

Abstract
Background
Over 40% of males and ~16% of female carriers of a premutation FMR1 allele (55-200 CGG repeats) will develop Fragile X associated Tremor/Ataxia Syndrome (FXTAS), an adult onset neurodegenerative disorder while, about 20% of female carriers will develop Fragile X-associated Primary Ovarian Insufficiency. Marked elevation in FMR1 mRNA transcript levels has been observed with premutation alleles, and RNA toxicity due to increased mRNA levels is the leading molecular mechanism proposed for these disorders. However, although the FMR1 gene undergoes alternative splicing, it is unknown if all or only some of the isoforms are overexpressed in premutation carriers and which isoforms may contribute to the premutation pathology.
Methods
To address this question we have applied a long-read sequencing approach using single molecule real time (SMRT) sequencing and qRT-PCR.
Results
Our SMRT sequencing analysis performed on peripheral blood mononuclear cells, fibroblasts and brain tissue samples derived from premutation carriers and controls revealed the existence of 16 isoforms out of 24 predicted variants. Although the relative abundance of all isoforms was significantly increased in the premutation group, as expected based on the bulk increase in mRNA levels; there was a disproportionate (4-6 fold) increase, relative to the overall increase in mRNA, in the abundance of isoforms spliced at both exons 12 and 14, specifically Iso10 and Iso10b, containing the complete exon 15 and differing only in splicing in exon 17.
Conclusions
These findings suggest that RNA toxicity may arise from a relative increase of all FMR1 mRNA isoforms. Interestingly, the Iso10 and Iso10b mRNA isoforms, lacking the C-terminal functional sites for FMRP function, are the most increased in premutation carriers relative to normal, suggesting a functional relevance in the pathology of FMR1 associated disorders.
Keywords: FMR1, isoforms, premutation, alternative splicing, RNA toxicity
INTRODUCTION
Carriers of premutation expansions (55-200 CGG repeats) of the fragile X mental retardation 1 (FMR1) gene can present with a range of clinical phenotypes, from mild cognitive and mood problems during childhood, and psychiatric and immune-mediated disorders in adulthood. Moreover, approximately 20% of the women will develop fragile X-associated primary ovarian insufficiency (FXPOI) by the age of 40 (reviewed in,[1]). Premutation carriers are also at high risk of developing the late onset neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome (FXTAS),[2]. With estimated prevalence of approximately 1:400 in males and 1:130 in females,[3 ,4], understanding the pathology of these disorders is of great societal importance.
Extensive alternative splicing of the FMR1 gene has been demonstrated by qRT-PCR analysis, and several FMR1 mRNA and FMRP isoforms have been observed in both human and mouse,[5-11]. The distribution and abundance of these isoforms may regulate the expression and functional properties of FMRP. Indeed, the FMR1 gene, which spans approximately 38Kb of genomic DNA and contains 17 exons, undergoes alternative splicing mainly involving inclusion or exclusion of exons 12 and 14 and the use of alternative splice acceptors in exons 15 and 17,[5 ,9] (Figure 1). The phosphorylation sites (Ser-499 in the mouse, Ser 500 in humans) mapping within exon 15, necessary for the translational repressor function of FMRP, are removed by the splicing of the first 36 nucleotides at the first acceptor site on exon 15. The second acceptor site removes an additional 39 nucleotides of exon 15, which results, in both the loss of the phosphorylation sites and the loss of the methylation sites, likely impacting the composition and/or abundance of FMRP-mRNP complexes and translation of FMRP mRNA targets,[12] (Figure 1). In addition, these splicing events in exon15, which occur in close proximity to the RGG box domain, modulate the RNA binding affinity of FMRP as evidenced by fluorescence spectroscopy analysis of the RNA binding properties of recombinant Iso1, Iso2 and Iso3 FMRP proteins, which differ only in the splicing at exon 15,[13]. Splicing of the full exon 12 shortens a loop (located from the 3’end of exon 10 through exon 12), within one of two high affinity RNA binding KH domains in FMRP,[14 ,15]. Splicing of exon 14 determines nuclear localization of FMRP isoforms,[7] and produces a +1 frame shift, which results in the loss of post-translational modification sites and loss of the RGG RNA binding domain of FMRP. The frame shift also results in several truncated FMRP variants with novel C-termini,[5] (Figure 1). The significance of a splice acceptor within exon 17 is currently unknown, although it has recently been demonstrated that a 17 amino acid stretch within this exon is important for the localization of FMRP nuclear isoforms within Cajal bodies,[16]. Thus, exons 12, 14, 15, and 17 are involved in splicing events that can generate a diversity of FMR1 isoforms, and likely with diverse biological properties.
