

Abstract
pH is an important factor that shapes the structure of bacterial communities. However, we have very limited information about the patterns and processes by which overall bacterioplankton communities assemble across wide pH gradients in natural freshwater lakes. Here, we used pyrosequencing to analyze the bacterioplankton communities in 25 discrete freshwater lakes in Denmark with pH levels ranging from 3.8 to 8.8. We found that pH was the key factor impacting lacustrine bacterioplankton community assembly. More acidic lakes imposed stronger environmental filtering, which decreased the richness and evenness of bacterioplankton operational taxonomic units (OTUs) and largely shifted community composition. Although environmental filtering was determined to be the most important determinant of bacterioplankton community assembly, the importance of neutral assembly processes must also be considered, notably in acidic lakes, where the species (OTU) diversity was low. We observed that the strong effect of environmental filtering in more acidic lakes was weakened by the enhanced relative importance of neutral community assembly, and bacterioplankton communities tended to be less phylogenetically clustered in more acidic lakes. In summary, we propose that pH is a major environmental determinant in freshwater lakes, regulating the relative importance and interplay between niche-related and neutral processes and shaping the patterns of freshwater lake bacterioplankton biodiversity.
INTRODUCTION
Bacterioplankton are a crucial component of aquatic ecosystems, and they play an important role in the ecological processes of freshwater lakes (1, 2). Understanding the mechanisms governing bacterioplankton community assembly (e.g., community composition, diversity, and phylogenetic structure) in natural freshwater lakes is a longstanding challenge in microbial community ecology. There is increasing evidence that the community structure of freshwater lake bacterioplankton is influenced by niche-related (deterministic) and neutral (stochastic) processes (2–4). Niche-related processes (5) include selection imposed by the abiotic environment (environmental filtering) and species interactions (competition, facilitation, mutualism, and predation), whereas neutral effects (6) include stochastic processes, such as unpredictable disturbances, probabilistic dispersal, and random birth/death events (7, 8). Neutral processes likely result in random fluctuations in community composition along a given environmental gradient (9). The observation that both niche and neutral processes are important in shaping bacterioplankton community assembly raises the question of what affects the relative contributions and interplay between stochastic and deterministic processes in structuring lacustrine bacterioplankton assembly.
Multiple environmental factors regulate the distribution of bacterioplankton communities in freshwater lakes (10–16). pH is proposed to be an important environmental factor influencing both overall bacterioplankton community composition (BCC) (2, 17) and the compositions of abundant bacterioplankton groups, including Polynucleobacter and Limnohabitans (e.g., 18, 19), in freshwater lakes. Newton et al. (20) performed clone library sequencing and observed that pH was also a key environmental determinant of the phylogenetic relatedness of the freshwater lake Actinobacteria acI lineage; acidic lakes (pH 5.1 to 6) contained more closely related acI clades than would be expected at random. However, so far, few papers have systematically discussed the assembly of overall lacustrine bacterioplankton communities across wide pH gradients with more detailed phylogenetic information, and therefore, we do not know much about the patterns of overall lacustrine bacterioplankton diversity and community composition across wide pH gradients. Furthermore, how pH influences the relative importance and interplay between niche-related and neutral processes in structuring overall communities is also largely unknown.
One promising approach to understanding the underlying niche-related versus neutral processes in shaping community assembly is the use of a community phylogenetic structure test (21–23). By comparing community phylogenetic structure with randomization procedures, known as null models, and estimating the deviations from null-model expectations, the changes in the relative influences of deterministic and stochastic processes across environmental gradients can be inferred (21–23). The foundation of this phylogenetic framework is the assumption that closely related taxa are more likely to have similar niches, often termed phylogenetic niche conservatism (21, 24). If the phylogenetic niche conservatism is supported, then a nonrandom community phylogenetic structure is expected to indicate the occurrence of niche-based processes, such as environmental filtering and competitive interactions (24). Although the phylogenetic framework has several notable advantages, it is rarely used to study bacterial communities in freshwater lakes, especially when the communities span a broad range of environmental characteristics (25). However, freshwater ecosystem studies have observed that freshwater bacterial communities tend to be more clustered in more “stressful” habitats; for example, there is increased clustering of Betaproteobacteria in less productive freshwater mesocosms (25) and increased clustering of the Actinobacteria acI lineage in more acidic lakes (20).
The intracellular pH of most microorganisms is usually within 1 pH unit of neutral pH (26). Therefore, we hypothesized that the “stress” habitats, such as acidic lakes in our study, might directly impose greater environmental filtering on the community assembly, thereby decreasing overall bacterioplankton diversity and shifting the community composition to taxa that can better adapt to the environments. Additionally, the increased pH filtering effects in acidic lakes might increase the phylogenetic clustering of the bacterioplankton communities. Although environmental filtering is an important determinant of community assembly (20, 25), other mechanisms, such as neutral processes, must also be considered. Recent researches have shown that neutral processes have a strong impact on the accumulation of rare taxa in a community, and the relative influence of neutral processes on community assembly may increase when community populations are small (6, 7). Therefore, we also hypothesized that the enhanced neutral assembly processes in small bacterioplankton communities might balance or weaken the relative importance of environmental filtering for overall bacterioplankton communities and would also alter the phylogenetic assembly patterns of overall communities across the pH gradient. To test these two hypotheses, we selected 25 discrete freshwater lakes in central Jutland, Denmark. The pH of freshwater lakes, including bog humic lakes, is generally between 4 and 9, but pH values can be lower than 4 in extreme volcanic or mining water (27). To sample a wider pH range, we included two lakes with pH values of ≤4 in our study; these lakes were formed from abandoned mining operations approximately 70 years ago. Using high-throughput pyrosequencing, we analyzed the compositions and diversity of the bacterioplankton communities in 25 discrete freshwater lakes with pH values ranging from 3.8 to 8.8. We also analyzed the bacterioplankton phylogenetic community structure across the pH gradient and studied the mechanisms shaping this structure.
MATERIALS AND METHODS
Sample collection and chemical analyses.
