

Abstract
Patients with breast cancer brain metastases have extremely limited survival and no approved systemic therapeutics. Triple negative breast cancer (TNBC) commonly metastasizes to the brain and predicts poor prognosis. TNBC frequently harbors BRCA mutations translating to platinum sensitivity potentially augmented by additional suppression of DNA repair mechanisms through poly(ADP-ribose)polymerase (PARP) inhibition. We evaluated brain penetrance and efficacy of Carboplatin +/− the PARP inhibitor ABT888, and investigated gene expression changes in murine intracranial (IC) TNBC models stratified by BRCA and molecular subtype status.
Athymic mice were inoculated intra-cerebrally with BRCA-mutant: SUM149 (basal), MDA-MB-436 (claudin-low), or BRCA-wild-type: MDA-MB-468 (basal), MDA-MB-231BR (claudin-low) TNBC cells and treated with PBS control (IP, weekly), Carboplatin (50mg/kg/week, IP), ABT888 (25mg/kg/day, OG), or their combination. DNA-damage (γ-H2AX), apoptosis (cleaved-Caspase-3(cC3)) and gene expression were measured in IC tumors.
Carboplatin+/−ABT888 significantly improved survival in BRCA-mutant IC models compared to control, but did not improve survival in BRCA-wild-type IC models. Carboplatin+ABT888 revealed a modest survival advantage versus Carboplatin in BRCA-mutant models. ABT888 yielded a marginal survival benefit in the MDA-MB-436, but not in the SUM149 model. BRCA-mutant SUM149 expression of γ-H2AX and cC3 proteins was elevated in all treatment groups compared to Control, while BRCA-wild-type MDA-MB-468 cC3 expression did not increase with treatment. Carboplatin treatment induced common gene expression changes in BRCA-mutant models.
Carboplatin+/−ABT888 penetrates the brain and improves survival in BRCA-mutant IC TNBC models with corresponding DNA damage and gene expression changes. Combination therapy represents a potential promising treatment strategy for patients with TNBC brain metastases warranting further clinical investigation.
Introduction
Brain metastases are a particularly devastating complication for patients with triple negative breast cancer (TNBC) with a limited survival and few therapeutic options (1). TNBC lacks expression of the estrogen receptor (ER), the progesterone receptor (PR), and amplification of the human epidermal growth factor receptor 2 (HER-2) gene (2). When compared to other breast cancer subtypes, TNBC has an increased potential to spread to the central nervous system (CNS) (3–5); approximately 50% of patients with advanced TNBC will recur in the brain (4). Despite local cranial therapies (i.e. radiation and/or neurosurgery), overall survival from detection of CNS metastases for patients with TNBC is less than 6 months (1). Currently, there are no FDA-approved systemic or targeted therapies for patients with TNBC brain metastases. The treatment of CNS metastases arising from TNBC is limited by both the lack of defined biologic targets and the inability of the majority of anti-cancer agents to penetrate the blood brain barrier (BBB). Thus, there is an urgent need to develop safe and effective treatments for this aggressive disease.
Although TNBC is a distinct clinical subtype, it is a molecularly heterogeneous disease. Given the lack of defined biologic targets in TNBC, it is important to study possible molecular drivers to develop successful therapies. Gene expression studies have shown that greater than 75% of TNBC are basal-like or claudin-low subtype (6, 7). With approximately 20% of TNBC patients harboring a mutation in either the BRCA1 or BRCA2 genes (8), exploitation of this molecular pathway with brain penetrant cytotoxic agents could be used as a therapeutic strategy to treat TNBC brain metastases.
The BRCA family is responsible for repairing DNA double-stranded breaks (DSB) via homologous recombination (9). Thus, tumors harboring BRCA mutations are sensitized to DNA damaging cytotoxic agents such as platinum derivatives (i.e. cisplatin or carboplatin) that bind to DNA and form DNA crosslinks, leading to DNA DSBs (10). Following platinum treatment of BRCA-mutated breast cancer cells, at least two possibilities may occur: (1) the cells are unable to repair widespread DSBs and undergo apoptosis, or (2) the cells rely on the base excision machinery to rescue DSBs via the enzyme poly(ADP-ribose)polymerase (PARP) (11, 12). One promising strategy to enhance the cytotoxic effect of platinum therapies in BRCA-mutated TNBC is concomitant PARP inhibition. This strategy may be particularly advantageous for TNBC BRCA-mutated brain metastases as several clinically-available PARP inhibitors cross the BBB (13).
Historically, platinums in combination with PARP inhibitors have shown activity in in vitro and in vivo models of BRCA-associated TNBC (14–16) and are brain penetrant. The combination of platinum/PARP inhibitor therapy has never been examined in breast cancer brain metastases. The novelty of this preclinical study is the exploration and efficacy of brain-permeable, clinically-available compounds in animal models of TNBC brain metastases, with the goal to translate these findings into the clinical setting. We present a novel study using preclinical murine models of intracranial TNBC, comparing both BRCA-mutant (mut) and wild-type (wt) models, as well as basal-like and claudin-low subtypes in response to systemic platinum +/− PARP inhibitor combination therapy. This study uniquely demonstrates brain penetration of both systemically administered Carboplatin and ABT888, improved survival in BRCA-mut intracranial TNBC models, and molecular mechanisms of DNA damage and increased apoptosis in response to therapy. Our results suggest that Carboplatin+/−ABT888 may be a viable therapeutic option for patients with TNBC brain metastases.
Materials and Methods
Cell Lines and Culture Conditions
SUM149 (BRCA1-mut, basal-like), MDA-MB-436 (BRCA1-mut, claudin-low), MDA-MB-468 (BRCA-wt, basal-like), and MDA-MB-231BR (BRCA-wt, claudin low) were selected for the study and obtained from the American Type Culture Collection unless otherwise specified. The identity of the cell lines was confirmed by gene expression (September 2010). All media and additives were purchased from Invitrogen (Grand Island, NY). SUM149 (17) was cultured in HuMEC with supplements and 5% fetal bovine serum (FBS). MDA-MB-468 was cultured in RPMI-1640 plus 10% FBS in a flask with plug seal cap (Corning, Tewksbury, MA). MDA-MB-231BR and MDA-MB-436 were maintained in DMEM (high glucose) plus 10% FBS. All cell lines except for MDA-MB-436 were transduced with pTK1261 vector carrying firefly Luciferase as previously described (18). Cell lines were maintained at 37°C and 5% carbon dioxide in the presence of Antibiotic-Antimycotic solution (Invitrogen, Grand Island, NY). Cells were harvested immediately prior to intracranial implantation.
