

Abstract
The synthesis of inositol provides precursors of inositol lipids and inositol phosphates that are pivotal for cell signaling. Mood-stabilizers lithium and valproic acid (VPA), used for treating bipolar disorder, cause cellular inositol depletion, which has been proposed as a therapeutic mechanism of action of both drugs. Despite the importance of inositol, the requirement for inositol synthesis in neuronal cells is not well understood. Here, we examined inositol effects on proliferation of SK-N-SH neuroblastoma cells. The essential role of inositol synthesis in proliferation is underscored by the findings that exogenous inositol was dispensable for proliferation, and inhibition of inositol synthesis decreased proliferation. Interestingly, the inhibition of inositol synthesis by knocking down INO1, which encodes inositol-3-phosphate synthase, the rate-limiting enzyme of inositol synthesis, led to inactivation of GSK-3α by increasing the inhibitory phosphorylation of this kinase. Similarly, the mood-stabilizer VPA effected transient decreases in intracellular inositol, leading to inactivation of GSK-3α. As GSK-3 inhibition has been proposed as a likely therapeutic mechanism of action, the finding that inhibition of inositol synthesis results in inactivation of GSK-3α suggests a unifying hypothesis for mechanism of mood-stabilizing drugs.
Keywords: Inositol, glycogen synthesis kinase, valproic acid, bipolar disorder, inositol depletion, myo-inositol-3-phosphate synthase
Introduction
Inositol, a six-carbon cyclitol, is an essential metabolite. It serves as the precursor of inositol lipids and inositol phosphates (Carman and Han, 2011; Henry et al., 2012; Michell, 2008; Michell, 2011), which play crucial roles in gene expression, signal transduction, lipid signaling, vesicle trafficking, and membrane biogenesis (De Camilli et al., 1996; Lemmon, 2003; Majerus and York, 2009; Michell, 2013; Shen et al., 2003; Steger et al., 2003; van Meer et al., 2008). Many types of cultured mammalian cells require supplementation of exogenous inositol for growth, and inositol-requiring mammalian cells and mutants of yeast undergo cell death in response to inositol deprivation (Culbertson and Henry, 1975; Eagle, 1955; Eagle et al., 1957; Kao and Puck, 1968; Keith et al., 1977).
Eukaryotic organisms can potentially obtain inositol from the environment, by the de novo synthesis of inositol from glucose, or via recycling of inositol by dephosphorylation of inositol phosphates. These processes are orchestrated to maintain intracellular inositol homeostasis. Inositol uptake in yeast (Lai et al., 1995; Lai and McGraw, 1994) and mammals (Wolfson et al., 2000; Wolfson et al., 1998), and inositol biosynthesis in yeast (Henry et al., 2014; Hirsch and Henry, 1986; Loewen et al., 2004) are affected by exogenous inositol. In mammals, inositol uptake is also regulated in response to glucose, pH, osmolality, growth factors, and other stimuli (Di Daniel et al., 2009; Fu et al., 2012; Miyakawa et al., 1999; Novak et al., 1999; Olgemoller et al., 1993; Spizz and Pike, 1992; Uldry et al., 2004; Yorek et al., 1998). Inositol de novo synthesis is a highly conserved pathway that is carried out in two steps, of which the conversion of glucose-6-phosphate to inositol-3-phosphate, catalyzed by the INO1 gene product inositol-3-phosphate synthase (EC 5.5.1.4), is rate-limiting (Eisenberg, 1967; Kindl and Hoffmann-Ostenhof, 1964; Loewus and Kelly, 1962a; Loewus and Kelly, 1962b; Strausberg et al., 2002). The regulation of inositol biosynthesis has been intensively studied in yeasts (Bachhawat et al., 1995; Carman and Han, 2011; Chen et al., 2007; Henry et al., 2012; Loewen et al., 2004; Ye et al., 2013). In addition to the transcriptional regulation of INO1 in response to exogenous inositol (Henry et al., 2014; Hirsch and Henry, 1986; Loewen et al., 2004), optimal inositol biosynthesis requires glycogen synthase kinase-3 (GSK-3) (Azab et al., 2007) and inositol pyrophosphates (Ye et al., 2013). Furthermore, Ino1 is posttranslationally regulated by phosphorylation (Deranieh et al., 2013), and enzyme activity is inhibited by the glycolysis intermediate dihydroxyacetone phosphate (DHAP) (Migaud and Frost, 1996; Shi et al., 2005). Mammalian INO1 expression is altered by estrogen, glucose, and lovastatin, and is regulated by the transcription factor E2F1 (Guan et al., 2003; Rivera-Gonzalez et al., 1998; Seelan et al., 2004; Seelan et al., 2011). Highly regulated inositol synthesis underscores the importance of maintaining inositol homeostasis.
The brain maintains a high level of free inositol (5–50 mM), which is about 100 times higher than that in blood and other tissues (Palmano et al., 1977; Sherman et al., 1977; Stokes et al., 1983; Wong et al., 1987). Altered inositol levels in the brain are associated with psychiatric and neurological problems (Seelan et al., 2009; Shi et al., 2006). For example, levels of inositol are altered in the brains of patients with Down syndrome (Acevedo et al., 1997; Berry et al., 1995), stroke (Rumpel et al., 2003), bipolar disorder (Belmaker et al., 2002; Shimon et al., 1997), and suicide victims (Shimon et al., 1997). Although dietary inositol can cross the blood-brain barrier and enter the cerebrospinal fluid and brain parenchyma, this process is very slow (Aukema, 1994; Spector and Lorenzo, 1975). Inositol levels in the brain primarily depend on inositol recycling and de novo synthesis (Williams et al., 2002). Interestingly, brain phosphatidylinositol levels are not affected when inositol uptake is blocked in inositol transporter-deficient mice (Berry et al., 2004), suggesting that inositol synthesis may be important for the synthesis of inositol lipids. However, the requirement of brain cells for inositol synthesis and the cellular consequences of perturbation of inositol synthesis in neuronal cells are not well studied.
Lithium, a mood-stabilizer used for the treatment of bipolar disorder, is an uncompetitive inhibitor of inositol monophosphatase and inositol polyphosphatase (Allison and Stewart, 1971; Berridge et al., 1989; Hallcher and Sherman, 1980; Pollack et al., 1994), and causes a decrease in intracellular inositol by blocking inositol recycling and synthesis. The mood-stabilizer valproic acid (VPA) inhibits inositol biosynthesis by indirectly decreasing activity of the rate-limiting enzyme Ino1 (Ju and Greenberg, 2003; Shaltiel et al., 2004; Vaden et al., 2001). Both drugs decrease cellular inositol and inositol 1,4,5-trisphosphate levels (Eickholt et al., 2005; Shimshoni et al., 2007; Williams et al., 2002), indicating that inositol depletion may attenuate inositol-dependent signaling. While inositol depletion may be therapeutically relevant (Berridge et al., 1989), another proposed target of mood-stabilizers is GSK-3 (Klein and Melton, 1996; Lucas and Salinas, 1997). The two major isoforms of GSK-3 (EC 2.7.11.1 and EC 2.7.11.26), GSK-3α and GSK-3β, share 85% sequence homology and are encoded by independent genes (Cohen and Frame, 2001). Both isoforms are expressed in the brain and have many regulatory functions in neural systems, including neurogenesis, neuronal structure, synaptic plasticity, and neuronal survival (Hur and Zhou, 2010). Lithium is a competitive inhibitor of GSK-3, and lithium treatment increases the inhibitory phosphorylation of this kinase (Klein and Melton, 1996; Lucas and Salinas, 1997; Ryves and Harwood, 2001; Zhang et al., 2003). Lithium-induced GSK-3 phosphorylation is caused by disrupting the signaling complex of Akt, β-arrestin 2, and protein phosphatase 2A (Beaulieu et al., 2008). Previous studies have not linked lithium-mediated inhibition of GSK-3 to inositol depletion. Interestingly, however, the inositol-depleting drug VPA has also been shown to inhibit GSK-3 activity (Chen et al., 1999; Chen et al., 2006; De Sarno et al., 2002; Kim et al., 2005). Common cellular effects of inositol depletion and GSK-3 inhibition in response to lithium and VPA treatment suggest that inositol metabolism and GSK-3 activity may be interdependent. It has not been determined if inositol metabolism affects GSK-3 activity in neuronal cells. Elucidating the interplay between inositol synthesis and GSK-3 in neuronal cells will have important implications for the pathophysiologic basis of a wide range of disorders in which inositol levels play a role.