During May and June 2012, we collected water samples from 25 discrete freshwater lakes in central Jutland, Denmark. These lakes are located in the same geographical area, and pairwise distances between the lakes are less than 40 km. They have no surface water inputs and span a pH range of 3.8 to 8.8 (see Table S1 in the supplemental material). Some acidic lakes, such as lakes No Name 1 and No Name 2 (see Table S1 in the supplemental material), were formed from mining activities conducted in the 1940s. Based on the lake shape and size (area < 0.76 km2; see Table S1 in the supplemental material), we selected six different sampling points in each lake. The sampling points were generally distributed uniformly. At each sampling point, bacterial samples (400 to 500 ml of water) were collected from surface waters (the top 50 cm) and filtered through 0.2-μm-pore-size Isopore filters (Millipore, Billerica, MA, USA). The filters were stored at −20°C. Water pH and conductivity were measured in the field at each location. Untreated surface water samples (approximately 300 ml) were collected from each sampling point for chemical analysis. Using standard methods (28), the total organic carbon (TOC), total nitrogen (TN), total phosphorus (TP), total iron (TFe), and nitrate nitrogen (NOx−) content of the water were determined. Additionally, historical data were obtained for most of the examined lakes and are cited in our study. The geographical, physical, and chemical characteristics of the examined lakes are shown in Table S1 in the supplemental material. Environmental variables, including conductivity, TOC, TN, TP, TFe, NOx−, and lake area, were transformed by log (x + 1) in all subsequent analyses.
DNA extraction, amplification, and 454 sequencing.
Genomic DNA was extracted from the biomass collected on the filters using a standard phenol-chloroform extraction method (11) and purified using the Wizard DNA cleanup kit (Promega, Madison, WI, USA). The bacterial 16S rRNA genes were amplified with the forward primer 341F (5′ CCT ACG GGA GGC AGC AG 3′) with the Roche 454 A pyrosequencing adapter and a unique 7-bp barcode sequence and with the reverse primer 1073R (5′ACG AGC TGA CGA CAG CCA TG 3′) with the Roche 454 B sequencing adapter at the 5′ end. Three replicates of each sample were amplified in 20-μl reaction mixtures under the following conditions: 24 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 5 min. The PCR products were visualized by electrophoresis, and the replicates were pooled. The purified PCR products were combined in equimolar ratios and then sequenced using a Roche 454 FLX+ pyrosequencing machine.
Pyrosequencing data analysis.
Raw reads of the 16S rRNA gene sequences were processed using the software package Mothur (v. 1.30.0) (http://www.mothur.org) according to the standard 454 operating procedure (SOP) (29). In brief, the raw reads were denoised, trimmed, quality filtered, and aligned to the Silva database (30) using Mothur. Clean sequences of six replicates for each lake were then aggregated into one group. To build the operational taxonomic unit (OTU) matrix, we first generated a distance matrix from the clean sequences using the dist.seqs command in Mothur. In the process, we excluded any pairwise distance larger than 0.15. Next, we clustered the clean sequences into OTUs based on the newly created distance matrix by using the cluster command and the furthest-neighbor algorithm method in Mothur. Finally, we generated an OTU-shared file at a cutoff value of 0.03 (OTU0.03s) using the make.shared command in Mothur. To correct for sequencing bias, we randomly subsampled the minimum number of sequences (i.e., 19,000 sequences) from each lake, and OTUs with fewer than 3 sequences were excluded from the OTU table using the Remove.rare command in Mothur. Representative sequences of OTU0.03s were generated within Mothur and then classified into taxonomic groups using the typical freshwater bacterial database provided by Newton et al. (2) at the recommended bootstrap threshold of 80% (31). Unclassified representative sequences of OTU0.03s were further identified using the Silva bacterial database and by BLAST against GenBank entries (http://blast.ncbi.nlm.nih.gov/Blast.cgi). A phylogenetic tree was constructed using the FastTree algorithm (v. 2.1.3) (32) (FastTree flagged conditions: -fastest, -gtr, and -nt). By matching with the corrected OTU table, a new phylogenetic tree was generated by using the match.phylo.comm command in the picante package in the R statistical language (R Development Core Team [2008]).
Diversity analyses.
Taxonomic richness (i.e., the number of OTU0.03s) (33) and Pielou's evenness (34) were calculated using the vegan package in R. Faith's phylogenetic diversity (PD) (35) was measured using the phylogenetic tree and the OTU0.03 table with the pd command in the picante package in R. The BCC dissimilarities between lakes (beta diversity) were examined using the taxonomy-based Bray-Curtis distance and the phylogeny-based UniFrac distance, which were calculated using the vegan package and the picante package, respectively, in R.
Community phylogenetic structure.
To exclude the impact of OTU richness on PD variation, we compared the PD of a particular community to those of randomized null communities of the same size and calculated the standardized-effect size of PD (SES.PD) using the ses.pd command in the picante package in R. The null model was performed by shuffling taxon labels 999 times across the tips of the phylogenetic tree using a given tree topology and branch lengths to randomize phylogenetic relationships among OTU0.03s. SES.PD equals the differences in PD between the observed communities and the mean value of the 999 null communities divided by the standardized deviation of the PD in the 999 null communities (21).
The phylogenetic signal was examined by conducting Mantel correlograms with 999 randomizations between the pairwise matrix of OTU0.03 phylogenetic distances and OTU0.03 ecological-niche distances (i.e., the difference in habitat requirements for each OTU0.03) (36, 37) as proposed by Wang et al. (37). The metrics measuring the mean phylogenetic distance to each taxon's closest relative (the mean nearest taxon distance [MNTD] and the standardized-effect size of the MNTD [SES.MNTD]) (22) were also examined in our study. Using the ses.mntd command in the picante package of R, the abundance-unweighted MNTD and SES.MNTD were calculated. The null model used for SES.PD was also used for SES.MNTD. SES.MNTD equals the differences in MNTD between the observed communities and the mean value of the 999 null communities divided by the standard deviation of the MNTD in the 999 null communities (21). Using a sample t test, the mean SES values of PD and MNTD across all lakes were compared with zero, which is the value expected when the average phylogenetic structure of local communities is the same as that of the null communities. SES values greater than zero indicate phylogenetic overdispersion (taxa more distantly related than would be expected at random), and SES values less than zero suggest clustering (taxa more closely related than would be expected at random).
Statistical analyses.
A heuristic method was used to estimate the relative weight of each environmental variable in relation to the relative abundances of the different phyla in multiple regression (38). Both the relative abundances and the numbers of OTUs of the main lineages or clades of bacterioplankton communities along the pH gradient were depicted in a heat map using the pheatmap command in the pheatmap (Pretty Heatmaps) package in R.
The taxonomy-based and phylogeny-based distances were visualized using nonmetric multidimensional scaling (NMDS) in the vegan package in R. By using the mantel.partial command in the vegan package in R, the partial Mantel statistic with 9,999 permutations (39) was performed to calculate the partial matrix correlation between three dissimilarity matrices: the BCC matrix, one of the examined environmental variables, and the conditioned matrix of the rest of the environmental variables. Analyses of similarity (ANOSIM) were also performed to test the differences in BCC across different pH categories (pH 3 to 4, 5 to 6, 6 to 7, and 7 to 9) using the anosim command in the vegan package of R. Additionally, multiple regression on distance matrices (MRM) with 9,999 permutations (40) was used to partition the variations in BCC into pure environment, pure space, mixed space/environment, and unexplained components by using the MRM command in the ecodist package of R. The best subset of environmental parameters applied in the MRM analysis was chosen by the BIOENV procedure using the vegan package in R (41).