Intracranial tumor implantation and animal handling
Ten week-old female Foxn1nu/nu mice (UNC Animal Studies Core) weighing at least 20 g before IC tumor implantation were used for all studies. IC tumor implantation was performed as previously described (18). All animals were handled and monitored for health conditions according to the Institutional Animal Care and Use Committee (IACUC) approved protocols and entered the study only if recovered following surgery. Bioluminescence imaging was performed weekly to monitor IC tumor growth as described previously (18).
Efficacy study design
For IC tumor models expressing Luciferase (SUM149, MDA-MB-468, MDA-MB-231BR), bioluminescence signal was used to evenly distribute tumor size among the treatment groups within each model. The average bioluminescence signal between treatment groups within each model was not statistically different prior to treatment (data not shown). MDA-MB-436 injected animals were assigned to treatment groups randomly.
At 14 days following tumor implantation for all models except for MDA-MB-231BR (7 days due to aggressive tumor phenotype), treatment started. Health and weight of the animals were monitored at least three times weekly until sacrifice due to clinical symptoms representing poor health condition (i.e. decreased response to stimuli, neurologic dysfunction, weight loss of 20%, and/or a Body Composition Score of 2 or less) or 12 weeks post-intracranial implantation as dictated by prespectified IACUC guidelines for humane reasons. Treatment doses were selected based on previous literature (16, 19, 20) and dose escalation studies (data not shown), and were: 100μl PBS control (IP, weekly), 50mg/kg/week Carboplatin (IP) [10 mg/mL sterile aqueous solution, University of North Carolina Hospital Pharmacy, Chapel Hill, NC], 25mg/kg/day ABT888 (oral gavage (OG)) [Chemistry Center for Integrative Chemical Biology and Drug Discovery, UNC] dissolved in PBS (Life Technologies, Grand Island, NY) immediately prior to OG, or combination 50 mg/kg/week Carboplatin+25 mg/kg/day ABT888 (IP and OG respectively). For the SUM149 model, survival data from three independent experiments were combined based on similar median survival for all control groups (data not shown, p=0.35). No toxicities were found throughout the survival studies in any model or treatment group.
Magnetic Resonance Imaging and Analysis of Blood Brain Barrier Permeability
Four mice from each IC model were used. Gadolinium enhanced brain magnetic resonance imaging (MRI) on a BioSpec 9.4 T small animal imaging system (Bruker Corporation, Billerica, MA) under isoflurane anesthesia began one week following MDA-MB-231-BR injection and two weeks following SUM149, MDA-MB-436, and MDA-MB-468 injection. Precontrast coronal T1 and T2 images were obtained for localization. Pre-contrast variable flip angle gradaint echo T1 sequences (TE=1.5 msec, TR 15 msec, flip angles 2, 5, 10, and 15 degrees) were performed followed by a dynamic scan using 15 degree flip angle was repeated for 130 scans (8.6 sec/repeat) for dynamic contrast enhancement. After the 5th scan, gadopentate dimeglumine (Magnevist, Bayer Healthcare) was administered at 0.1 mmol/kg followed by saline flush via teil vein catheter. Coronal post-contrast T1 images were obtained. Standard physiologic monitoring occurred during imaging procedure. Animals were recovered and returned to standard housing. Mice were reimaged each week until time of sacrifice due to 20% weight loss or signs of tumor burden.
Utilizing the T1 MR 20th and 3rd scan of the dynamic sequence, a difference map was generated and Relative Enhancement was calculated as follows:
where Δ[Gd]T and Δ[Gd]P represent contrast concentration changes in tissue and plasma during time zero and t, respectively, ST(t) is signal intensity in selected tissue (tumor, contralateral normal appearing brain tissue or left lateral ventricular choroid plexus) at time point t, SP(t) is signal intensity in plasma at time point t and ST0 and SP0 are pre-contrast signal intensity in selected tissue or plasma, respectively.
Multiple regions of interest (ROIs) sized 10–15 pixel (0.1 mm2) were manually drawn by one blinded reader on T2 images, guided by the lesion shape, avoiding macroscopic vessels, necrosis, and areas of hemorrhage. Region with greatest signal change was selected as the tumor ROI. ROIs were propogated to the subtracted T1 DCE MR images, acquired in anatomic registration. The same size ROIs were drawn on contralateral amygdala, choroid plexus of the contralateral lateral ventricle, and contralateral posterior facial vein lumen.
Pharmacodynamic study design
In vivo PAR Pharmacodynamic Study
An in vivo PAR assay was performed using intracranial tissue from the TNBC SUM149 IC murine model. Animals were divided into treatment groups, and drug was administered using doses specified in efficacy studies. At harvest, tumors were flash frozen in liquid nitrogen, and stored at −80°C until further analysis. Net PAR levels were determined in SUM149 IC tumors collected immediately following 14 days of treatment with either control or ABT888. To determine net PAR levels in the protein extracts from intracranial tumors, a high throughput chemiluminescent ELISA PARP in vivo Pharmacodynamic Assay was performed (Trevigen, Gatherburg, MD). Frozen SUM149 IC tumors from Control (n=11) or ABT888 (n=8) were homogenized in Lysis buffer, sonicated 3 times for 10 seconds each, and assayed for PAR according to the manufacturer’s protocol. Luminescence signal of the experimental samples and PAR standards were recorded in photons/sec using an IVIS Lumina camera (Caliper Life Sciences, Hopkinton, MA) and corrected for background. PAR concentrations in the experimental samples were calculated using a standard curve, and PAR levels were determined as pg/100ug of total protein. Experimental samples and freshly made PAR standards were assayed in triplicate. The assay was repeated once for samples with sufficient amount of protein extract. Concentration of the total protein in the extract was measured using the BCA Assay (Thermo Fisher Scientific, Rockford, IL).
In vitro PAR Pharmacodynamic Assay
To examine the ability of ABT888 to inhibit PAR independently of BRCA status, in vitro PAR levels of BRCA-mut SUM149 and BRCA-wt MDA-MB-231BR was examined at the IC50 of the SUM149 cell line (60 uM). SUM149 and MDA-MB-231BR cells were plated at 1.5 and 1.0 million cells/6 cm dish respectively in triplicate. After 24 hours, media was changed to either ABT888 (60 uM) or 0.1% DMSO vehicle control. After 2 hours of inhibition, cells were harvested, and PAR was measured according to ELISA PARP in vivo Pharmacodynamic Assay protocol (Trevigen, Gatherburg, MD).