In the current study, we characterized the role of inositol synthesis in proliferation of SK-N-SH neuroblastoma cells. We found that INO1 expression is essential for cell proliferation and neurite outgrowth. We further showed that inositol synthesis regulates GSK-3α activation, as inhibition of inositol synthesis by knocking down INO1 expression or exposure to VPA leads to increased phosphorylation at Ser21 of GSK-3α, which inactivates the kinase. Interestingly, inositol depletion caused by starving for exogenous inositol did not affect proliferation and GSK-3 phosphorylation. This is the first demonstration in neuronal cells of the importance of inositol de novo synthesis, and the first report showing that inositol synthesis affects GSK-3α activation. These findings have implications for the therapeutic mechanisms of mood-stabilizers and suggest that inositol synthesis and GSK-3 activity are intrinsically related.
Materials and Methods
Materials
Dulbecco’s Modified Eagle Medium (DMEM), Medium 199, and penicillin-streptomycin solution (100X) were purchased from Invitrogen. Fetal bovine serum (FBS) and dialyzed FBS were purchased from Hyclone. Inositol, lithium chloride, valproic acid, glucose-6-phosphate, NAD+, inositol dehydrogenase, and diaphorase were purchased from Sigma. Control shRNA lentiviral particles and INO1 (also named ISYNA1) shRNA lentiviral particles, puromycin dihydrochloride, and polybrene were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The protease inhibitors and phosphatase inhibitors were purchased from Roche.
The rabbit polyclonal IgG against the Ino1 protein (H-300), mouse monoclonal IgG against actin (C-2), and anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit monoclonal IgG against phospho-GSK-3α (ser21) (36E9), rabbit monoclonal IgG against GSK-3α (D80E6), rabbit monoclonal IgG against phospho-GSK-3β (Ser9) (D85E12), rabbit monoclonal IgG against GSK-3β (27C10), and anti-rabbit IgG were purchased from Cell Signaling Technology. All the primary antibodies were diluted 1:1000 and secondary antibodies 1:5000 in dilution buffer containing 1X tris-buffered saline (TBS), 0.1% Tween-20 with 5% nonfat dry milk.
Cell culture
SK-N-SH neuroblastoma cells were obtained from ATCC. Cells were regularly cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. For experiments to test effects of inositol deficiency, Medium 199 supplemented with 10% dialyzed FBS was used as inositol-deficient media. Inositol was added to this medium where indicated for inositol-rich media. All cells were cultured at 37°C in 95% air and 5% CO2. Of note, DMEM contains 40 μM inositol, and Medium 199 has 0.28 μM inositol. The dialyzed FBS contains trace amounts of small molecules.
Establishment of stable cell lines
SK-N-SH cells were transduced with lentiviral particles that contain 3 specific constructs targeting human INO1, or with control lentiviral particles containing non-specific scrambled shRNA. Cells were grown for 24 hours after the transduction, and stable cells were selected after culture in fresh media with 5 μg/ml puromycin for 2 weeks. The knockdown efficiency was determined by measuring Ino1 protein levels in the knockdown cells compared to levels in the control cells.
Proliferation assay
Cell proliferation was carried out using a proliferation assay kit (CellTiter 96 AQueous One Solution cell proliferation assay, Promega) and following the manufacturer’s instruction. Cells were inoculated at a concentration of 5,000 cells per well in 96-well plates. On the indicated days, the assay reagent containing tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was added and incubated at 37°C for 4 hours. Relative cell numbers were quantified by absorbance at 490 nm. The quantity of MTS formazan produced from MTS tetrazolium is directly proportional to the number of living cells (Cory et al., 1991).
RNA isolation and real-time quantitative PCR (RT-qPCR)
SK-N-SH cells were cultured in 6-well plates. Total RNA was extracted using the RNeasy Mini Plus kit (QIAGEN, Valencia, CA). Complementary DNA (cDNA) was synthesized using the First Strand cDNA synthesis Kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s manuals. RT-qPCR reactions were performed in a 20 μL volume using Brilliant III Ultra-Faster SYBR Green QPCR Master Mix (Agilent Technologies, Santa Clara, CA). Triplicates were included for each reaction. The primers for RT-qPCR are listed in Table 1. RNA levels were normalized to succinate dehydrogenase, SDHA. Relative values of mRNA transcripts are shown as fold change relative to indicated controls.
Table 1.
Real-time PCR primers used in this study.
| Gene | Primers | Sequence (5′ to 3′) |
|---|---|---|
| SDHA | Forward | CGAACGTCTTCAGGTGCTTT |
| Reverse | AAGAACATCGGAACTGCGAC | |
| INO1 | Forward | CTGCATCGAGAACATCCTCAG |
| Reverse | GTTCAACATAGGGTAGGTGGC | |
| SMIT1 | Forward | AAGGTGGTGGTTCGAATCTG |
| Reverse | CCACAGGATTGTTTTGGGTC | |
| HMIT | Forward | CATCTGCAGAATGGTTGCAC |
| Reverse | AACTCGCCGAGCTTTAATTG |
SDS-PAGE and Western blots
Cell extracts were obtained by breaking cells in lysis buffer containing 50 mM Tris, 125 mM sodium chloride, 1% NP-40, 2 mM EDTA, 1x protease inhibitor cocktail, and 1× phosphatase inhibitor cocktail and were clarified twice by 10 min-centrifugation at 13,000 g at 4°C to remove cell debris. Protein concentration was determined using the BCA™ protein assay (Pierce Protein), with bovine serum albumin as the standard. Cell extracts containing 20 μg protein were boiled with protein gel sample buffer, separated on 10% SDS-PAGE, and electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). The membrane was incubated with antibodies and visualized using ECL substrate (Pierce Protein). ImageJ software was used to quantify the intensities of bands.
Measurement of intracellular inositol
Intracellular inositol was measured as described previously (Ju and Greenberg, 2003; Ye et al., 2013) with minor modifications. Briefly, cell extracts were obtained in lysis buffer containing 50 mM Tris, 125 mM sodium chloride, 1% NP-40, 2 mM EDTA and were clarified twice by 10 min-centrifugation at 13,000 g at 4°C to remove cell debris. Cell extracts containing 50 μg protein were used to measure intracellular inositol. Protein was precipitated using ice-cold 7.5% perchloric acid. After centrifugation, perchloric acid in the supernatants was removed by titration to pH 7.0 with ice-cold 10 M potassium hydroxide. The cell extracts were again clarified by centrifugation for 5 min at 2,000 g at 4°C. The supernatants were collected, and intracellular inositol was measured by enzyme-coupled fluorescence assay (Maslanski and Busa, 1990). Inositol content in cell extracts of 50 μg protein was normalized to the indicated controls.