We also performed univariate models to predict bacterioplankton alpha diversity and the metrics of community phylogenetic structure with various environmental characteristics by using the lm command in the stats package of R. In each case, we fitted a linear and a quadratic model, and the results are shown for the model with the lowest Akaike information criterion (AIC) value.
Nucleotide sequence accession numbers.
The sequence data were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under the study accession number SRP034651. The accession numbers for each lake are listed in Table S1 in the supplemental material.
RESULTS
From the entire sample set (see Table S1 in the supplemental material), we obtained 638,782 quality sequences with an average read length of 566 bp. The number of quality sequences per lake ranged from 19,168 to 34,881 (see Table S1 in the supplemental material). After rarefaction to an equal sequencing depth (19,000 sequences per lake), we obtained 9,401 OTUs with 97% similarity across the entire sample set, with an average of 780 OTUs per sample. To avoid possible sequencing bias, OTUs with fewer than 3 sequences were excluded from the OTU table. We then obtained a total of 3,168 OTUs; 5 OTUs contained more than 10,000 sequences, with a maximum value of 18,098 sequences. In addition, the numbers of OTUs with sequence counts ranging from 1,000 to 10,000, 100 to 1,000, 10 to 100, and 3 to 10 were 73, 333, 915, and 1,842, respectively. Among the quality sequences, 92% were classifiable at the phylum level, and a total of 28 phyla (subphyla) were identified. The predominant phyla (subphyla) across all lakes were Betaproteobacteria, Actinobacteria, Gammaproteobacteria, Bacteroidetes, and Alphaproteobacteria, representing approximately 26.9%, 25.8%, 12.0%, 10.5%, and 10.2% of all sequences, respectively (see Fig. S1 in the supplemental material). In addition, Firmicutes (4.7%), Verrucomicrobia (1.3%), Deltaproteobacteria (0.25%), TM7 (0.10%), and Acidobacteria (0.06%) were present in most lakes, but at relatively low abundances (see Fig. S1 in the supplemental material).
We observed that the relative abundances of some phyla, such as Actinobacteria, Bacteroidetes, Alphaproteobacteria, Betaproteobacteria, and Acidobacteria, were best explained by pH among all of the environmental variables included in our study (see Table S2 in the supplemental material). The relative abundances of Actinobacteria (Fig. 1a) and Bacteroidetes (Fig. 1b) were positively correlated with pH, and the abundances of Alphaproteobacteria (Fig. 1c), Betaproteobacteria (Fig. 1d), and Acidobacteria (Fig. 1e) decreased with pH. However, the abundance of Gammaproteobacteria showed no obvious relationship with any environmental variable (Fig. 1f; see Table S2 in the supplemental material). The impact of pH on the BCC was also evident at the lineage and clade levels. In the two most acidic lakes, the main lineages or clades were mostly composed of the specific bacteria Ferrovum (on average, 65% of all sequences; Betaproteobacteria) and Acidocella (20%; Alphaproteobacteria) (Fig. 2). Furthermore, we observed that the relative abundances of acI-A1, acI-B2, and acI-B4 of Actinobacteria; alfI of Alphaproteobacteria; and PnecC of Betaproteobacteria were greater in moderately acidic lakes (pH 5 to 7), whereas the abundances of acI-B1, acIV-A, acIV-B, Luna1, and acSTL of Actinobacteria; LD12 of Alphaproteobacteria; Lhab-A1 and PnecB of Betaproteobacteria; and Acin of Gammaproteobacteria were greater in neutral and alkaline lakes (Fig. 2).
FIG 1.

Relative abundances of Actinobacteria (a), Bacteroidetes (b), Alphaproteobacteria (c), Betaproteobacteria (d), Acidobacteria (e), and Gammaproteobacteria (f) across the pH gradient. The relative abundances of Actinobacteria and Bacteroidetes positively correlated with pH; the abundances of Alphaproteobacteria, Betaproteobacteria, and Acidobacteria decreased with pH; Gammaproteobacteria showed no significant relationship with pH. The formulas, R2 values, and significances are given in each panel.
FIG 2.

Heat map describing the relative abundances of the main lineages or clades of bacterioplankton communities (y axis) across the pH gradient (x axis). High heterogeneity in community composition was also evident at the lineage and clade levels across the pH gradient.
The bacterioplankton community composition was also strongly influenced by the water pH at the species-like level. An NMDS plot revealed that both the taxonomic (OTU) and phylogenetic compositions of different pH groups (pH 3 to 4, 5 to 6, 6 to 7, and 7 to 9) aligned along NMDS axis 1 (Fig. 3a and b), suggesting the existence of strong linkages between water pH and community structure. Furthermore, we observed that the bacterioplankton communities from different lakes within the same pH group (pH 3 to 4, 5 to 6, 6 to 7, and 7 to 9) tended to be similar in composition (Fig. 3a and b; see Table S1 in the supplemental material). Conversely, substantial heterogeneity in community composition was detected between different pH groups (ANOSIM analyses; R = 0.63 [P < 0.001] for taxonomic composition [Fig. 3a] and R = 0.61 [P < 0.001] for phylogenetic composition [Fig. 3b]). The taxonomic and phylogenetic distances between bacterial communities (beta diversity) were significantly correlated with the pH differences between lakes (Fig. 3c and d), and they increased with increasing pH differences (R2 = 0.54 [P < 0.001] for taxonomic composition; R2 = 0.50 [P < 0.001] for phylogenetic composition [Fig. 3c and d]). A partial Mantel test indicated that, among the environmental characteristics examined, pH was the best predictor of bacterioplankton community composition (Spearman's R = 0.725 [P < 0.001] for taxonomic composition; R = 0.789 [P < 0.001] for phylogenetic composition [Table 1]), followed by TOC (Spearman's R = 0.340 [P < 0.05] for taxonomic composition; R = 0.328 [P < 0.05] for phylogenetic composition [Table 1]). MRM analysis revealed that the best subset of environmental variables (including pH and TOC) selected by the BIOENV procedure explained 38% of the taxonomic composition and 46% of the phylogenetic composition (see Table S3 in the supplemental material). The geographical distances between lakes also displayed significant correlation with community composition, but the effect of pure geographical distance was much weaker than that of pure environmental variables (see Table S3 in the supplemental material).
FIG 3.