Immunohistochemistry Study Design and Tissue Preparation
For immunohistochemistry, we collected tissue from BRCA-mut SUM149 and BRCA-wt MDA-MB-468 TNBC intracranial murine models. Animals were divided into treatment groups within each model and treated for 14 days after tumor injection with 3 doses of Carboplatin, 15 doses of ABT888, or Carboplatin+ABT888 in combination. Brain tissues from 3 animals per group were collected in formalin the day following last dose of Carboplatin and/or immediately after the last dose of ABT888. Whole mouse brains fixed in formalin were cut parasagittaly approximately 2mm of midline, incubated in 70% Ethanol, embedded in paraffin, and sectioned immediately prior to staining.
Immunohistochemical Staining for γ-H2AX and cleaved Caspase-3
The following antibodies were used to stain 4 μm sections placed on coated glass slides: Rabbit monoclonal anti-phospho-ser139-H2AX [γ-H2AX] (1:2000, 2h, Cell Signaling Technology, Danvers, MA), rabbit polyclonal Cleaved Caspase-3 [cC3] (1:50, 1h, Biocare Medical, Concord, CA). Staining was performed as previously described (21, 22). IHC was carried out on the Bond Autostainer, and all solutions were from Leica Microsystems (Norwell, MA). Briefly, slides were deparaffinized in Bond dewax solution (AR9222) and hydrated in Bond wash solution (AR9590). Antigen retrieval of γ-H2AX was performed for 30 min at 100°C in Bond-epitope retrieval solution 1 pH-6.0 (AR9961) and in solution 2 pH-9.0 (AR9640) for cC3. After incubation with the appropriate antibody, detection was performed with the Bond Polymer Refine Detection System (DS9800). Stained slides were dehydrated and coverslipped. Positive and negative (no primary antibody) controls were included for each antibody. Sections from 2–3 biological replicas and 2–3 technical replicas (total 6 sections) per treatment group were stained.
Digital imaging and image analysis of immunohistochemical staining
H&E and IHC stained sections were digitally imaged (20× objective) using Aperio ScanScope XT (Aperio Technologies, Vista, CA),and analyzed within the Aperio Spectrum Database. Folded tissues, non-tumor areas, and artifacts were excluded from the analysis using a negative pen. Tumor area identification was guided by H&E staining of an adjacent section. The expression of γ-H2AX and cleaved Caspase-3 (cC3) was measured using the Aperio Nuclear V9 (cell quantification) algorithm. Positive and negative (no primary antibody) controls were included for each antibody. The intensity score and percentage of γ-H2AX-positive nuclei in the tumor area obtained with the modified nuclear v9 algorithm at each intensity level were used to calculate the H-Score using the formula: . The H-Score was normalized to a γ-H2AX positive SK-MEL 181 cell line control included in each staining. Expression of cC3 protein was calculated using the percentage of cC3-positive nuclei.
Microarrays and RNA purification
RNA from BRCA-mutant intracranial SUM149 and MDA-MB-436 tumors was purified using the RNeasy Mini kit (Qiagen, Germantown, MD) following the manufacturer’s protocol, labeled as previously described (23), amplification incorporating cyanine-5 (Cy5) dye for the tumor samples and cyanine-3 (Cy3) dye for the reference (24) using Agilent Low RNA Input Fluorescent Linear Amplification Kit (Agilent Technologies, Santa Clara, CA), and hybridized to 4×44K Customized Human Oligomicroarrays (Agilent Technologies, Santa Clara, CA). Arrays were scanned with either the Agilent Scanner (Agilent Technologies, Santa Clara, CA) or the GenePix Scanner (Molecular Devices, Sunnyvale, CA) as previously described (25). All microarray data has been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE55399.
SUM149 arrays scanned with the GenePix scanner were normalized to be comparable to the Agilent scanner were adjusted according to Supplemental Methods (Supplemental Figure S1). Principle component analysis demonstrates effective removal of scanner bias (Supplemental Figure S2). Treatment specific gene sets were identified using R v3.0.2 and the two class unpaired significance analysis of microarray (SAM) (v3.11) (26) and as further described in Supplemental Methods (Supplemental Figure S3). The following comparisons were conducted within each model: Control versus Carboplatin, Control versus ABT888, Control versus Carboplatin+ABT888, and Carboplatin versus Carboplatin+ABT888 (Supplemental Tables S1b-S1i). All genes with an FDR = 0 in control versus Carboplatin intracranial SUM149 or MDA-MB-436 tumors were analyzed with DAVID (27, 28). Pathways are reported with a Bonferroni corrected p value of < 0.05 (Supplemental Tables S1j–S1k). For comparative visualization, SUM149 arrays were scaled to the same dynamic range as the MDA-MB-436 arrays (Supplemental Figure S4). Unsupervised and supervised hierarchical clustering were clustered using centroid linkage in Gene Cluster 3.0 v1.52 (29) and viewed with Java Treeview v1.1.6r4 (30).
Statistical Considerations
Results for BBB permeability are reported as mean relative enhancement ± Standard Error of Means (SEM). PAR assay results were normalized to the percent of the respective control group’s median and are presented as mean of the normalized values ± SEM. IHC to evaluate DNA damage and apoptosis is reported as mean values ± SEM. One-way ANOVA tests were used to compare mean values between treatment groups using GraphPad Prism 6.04. Unpaired t-tests were performed for all pairwise comparisons, with Bonferroni correction for multiple comparisons when applicable.
The Kaplan-Meier method and Log-rank tests were used to compare overall survival among treatment groups. Mice were censored at pre-specified study endpoints if they had not been previously sacrificed. Overall and pair-wise tests were completed, and unadjusted p-values are reported. For bioluminescence imaging, fold changes were calculated relative to the start date of treatment, and if present, negative imaging values (due to correction for background) were recorded and set to zero. The common logarithm of these values were used for this analysis. For every time point where at least two animals were alive in the treatment group, the median level and interquartile range (25th–75th percentile) for bioluminescence imaging were calculated. For gene expression analysis, significance analysis of microarray was employed through R v. 2.15.0 (31).
Results
Our preclinical study evaluates efficacy of Carboplatin therapy as a single agent and in combination with the PARP inhibitor, ABT888, in intracranial (IC) orthotopic murine models of BRCA-mut and BRCA-wild-type TNBC.
