.Sen2069ORF4005P | II | alpha | G6mATC | |
| M.Sen2069ORF23325P | II | beta | - | |
| Sen2069ORF9735P | II | G,S | GATC6mAG | |
| S. Javiana CFSAN001992 | M.SenJORF11520P | I | gamma | - |
| M.SenJDamP | orphan | alpha | G6mATC | |
| M.SenJORF19790P | II | alpha | G6mATC | |
| M.SenJORF20475P | II | alpha | G6mATC | |
| M.SenJORF6415P | II | G6mATC | ||
| S. Montevideo CFSAN000255 | M.Sen255DamP | Orphan | alpha | G6mATC |
| M.Sen255ORF17075P | II | alpha | G6mATC | |
| M.Sen255ORF20925P | I | gamma | - | |
| M.Sen255ORF5995P | I | gamma | - | |
| S. Enteritidis CSFAN000111 | M.Sen1427DamP | Orphan | alpha | G6mATC |
| M.Sen1427ORF7380P | I | gamma | - | |
| M.Sen1427ORF7910P | II | alpha | G6mATC | |
| S. Enteritidis CFSAN000158 | M.Sen158DamP | Orphan | alpha | G6mATC |
| M.Sen158ORFDP | II | alpha | G6mATC | |
| M.Sen158ORF20930P | I | gamma | - | |
| S. Typhimurium CFSAN001921 | M.SenTFDamP | Orphan | alpha | G6mATC |
| M.SenTFORF6885P | II | G6mATC | ||
| M.SenTFORF23885P | II | beta | Cm6AGCTG | |
| M.SenTFORF24320P | II | - | ||
| M.SenTFORF3520P | III | beta | - |
am5C MTases not included.
Other observed motifs were common among a subsection of the serovars examined. For example S. Typhimurium and both isolates of S. Heidelberg contained the common motif Gm6AGN6RTAYG that is methylated by a Type I MTase. Six of the nine serovars, S. Bareilly, S. Abony, S. Cubana, S. Javiana, S. Montevideo, and S. Anatum, contained a motif not found in the other serovars tested (Fig 1). For example, in S. Anatum, we observed the motif CCm6AN7 TGAG. Fig 2 shows the kinetic signals of three of these motifs. In most cases these unique motifs were strongly methylated. Several novel motifs were not matched to any MTases including GGm6AN6ATTA and RAm6ACN5 TGA in S. Cubana, and CGm6AYN7RTRTC in S. Montevideo.
Fig 2. Diagram of the interpulse duration (IPD) ratio of three novel motifs identified in three Salmonella serovars.

Vertical axis = IPD ratio, horizontal axis = genome position. IPD ratio listed next to bar. A. Motif CCm6AN8 TGAG in S. Anatum. B. Motif RAm6ACN5 TGA in S. Cubana. C. Motif CGm6AYN6RTRTC in S. Montevideo.
Several observed motifs could not be assigned to a single MTase. In some cases, there were multiple MTases with predicted specificities that matched that of an observed motif. In these cases, it was not possible to predict which enzyme was responsible for the methylation of the observed motifs, and thus no enzyme was assigned. Furthermore, we could not rule out the possibility that multiple enzymes methylated the same motif, as has been observed with Gm6ATC [28]. MTases may also be promiscuous [29], i.e., they methylate multiple motifs, making a match to any single motif unrealistic. In some cases, there was no MTase present in the genome with a specificity predicted to recognize an observed methylated motif.
On other occasions, we did not observe the methylation of a motif that we predicted would be present based on a putative MTase identification. For example, in S. Heidelberg CFSAN002064, we detected the gene for the putative methyltransferase Sen2064ORF15615P, and predicted that it would be responsible for GATCm6AG methylation. However, we did not observe the activity of this methyltransferase in S. Heidelberg, which means the enzyme is inactive. Inactivity can be the result of a mutation in the enzyme which renders it inactive, or, the enzyme may be functional, but not at the time of analysis. For example, some MTases may be inactive due to transcriptional silencing as is often found when the genes are present as part of a prophage [30]. Furthermore, an MTase may be transcribed, but for unknown reasons, may not routinely modify its’ target motif [12]. Cloning MTase genes has shown to be a useful approach for their characterization [6], and may help to match motifs to predicted MTases in cases where bioinformatics alone was insufficient. This approach should be incorporated into future studies that target particular MTases. For example, the cloning of Sen2064ORF15615P in an expression vector would resolve whether the enzyme is inactive or not functional in S. Heidelberg at the time of analysis.