(a and b) NMDS plots derived from the taxonomy-based Bray-Curtis distance (a) and the phylogeny-based UniFrac distance (b), with symbols showing pH categories. (c and d) Relationships between pH differences and the taxonomic (Bray-Curtis) (c) and phylogenetic (UniFrac) (d) dissimilarities of lakes compared pairwise. Substantial heterogeneity in BCC was observed between different pH groups (pH 3 to 4, 5 to 6, 6 to 7, and 7 to 9). The taxonomic and phylogenetic BCC dissimilarities between lakes increased with increasing pH differences. The formulas, R2 values, and significances are given in each panel.
TABLE 1.
Spearman's correlations (R values) between transformed environmental variables and taxonomic (Bray-Curtis distance) and phylogenetic (UniFrac distance) community compositions as determined via partial Mantel tests
| Factor | Taxonomic composition |
Phylogenetic composition |
||
|---|---|---|---|---|
| R | P | R | P | |
| pH | 0.725 | 0.001a | 0.789 | 0.001a |
| TOC (mg · liter−1) | 0.340 | 0.010b | 0.328 | 0.016b |
| TFe (mg · liter−1) | −0.020 | 0.563 | −0.059 | 0.736 |
| Area (km2) | −0.164 | 0.926 | −0.197 | 0.967 |
| NOx− (mg · liter−1) | 0.223 | 0.076 | 0.187 | 0.077 |
| TP (mg · liter−1) | −0.115 | 0.783 | −0.111 | 0.811 |
| Conductivity (μS · cm−1) | −0.114 | 0.862 | −0.068 | 0.733 |
| TN (mg · liter−1) | −0.168 | 0.967 | −0.164 | 0.960 |
P < 0.01.
P < 0.05.
We observed that pH was the best predictor of community composition at different taxonomic resolution levels; pH was also the strongest predictor of overall bacterioplankton alpha diversity (Fig. 4a). We observed that bacterioplankton OTU richness (at 97% similarity) varied across the lakes (Fig. 4a) and that water pH was the best predictor of OTU richness among all of the examined environmental parameters (Table 2). The best model relating OTU richness and pH followed a quadratic (hump-shaped) pattern (R2 = 0.78; P < 0.001), with peak diversity in nearly neutral lakes and the lowest diversity in the most acidic lakes (Fig. 4a). Furthermore, we observed that among all the environmental variables examined, pH best explained Pielou's evenness of bacterioplankton communities (Table 2). This variable had a quadratic relationship with pH (R2 = 0.79; P < 0.001); the evenness peaked in nearly neutral lakes and reached a minimum in the two most acidic lakes (Fig. 4b).
FIG 4.
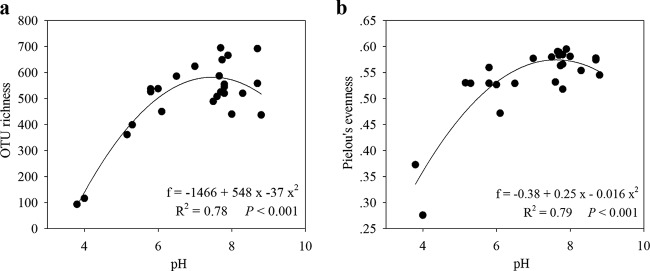
Bacterioplankton OTU richness (a) and community equitability, as indicated by Pielou's evenness (b), along the pH gradient. OTU richness and Pielou's evenness showed quadratic (hump-shaped) relationships with pH; the OTU diversity and Pielou's evenness peaked in nearly neutral lakes and were lowest in the most acidic lakes. The formulas, R2 values, and significances are given in each panel.
TABLE 2.
Univariate models predicting bacterioplankton alpha diversity as a function of various environmental characteristics in freshwater lakesa
| Characteristic | OTU richness |
Pielou's evenness |
Faith's PD |
SES.PD |
MNTD |
SES.MNTD |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model type | AIC value | R2 | Model type | AIC value | R2 | Model type | AIC value | R2 | Model type | AIC value | R2 | Model type | AIC value | R2 | Model type | AIC value | R2 | |
| pH | Quadratic | 290 | 0.78 | Quadratic | −93 | 0.79 | Quadratic | 169 | 0.74 | Linear | 108 | 0.66 | Quadratic | −130 | 0.86 | Linear | 103 | 0.64 |
| TOC (mg · liter−1) | Quadratic | 313 | 0.43 | Quadratic | −62 | 0.28 | Quadratic | 191 | 0.39 | Quadratic | 131 | 0.21 | Quadratic | −92 | 0.34 | Quadratic | 125 | 0.20 |
| TFe (mg · liter−1) | Linear | 325 | 0.01 | Linear | −56 | 0.02 | Linear | 200 | 0.06 | Quadratic | 129 | 0.29 | Quadratic | −86 | 0.15 | Quadratic | 124 | 0.26 |
| Area (km2) | Linear | 323 | 0.08 | Linear | −58 | 0.08 | Linear | 200 | 0.06 | Linear | 133 | 0.08 | Linear | −86 | 0.06 | Linear | 128 | 0.05 |
| NOx− (mg · liter−1) | Linear | 325 | 0.01 | Quadratic | −57 | 0.14 | Quadratic | 199 | 0.17 | 135 | 0.01 | Quadratic | −87 | 0.18 | 128 | 0.03 | ||
| TP (mg · liter−1) | Linear | 325 | 0.02 | Linear | −57 | 0.04 | Linear | 201 | 0.01 | Quadratic | 133 | 0.12 | Quadratic | −85 | 0.12 | Quadratic | 128 | 0.12 |
| Conductivity (μS · cm−1) | Linear | 325 | 0.01 | Linear | −56 | 0.01 | Linear | 202 | 0.01 | Linear | 134 | 0.05 | Linear | −85 | 0.04 | Linear | 128 | 0.05 |
| TN (mg · liter−1) | Linear | 324 | 0.03 | Linear | −56 | 0.02 | Linear | 201 | 0.01 | Linear | 135 | 0.02 | Linear | −85 | 0.02 | Linear | 129 | 0.02 |
In each case, we fitted a linear and a quadratic model; results are shown for the model with the lowest AIC value. Lower AIC values indicate stronger support for the model, balancing model fit and parsimony.
To refine our quantification of alpha diversity by adding an evolutionary dimension, we calculated Faith's PD, which focuses on summing the total phylogenetic branch length contained in a community. We observed that PD had a significantly positive correlation with OTU richness (R = 0.902; P < 0.001; n = 25) and also showed the best relationship with pH (quadratic model; R2 = 0.74; P < 0.001) (Fig. 5a) among all the environmental variables examined (Table 2). However, this quadratic pattern of PD across the pH gradient might simply be due to differences in OTU richness, as the PD increases as species (OTUs) are added to a community. Therefore, to exclude the effect of OTU richness on PD variation, standardized PD was examined using a null-model analysis. Notably, we observed that, in contrast to PD, the SES.PD showed extremely different patterns across the pH gradient (Fig. 5b). The SES.PD decreased linearly with increasing pH (R2 = 0.66; P < 0.001) (Fig. 5b), suggesting that the overall degree of phylogenetic relatedness of phylogenetic relatives in communities was lower in more acidic lakes. Furthermore, a sample t test revealed that, across all local lakes, the mean of the SES.PD (mean = −10.8; t = −16.6; P < 0.001) was significantly less than zero, indicating that the overall bacterioplankton communities across all the lakes tended to be more phylogenetically clustered than would be expected by random dispersion.