Statistical Analysis
The data were presented as the mean ± SD. The p value was calculated by two-tailed Student’s t-test, and differences were considered significant when p < 0.05. All experiments were carried out in at least triplicates.
RESULTS
Exogenous inositol is not essential for cell proliferation or maintaining inositol homeostasis in SK-N-SH cells
Inositol is an essential growth factor that is required for survival and proliferation of many types of cultured cells (Eagle et al., 1957). Inositol deficiency in these cells causes an arrest of cell growth, cytopathogenic defects, and cell death. However, some cells are able to proliferate in inositol-free or inositol-deficient culture media due to active inositol biosynthesis (Eagle et al., 1957). To determine if inositol is essential for SK-N-SH neuronal cells, we assayed cell growth in inositol-deficient media (Medium 199 with 10% dialyzed serum). Medium 199 contains only 0.28 μM inositol, which is significantly less than the minimal requirement (1 μM) reported for most types of cells (Eagle et al., 1957). As seen in Fig. 1A, proliferation of cells cultured in the inositol-deficient media was similar to that of cells grown in media supplemented with exogenous inositol. The ability of SK-N-SH cells to grow in inositol-deficient media indicates that the de novo synthesis of inositol provides sufficient inositol for cell proliferation. Interestingly, cells cultured in inositol-deficient media exhibited levels of intracellular inositol similar to those of cells grown in media supplemented with 0.5–10 mM inositol (Fig. 1B). The homeostatic inositol pool in SK-N-SH cells suggests that inositol biosynthesis may be upregulated in inositol-deficient media.
Fig. 1. Exogenous inositol is not essential for cell proliferation or maintaining inositol homeostasis in SK-N-SH cells.
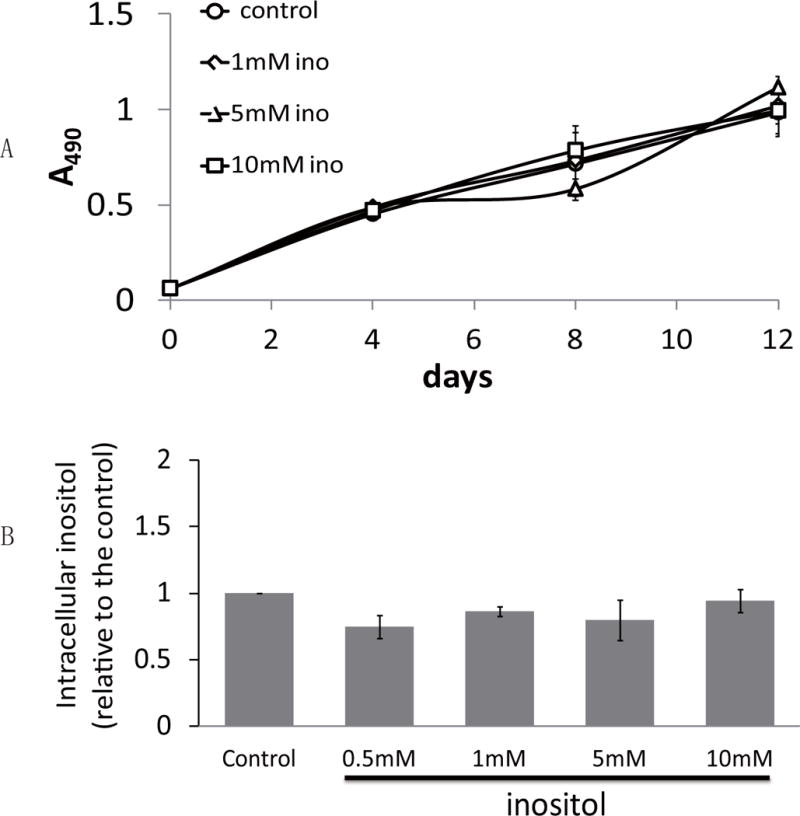
(A) SK-N-SH cells were inoculated at a concentration of 5,000 cells per well in 96-well plates at day 0, and cell numbers were estimated by the proliferation assay described under “Materials and Methods.” (B) Intracellular inositol levels were assayed in cells cultured in inositol-deficient media without (control) or with inositol supplement (0.5, 1, 5, 10 mM). The data shown in A and B are the average of at least three experiments ± S.D, n≥3.
Inositol biosynthesis is essential for cell proliferation
To understand if inositol biosynthesis is required for SK-N-SH cell growth, we decreased inositol biosynthesis by stably knocking down expression of the gene encoding inositol-3-phosphate synthase, INO1, which encodes the rate-limiting enzyme of inositol biosynthesis. As shown in Fig. 2A, INO1 knockdown cells exhibited a 90% decrease in Ino1 protein levels. As expected, INO1 knockdown resulted in a profound decrease in intracellular inositol levels (Fig. 2B). Cell proliferation was significantly decreased in INO1 knockdown cells (Fig. 2C). In addition, neurite outgrowth was remarkably inhibited in cells in which INO1 expression was decreased (Fig. 2D). These findings indicate that de novo inositol synthesis is essential for cell proliferation and neurite outgrowth in SK-N-SH cells.
Fig. 2. Inositol biosynthesis is essential for cell proliferation.
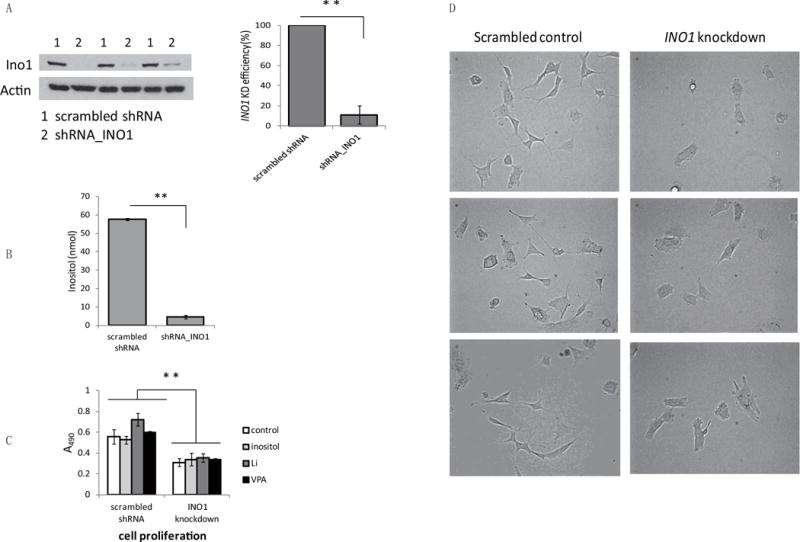
(A) Western blot analysis of Ino1 protein levels (left) and quantitative analysis of INO1 knockdown efficiency (right). (B) Inositol levels were measured in control and INO1 knockdown cells cultured in DMEM supplemented with 10% serum. (C) Cell proliferation was assayed as described in Fig. 1A, and cell numbers were estimated 4 days after inoculation. (D) Control and INO1 knockdown cells (about 1×106) were plated in 100-mm dishes and photographed at day 2 using a microscope at 200 X magnification. The data in A, B, and C are presented as the mean ± S.D, n=3, and **p< 0.01.