We cannot completely rule out the possibility that DNA MTase genes exist that show no similarity to characterized MTase genes. However, with methylation data from more than 700 genomes available and almost 2,500 characterized and 50,000 putative MTase genes identified in REBASE, the chances of finding a completely new way of methylating DNA are getting increasingly smaller. In particular, we rarely come across a case where we can be certain that there are insufficient MTases to account for the observed patterns of methylation. However, in Salmonella enterica subsp. enterica serovar Heidelberg CFSAN002064, the methylated motif ACCm6ANCC occurs, which may indicate a plasmid is missing. This contrasts with CFSAN002069, which also has this motif, but does have a potential plasmid-encoded MTase. In other cases we have observed this motif is present in strains containing plasmids (R.J. Roberts, unpublished). Furthermore, as more genome sequence data and PacBio methylation data appear, our ability to predict recognition sequences from sequence data alone is growing. Already, rules are becoming apparent for predicting the specificity of Type IIG enzymes [31].
Most of the novel motifs observed in each serovar were modified by Type I RM systems (Fig 1). Type I systems have a modular structure that may allow sequence specificities to diversify more easily than the structures of other RM types (for review, see [32]). Each system consists of two methylase (M) units, two restriction endonuclease (R) units, and one sequence specificity (S) subunit [33, 34]. The S subunit has two TRDs, each of which recognizes one half of the target motif. Recombination events may occur on the S subunit, either within a single TRD or within the sequence that joins the two, resulting in novel specificity. Also, R and M subunits may interact with foreign S subunits entering the cell, also resulting in novel specificity. This has been observed in Lactococcus [35]. One interesting Type I motif, Gm6ATGN5G4m GC, is exhibited by the specificity subunit of the SenAboIII system. This example of cooperation between an m6A methylase and an m4C methylase is quite rare and has only been infrequently observed previously (R. Morgan, unpublished observations).
Unique motifs found among closely related taxa may be the result of horizontal gene transfer (HGT). Studies have demonstrated that HGT accounts for the movements of RM systems based on evidence of codon usage bias [36] and differential GC content of RM genes [37]. We identified several MTases that are located on prophages and plasmids, indicating possible mechanisms of transfer (Table 1). Also, through BLAST similarity searches against REBASE we found that several MTase sequences are most similar, or highly similar, to enzymes in Enterobacteriaceae genera other than Salmonella, suggesting that these systems may have been acquired via HGT. For example, M.SbaUII from S. Bareilly, which methylates the motif Cm6AGCTG, is most similar to an MTase found in Pectobacterium. Currently, we are building a robust Salmonella phylogeny, including representatives of other Enterobacteriaceae genera, to test these and other evolutionary hypotheses.
In some taxa, we detected a proportion of motifs that were not fully methylated within the genome. In particular, only 38–78.5% of ATGCm6AT sites across the genome were methylated, and 89.3–100% of CAGm6AG sites were methylated (Table 2). Orphan MTases or RM systems with an inactive REase often do not methylate all sites in the genome, as complete methylation at all sites to protect from cleavage is usually unnecessary. Incomplete methylation may also be due to the fact that cells are analyzed at different times during the cell cycle, or methylation at certain sites may be inhibited by DNA binding proteins [38]. Environmental factors, including culture conditions, may also affect the frequency of methylation [9, 39]. Incomplete methylation may play a role in the regulation of gene expression. Thus, studies examining the functional implications of ATGCm6AT and CAGm6AG methylation will be particularly interesting.
In several of the genomes, ATGCm6AT methyltransferases are biased towards preferentially methylating this motif when preceded by a cytosine, a thymine, or both. For example, in S. Heidelberg CFSAN002069, AATGCm6AT and GATGCm6AT are methylated at lower frequencies than TATGCm6AT and CATGCm6AT. All four motifs are found in a roughly 1:1:1:1 ratio throughout the genome, indicating a true bias in methyltransferase activity. Currently, we are investigating the biological significance of these observations. Interestingly, 20 ATGCm6AT motifs are present in a collection of 101 previously characterized Salmonella virulence genes [40], and ten of these are AATGCm6ATs, a much higher proportion than what is expected by chance.