FIG 5.

Phylogenetic structures of bacterioplankton communities along the pH gradient. (a) PD. (b) SES.PD. (c) MNTD. (d) SES.MNTD. In contrast to PD and MNTD, SES.PD and SES.MNTD decreased linearly with increasing pH, suggesting that phylogenetic clustering of both overall phylogenetic relatives and the most closely related taxa was lower in more acidic lakes. The formulas, R2 values, and significances are given in each panel.
Our results also revealed a significantly positive relationship between OTU phylogenetic distances and OTU ecological-niche distances across relatively short phylogenetic distances (<13% of the maximum phylogenetic distance) (see Fig. S2 in the supplemental material). This relationship indicates that closely related taxa (OTUs) possess niche conservatism (24), and hence, there might be strong interactions between closely related taxa (36, 37). We therefore calculated the MNTD metric and observed that water pH was the strongest predictor of MNTD among all the environmental characteristics examined (Table 2). However, unlike OTU richness and PD, MNTD showed a U-shaped quadratic pattern across the pH gradient (R2 = 0.86; P < 0.001) (Fig. 5c), with the lowest MNTD in nearly neutral lakes and the highest MNTD in the most acidic lakes (Fig. 5c). To exclude the potential impact of OTU richness on MNTD changes, we performed a null-model analysis to estimate the standardized MNTD. A sample t test revealed that, similar to SES.PD, the mean of the SES.MNTD across all the lakes was also significantly negative (mean = −9.6; t = −16.5; P < 0.001), suggesting that niche-related processes were more important than neutral effects in shaping bacterioplankton community assembly in these lakes, and the terminal phylogenetic dispersion of the overall bacterioplankton communities across all lakes was more phylogenetically clustered than would be expected by chance. However, we observed that, similar to SES.PD, the SES.MNTD also decreased linearly with increasing pH along the pH gradient (R2 = 0.64; P < 0.001) (Fig. 5d). Therefore, although environmental filtering was determined to be the most important determinant of bacterioplankton community assembly in these lakes, the relative importance of neutral community assembly processes increased in more acidic lakes, and the most closely related bacterioplankton OTUs were more phylogenetically distant from each other in more acidic lakes.
DISCUSSION
In this study, we examined changes in freshwater bacterioplankton community assembly and the mechanisms underlying these changes, including the relative importance of and interplay between niche-related and neutral processes across the pH gradient. We observed that pH was the key environmental determinant of the assembly processes in bacterioplankton communities in freshwater lakes, and apparent pH-related patterns of composition, diversity, and phylogenetic structure were observed across the pH gradient.
pH acts as a major environmental determinant of bacterial community assembly in freshwater lakes.
Recent studies have shown that both niche and neutral processes are important in shaping freshwater lake bacterial community assembly (2, 3). Our study also revealed that both environmental variables and geographical distance were significant for explaining variations in the BCC; however, environmental variables had a much stronger effect than geographical distance. This finding suggests that niche-based assembly has an important role in generating spatial patterns of the lacustrine BCC across pH gradients. The potential but weak effect of geographical factors suggests that air, dust, rain, and seepage do not serve as strong vectors of bacterioplankton dispersal in discrete lakes (42–44). Furthermore, we observed similar compositions in geographically distant lakes with similar pHs (Fig. 3; see Table S1 in the supplemental material); therefore, dispersal limitation might be impossible within the narrow region included in this study. Among all the environmental variables examined, we observed that pH was the strongest determinant of community assembly, including its composition, diversity, and phylogenetic structure. This result is consistent with those of Newton et al. (2), who used detrended correspondence analysis (DCA) and observed that, compared to the ratio of TP to dissolved organic carbon (DOC) concentrations and to the latitudinal positions of the lakes, pH showed the best correlation with the axis that explained the major fraction of freshwater lake bacterial community distribution.
High heterogeneity in community composition across the pH gradient.
Similar to the results for soil bacterial communities (45, 46), our results revealed that both BCC taxonomic and phylogenetic dissimilarities increased with increasing pH differences. This powerful effect of pH was also evident at coarse levels of taxonomic resolution. For instance, we observed that, in line with the findings of Kampe et al. (47), bacterioplankton communities in the two most acidic mining lakes were mostly dominated by Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria, whereas Actinobacteria, and Bacteroidetes were rarely present (see Fig. S1 in the supplemental material). At the lineage or clade level, we also observed large shifts in BCC in the two acidic mining lakes compared with other lakes with pH values over 5, suggesting a potential threshold effect or regime shift in the filtering impact of pH on BCC. This hypothesis is supported by the observation that the bacterioplankton communities in the two mining lakes were predominantly composed of the specific bacteria Ferrovum and Acidocella rather than the common lineages or clades in the other lakes. These two clades, Ferrovum and Acidocella, comprise acidophilic iron-oxidizing (48) and acidophilic iron-reducing (49) bacteria, respectively. Both types of bacteria can tolerate high proton concentrations in the surrounding water (47); the dominance of Ferrovum and Acidocella has been observed in other mining lakes with pH values below 4 (47, 49, 50). Except in the most acidic mining lakes, high composition heterogeneity was observed in lakes belonging to different pH categories (pH 5 to 6, 6 to 7, and 7 to 9). As shown previously (2, 18, 19), Pnec (Betaproteobacteria) was abundant in moderately acidic lakes; within this clade, PnecB preferred neutral and alkaline lakes, whereas PnecC was present in all lakes but maximally abundant in moderately acidic lakes. Additionally, we found, consistent with the research of Newton et al. (20), that there was potential ecological diversification within the Actinobacteria acI lineage: acI-A1, acI-B2, and acI-B4 prefer moderately acidic lakes, whereas acI-B1 favors more alkaline lakes.
Bacterioplankton OTU richness peaked in nearly neutral lakes and decreased in more acidic lakes.
We observed that the OTU richness of the bacterioplankton communities shared a quadratic (hump-shaped) relationship with pH, with peak diversities occurring in nearly neutral pH lakes and the lowest diversities occurring in the most acidic mining lakes. A hump-shaped relationship between pH and bacterial alpha diversity has rarely been observed in lake ecosystems, possibly because earlier studies spanned a narrower pH range. However, a similar pattern was observed in soil ecosystems (51, 52). Our results are also consistent with those of previous studies on acidification-impacted lakes that show low diversity with respect to bacteria (53) and higher organisms (54, 55) because of their acidification.