Blood brain barrier permeability across models
To assess the inherent differences in therapy accessibility due to the vasculature across all IC murine models, we performed MRI with Gadolinium contrast. Because contrast can only enter brain tissue where the BBB is disrupted, relative enhancement (RE) estimates the BBB permeability. Tumor mean RE for each IC model was not significantly different across models (SUM149: 1.081±0.1084; MDA-MB-436: 0.8711±0.1788; MDA-MB-468: 0.7683±0.2210; MDA-MB-231BR: 0.6835±0.2052; p=.48). In addition, RE was significantly higher in all tumors compared to matched normal brain tissue across all models (p<0.001; Figure 1A), indicating a similarly compromised, leaky BBB within all IC tumor models. There was no significant difference across models within the normal (p=.14), tumor (p=.48), and choroid plexus (p=.82) tissues (Figure 1A). The small amount of RE in normal tissues is in agreement with previous studies, representing normal cerebral blood volume (32). Representative MRI images are shown for each model and demonstrate that the morphologic appearance of the IC tumors formed by each model is a similar, focal, concentrated mass with defined borders in the right hemisphere in vivo (Figure 1B).
Figure 1. Blood brain barrier (BBB) permeability studies demonstrate compromised BBB in tumors as compared to normal brain and no significant differences in BBB permeability between models.

(A) Relative enhancement of four tumor cell lines with matched normal, tumor, and choroid plexus as determined from T1 DCE MR images, formed from digitally subtracting pre-contrast T1-DCE-weighted sequence from the identical sequence performed after gadolinium administration (20th sequence). Significant difference was observed between tumor and normal brain tissue (p<0.001), and no significant difference was seen between models within each tissue type (p>.14 within each tissue). (B) Brain MR imaging of a preresentative intracranial tumor from each of the four intracranial models.
ABT888 inhibits the function of PARP in SUM149 IC tumors
PARP catalyzes the formation of poly(ADP-ribose) (PAR) chains on a variety of proteins including PARP itself. To assess penetration of the PARP inhibitor ABT888 across the BBB and monitor inhibitory activity, we measured net PAR levels (± SEM) in SUM149 intracranial tumors following14 days of ABT888 treatment. PAR levels in ABT888-treated SUM149 IC tumors (18.76±7.566) were significantly decreased as compared to control tumors (119.8±30.37; p=0.0091), indicating 84.3% inhibition and IC tumor exposure with ABT888 treatment (Figure 2A).
Figure 2. Formation of poly(ADP-ribose) chain fragments [PAR] indicates intracranial exposure for ABT888 and significant in vitro inhibition regardless of BRCA status.
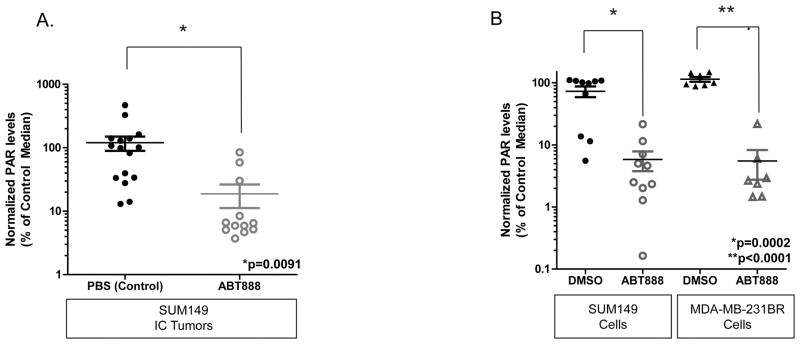
A. Relative net PAR levels measured in the SUM149 intracranial tumor tissue after treatment with PBS control or ABT888 for 14 days. PAR levels decreased from 119.8±30.37 to 18.76±7.57 following ABT888 treatment (p<0.0001). Data are expressed as the percentage of the mean control tumor mean PAR values (±SEM), normalized to the total amount of protein in the sample protein extract (pg/ml per 100ug total protein).
B. PAR levels measured in cell culture of BRCA-mut SUM149 and BRCA-wt MDA-MB-231BR following 2h ABT888 treatment. SUM149 control treated (72.89±14.25) as compared to ABT888 treated (5.820±2.044) and MDA-MB-231BR control (114.1±10.01) as compared to ABT888 treated (5.518±2.763) are significantly decreased (SUM149: p<0.0002; MDA-MB-231BR: p<0.0001).
To assess differences in the ability to form PAR chains in BRCA-mut as compared to BRCA-wt models, in vitro PAR assays of BRCA-mut SUM149 and BRCA-wt MDA-MB-231BR models were performed following 2 hrs of treatment with 60 uM of ABT888. PAR levels in SUM149 cells were significantly decreased with ABT888 treatment (5.820±2.044) as compared to control (72.89± 14.25; p=0.0002). In parallel, PAR levels in MDA-MB-231BR were significantly decreased in ABT888 treated cells (5.518± 2.763) as compared to control cells (114.1± 10.01; p<0.0001). Similar percent inhibition was observed between the BRCA-mut and BRCA-wt cell lines (SUM149: 92.0% inhibition; MDA-MB-231BR: 95.2%) (Figure 2B). Taken together, these data demonstrate effective ABT888 penetration into intracranial tumor and similar relative inhibition in vitro between a BRCA-mut and BRCA-wt model.
Treatment with Carboplatin alone and in combination with ABT888 improves survival of BRCA-mut, but not BRCA-wt, TNBC intracranial models
Recognizing the BBB permeability of Carboplatin and ABT888, the efficacy of platinum agents and PARP inhibitors in advanced extracranial BRCA-mutant TNBC, and the potential rational therapeutic combination to treat breast cancer brain metastases, we evaluated the efficacy of Carboplatin and ABT888 as single agents and in combination in both BRCA-mut and BRCA-wt TNBC IC murine models. Median survivals are reported for each model by treatment group (Table 1).
Table 1.
Median Survival of Intracranial TNBC Murine Models treated with Carboplatin with or without ABT888 in comparison to Control.
| BRCA-mutant IC TNBC Murine Models | ||||||
|---|---|---|---|---|---|---|
| SUM149 (BRCA1mut, pTEN-, basal) | MDA-MB436 (BRCA1mut, pTEN-, claudin-low) | |||||
| Treatment Group | Mice per group, n | Median Survival and 95% CI, days | pValue* | Mice per group, n | Median Survival and 95% CI, days | pValue* |
| PBS, Control | 17 | 36 (34, 40) | N/A | 7 | 44 (35–47) | N/A |
| ABT888 | 10 | 39 (30, 46) | p=0.2171 | 5 | 57 (44–74) | p=0.0286 |
| Carboplatin | 16 | 58 (47, 67) | p <0.0001 | 5 | 86 (61, undefined) | p=0.0011 |
| Carboplatin+ABT888 | 17 | 64 (59, 75) | p <0.0001 | 5 | Not reached (65, undefined) | p=0.0011 |
| BRCA-wild-type IC TNBC Murine Models | ||||||
|---|---|---|---|---|---|---|
| MDA-MB468 (pTEN-, basal) | MDA-MB231BR (claudin-low) | |||||
| Treatment Group | Mice per group, n | Median Survival and 95% CI, days | pValue | Mice per group, n | Median Survival and 95% CI, days | pValue |
| PBS, Control | 5 | 35 (29–57) | p=0.81** | 9 | 25 (12–30) | p=0.104** |
| ABT888 | 5 | 44 (33–57) | N/A | 6 | 33.5 (18–42) | N/A |
| Carboplatin | 5 | 37 (34–54) | N/A | 5 | 29 (14–40) | N/A |
| Carboplatin+ABT888 | 5 | 37 (33–50) | N/A | 7 | 26 (23–27) | N/A |
CI=Confidence Interval; N/A=Not Applicable; * Individual treatment group in comparison to PBS (Control); ** All treatments compared to PBS (Control).