Conclusions
In total, we observed 18 motifs among the nine Salmonella serovars, eight of which are novel. These findings indicate the diversity of motifs present in Salmonella enterica. The functions of the observed motifs are unknown, except for Gm6ATC, which has been well studied and is involved in a variety of biological processes including virulence [15]. In E. coli, methylation of CTGCm6AG by the MTase M.EcoGIII, is shown to affect the transcription of over 30% of genes [12]. It is possible that the methylation of motifs in Salmonella described here may also play a role in virulence and other cell functions, and thus merit further study. Future studies should also continue to explore how methylation patterns vary across serovars, and examine within-serovar variation. Methylation may be useful as a typing marker, as closely related taxa are often difficult to differentiate using morphological and molecular markers. The reconstruction of a Salmonella phylogeny, along with the analysis of the methylomes will allow us to address these issues and gain a more broad view of the evolutionary history and functional significance of methylation within the genus.
Supporting Information
The line indicates the QV cutoff used for MTase specificity determination.
(TIF)
(XLSX)
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Center for Food Safety and Applied Nutrition at the US Food and Drug Administration, http://www.fda.gov/AboutFDA/CentersOffices/OfficeofFoods/CFSAN/; the National Institutes of Health (grant number 1R44GM105125 to RJR), www.nih.gov; and the Research Fellowship Program for Center for Food Safety and Applied Nutrition, administered by the Oak Ridge Associated Universities to CPD. New England Biolabs and Pacific Biosciences provided support in the form of salaries for authors RJR, KL, YS, YCT, MB, TAC, and JK, but did not have any additional role in the study design, data collection, decision to publish, or preparation of the manuscript. However, they did participate in analysis of some of the data; RJR identified methyltransferases in the genomes sequenced, and KL, YS, YCT, MB, TAC, and JK helped assemble several of the genomes. The specific roles of these authors are articulated in the ‘author contributions’ section.
References
- 1. Ooi SK, O'Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci. 2009;122:2787–2791. 10.1242/jcs.015123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davis BM, Chao MC, Waldor MK. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. Curr Opin Microbiol. 2013;16:192–198. 10.1016/j.mib.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70:830–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He XJ, Chen T, Zhu JK. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011;21:442–465. 10.1038/cr.2011.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts RJ. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Research. 2003;31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, et al. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012;40:e29 10.1093/nar/gkr1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. 10.1038/nmeth.1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lluch-Senar M, Luong K, Llorens-Rico V, Delgado J, Fang G, Spittle K, et al. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 2013;9:e1003191 10.1371/journal.pgen.1003191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bendall ML, Luong K, Wetmore KM, Blow M, Korlach J, Deutschbauer A, et al. Exploring the roles of DNA methylation in the metal-reducing bacterium Shewanella oneidensis MR-1. J Bacteriol. 2013;195:4966–4974. 10.1128/JB.00935-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, et al. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–11462. 10.1093/nar/gks891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann M, Muruvanda T, Allard MW, Korlach J, Roberts RJ, Timme R, et al. Complete Genome Sequence of a Multidrug-Resistant Salmonella enterica Serovar Typhimurium var. 5- Strain Isolated from Chicken Breast. Genome Announc. 2013;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol. 2012;30:1232–1239. 10.1038/nbt.2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–236. 10.1093/nar/gkp874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, et al. Foodborne Illness Acquired in the United States—Major Pathogens. Emerging Infectious Diseases. 2011;17:7–15. 10.3201/eid1701.091101p1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heithoff DM. An Essential Role for DNA Adenine Methylation in Bacterial Virulence. Science. 1999;284:967–970. [DOI] [PubMed] [Google Scholar]
- 16. Nicolson B, Low D. DNA methylation-dependent regulation of Pef expression in Salmonella typhimurium. Molecular Microbiology. 2000;35:728–742. [DOI] [PubMed] [Google Scholar]