There are several potential explanations for the observed alpha diversity pattern. The intracellular pH of most microorganisms is usually within 1 pH unit of neutral (26); therefore, acidic habitats may directly impose a strong physiological stress on bacteria, alter competitive outcomes, or restrict the net growth of bacterial populations. Some taxa may be unable to survive or actively reproduce when the pH is outside a certain range (26). pH also affects other factors, including nutrient availability (e.g., decreased labile organic carbon in acidic mining waters [47, 56]) and cationic metal solubility (e.g., increased toxic metal solubility in acidic lakes [49]), that are crucial for the maintenance of bacterial diversity. In addition, the idea that higher environmental heterogeneity (such as a complex food web and complex biotic interactions) supports higher biodiversity may be applied to bacterioplankton, as the food web is much simpler in acidic lakes (57, 58).
Less phylogenetic clustering and increased neutral assembly processes of bacterioplankton communities in more acidic lakes.
We performed a community phylogenetic-structure test to incorporate phylogenetic data into analyses of community assembly (22, 23). This approach allows us to obtain insight into the potential processes underlying the patterns of phylogenetic structure (22). Our study revealed that, among the environmental variables examined, the patterns of community phylogenetic structure (SES.PD and SES.MNTD) correlated best with pH, and phylogenetic clustering of both overall phylogenetic relatives and the most closely related OTUs decreased linearly with decreasing pH. This observation raised the following question: why is the phylogenetic dispersion of bacterioplankton communities less phylogenetically clustered in more acidic lakes? In our analyses of bacterioplankton diversity and composition, we observed that pH was a key environmental determinant that restricted the composition and decreased the taxonomic richness of bacterioplankton communities in more acidic lakes. Only those taxa that can adapt well to habitats with high proton concentrations are able to actively reproduce and reach prevalence in acidic lakes (26). We therefore propose that the direct or indirect filtering effect of pH must increase in more acidic lakes. Under these conditions, pH likely determines whether the niche-conservative taxa are good competitors, fugitives, or dormant in a habitat with a certain pH; therefore, a clade of good competitors is likely predominant and overrepresented in competitive communities, as observed for the most abundant acidophilic bacteria, Ferrovum (Betaproteobacteria; the highest number of OTUs [5 OTUs] per lake) and Acidocella (Alphaproteobacteria; 3 OTUs), in the most acidic mining lakes and for the thriving clade PnecC (Betaproteobacteria; 6 OTUs) in moderately acidic lakes. This filtering effect of pH may have caused the increased phylogenetic clustering of overall bacterioplankton communities in more acidic lakes, as shown by Newton et al. (20), who performed clone library sequencing and observed that acidic lakes (pH 5.1 to 7) tended to contain more closely related Actinobacteria acI clades. However, in our study, overall bacterioplankton communities tended to be less phylogenetically clustered in more acidic lakes.
Our contradictory results might be due to the numbers of rare taxa (OTUs) observed in our study using deep pyrosequencing, and these rare taxa were as important as the abundant taxa in the calculations of SES.PD and abundance-unweighted SES.MNTD. Most of these rare OTUs arose by neutral assembly, such as unpredictable disturbances, probabilistic dispersal, and random birth/death (7). Previous studies have suggested that neutral processes have a stronger influence on community assembly when community populations are small (6, 7). In our study, we also found that the relative importance of neutral community assembly processes increased in more acidic lakes, where the OTU richness was low (Fig. 4a). Thus, the rare OTUs found in the most acidic mining lakes and the moderately acidic lakes were most likely caused by the enhanced neutral processes, and many of them accumulate over time in communities (7). Because of the rigorous physiological stress of low pH, these rare OTUs in acidic lakes might have low metabolic or reproductive activity (probably dormant taxa), and thus, many of them might have fewer congeners (59). Such taxa in the two most acidic mining lakes probably included some of the common lineages or clades in the lakes with pH values above 5.0, such as acIV-C (with the highest number of OTUs [1 OTU] per lake), acIV-D (1 OTU), Luna1 (1 OTU), and acSTL (2 OTUs) of Actinobacteria; bacIII (1 OTU) and Muci (1 OTU) of Bacteroidetes; Brev (3 OTUs), alfIII (2 OTUs), LD12 (1 OTU), and alfVI (1 OTU) of Alphaproteobacteria; Lhab-A4 (2 OTUs), Rhodo (1 OTU), PnecC (1 OTU), and betVII-A1 (2 OTUs) of Betaproteobacteria; and gamII-A1 (2 OTUs), gamII-A2 (1 OTU), Acin (6 OTUs), Pseudo-A1 (7 OTUs), and gamV (4 OTUs) of Gammaproteobacteria (see Fig. S3 in the supplemental material), while such taxa in the moderately acidic lakes likely included some of the common lineages or clades in the neutral and alkaline lakes, such as acI-A7 (1 OTU), acI-B1 (7 OTUs), acIV-A (3 OTUs), and acIV-B (11 OTUs) of Actinobacteria; bacIV (1 OTU) and bacV (5 OTUs) of Bacteroidetes; and alfVI (2 OTUs) of Alphaproteobacteria (see Fig. S3 in the supplemental material). Besides the good competitors and seemingly dormant taxa, many of the taxa that arose by neutral assembly in acidic lakes might be unable to survive or actively reproduce (fugitives), like some of the absent taxa in the two most acidic mining lakes, which probably included some of the common lineages or clades in the lakes with pH values above 5.0, such as acI-A1, acI-A7, acI-B1, acIV-A, acIV-B, and acTH1 of Actinobacteria; bacIV and bacV of Bacteroidetes; alfI and Pyxis of Alphaproteobacteria; Lhab-A1, Lhab-A2, Lhab-A3, PnecB, LD28, and betVII-A1 of Betaproteobacteria; and gamI of Gammaproteobacteria (see Fig. S3 in the supplemental material). Altogether, we found that the overall bacterioplankton communities in more acidic lakes were composed of fewer good competitors, which also involved fewer phylogenetic relatives and closely related OTUs, and that more dormant taxa, as well as more fugitive bacterioplankton taxa, arose by neutral assembly. Thus, both overall phylogenetic relatives (SES.PD) and the most closely related OTUs (SES.MNTD) were less phylogenetically clustered in more acidic lakes.
Conclusion.