Because INO1 knockdown causes inositol depletion leading to decreased proliferation, we questioned if the defect might be rescued by exogenous inositol or exacerbated by the inositol-depleting drugs, lithium and VPA. Unexpectedly, neither exogenous inositol nor lithium and VPA affected proliferation in control and INO1 knockdown cells (Fig. 2C). To determine if exogenous inositol restores the decreased intracellular inositol in INO1 knockdown cells, control and INO1 knockdown cells cultured in DMEM supplemented with 10% serum were replenished with inositol-rich (containing 1 mM inositol) or inositol-deficient media for 5 hours. As shown in Fig. 3A, in inositol-deficient media, inositol levels in INO1 knockdown cells were only 20% of those in control cells, whereas inositol in INO1 knockdown cells cultured in inositol-rich media was similar to the control levels. While exogenous inositol was able to restore the inositol depletion caused by INO1 knockdown, it was not able to rescue the proliferation defect (Fig. 2C). It is likely that the regulation of inositol synthesis may provide a pool of inositol functionally distinct from imported inositol, which is important for proliferation.
Fig. 3. Exogenous inositol does not regulate transcription of the genes for inositol biosynthesis or uptake.
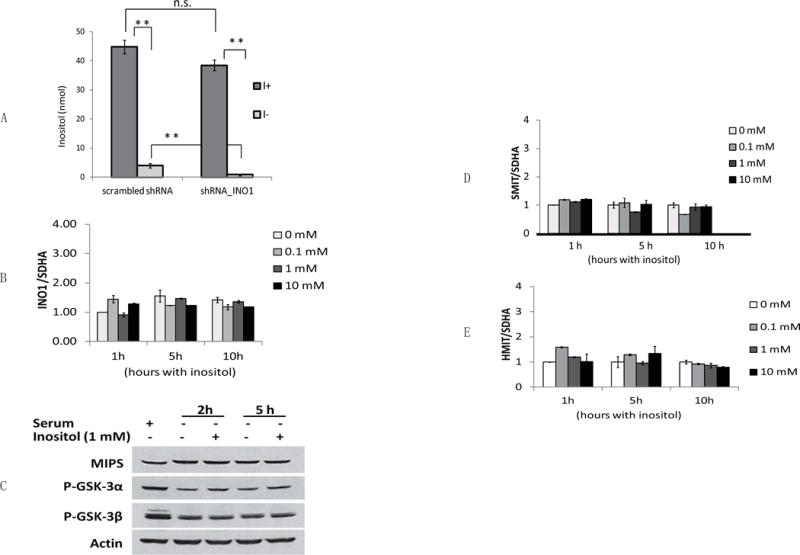
(A) Inositol levels were measured in control or INO1 knockdown cells in inositol-rich (I+) and inositol-deficient (I-) media. Cells were cultured in DMEM with 10% serum to reach 70% confluence, washed twice with PBS, and replenished with media as indicated. The data are presented as the mean ± S.D, n=3, and **p<0.01. (B) mRNA levels of INO1 were measured in SK-N-SH cells incubated in the presence of inositol (0, 0.1, 1, 10 mM) for the indicated times (1, 5, and 10 hours) after growth in inositol-deficient media. Values were normalized to the internal control SDHA (succinate dehydrogenase complex, subunit A). INO1 mRNA levels normalized to SDHA were represented as fold change relative to cells exposed to 0 mM inositol for 1 hour. (C) Western blot analysis of Ino1 protein levels. SK-N-SH cells were cultured to reach about 70% confluence and then were incubated with either serum or inositol for indicated times. Cells were harvested and lysed for Western blot analysis as described under “Materials and Methods.” Actin was used as the loading control. The figure is representative of experiments in triplicate. (D) mRNA levels of Na+/inositol transporter SMIT1 and (E) H+/inositol transporter HMIT were measured as described above. The data in B, D, and E are presented as the mean ± S.D, n≥3.
Exogenous inositol does not regulate transcription of genes for inositol synthesis and uptake, INO1, SMIT1, and HMIT
In yeast cells, exogenous inositol modulates the biosynthesis and uptake of inositol by controlling transcription of the inositol biosynthetic gene INO1 (Henry et al., 2014; Hirsch and Henry, 1986; Loewen et al., 2004) and the genes encoding inositol transporters (Lai et al., 1995; Lai and McGraw, 1994). To ascertain if this is a conserved mechanism regulating inositol metabolism in SK-N-SH cells, we determined the effects of exogenous inositol on mRNA levels of INO1, Na+/inositol transporter SMIT1, and H+/inositol transporter HMIT in these cells. Cells were initially cultured in inositol-deficient media to deplete inositol. After supplementation with inositol (0.1, 1, 10 mM) for the indicated times, cells were harvested for mRNA analysis. As shown in Fig. 3B, mRNA levels of human INO1 were not affected by the addition of inositol. Consistent with this finding, INO1 protein levels were also not altered by exogenous inositol (Fig. 3B). In addition, the genes encoding inositol transporters were not regulated by inositol, as mRNA levels of SMIT1 (Fig. 3C) and HMIT (Fig. 3D) were not significantly changed in response to exogenous inositol. Therefore, in contrast to regulation of inositol biosynthesis in yeast cells, the biosynthesis and uptake of inositol were not transcriptionally regulated in response to exogenous inositol in SK-N-SH cells.
Decreased Ino1 leads to inactivation of GSK3α
Previous studies showed that lithium and VPA inhibit inositol synthesis (Allison and Stewart, 1971; Berridge et al., 1989; Hallcher and Sherman, 1980; Ju and Greenberg, 2003; Pollack et al., 1994; Shaltiel et al., 2004; Vaden et al., 2001) and GSK-3 activity (Chen et al., 1999; Chen et al., 2006; De Sarno et al., 2002; Kim et al., 2005; Klein and Melton, 1996; Lucas and Salinas, 1997). To address the possibility that inositol synthesis affects GSK-3 activity, we measured levels of phosphorylation at Ser21 of GSK-3α and at Ser9 of GSK-3β in SK-N-SH cells as a function of inositol depletion. Phosphorylation of these sites inactivates the kinase activity of GSK-3 (Cross et al., 1995; Srivastava and Pandey, 1998). As seen in Fig. 4A, GSK-3α phosphorylation was increased in response to inhibition of inositol synthesis, while GSK3β was not significantly altered. GSK-3α phosphorylation was dependent on the level of Ino1 knockdown. Thus, an 82% decrease in Ino1 protein in shRNA-INO1–2 cells led to a 2.4-fold increase in GSK-3α phosphorylation, while a 60% decrease in shRNA-INO1–1 led to a 1.5-fold increase. These results indicated that decreasing the de novo synthesis of inositol led to GSK-3α inactivation.
Fig. 4. INO1 knockdown and VPA treatment lead to inactivation of GSK3α.
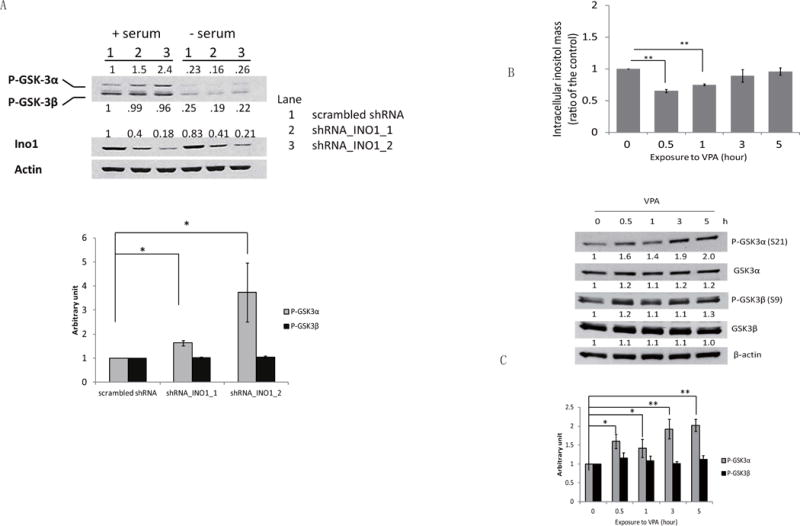
(A) Western blot analysis and quantification of Ino1 and inhibitory phosphorylation levels of GSK-3α (Ser21) and GSK-3β (Ser9). Actin was used as the loading control. Scrambled control and Ino1 knockdown (shRNA_INO1_1 and shRNA_INO1_2) SK-N-SH cells were cultured to about 70% confluence, and cells were refreshed with media containing 10% serum (+serum) or no serum (-serum) for 4 hours. Cells were harvested and lysed for Western blot analysis as described under “Materials and Methods.” The figure is representative of experiments in triplicate. The quantification data are presented as the mean ± S.D, n=3, and *p< 0.05. (B) Intracellular inositol levels were measured in SK-N-SH cells after exposure to VPA for the indicated times. The data are presented as the mean ± S.D, n=3, and **p< 0.01. (C) Western blot analysis and quantification of the protein levels of GSK-3α (Ser21) and GSK-3β (Ser9) and total protein levels of GSK-3α and GSK-3β. Actin was used as the loading control. The figure is representative of at least three independent experiments. The quantification data are presented as the mean ± S.D, n≥4, and *p< 0.05, **p<0.01.