- 17. Jakomin M, Chessa D, Baumler AJ, Casadesus J. Regulation of the Salmonella enterica std fimbrial operon by DNA adenine methylation, SeqA, and HdfR. J Bacteriol. 2008;190:7406–7413. 10.1128/JB.01136-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cota I, Blanc-Potard AB, Casadesus J. STM2209-STM2208 (opvAB): a phase variation locus of Salmonella enterica involved in control of O-antigen chain length. PLoS One. 2012;7:e36863 10.1371/journal.pone.0036863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Broadbent SE, Davies MR, van der Woude MW. Phase variation controls expression of Salmonella lipopolysaccharide modification genes by a DNA methylation-dependent mechanism. Mol Microbiol. 2010;77:337–353. 10.1111/j.1365-2958.2010.07203.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allard MW, Muruvanda T, Strain E, Timme R, Luo Y, Wang C, et al. Fully assembled genome sequence for Salmonella enterica subsp. enterica Serovar Javiana CFSAN001992. Genome Announc. 2013;1:e0008113 10.1128/genomeA.00081-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann M, Muruvanda T, Pirone C, Korlach J, Timme R, Payne J, et al. First Fully Assembled Genome Sequence of Salmonella enterica Serovar Cubana associated with a Multistate Outbreak in the United States. Genome Announcements. in press. [DOI] [PMC free article] [PubMed]
- 22. Evans PS, Luo Y, Muruvanda T, Ayers S, Hiatt B, Hoffman M, et al. Complete Genome Sequences of Salmonella enterica Serovar Heidelberg Strains Associated with a Multistate Food-Borne Illness Investigation. Genome Announc. 2014;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- 24. Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23:1026–1028. [DOI] [PubMed] [Google Scholar]
- 25. Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, et al. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 2009;37:D216–223. 10.1093/nar/gkn734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Broadbent SE, Balbontin R, Casadesus J, Marinus MG, van der Woude M. YhdJ, a nonessential CcrM-like DNA methyltransferase of Escherichia coli and Salmonella enterica. J Bacteriol. 2007;189:4325–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lobner-Olesen A, Skovgaard O, Marinus MG. Dam methylation: coordinating cellular processes. Curr Opin Microbiol. 2005;8:154–160. [DOI] [PubMed] [Google Scholar]
- 29. Woodbury CP, Downey RL, von Hippel PH. DNA site recognition and overmethylation by the Eco RI methylase. Journal of Biological Chemistry. 1980;255:11526–11533. [PubMed] [Google Scholar]
- 30. Ventura M, Canchaya C, Bernini V, Altermann E, Barrangou R, McGrath S, et al. Comparative genomics and transcriptional analysis of prophages identified in the genomes of Lactobacillus gasseri, Lactobacillus salivarius, and Lactobacillus casei. Appl Environ Microbiol. 2006;72:3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morgan RD, Luyten YA. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res. 2009;37:5222–5233. 10.1093/nar/gkp535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blumenthal RM, Cheng X. Restriction-Modification Systems Modern Microbial Genetics: John Wiley & Sons, Inc.; 2002. p. 177–225. [Google Scholar]
- 33. Davies GP, Martin I, Sturrock SS, Cronshaw A, Murray NE, Dryden DT. On the structure and operation of type I DNA restriction enzymes. J Mol Biol. 1999;290:565–579. [DOI] [PubMed] [Google Scholar]
- 34.Loenen WA, Dryden DT, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2013. [DOI] [PMC free article] [PubMed]
- 35. Schouler C, Gautier M, Ehrlich SD, Chopin MC. Combinational variation of restriction modification specificities in Lactococcus lactis. Mol Microbiol. 1998;28:169–178. [DOI] [PubMed] [Google Scholar]
- 36. Jeltsch A, Pingoud A. Horizontal gene transfer contributes to the wide distribution and evolution of type II restriction-modification systems. J Mol Evol. 1996;42:91–96. [DOI] [PubMed] [Google Scholar]
- 37. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. [DOI] [PubMed] [Google Scholar]
- 38. Tavazoie S, Church GM. Quantitative whole-genome analysis of DNA-protein interactions by in vivo methylase protection in E. coli. Nat Biotechnol. 1998;16:566–571. [DOI] [PubMed] [Google Scholar]
- 39. Hale WB, van der Woude MW, Low DA. Analysis of nonmethylated GATC sites in the Escherichia coli chromosome and identification of sites that are differentially methylated in response to environmental stimuli. J Bacteriol. 1994;176:3438–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huehn S, Bunge C, Junker E, Helmuth R, Malorny B. Poultry-Associated Salmonella enterica subsp. enterica Serovar 4,12:d:-Reveals High Clonality and a Distinct Pathogenicity Gene Repertoire. Applied and Environmental Microbiology. 2009;75:1011–1020. 10.1128/AEM.02187-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The line indicates the QV cutoff used for MTase specificity determination.
(TIF)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.