This study systematically demonstrated that pH is the key environmental factor in determining the patterns and processes of freshwater lake bacterioplankton community assembly. More acidic lakes imposed stronger environmental filtering, which decreased the richness and evenness of overall bacterioplankton OTUs and largely shifted the community composition. Although environmental filtering was the most important determinant of bacterioplankton community assembly in our studied lakes, the importance of neutral processes must also be considered, notably in more acidic lakes, where the species (OTU) diversity was low. We propose that enhanced relative importance of random community assembly processes in more acidic lakes weakened the strong effect of environmental filtering on overall communities, resulting in more distantly related bacterioplankton communities in more acidic lakes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (31225004 and U1202231) and a project of the Chinese Academy of Sciences (KZZD-EW-TZ-08), as well as the EU REFRESH and MARS projects.
We thank Lissa Skov Hansen for her excellent work with the chemical analyses and Anne Mette Poulsen for English language assistance. We also acknowledge Te Cao, Femke van Beersum, Zhen Chen, Jin Zeng, Huabing Li, Xue Wang, Yujin Wang, and Biying Zhao for their assistance in the experiments and data analyses. We especially thank Ryan J. Newton for providing us with the typical freshwater bacterial database.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04042-14.
REFERENCES
- 1.Grossart HP, Ploug H. 2001. Microbial degradation of organic carbon and nitrogen on diatom aggregates. Limnol Oceanogr 46:267–277. doi: 10.4319/lo.2001.46.2.0267. [DOI] [Google Scholar]
- 2.Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S. 2011. A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75:14–49. doi: 10.1128/MMBR.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Logares R, Lindström ES, Langenheder S, Logue JB, Paterson H, Laybourn-Parry J, Rengefors K, Tranvik L, Bertilsson S. 2013. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J 7:937–948. doi: 10.1038/ismej.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He D, Ren L, Wu QL. 2014. Contrasting diversity of epibiotic bacteria and surrounding bacterioplankton of a common submerged macrophyte, Potamogeton crispus in freshwater lakes. FEMS Microbiol Ecol 90:551–562. doi: 10.1111/1574-6941.12414. [DOI] [PubMed] [Google Scholar]
- 5.Cavender-Bares J, Kozak KH, Fine PVA, Kembel SW. 2009. The merging of community ecology and phylogenetic biology. Ecol Lett 12:693–715. doi: 10.1111/j.1461-0248.2009.01314.x. [DOI] [PubMed] [Google Scholar]
- 6.Hubbell SP. 2001. The unified neutral theory of biodiversity and biogeography. Princeton University Press, Princeton, NJ. [DOI] [PubMed] [Google Scholar]
- 7.Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JB. 2012. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10:497–506. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Liu W, Deng Y, Jiang YH, Xue K, He Z, Van Nostrand JD, Wu L, Yang Y, Wang A. 2013. Stochastic assembly leads to alternative communities with distinct functions in a bioreactor microbial community. mBio 4:e00584–12. doi: 10.1128/mBio.00584-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brendan Logue J, Lindström ES. 2008. Biogeography of bacterioplankton in inland waters. Freshwater Rev 1:99–114. doi: 10.1608/FRJ-1.1.9. [DOI] [Google Scholar]
- 10.Bertilsson S, Eiler A, Nordqvist A, Jørgensen NOG. 2007. Links between bacterial production, amino-acid utilization and community composition in productive lakes. ISME J 1:532–544. doi: 10.1038/ismej.2007.64. [DOI] [PubMed] [Google Scholar]
- 11.Wu QL, Zwart G, Schauer M, Kamst-van Agterveld MP, Hahn MW. 2006. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl Environ Microbiol 72:5478–5485. doi: 10.1128/AEM.00767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu QL, Zwart G, Wu J, Kamst-van Agterveld MP, Liu S, Hahn MW. 2007. Submersed macrophytes play a key role in structuring bacterioplankton community composition in the large, shallow, subtropical Taihu Lake, China. Environ Microbiol 9:2765–2774. doi: 10.1111/j.1462-2920.2007.01388.x. [DOI] [PubMed] [Google Scholar]
- 13.Bouvy M, Bettarel Y, Bouvier C, Domaizon I, Jacquet S, Le Floc'h E, Montanié H, Mostajir B, Sime-Ngando T, Torréton JP, Vidussi F, Bouvier T. 2011. Trophic interactions between viruses, bacteria and nanoflagellates under various nutrient conditions and simulated climate change. Environ Microbiol 13:1842–1857. doi: 10.1111/j.1462-2920.2011.02498.x. [DOI] [PubMed] [Google Scholar]
- 14.Lindström ES, Langenheder S. 2012. Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4:1–9. doi: 10.1111/j.1758-2229.2011.00257.x. [DOI] [PubMed] [Google Scholar]
- 15.Ren L, He D, Zeng J, Wu QL. 2013. Bacterioplankton communities turn unstable and become small under increased temperature and nutrient-enriched conditions. FEMS Microbiol Ecol 84:614–624. doi: 10.1111/1574-6941.12089. [DOI] [PubMed] [Google Scholar]
- 16.Eigemann F, Hilt S, Salka I, Grossart HP. 2013. Bacterial community composition associated with freshwater algae: species specificity vs. dependency on environmental conditions and source community. FEMS Microbiol Ecol 83:650–663. doi: 10.1111/1574-6941.12022. [DOI] [PubMed] [Google Scholar]
- 17.Lindström ES, Kamst-Van Agterveld MP, Zwart G. 2005. Distribution of typical freshwater bacterial groups is associated with pH, temperature, and lake water retention time. Appl Environ Microbiol 71:8201–8206. doi: 10.1128/AEM.71.12.8201-8206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jezbera J, Jezberová J, Brandt U, Hahn MW. 2011. Ubiquity of Polynucleobacter necessarius subspecies asymbioticus results from ecological diversification. Environ Microbiol 13:922–931. doi: 10.1111/j.1462-2920.2010.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jezbera J, Jezberová J, Koll U, Horňák K, Šimek K, Hahn MW. 2012. Contrasting trends in distribution of four major planktonic betaproteobacterial groups along a pH gradient of epilimnia of 72 freshwater habitats. FEMS Microbiol Ecol 81:467–479. doi: 10.1111/j.1574-6941.2012.01372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton RJ, Jones SE, Helmus MR, McMahon KD. 2007. Phylogenetic ecology of the freshwater Actinobacteria acI lineage. Appl Environ Microbiol 73:7169–7176. doi: 10.1128/AEM.00794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb CO. 2000. Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am Nat 156:145–155. doi: 10.1086/303378. [DOI] [PubMed] [Google Scholar]