Figure 1.

Top: Diagram of the exon structure of the FMR1 gene. A CGG expansion in the premutation allele is located in the 5’UTR of the gene. Seventeen exons are present and include functional domains, which are localized to different exon regions. Two Tudor motifs and a nuclear localization signal are localized to the first half of the FMR1 gene. Two K-homology RNA binding domains are encoded by exons 8-11 and exon 13 ,[15]. An Arginine-Glycine-Glycine (RGG) high affinity RNA binding domain, sites of phosphorylation,[12] and methylation are encoded by exon 15. A nuclear export signal is localized in exon 14,[7]. A) The diagram illustrates the amino acid sequence generated by the full-length and by the presence of two alternative splice sites within exon 15 (splice acceptor 1 and 2) in the presence of exon 14. The sites of post-translational phosphorylation and RGG box domain are indicated in bold. B) In those FMR1 isoforms lacking exon 14, a frame shift results in the loss of the phosphorylation sites and of the RGG box thus generating truncated proteins for the exon 15 full length (Iso10) and for the isoform resulting from the use of splice acceptor 1 of exon 17 (Iso10b). Truncated proteins contain potential phosphorylation sites (*) as predicted by the Group-based Phosphorylation Scoring algorithm, GPS 2.0,[39].
Despite their potential impact on FMRP function, the expression profiles of the different FMR1 isoforms have not been characterized in either normal or premutation carriers. Although elevated FMR1 mRNA levels have been observed for FMR1 mRNA expanded alleles ranging from the premutation to the full mutation range,[17-21], it is currently unknown whether all or only specific FMR1 mRNA isoforms are differentially over-expressed or distributed within cellular compartments as a function of CGG repeat number. As alternative protein isoforms are likely to have differing functions, alteration in their relative abundances could have profound biological consequences.
To address these questions we have used single molecule real time (SMRT) sequencing technology and qRT-PCR to determine which of the 24 or more predicted isoforms are actually expressed. Relative expression levels were measured by qRT-PCR in peripheral blood mononuclear cells (PBMCs) and brain tissue derived from premutation carriers and compared to age matched typically developing controls. The relative distribution and abundance of the isoforms was measured by SMRT sequencing in three different tissues (PBMCs, primary fibroblast cells and post-mortem cerebellar tissue of the brain), derived from premutation carriers, and compared to age matched typically developing controls.
RESULTS
Expression levels of all FMR1 mRNA isoforms are elevated for alleles in the premutation range
The mRNA expression levels of those FMR1 isoforms within Group A (Iso1, Iso2, Iso3, Iso13, Iso14, and Iso15), of those missing exon 14 within group B (specifically Iso 4 and Iso 4b), of those missing exon 12 within group C (Iso7, Iso8, Iso9, Iso17, Iso18, and Iso19), of Iso7 and Iso17 combined, as well as of those simultaneously missing exons 12 and 14, containing the full exon 15 and differing in alternative splicing at exon 17 (specifically Iso10 and Iso10b), were quantified by qRT-PCR in total RNA isolated from PBMCs (premutations n=70, controls n=40) and from post-mortem cerebellum brain tissue (premutations n=22, and controls n=7) of male subjects.
Significant increases in expression levels were observed for all groups of isoforms, in both PBMCs and brain tissue. However, while the expression levels in PBMCs of isoforms from group A (p<0.02), B (p<0.023) and C (p<0.031) were approximately 2-fold higher in premutations compared to controls, the expression levels of Iso10 and Iso10b were disproportionately elevated, from 4 to 6-fold, in premutation samples (p<0.001) (Figure 2a, b, c, d). In brain tissue, the increases in expression levels of the total FMR1 mRNA (p=0.233) and of the isoforms from group A (p=0.615), C (p<0.05) and of Iso10 and Iso10b (p<0.05) were marginally significant, between premutations and controls, in agreement with previous reports,[22 ,23] (Figure 2e, f, g, h). As expected, transcript levels of the various isoforms positively correlated with a longer CCG repeat tract in both tissues examined (Figure 2i, j, k, l).