To investigate if GSK-3α inactivation resulting from INO1 knockdown is due to inositol depletion, we determined the effects of exogenous inositol on GSK-3 phosphorylation. While intracellular inositol was significantly decreased by starving for exogenous inositol (Fig. 3A), GSK-3 phosphorylation remained similar in cells cultured in the absence or presence of inositol (Fig. 3C). Therefore, it is the inhibition of Ino1-catalyzed inositol synthesis, but not the resulting inositol depletion, that leads to the inactivation of GSK-3α.
VPA-induced transient decrease in inositol is associated with GSK-3α inactivation
We have previously shown that the mood stabilizer VPA inhibits inositol synthesis in both yeast and human cells (Ju and Greenberg, 2003; Shaltiel et al., 2004; Vaden et al., 2001). If inhibition of inositol synthesis was the cause of inhibitory phosphorylation of GSK-3α as seen in Fig. 4A, VPA may be expected to cause a similar effect. To address this possibility, we assayed GSK-3 phosphorylation in cells treated with VPA. As seen in Fig. 4B, VPA caused a significant decrease in inositol levels in the first hour of exposure, after which inositol levels were restored, probably as a result of recycling from inositol phosphates. As predicted, GSK-3α phosphorylation was increased in response to VPA (Fig. 4C), while GSK-3β phosphorylation was not affected (Fig. 4C). However, GSK-3α phosphorylation continued to increase, even after inositol levels were restored (Figs. 4B and 4C), suggesting that inhibition of inositol synthesis, but not inositol depletion, is responsible for inactivation of GSK-3α.
DISCUSSION
Despite the importance of inositol, there are very few reported studies of the consequences of inhibition of inositol synthesis in human cells. Here, we report that inositol synthesis is essential for proliferation and neurite outgrowth of SK-N-SH human neuroblastoma cells, and that inhibition of inositol synthesis leads to GSK-3α inactivation.
The interplay between inositol biosynthesis and GSK-3 activity reported in the current study has implications for understanding the therapeutic mechanisms of the mood-stabilizers used to treat bipolar disorder. Lithium and VPA are mood-stabilizers with disparate chemical properties. Interestingly, both drugs have been shown to decrease cellular inositol content by blocking inositol biosynthesis, and both drugs also inhibit GSK-3 activity. Consistent with these findings, two prevailing hypotheses for the therapeutic mechanisms of action of these drugs are inositol depletion (Berridge et al., 1989) and GSK-3 inhibition (Klein and Melton, 1996). While the common effects of lithium and VPA on inositol signaling and GSK-3 highlight the potential interplay between inositol metabolism and GSK-3 regulation, several studies reported different effects of these drugs on inositol signaling and GSK-3 (Eickholt et al., 2005; Jin et al., 2005; Whitworth et al., 1990), which complicates our understanding of the mechanism of action of these drugs. The drug-induced perturbation of inositol signaling and GSK-3 activity may be dependent on cell type and study model, the pattern of treatment, and experimental procedures. A potential link between inositol depletion and GSK-3 inhibition has not been previously tested in human cells. While the inositol-depleting drug lithium leads to increased phosphorylation of GSK-3β (Beaulieu et al., 2008; Zhang et al., 2003), we showed that VPA causes transient inositol depletion leading to increased phosphorylation of GSK-3α. Interestingly, during VPA treatment, GSK-3α phosphorylation continued to increase even after inositol levels were restored, suggesting that inhibition of inositol synthesis, but not inositol depletion, is sufficient to inactivate GSK-3α. We further report a direct link between inositol synthesis and GSK-3 activity in neuronal cells. Specifically, inositol synthesis inhibited by knocking down INO1 expression results in GSK-3α inactivation.
While GSK-3α phosphorylation was increased in response to inhibition of inositol synthesis, GSK-3β was not significantly altered. Previous studies have shown that inhibitory phosphorylation and kinase activity of GSK-3 are affected by exposure to lithium and VPA (Beaulieu et al., 2008; Chen et al., 1999; Chen et al., 2006; De Sarno et al., 2002; Kim et al., 2005; Kim et al., 2013; Klein and Melton, 1996; Lucas and Salinas, 1997; Phiel et al., 2003; Ryves and Harwood, 2001; Zhang et al., 2003). While some studies characterized the inhibitory effects of the drugs on both GSK-3α and GSK-3β, most have focused on the effects on GSK-3β. This is the first demonstration in SK-N-SH cells that a decrease in inositol biosynthesis led to the preferential inactivation of GSK-3α. Further studies are needed to elucidate the significance of the differential inhibition of GSK-3α and GSK-3β, which do not have identical functions.
Inactivation of GSK-3α in SK-N-SH cells was dependent on the degree of Ino1 knockdown (Fig. 4A). The degree of inactivation of GSK-3α also correlated with exposure times to VPA (Fig. 4C). While inhibition of inositol synthesis potently increases GSK-3α phosphorylation, inositol homeostasis is highly maintained, as intracellular inositol levels were restored upon prolonged exposure to VPA (Fig. 4B). A similar VPA effect was observed in our previous study in mice, in which mouse brain inositol was only depleted following acute, but not chronic, VPA administration (Shaltiel et al., 2004). It is likely that turnover of phosphatidylinositol and/or recycling of inositol phosphates are increased to compensate for decreased inositol biosynthesis when subjects are exposed to VPA. However, the mechanism whereby inhibition of inositol synthesis causes inactivation of GSK-3α activity (by increasing phosphorylation of this kinase) remains unclear. Perturbation of inositol homeostasis resulting from disrupted inositol synthesis may modulate PI3K/AKT signaling. We speculate that inhibition of inositol synthesis activates the synthesis of PI3,4,5P3. The synthesis of PI3,4,5P3 is required for recruiting AKT, and the subsequent activation of AKT on the plasma membrane potently inhibits GSK-3 by phosphorylation (Cantley, 2002; Czech, 2003; Di Paolo and De Camilli, 2006). The inactivation of GSK-3α in response to inhibition of inositol synthesis suggests that GSK-3α may have a unique role in regulating cellular activity. GSK-3α has been shown to regulate amyloid-β production by processing the amyloid precursor protein in cultured cells and in mouse brain, suggesting that GSK-3α may be a valuable target for the treatment of Alzheimer’s disease (Phiel et al., 2003). We speculate that GSK-3α may regulate proliferation. Therefore, inactivation of GSK-3α by inhibition of inositol synthesis may lead to decreased proliferation.