In the basal-like BRCA1-mut SUM149 IC model, the median survival of animals treated with Carboplatin±ABT888 were both significantly longer then control (Carboplatin: 58 days (95% CI 47 to 67 days); Carboplatin+ABT888: 64 days (95% CI 59 to 75 days); Control: 36 days (95% CI 34 to 40 days); both treatment groups p < 0.0001 relative to control) (Table 1 and Figure 3A). The difference between median survival of control and single agent ABT888 treated animals (39 days (95% CI 30 to 46 days)) was not statistically significant (p=0.2). In addition, ABT888 alone was inferior compared to therapies with Carboplatin as a single agent (p=0.007) or Carboplatin+ABT888 combination (p=0.0012). Despite theoretical promise, the Carboplatin+ABT888 combination therapy exhibited modest improvement of survival in comparison to single agent Carboplatin and was not statistically significant (p=0.4; Table 1 and Figure 3A). Of note, 2/16 and 2/17 animals from the Carboplatin and Carboplatin+ABT888 groups, respectively, remained alive at the pre-specified study completion date (12 weeks post-intracranial implantation of tumor cells).
Figure 3. Treatment with Carboplatin +/− ABT888 improves survival in BRCA-mutant intracranial TNBC.
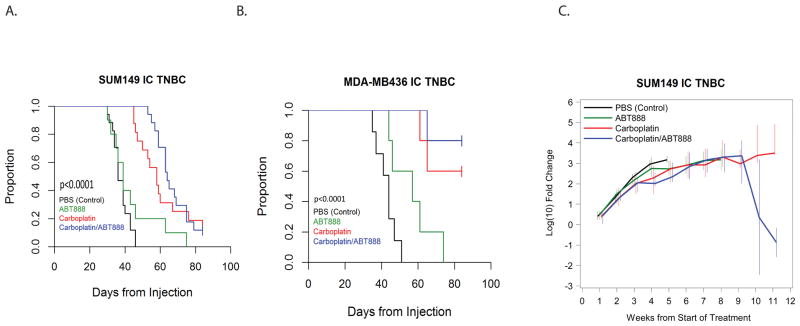
Animals in both models were treated with PBS (control), Carboplatin 50mg/kg/week (IP), ABT888 25mg/kg/day (OG) or combination Carboplatin+ABT888 (doses as in single agent therapy). Treatment began 14 days after intracranial implantation. A total of 4 animals in the SUM149 model and a total of 6 animals in the MDA-MB436 model treated with Carboplatin and Carboplatin+ABT888 were sacrificed 12 weeks post-intracranial implantation, as defined by IAUCUC protocol guidelines, and were censored for the purpose of the analysis.
A. Median survival of the SUM149 intracranial model.
B. Median survival of the MDA-MB-436 intracranial model.
C. Dynamic changes in intracranial tumor growth measured by median fold change of the bioluminescence signal intensity from the start of the treatment in SUM149 TNBC murine model. The median log fold changes for each treatment group were calculated weekly. Data for each time point were plotted if at least 2 animals per treatment group remained alive. The vertical bars represent the interquartile ranges (25th–75th percentiles).
Recognizing the heterogeneity of TNBC, we investigated response to Carboplatin and Carboplatin+ABT888 in a second BRCA-mut IC TNBC model, MDA-MB-436. Median survival following treatment with Carboplatin alone (86 days (95% CI 61 to undefined)) and with combination Carboplatin+ABT888 (Not reached, ≥ 65 days) were both significantly longer (both p=0.001) as compared to control (44 days (95% CI 35 to 47 days); Table 1, Figure 3B). Single agent ABT888 resulted in a modest survival advantage compared to control (57 days (95% CI 44 to 74 days), p=0.03). The addition of ABT888 to Carboplatin did not yield a significant improvement in survival in the MDA-MB-436 model compared to Carboplatin alone (p=0.2); however, 4/5 animals in the combination group versus 2/5 animals in the Carboplatin alone group remained alive 12 weeks post cell injection, suggesting benefit of the addition of ABT888 to Carboplatin.
To examine the efficacy of Carboplatin and Carboplatin+ABT888 in BRCA-wt intracranial TNBC models, we applied the same experimental design to BRCA-wt intracranial TNBC models, basal-like MDA-MB-468 and claudin-low MDA-MB-231BR. In contrast to the BRCA-mut TNBC intracranial models, neither single agent Carboplatin, ABT888, nor combination Carboplatin+ABT888 resulted in an improved median survival compared to control in either BRCA-wt TNBC intracranial model (Supplemental Figure S5A and S5B) (MDA-MB-468, p=0.8; MDA-MB-231BR, p=0.1).
Treatment with Carboplatin alone or in combination with ABT888 impairs tumor growth in BRCA-mut basal-like TNBC intracranial murine model
In addition to the survival analysis, we examined the effect of treatment with Carboplatin alone or in combination with ABT888 on intracranial tumor growth via bioluminescence in the BRCA-mut SUM149 model. Consistent with overall survival results, dynamic changes in the intracranial tumor growth as measured by median log fold change of the bioluminescence signal intensity (photons/second) from the start of the treatment were lowest following treatment with Carboplatin+/− ABT888 (Figure 3C). The median log fold-change in IC tumors treated with either Carboplatin or Carboplatin+ABT888 was similar at 8 weeks of treatment (median 3.3, Interquartile Range (IQR) 2.85 to 4.14 versus median 3.32, IQR 2.56 to 3.97, respectively). Although based on small numbers of animals remaining, at 10 weeks post treatment, the median log fold change for the Carboplatin+ABT888 group remained low until the end of the study, while median fold change for the Carboplatin single agent group continued to increase over time (Figure 3C).