- 22.Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. 2002. Phylogenies and community ecology. Annu Rev Ecol Syst 33:475–505. doi: 10.1146/annurev.ecolsys.33.010802.150448. [DOI] [Google Scholar]
- 23.Cavender-Bares J, Ackerly DD, Baum DA, Bazzaz FA. 2004. Phylogenetic overdispersion in Floridian oak communities. Am Nat 163:823–843. doi: 10.1086/386375. [DOI] [PubMed] [Google Scholar]
- 24.Wiens JJ, Ackerly DD, Allen AP, Anacker BL, Buckley LB, Cornell HV, Damschen EI, Jonathan Davies T, Grytnes JA, Harrison SP, Hawkins BA, Holt RD, McCain CM, Stephens PR. 2010. Niche conservatism as an emerging principle in ecology and conservation biology. Ecol Lett 13:1310–1324. doi: 10.1111/j.1461-0248.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 25.Horner-Devine MC, Bohannan BJM. 2006. Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87:S100–S108. doi: 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 26.Madigan MT, Martinko JM, Stahl DA, Clark DP. 2011. Brock biology of microorganisms, 13th ed. Pearson Benjamin Cummings, San Francisco, CA. [Google Scholar]
- 27.Kalff J. 2002. Limnology: inland water ecosystem. Prentice Hall, Upper Saddle River, NJ. [Google Scholar]
- 28.Søndergaard M, Jeppesen E, Mortensen E, Dall E, Kristensen P, Sortkjær O. 1990. Phytoplankton biomass reduction after planktivorous fish reduction in a shallow, eutrophic lake: a combined effect of reduced internal P-loading and increased zooplankton grazing. Hydrobiologia 200-201:229–240. doi: 10.1007/BF02530342. [DOI] [Google Scholar]
- 29.Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price MN, Dehal PS, Arkin AP. 2009. Fasttree: computing large minimum-evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 34.Hurlbert SH. 1971. The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577–586. doi: 10.2307/1934145. [DOI] [PubMed] [Google Scholar]
- 35.Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biol Conserv 61:1–10. doi: 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- 36.Andersson AF, Riemann L, Bertilsson S. 2010. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J 4:171–181. doi: 10.1038/ismej.2009.108. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, He J, Liu X, Zhang L, Zhang E. 2013. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7:1310–1321. doi: 10.1038/ismej.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson JW. 2000. A heuristic method for estimating the relative weight of predictor variables in multiple regression. Multivar Behav Res 35:1–19. doi: 10.1207/S15327906MBR3501_1. [DOI] [PubMed] [Google Scholar]
- 39.Legendre P, Legendre L. 2012. Numerical ecology, 3rd English ed. Elsevier Science, Amsterdam, The Netherlands. [Google Scholar]
- 40.Lichstein JW. 2007. Multiple regression on distance matrices: a multivariate spatial analysis tool. Plant Ecol 188:117–131. doi: 10.1007/s11258-006-9126-3. [DOI] [Google Scholar]
- 41.Clarke KR, Ainsworth M. 1993. A method of linking multivariate community structure to environmental variables. Mar Ecol Prog Ser 92:205–219. doi: 10.3354/meps092205. [DOI] [Google Scholar]
- 42.Östman Ö, Drakare S, Kritzberg ES, Langenheder S, Logue JB, Lindström ES. 2010. Regional invariance among microbial communities. Ecol Lett 13:118–127. doi: 10.1111/j.1461-0248.2009.01413.x. [DOI] [PubMed] [Google Scholar]
- 43.Jones SE, McMahon KD. 2009. Species-sorting may explain an apparent minimal effect of immigration on freshwater bacterial community dynamics. Environ Microbiol 11:905–913. doi: 10.1111/j.1462-2920.2008.01814.x. [DOI] [PubMed] [Google Scholar]
- 44.Lindström ES, Östman Ö. 2011. The importance of dispersal for bacterial community composition and functioning. PLoS One 6:e25883. doi: 10.1371/journal.pone.0025883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351. doi: 10.1038/ismej.2010.58. [DOI] [PubMed] [Google Scholar]
- 46.Xiong J, Liu Y, Lin X, Zhang H, Zeng J, Hou J, Yang Y, Yao T, Knight R, Chu H. 2012. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ Microbiol 14:2457–2466. doi: 10.1111/j.1462-2920.2012.02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kampe H, Dziallas C, Grossart HP, Kamjunke N. 2010. Similar bacterial community composition in acidic mining lakes with different pH and lake chemistry. Microb Ecol 60:618–627. doi: 10.1007/s00248-010-9679-5. [DOI] [PubMed] [Google Scholar]
- 48.Tischler JS, Wiacek C, Janneck E, Schlömann M. 2013. Microbial abundance in the schwertmannite formed in a mine water treatment plant. Mine Water Environ 32:258–265. doi: 10.1007/s10230-013-0250-8. [DOI] [Google Scholar]
- 49.Lu S, Gischkat S, Reiche M, Akob DM, Hallberg KB, Küsel K. 2010. Ecophysiology of Fe-cycling bacteria in acidic sediments. Appl Environ Microbiol 76:8174–8183. doi: 10.1128/AEM.01931-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wenderoth DF, Abraham WR. 2005. Microbial indicator groups in acidic mining lakes. Environ Microbiol 7:133–139. doi: 10.1111/j.1462-2920.2004.00677.x. [DOI] [PubMed] [Google Scholar]
- 51.Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community composition at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Percent SF, Frischer ME, Vescio PA, Duffy EB, Milano V, McLellan M, Stevens BM, Boylen CW, Nierzwicki-Bauer SA. 2008. Bacterial community structure of acid-impacted lakes: what controls diversity? Appl Environ Microbiol 74:1856–1868. doi: 10.1128/AEM.01719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts DA, Singer R, Boylen CW. 1985. The submersed macrophyte communities of Adirondack lakes (New York, USA) of varying degrees of acidity. Aquat Bot 21:219–235. doi: 10.1016/0304-3770(85)90050-6. [DOI] [Google Scholar]
- 55.Findlay DL. 2003. Response of phytoplankton communities to acidification and recovery in Killarney Park and the Experimental Lakes Area, Ontario. Ambio 32:190–195. doi: 10.1579/0044-7447-32.3.190. [DOI] [PubMed] [Google Scholar]
- 56.Olsson H, Pettersson A. 1993. Oligotrophication of acidified lakes: a review of hypotheses. Ambio 22:312–317. [Google Scholar]
- 57.Havens KE, Carlson RE. 1998. Functional complementarity in plankton communities along a gradient of acid stress. Environ Pollut 101:427–436. doi: 10.1016/S0269-7491(98)00028-1. [DOI] [Google Scholar]
- 58.Gaedke U, Kamjunke N. 2006. Structural and functional properties of low and high diversity planktonic food webs. J Plankton Res 28:707–718. doi: 10.1093/plankt/fbl003. [DOI] [Google Scholar]
- 59.Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9:119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.