Surprisingly, exogenous inositol did not affect proliferation or GSK-3α phosphorylation, suggesting that exogenous inositol does not regulate inositol synthesis. Indeed, exogenous inositol did not affect inositol biosynthesis or uptake by controlling expression of the inositol biosynthetic gene INO1 or the genes encoding inositol transporters SMIT1 and HMIT. In contrast, the transcription of INO1 and the inositol transporter genes in yeast cells is highly regulated in response to exogenous inositol, and inositol synthesis and uptake are modulated by this regulation (Henry et al., 2014; Hirsch and Henry, 1986; Lai et al., 1995; Lai and McGraw, 1994; Loewen et al., 2004). This indicates that neuronal cells have evolved different mechanisms to regulate inositol metabolism. For example, inositol uptake in mammals is regulated by glucose, pH, osmolality, and growth factors (Di Daniel et al., 2009; Fu et al., 2012; Miyakawa et al., 1999; Novak et al., 1999; Olgemoller et al., 1993; Spizz and Pike, 1992; Uldry et al., 2004). While the genes encoding inositol transporters are not transcriptionally regulated in response to exogenous inositol in SK-N-SH cells, the activity of inositol uptake may be controlled by different mechanisms. Inositol synthesis in yeast is also regulated by the synthesis of inositol pyrophosphates (Ye et al., 2013) and the glycolysis intermediate dihydroxyacetone phosphate (DHAP) (Migaud and Frost, 1996; Shi et al., 2005), and requires GSK-3 (Azab et al., 2007). One or more of these mechanisms identified in yeast may also control inositol biosynthesis in mammalian cells.
Interestingly, the effects of exogenous inositol on intracellular inositol vary with the experimental conditions. When cells were inoculated and continuously grown to about 70% confluence for about 5 days in inositol-deficient media or inositol-containing media, intracellular inositol levels in cells grown in inositol-deficient media were similar to those grown in inositol-containing media. However, intracellular inositol levels were profoundly decreased in cells that were shifted to inositol-deficient media for 5 hours compared to those shifted to inositol-rich media after they reached about 70% confluence in DMEM supplemented with regular serum. While intracellular inositol levels were greatly altered in response to an acute manipulation of exogenous inositol, it is curious that those levels are similar in cells grown in media with or without inositol supplementation. We speculate that inositol synthesis is upregulated and/or utilization of inositol for phosphatidylinositol synthesis is decreased in cells grown in inositol-deficient media, thus these cells manifest similar inositol levels and are able to grow similar to cells grown in inositol-containing media.
In summary, we showed that de novo inositol synthesis catalyzed by Ino1 is required for proliferation of SK-N-SH cells during inositol-deficient conditions and for GSK-3α activation. These findings have implications for understanding the therapeutic mechanisms of the mood-stabilizers used for treatment of bipolar disorder.
Acknowledgments
We thank Dr. Xiang-Dong Zhang, Dr. Karen A. Beningo, and William D. Lyman for sharing cell culture facilities. This work was funded by grant R01 DK081367 from the National Institutes of Health (to M.L.G.), and the WSU Thomas C. Rumble University Fellowship and Dissertation Fellowship, WSU graduate enhancement research funds (to C.Y.).
Abbreviations
- Ino1
inositol-3-phosphate synthase
- GSK-3
glycogen synthase kinase-3
- DHAP
dihydroxyacetone phosphate
- VPA
valproic acid
- SMIT1
Na+/inositol transporter
- HMIT
H+/inositol transporter
Footnotes
conflict of interest disclosure
The authors have no conflicts of interest to declare.
References
- Acevedo LD, Holloway HW, Rapoport SI, Shetty HU. Application of stable isotope tracer combined with mass spectrometric detection for studying myo-inositol uptake by cultured neurons from fetal mouse: effect of trisomy 16. J Mass Spectrom. 1997;32:395–400. doi: 10.1002/(SICI)1096-9888(199704)32:4<395::AID-JMS487>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Allison JH, Stewart MA. Reduced brain inositol in lithium-treated rats. Nature: New biology. 1971;233:267–268. doi: 10.1038/newbio233267a0. [DOI] [PubMed] [Google Scholar]
- Aukema HM, Holub BJ. Inositol and pyrroloquinoline quinone. A: inositol. Lea and Febiger; Philadelphia: 1994. [Google Scholar]
- Azab AN, He Q, Ju S, Li G, Greenberg ML. Glycogen synthase kinase-3 is required for optimal de novo synthesis of inositol. Mol Microbiol. 2007;63:1248–1258. doi: 10.1111/j.1365-2958.2007.05591.x. [DOI] [PubMed] [Google Scholar]
- Bachhawat N, Ouyang Q, Henry SA. Functional characterization of an inositol-sensitive upstream activation sequence in yeast. A cis-regulatory element responsible for inositol-choline mediated regulation of phospholipid biosynthesis. J Biol Chem. 1995;270:25087–25095. doi: 10.1074/jbc.270.42.25087. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- Belmaker RH, Shapiro J, Vainer E, Nemanov L, Ebstein RP, Agam G. Reduced inositol content in lymphocyte-derived cell lines from bipolar patients. Bipolar disorders. 2002;4:67–69. doi: 10.1034/j.1399-5618.2002.00108.x. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Berry GT, Buccafusca R, Greer JJ, Eccleston E. Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Molecular genetics and metabolism. 2004;82:87–92. doi: 10.1016/j.ymgme.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Berry GT, Mallee JJ, Kwon HM, Rim JS, Mulla WR, Muenke M, Spinner NB. The human osmoregulatory Na+/myo-inositol cotransporter gene SLC5A3: molecular cloning and localization to chromosome 21. Genomics. 1995;25:507–513. doi: 10.1016/0888-7543(95)80052-n. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Carman G, Han GS. Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annual review of biochemistry. 2011;80:859–883. doi: 10.1146/annurev-biochem-060409-092229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Chen M, Hancock LC, Lopes JM. Transcriptional regulation of yeast phospholipid biosynthetic genes. Biochimica et biophysica acta. 2007;1771:310–321. doi: 10.1016/j.bbalip.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Molecular psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nature reviews Molecular cell biology. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer communications. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Culbertson MR, Henry SA. Inositol-requiring mutants of Saccharomyces cerevisiae. Genetics. 1975;80:23–40. doi: 10.1093/genetics/80.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP. Dynamics of phosphoinositides in membrane retrieval and insertion. Annual review of physiology. 2003;65:791–815. doi: 10.1146/annurev.physiol.65.092101.142522. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Deranieh RM, He Q, Caruso JA, Greenberg ML. Phosphorylation regulates myo-inositol-3-phosphate synthase: a novel regulatory mechanism of inositol biosynthesis. J Biol Chem. 2013;288:26822–26833. doi: 10.1074/jbc.M113.479121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Daniel E, Mok MH, Mead E, Mutinelli C, Zambello E, Caberlotto LL, Pell TJ, Langmead CJ, Shah AJ, Duddy G, Kew JN, Maycox PR. Evaluation of expression and function of the H+/myo-inositol transporter HMIT. BMC cell biology. 2009;10:54. doi: 10.1186/1471-2121-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Eagle H. The specific amino acid requirements of a human carcinoma cell Stain HeLa in tissue culture. The Journal of experimental medicine. 1955;102:37–48. doi: 10.1084/jem.102.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H, Oyama VI, Levy M, Freeman AE. Myo-Inositol as an essential growth factor for normal and malignant human cells in tissue culture. J Biol Chem. 1957;226:191–205. [PubMed] [Google Scholar]