Treatment with Carboplatin and Carboplatin+ABT888 induces DNA damage and apoptosis in BRCA-mutant intracranial tumor but not BRCA-wt intracranial tumors
To investigate whether processes of DNA damage and apoptosis are activated in intracranial tumors in response to treatment, we evaluated expression of γ-H2AX protein, a known biomarker of DNA damage, and cC3, the central protein in the terminal phase of apoptosis (15, 33–35).
Expression of γ-H2AX was elevated in all treatment groups compared to control following 14 days of treatment in BRCA-mut SUM149 intracranial tumors (Figure 4A, p=0.0106). DNA damage was highest in the Carboplatin group (91.0 ± 3.7, p=0.001) followed by combination Carboplatin+ABT888 (76.6 ± 8.8, p=0.12), which were both higher than control (58.5 ± 6.0). ABT888 treatment resulted in an γ-H2AX H score (75.4 ± 4.3) that was higher than control (p=0.045).
Figure 4. Immunohistochemical staining to evaluate DNA damage and apoptosis via γ-H2AX and Cleaved Caspase-3 Protein Expression in BRCA-mutant and BRCA-wild-type intracranial TNBC.

Gamma-H2AX [γ-H2AX] (A) and cleaved Caspase-3 [cC3] (B) protein expression in BRCA-mutant SUM149 and BRCA wild-type MDA-MB-468 intracranial tumors evaluated after 14 days of treatment in PBS control, ABT888, Carboplatin, Carboplatin+ABT888 groups by IHC staining. Data are presented as H-Score mean values (±SEM) for γ-H2AX protein expression, and as mean percentage (±SEM) of cC3 positive cells to reflect cC3 protein expression. p values of all treatment groups compared to Control are presented for each model. Representative sections stained with γ-H2AX after treatment with Carboplatin are displayed for SUM149 model (C) and MDA-MB468 model (D), magnification 10×.
Representative sections stained with cC3 after treatment with PBS (control, E) or Carboplatin (F) are displayed for SUM149 model (magnification 20×, zoom 10×).
With similar BBB permeability observed between the BRCA-wt and BRCA-mut models, significant differences in DNA damage were also observed in the BRCA-wt MDA-MB-468 IC model for both Carboplatin and Carboplatin+ABT888 treated groups as compared to control (p=0.0015). Expression of γ-H2AX, however, was notably weaker in the MDA-MB-468 IC tumors as compared to the SUM149 IC model (p<0.0001 MDA-MB-468 Control vs SUM149 Control, Figure 4A, 4C, and 4D). Attenuated DNA damage after DNA adduct formation rather than differences in exposure to Carboplatin may provide an explanation as to why Carboplatin treatment +/− ABT888 was effective in the BRCA-mut and not in the BRCA-wt intracranial models.
In addition to γH2AX expression, the same tumor samples from BRCA-mut SUM149 and BRCA-wt MDA-MB-468 intracranial models were analysed to determine expression levels of cC3 protein. In BRCA-mut SUM149 intracranial tumors, mean percentages of cC3 positive cells for all treatments were significantly higher in comparison to Control (7.7±0.4, p=0.0297, Figure 4B, 4E, and 4F); however, mean percentages of cC3 positive cells (± SEM) by treatment group were not significantly different from each other (ABT888 15.5 ± 2.6, Carboplatin 12.8 ± 1.5, and Carboplatin+ABT888 12.2 ± 1.5; p=0.46).
In the BRCA-wt MDA-MB-468 intracranial model, mean values of percent cC3 positive cells for ABT888 (14.5±2.7), Carboplatin (7.0±2.0), and Carboplatin/ABT888 (13.0±2.0) exhibited overall borderline significance when compared to control (8.2±1.3; p=0.051; Figure 4B, Supplemental Figure S6).
Taken together, these data support previous findings that BRCA status has a significant impact on sensitivity to Carboplatin treatment through increased DNA damage and apoptosis levels, and provides a possible explanation as to why BRCA-wt intracranial tumors are less responsive to Carboplatin treatment +/− ABT888.
Intracranial TNBC Gene Expression Changes in Response to Carboplatin with and without ABT888
To examine the dynamic transcriptional changes that occur in IC tumors in response to treatment, we performed gene expression analysis of BRCA-mut IC tumors from mice treated with control, ABT888, Carboplatin, and Carboplatin+ABT888 in the BRCA-mut basal-like SUM149 and the BRCA-mut claudin-low MDA-MB-436 intracranial models.
Unsupervised hierarchical clustering of all samples identified cell line specific delineations as well as distinct clusters defined by Carboplatin treatment +/− ABT888 (Supplemental Figure S7). Independent analyses within each model further demonstrated little to no contribution with the addition of ABT888 to control or Carboplatin treatments (Figure 5A, 5B).
Figure 5. Gene expression changes of Carboplatin-sensitive SUM149 and MDA-MB-436 TNBC intracranial tumors.

Measured expression patterns of tumors treated within the BRCA-mutant models, with up-regulated genes in red and down-regulated genes in green. Unsupervised hierarchical clustering of the most highly differentiating genes within the (A) MDA-MB-436 (purple) and (B) SUM149 (yellow) tumors stratified by Carboplatin treatment (red), Carboplatin+ABT888 (blue), ABT888 alone (green), or Control (black). (C) Supervised clustering using the 38 commonly altered genes with Carboplatin treatment across all tumors. Color bars mark oncogenes (orange), therapeutic targets (gray), and nervous system genes (light blue).
To quantitate gene expression changes that occurred with treatment, each treatment group was compared to control within SUM149 and separately within MDA-MB-436 tumors (Supplemental Tables S1b–S1i). Treatment with ABT888 as compared to control (Supplemental Table S1b, S1c) resulted in 19 differentially expressed genes for the SUM149 model, and 153 differentially expressed genes for the MDA-MB436 model. In contrast, comparison of Carboplatin treatment to control (Supplemental Table S1d, S1e) demonstrated a substantially higher number of genes differentially expressed within each model (Carboplatin vs. Control: SUM149 = 176; MDA-MB-436 = 5125). Supervised clustering of these genes reveals consistent gene expression patterns throughout each cluster of samples (Supplemental Figure S8A and S8B). Similar results were seen in comparing Carboplatin+ABT888 to Control, with 53 genes differentially regulated in SUM149 tumors, and 4183 genes altered in MDA-MB-436 tumors (Supplemental Tables S1f, S1g). When Carboplatin was compared to Carboplatin+ABT888 treatment, less than 10 differentially expressed transcripts were identified, and none were shared between the two models (SUM149: n=9; MDA-MB-436 n=2) (Supplemental Tables S1h, S1i). Thus, while not inhibiting the ability of Carboplatin to induce dynamic transcriptional changes, ABT888 minimally changes gene expression when combined with Carboplatin treatment.
To annotate the transcriptional changes occurring with Carboplatin treatment, pathway analysis of genes altered with Carboplatin treatment within each model was analyzed with DAVID (27, 28). Up-regulated genes in both SUM149 and MDA-MB-436 were enriched for ‘signal peptide’, ‘glycopeptide’, and ‘SH3 domain’ (Supplemental Table S1j). Interestingly, in MDA-MB-436, neuronal development, neuronal differentiation, and neuronal morphogenesis were significantly up-regulated (Supplemental Table S1j). Down-regulated pathways in DAVID were only significant in the MDA-MB-436 model, with 16 of the top 30 most significant terms involving processes in the mitochondria including ‘electronic chain transport’, ‘respiratory chain’, and ‘mitochondrial inner membrane’ (Supplemental Table S1k).
To define the common transcriptional program that is altered with Carboplatin treatment, the intersection of up-regulated and down-regulated genes common to both models was used. 37 genes were commonly up-regulated (Supplemental Figure S9A), and 1 gene was commonly down-regulated (Supplemental Figure S9B) with Carboplatin treatment in SUM149 and MDA-MB-436 tumors (Supplemental Figure S9C). Interestingly, we found three oncogenes upregulated with Carboplatin treatment, including N-myc (NDRG4), Ras (RAB39B), and Kit, and genes previously demonstrated to be potential therapeutic targets in TNBC, including αB-crystallin (CRYAB) and folate receptor (FOLR3) (Figure 5C) (36, 37). Additionally, multiple neurological genes were up-regulated including CNTNAP2, a member of the neurexin family that provides adhesion in the central nervous system (CNS), CES1, a possible member of the blood-brain barrier, and two CNS specific G-protein coupled receptors, GPR98 and NPY1R (38, 39).
Discussion
This study has demonstrated efficacy of Carboplatin+/−ABT888 treatment in two murine models of BRCA-mut intracranial TNBC with little to no efficacy in models of intracranial BRCA-wt TNBC despite similar BBB permeability and relative PAR inhibition across both BRCA-mut and BRCA-wt models. With confirmed brain penetrance, the addition of PARP inhibition to platinum therapy via ABT888 exhibited only modest impact on efficacy as measured by tumor burden, survival, and molecular changes within the intracranial tumors themselves.
In BRCA-mut intracranial TNBC models, Carboplatin treatment not only resulted in reduction of intracranial tumor volume, but in several cases, in growth arrest of intracranial tumors as evidenced by bioluminescence imaging. The mechanism of increased cytotoxicity within BRCA-mut intracranial TNBC is hypothesized to be a failure to effectively repair DNA damage induced by Carboplatin due to impaired homologous recombination (HR) repair pathways with BRCA loss of function (9). This hypothesis is confirmed by augumentation of γ-H2AX activation and apoptosis mediated by increased levels of cC3 following treatment with Carboplatin. Interestingly, the addition of a PARP inhibitor, which impairs base excision repair, failed to yield a significant survival benefit as a single agent in these model systems. Moreover, the PARP inhibitor did not induce widespread or common gene expression alterations in the BRCA-mut models either alone or when combined with Carboplatin. In contrast, Carboplatin treatment led to substantial gene expression changes observed in BRCA-mut SUM149 and BRCA-mut MDA-MB-436 intracranial models with 37 genes commonly up-regulated, suggesting possible compensatory pathways that are activated upon treatment with Carboplatin. These included oncogenes, previously described brain metastasis therapeutic targets, and neural genes. Interestingly, several recent studies report similar upregulation of neural genes in breast cancer brain metastases including genes related to GABA (40), cellular adhesion (41), and several neurobiological functions (42, 43). While our results could be due in part to normal mouse brain contamination, these complementary and recent studies have excluded this caveat by assessing expression in purified tumor cells (40), passaging metastases cells in vitro (42), or by using species-specific microarrays (44). Exploration of potentially targetable upregulated genes in combination with Carboplatin treatment that are low or absent in normal brain tissue will be critical to future rational combination therapies to avoid neurological side-effects.
Our results demonstrate efficacy of well-tolerated therapeutics in model systems for a patient population with few treatment options. Currently in the field of TNBC brain metastases, a lack of therapeutic targets leaves clinicians with few beneficial choices aside from neurosurgical resection or crainal radiation. In addition, TNBC brain metastases are commonly (>80% of the time) accompanied by extracranial metastases, warranting effective systemic therapies capable of controlling both CNS and non-CNS disease (4). Other studies have demonstrated that irinotecan and iniparib yield an approximately 30% clinical benefit rate in patients with progressive TNBC brain metastases (45). In addition, a recent phase II study showed an approximately 65% intracranial response rate as measured by volumetric imaging following treatment with Carboplatin and Bevacizumab in patients with progressive HER2 negative brain metastases (46). Despite these promising results, there remains no FDA-approved systemic therapy for TNBC brain metastases. Our data continues to support the role of Carboplatin for the treatment of breast cancer brain metastases, specifically in TNBC with BRCA mutations, and the rationale of using Carboplatin±ABT888 combination in the clinical setting to determine whether ABT888 provides an additional benefit in patients with brain metastases.
While our study adds to the literature surrounding TNBC brain metastases treatment, there are several limitations to our study. Direct intracranial implantation, as opposed to hematogenous spread of tumor cells to the CNS, may not recapitulate human spread of metastases; however, the purpose of this study was to evaluate established brain metastases, not prevention, in response to drug delivery and its impact on the intracranial tumor. Additionally, while ABT888 reduced PAR levels in intracranial tumors, suggesting effective BBB penetration, limited efficacy and few gene expression changes were observed. Future studies may aim to investigate other PARP inhibitors with varying degrees of catalytic activity, and thus may be more effective and synergistic with Carboplatin treatment in brain metastases (47).
In conclusion, to our knowledge this study is the first to demonstrate a significant survival advantage in response to systemic Carboplatinn+/−ABT888 treatment in BRCA-mut TNBC intracranial murine models, as well as a mechanistic explanation of these results. Moreover, we have shown that PARP inhibition as a single agent, while effectively penetrating the BBB, does not yield significant improvement in survival or have a dynamic impact on gene expression. These results provide strong rationale to translate our findings into the design of early phase clinical trials for patients with BRCA-mut TNBC brain metastases testing Carboplatin+/−ABT888, with the ultimate goal of improving the survival of patients with an incurable disease who, at present, have limited systemic therapeutic options.