- Eickholt BJ, Towers GJ, Ryves WJ, Eikel D, Adley K, Ylinen LM, Chadborn NH, Harwood AJ, Nau H, Williams RS. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, glycogen synthase kinase-3beta inhibition, and viral replication: a screening approach for new bipolar disorder drugs derived from the valproic acid core structure. Molecular pharmacology. 2005;67:1426–1433. doi: 10.1124/mol.104.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg F., Jr D-myoinositol 1-phosphate as product of cyclization of glucose 6-phosphate and substrate for a specific phosphatase in rat testis. J Biol Chem. 1967;242:1375–1382. [PubMed] [Google Scholar]
- Fu H, Li B, Hertz L, Peng L. Contributions in astrocytes of SMIT1/2 and HMIT to myo-inositol uptake at different concentrations and pH. Neurochemistry international. 2012;61:187–194. doi: 10.1016/j.neuint.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Guan G, Dai P, Shechter I. cDNA cloning and gene expression analysis of human myo-inositol 1-phosphate synthase. Arch Biochem Biophys. 2003;417:251–259. doi: 10.1016/s0003-9861(03)00388-6. [DOI] [PubMed] [Google Scholar]
- Hallcher LM, Sherman WR. The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. The Journal of biological chemistry. 1980;255:10896–10901. [PubMed] [Google Scholar]
- Henry S, Kohlwein S, Carman G. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics. 2012;190:317–349. doi: 10.1534/genetics.111.130286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SA, Gaspar ML, Jesch SA. The response to inositol: Regulation of glycerolipid metabolism and stress response signaling in yeast. Chemistry and physics of lipids. 2014 doi: 10.1016/j.chemphyslip.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JP, Henry SA. Expression of the Saccharomyces cerevisiae inositol-1-phosphate synthase INO1 gene is regulated by factors that affect phospholipid synthesis. Molecular and cellular biology. 1986;6:3320–3328. doi: 10.1128/mcb.6.10.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nature reviews Neuroscience. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Kovacs AD, Sui Z, Dewhurst S, Maggirwar SB. Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death. Neuropharmacology. 2005;48:576–583. doi: 10.1016/j.neuropharm.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Ju S, Greenberg ML. Valproate disrupts regulation of inositol responsive genes and alters regulation of phospholipid biosynthesis. Molecular microbiology. 2003;49:1595–1603. doi: 10.1046/j.1365-2958.2003.03641.x. [DOI] [PubMed] [Google Scholar]
- Kao FT, Puck TT. Genetics of somatic mammalian cells, VII. Induction and isolation of nutritional mutants in Chinese hamster cells. Proc Natl Acad Sci U S A. 1968;60:1275–1281. doi: 10.1073/pnas.60.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith AD, Pollard EC, Snipes W. Inositol-less death in yeast results in a simultaneous increase in intracellular viscosity. Biophysical journal. 1977;17:205–212. doi: 10.1016/S0006-3495(77)85650-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. Journal of cell science. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- Kim J, Yang M, Kim SH, Kim JC, Wang H, Shin T, Moon C. Possible role of the glycogen synthase kinase-3 signaling pathway in trimethyltin-induced hippocampal neurodegeneration in mice. PloS one. 2013;8:e70356. doi: 10.1371/journal.pone.0070356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindl H, Hoffmann-Ostenhof O. Studies on the Biosynthesis of Cyclitols. Ii. Formation of Meso-Inositol from C14–1-Glucose in Sinapis Alba and Selective Degradation of the Resulting Product. Biochem Z. 1964;339:374–381. [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Bolognese CP, Swift S, McGraw P. Regulation of inositol transport in Saccharomyces cerevisiae involves inositol-induced changes in permease stability and endocytic degradation in the vacuole. J Biol Chem. 1995;270:2525–2534. doi: 10.1074/jbc.270.6.2525. [DOI] [PubMed] [Google Scholar]
- Lai K, McGraw P. Dual control of inositol transport in Saccharomyces cerevisiae by irreversible inactivation of permease and regulation of permease synthesis by INO2, INO4, and OPI1. J Biol Chem. 1994;269:2245–2251. [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Gaspar ML, Jesch SA, Delon C, Ktistakis NT, Henry SA, Levine TP. Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science. 2004;304:1644–1647. doi: 10.1126/science.1096083. [DOI] [PubMed] [Google Scholar]
- Loewus F, Kelly S. Conversion of glucose to inositol in parsley leaves. Biochemical and biophysical research communications. 1962a;7:204–208. doi: 10.1016/0006-291x(62)90175-4. [DOI] [PubMed] [Google Scholar]
- Loewus FA, Kelly S. Conversion of glucose to inositol in parsley leaves. Biochem Biophys Res Commun. 1962b;7:204–208. doi: 10.1016/0006-291x(62)90175-4. [DOI] [PubMed] [Google Scholar]
- Lucas FR, Salinas PC. WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Developmental biology. 1997;192:31–44. doi: 10.1006/dbio.1997.8734. [DOI] [PubMed] [Google Scholar]
- Majerus PW, York JD. Phosphoinositide phosphatases and disease. Journal of lipid research. 2009;50(Suppl):S249–254. doi: 10.1194/jlr.R800072-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslanski JA, Busa WB. A sensitive and sepecific mass assay for myo-inositol and inositol phosphate. In: Irvine RF, editor. Methods in Inositide Research. 1990. pp. 113–126. [Google Scholar]
- Michell RH. Inositol derivatives: evolution and functions. Nature reviews Molecular cell biology. 2008;9:151–161. doi: 10.1038/nrm2334. [DOI] [PubMed] [Google Scholar]
- Michell RH. Inositol and its derivatives: their evolution and functions. Adv Enzyme Regul. 2011;51:84–90. doi: 10.1016/j.advenzreg.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Michell RH. Inositol lipids: from an archaeal origin to phosphatidylinositol 3,5-bisphosphate faults in human disease. The FEBS journal. 2013;280:6281–6294. doi: 10.1111/febs.12452. [DOI] [PubMed] [Google Scholar]
- Migaud ME, Frost JW. Elaboration of a general strategy for inhibition of myo-inositol 1-phosphate synthase: Active site interactions of analogues possessing oxidized reaction centers. J Am Chem Soc. 1996;118:495–501. [Google Scholar]
- Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci U S A. 1999;96:2538–2542. doi: 10.1073/pnas.96.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JE, Turner RS, Agranoff BW, Fisher SK. Differentiated human NT2-N neurons possess a high intracellular content of myo-inositol. J Neurochem. 1999;72:1431–1440. doi: 10.1046/j.1471-4159.1999.721431.x. [DOI] [PubMed] [Google Scholar]
- Olgemoller B, Schwaabe S, Schleicher ED, Gerbitz KD. Upregulation of myo-inositol transport compensates for competitive inhibition by glucose. An explanation for the inositol paradox? Diabetes. 1993;42:1119–1125. doi: 10.2337/diab.42.8.1119. [DOI] [PubMed] [Google Scholar]