Supplementary Material
Acknowledgments
We kindly acknowledge Toshiyuki Yoneda, PhD as the source of the MDA-MD-231-BR cell line, and Dr. Nancy Thomas and Dr. Bill Kaufmann for providing SK-MEL cell line for IHC staining control. We thank Yongjuan Xia in the UNC Translational Pathology Laboratory (TPL) for expert technical assistance. The UNC Translational Pathology Laboratory is supported in part, by grants from the National Cancer Institute (3P30CA016086), the UNC University Cancer Research Fund (UCRF). We thank the Small Animal Imaging Core facility at the UNC Biomedical Imaging Research Center for providing the magnetic resonance imaging service, and the imaging core is supported in part by an NCI cancer center Grant #P30-CA016086-35-37.
Financial Information: This research is supported by the 2010 Breast Cancer Research Foundation-AACR Grant for Translational Breast Cancer Research, Grant Number 10-60-26-ANDE (C.K. Anders) as well as NIH K23157728 (C.K. Anders). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number K23CA157728. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. C.K. Anders, MD and C.R. Miller, MD PhD are Damon Runyon Clinical Investigators supported (in part) by the Damon Runyon Cancer Research Foundation (CI-64-12 C.K. Anders, CI-45-09 C.R. Miller).
Footnotes
Conflict of interest statement: No conflicts of interest for all authors.
References
- 1.Niwinska A, Murawska M, Pogoda K. Breast cancer brain metastases: differences in survival depending on biological subtype, RPA RTOG prognostic class and systemic treatment after whole-brain radiotherapy (WBRT) Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2010;21:942–8. doi: 10.1093/annonc/mdp407. [DOI] [PubMed] [Google Scholar]
- 2.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9(Suppl 2):S73–81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:2329–34. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 4.Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113:2638–45. doi: 10.1002/cncr.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132:523–35. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. The oncologist. 2013;18:123–33. doi: 10.1634/theoncologist.2012-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:1082–9. doi: 10.1158/1078-0432.CCR-10-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 10.Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast cancer research : BCR. 2012;14:R110. doi: 10.1186/bcr3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin. 2011;61:31–49. doi: 10.3322/caac.20095. [DOI] [PubMed] [Google Scholar]
- 12.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 13.Freedman RA, Anders CK. Treatment of breast cancer brain metastases. Current Breast Cancer Reports. 2012;4:1–9. doi: 10.1007/s12609-011-0061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The lancet oncology. 2011;12:852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 15.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 16.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clinical Cancer Research. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 17.Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anders CK, Adamo B, Karginova O, Deal AM, Rawal S, Darr D, et al. Pharmacokinetics and Efficacy of PEGylated Liposomal Doxorubicin in an Intracranial Model of Breast Cancer. PLoS ONE. 2013;8:e61359. doi: 10.1371/journal.pone.0061359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts PJ, Usary JE, Darr DB, Dillon PM, Pfefferle AD, Whittle MC, et al. Combined PI3K/mTOR and MEK Inhibition Provides Broad Antitumor Activity in Faithful Murine Cancer Models. Clinical Cancer Research. 2012;18:5290–303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Usary J, Zhao W, Darr D, Roberts PJ, Liu M, Balletta L, et al. Predicting drug responsiveness in human cancers using genetically engineered mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:4889–99. doi: 10.1158/1078-0432.CCR-13-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad RY, Chastain PD, Nikolaishvili-Feinberg N, Smeester L, Kaufmann WK, Fry RC. Titanium dioxide nanoparticles activate the ATM-Chk2 DNA damage response in human dermal fibroblasts. Nanotoxicology. 2013;7:1111–9. doi: 10.3109/17435390.2012.710659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlegel J, Sambade MJ, Sather S, Moschos SJ, Tan A-C, Winges A, et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. The Journal of Clinical Investigation. 2013;123:2257–67. doi: 10.1172/JCI67816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome biology. 2013;14:R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Z, Troester M, Perou CM. High reproducibility using sodium hydroxide-stripped long oligonucleotide DNA microarrays. BioTechniques. 2005;38:121–4. doi: 10.2144/05381MT02. [DOI] [PubMed] [Google Scholar]
- 25.Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics (Oxford, England) 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 30.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics (Oxford, England) 2004;20:3246–8. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 31.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:5664–78. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huppertz B, Frank HG, Kaufmann P. The apoptosis cascade--morphological and immunohistochemical methods for its visualization. Anat Embryol (Berl) 1999;200:1–18. doi: 10.1007/s004290050254. [DOI] [PubMed] [Google Scholar]
- 34.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–68. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–86. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 36.Herold CI, Anders CK. New targets for triple-negative breast cancer. Oncology (Williston Park, NY) 2013;27:846–54. [PubMed] [Google Scholar]
- 37.Malin D, Strekalova E, Petrovic V, Deal AM, Al Ahmad A, Adamo B, et al. alphaB-Crystallin: A Novel Regulator of Breast Cancer Metastasis to the Brain. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:56–67. doi: 10.1158/1078-0432.CCR-13-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maglott D, Pruitt K, Tatusova T, Murphy T. The NCBI Handbook [Internet] 2. Bethesda, MD: National Center for Biotechnology Information (US); 2013. [Google Scholar]
- 39.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic acids research. 2011;39:D52–7. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:984–9. doi: 10.1073/pnas.1322098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rondeau G, Abedinpour P, Desai P, Baron VT, Borgstrom P, Welsh J. Effects of different tissue microenvironments on gene expression in breast cancer cells. PloS one. 2014;9:e101160. doi: 10.1371/journal.pone.0101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17456–61. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nature cell biology. 2014;16:876–88. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders CK, Deal AM, Dees EC, Irvin WJ, Muss HB, Noe J, et al. LCCC 1025: A phase II study evaluating the mTOR inhibitor everolimus with trastuzumab and vinorelbine to treat progressive HER2-positive breast cancer brain metastases. Journal of Clinical Oncology. 2012;30:2012. ASCO Annual Meeting Abstracts:TPS656. [Google Scholar]
- 46.Lin NU, Gelman RS, JYW, Sohl J, Freedman RA, GSA, et al. Phase II trial of carboplatin (C) and bevacizumab (BEV) in patients (pts) with breast cancer brain metastases (BCBM) Journal of Clinical Oncology. 2013;31:2013. ASCO Annual Meeting Abstracts. [Google Scholar]
- 47.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.