- Palmano KP, Whiting PH, Hawthorne JN. Free and lipid myo-inositol in tissues from rats with acute and less severe streptozotocin-induced diabetes. The Biochemical journal. 1977;167:229–235. doi: 10.1042/bj1670229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Pollack SJ, Atack JR, Knowles MR, McAllister G, Ragan CI, Baker R, Fletcher SR, Iversen LL, Broughton HB. Mechanism of inositol monophosphatase, the putative target of lithium therapy. Proc Natl Acad Sci U S A. 1994;91:5766–5770. doi: 10.1073/pnas.91.13.5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Gonzalez R, Petersen DN, Tkalcevic G, Thompson DD, Brown TA. Estrogen-induced genes in the uterus of ovariectomized rats and their regulation by droloxifene and tamoxifen. The Journal of steroid biochemistry and molecular biology. 1998;64:13–24. doi: 10.1016/s0960-0760(97)00142-8. [DOI] [PubMed] [Google Scholar]
- Rumpel H, Lim WE, Chang HM, Chan LL, Ho GL, Wong MC, Tan KP. Is myo-inositol a measure of glial swelling after stroke? A magnetic resonance study. J Magn Reson Imaging. 2003;17:11–19. doi: 10.1002/jmri.10233. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Seelan RS, Lakshmanan J, Casanova MF, Parthasarathy RN. Identification of myo-inositol-3-phosphate synthase isoforms: characterization, expression, and putative role of a 16-kDa gammac isoform. J Biol Chem. 2009;284:9443–9457. doi: 10.1074/jbc.M900206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelan RS, Parthasarathy LK, Parthasarathy RN. E2F1 regulation of the human myo-inositol 1-phosphate synthase ISYNA1 gene promoter. Arch Biochem Biophys. 2004;431:95–106. doi: 10.1016/j.abb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Seelan RS, Pisano MM, Greene RM, Casanova MF, Parthasarathy RN. Differential methylation of the gene encoding myo-inositol 3-phosphate synthase Isyna1 in rat tissues. Epigenomics. 2011;3:111–124. doi: 10.2217/epi.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel G, Shamir A, Shapiro J, Ding D, Dalton E, Bialer M, Harwood AJ, Belmaker RH, Greenberg ML, Agam G. Valproate decreases inositol biosynthesis. Biol Psychiatry. 2004;56:868–874. doi: 10.1016/j.biopsych.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Shen X, Xiao H, Ranallo R, Wu WH, Wu C. Modulation of ATP-dependent chromatin-remodeling complexes by inositol polyphosphates. Science (New York, N.Y) 2003;299:112–114. doi: 10.1126/science.1078068. [DOI] [PubMed] [Google Scholar]
- Sherman WR, Packman PM, Laird MH, Boshans RL. Measurement of myo-inositol in single cells and defined areas of the nervous system by selected ion monitoring. Analytical biochemistry. 1977;78:119–131. doi: 10.1016/0003-2697(77)90015-x. [DOI] [PubMed] [Google Scholar]
- Shi Y, Azab AN, Thompson MN, Greenberg ML. Inositol phosphates and phosphoinositides in health and disease. Sub-cellular biochemistry. 2006;39:265–292. doi: 10.1007/0-387-27600-9_11. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vaden DL, Ju S, Ding D, Geiger JH, Greenberg ML. Genetic perturbation of glycolysis results in inhibition of de novo inositol biosynthesis. J Biol Chem. 2005;280:41805–41810. doi: 10.1074/jbc.M505181200. [DOI] [PubMed] [Google Scholar]
- Shimon H, Agam G, Belmaker RH, Hyde TM, Kleinman JE. Reduced frontal cortex inositol levels in postmortem brain of suicide victims and patients with bipolar disorder. Am J Psychiatry. 1997;154:1148–1150. doi: 10.1176/ajp.154.8.1148. [DOI] [PubMed] [Google Scholar]
- Shimshoni JA, Dalton EC, Jenkins A, Eyal S, Ewan K, Williams RS, Pessah N, Yagen B, Harwood AJ, Bialer M. The effects of central nervous system-active valproic acid constitutional isomers, cyclopropyl analogs, and amide derivatives on neuronal growth cone behavior. Molecular pharmacology. 2007;71:884–892. doi: 10.1124/mol.106.030601. [DOI] [PubMed] [Google Scholar]
- Spector R, Lorenzo AV. Myo-inositol transport in the central nervous system. The American journal of physiology. 1975;228:1510–1518. doi: 10.1152/ajplegacy.1975.228.5.1510. [DOI] [PubMed] [Google Scholar]
- Spizz G, Pike LJ. Growth factors promote inositol uptake in BC3H1 cells. Biochem Biophys Res Commun. 1992;182:1008–1015. doi: 10.1016/0006-291x(92)91832-b. [DOI] [PubMed] [Google Scholar]
- Srivastava AK, Pandey SK. Potential mechanisms involved in the regulation of glycogen synthesis by insulin. Molecular and cellular biochemistry. 1998;182:135–141. [PubMed] [Google Scholar]
- Steger D, Haswell E, Miller A, Wente S, O’Shea E. Regulation of chromatin remodeling by inositol polyphosphates. Science (New York, N.Y) 2003;299:114–116. doi: 10.1126/science.1078062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes CE, Gillon KR, Hawthorne JN. Free and total lipid myo-inositol concentrations decrease with age in human brain. Biochimica et biophysica acta. 1983;753:136–138. doi: 10.1016/0005-2760(83)90108-x. [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra MA. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Steiner P, Zurich MG, Beguin P, Hirling H, Dolci W, Thorens B. Regulated exocytosis of an H+/myo-inositol symporter at synapses and growth cones. The EMBO journal. 2004;23:531–540. doi: 10.1038/sj.emboj.7600072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaden DL, Ding D, Peterson B, Greenberg ML. Lithium and valproate decrease inositol mass and increase expression of the yeast INO1 and INO2 genes for inositol biosynthesis. J Biol Chem. 2001;276:15466–15471. doi: 10.1074/jbc.M004179200. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker D, Feigenson G. Membrane lipids: where they are and how they behave. Nature reviews Molecular cell biology. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth P, Heal DJ, Kendall DA. The effects of acute and chronic lithium treatment on pilocarpine-stimulated phosphoinositide hydrolysis in mouse brain in vivo. British journal of pharmacology. 1990;101:39–44. doi: 10.1111/j.1476-5381.1990.tb12085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Cheng L, Mudge AW, Harwood AJ. A common mechanism of action for three mood-stabilizing drugs. Nature. 2002;417:292–295. doi: 10.1038/417292a. [DOI] [PubMed] [Google Scholar]
- Wolfson M, Bersudsky Y, Zinger E, Simkin M, Belmaker RH, Hertz L. Chronic treatment of human astrocytoma cells with lithium, carbamazepine or valproic acid decreases inositol uptake at high inositol concentrations but increases it at low inositol concentrations. Brain research. 2000;855:158–161. doi: 10.1016/s0006-8993(99)02371-9. [DOI] [PubMed] [Google Scholar]
- Wolfson M, Hertz E, Belmaker RH, Hertz L. Chronic treatment with lithium and pretreatment with excess inositol reduce inositol pool size in astrocytes by different mechanisms. Brain research. 1998;787:34–40. doi: 10.1016/s0006-8993(97)00775-0. [DOI] [PubMed] [Google Scholar]
- Wong YH, Kalmbach SJ, Hartman BK, Sherman WR. Immunohistochemical staining and enzyme activity measurements show myo-inositol-1-phosphate synthase to be localized in the vasculature of brain. J Neurochem. 1987;48:1434–1442. doi: 10.1111/j.1471-4159.1987.tb05682.x. [DOI] [PubMed] [Google Scholar]
- Ye C, Bandara WM, Greenberg ML. Regulation of inositol metabolism is fine-tuned by inositol pyrophosphates in Saccharomyces cerevisiae. J Biol Chem. 2013;288:24898–24908. doi: 10.1074/jbc.M113.493353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorek MA, Dunlap JA, Lowe WL., Jr Opposing effects of tumour necrosis factor alpha and hyperosmolarity on Na+/myo-inositol co-transporter mRNA levels and myo-inositol accumulation by 3T3-L1 adipocytes. The Biochemical journal. 1998;336(Pt 2):317–325. doi: 10.1042/bj3360317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 GSK-3 in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]