Figure 2.

Relative mRNA expression levels of FMR1 isoforms by qRT-PCR. Box plots show mRNA expression levels between the control and the premutation groups in PBMC (a, b, c, d) and brain tissue (e, f, g, h). As expected increased levels of FMR1 mRNA were observed in premutation PBMCs (a), (p<0.001) compared to controls. Statistical significant differences were also observed between premutations and controls in the expression levels of FMR1 isoforms included in Group A (b), in Group B (not showed), in Group C (not showed), in the expression levels of Iso7 and Iso 17 (part of Group C) (c), and of Iso10 and Iso10b (d). In brain tissue statistical significant differences were observed between the premutation and the control group in the expression levels of Iso7 and Iso17 (g) and of Iso10 and Iso10b (h). Linear regression plots show a significant correlation between total FMR1 mRNA expression levels and CGG repeat length in PBMCs (i) (p<0.001) and in brain tissue (k). A significant correlation between FMR1 variants Iso10 and Iso10b expression levels and CGG repeat number was also observed in PBMCs (j) (p<0.001) and in brain tissue (l) P<0.001.
Base-resolution splicing patterns observed with single molecule, long-read sequencing
Accurate consensus calls of full-length isoforms were obtained at a single molecule level,[24]. The reads were aligned to our reference sequence, the full length FMR1 or Iso1 (NM_002024.5), by using MUMer, a system for rapidly aligning entire genomes, with a low setting for the break-length variable. The resulting alignment data were then parsed to produce the normalized count breakdown by tissue (PBMCs, cerebellum brain tissue and primary fibroblasts) and category (premutation or control) as well as the isoform breakpoints at single base resolution (Figure 3).
Figure 3.

Sequencing analysis overview. Circular consensus sequencing (CCS) was performed on the Pacific Biosciences platform resulting in accurate consensus calls of full-length isoforms on a single molecule level. The reads were then aligned to the full-length reference by using MUMer with a low setting for the break length variable. The resulting alignment data was then parsed using R to produce the normalized count breakdown by tissue and group category as well as the isoform breakpoints at single base resolution.
For each of the six cases (see study subject section), a SMRTbell™ sequencing library was constructed from PCR products generated using primers complementary to the start of exon 9 and the 3’ UTR of the FMR1 mRNA. SMRT® sequencing was performed to generate exon count analysis, by alignment to the reference exons 9 through the 3’ UTR in either the forward or reverse directions. A total of 108,126 reads was obtained for the six samples (~18,000 reads per sample), with a bimodal read-length distribution and median read-length of 1,053 nucleotides (nt). The main peak of the distribution was centered at ~1,100 nt (the reference length of exons 9 through the 3’ UTR is expected to be 1,095 nt) with a smaller peak at about 500 nt. Of the total, 14% of the reads did not align to any exon and belonged to the shorter read-length peak. A filter step, requiring a match to both exon 9 and the 3’ UTR removed 26% of the sequences. A total of 16 isoforms, comprising 57% of the total sequenced pool, had at least 350 reads in agreement which verified the existence of 16 isoforms in PBMCs, in brain tissue and in fibroblast cells derived from premutations and controls, and showed complete exon sequence fidelity with >90% CCS accuracy.
Analysis of the read alignment generated by SMRT sequencing revealed that of all exons, only exons 15 and 17 exhibited alternative start sites (Figure 4). In addition, analysis of the relative abundance of the various isoforms led to the following findings. First, the isoforms missing only exon 12 regardless of splicing in exon 15 and 17 (Iso7, Iso8, Iso9, Iso17, Iso18, and Iso19) were collectively the most abundant isoforms in both premutations and controls, with similar representation in all three tissues examined (Figure 5; Supplementary Figure 1). However, the relative proportion of Iso10 and Iso10b was higher within the premutation samples. Furthermore, the percent of reads for Iso10 and Iso10b, lacking exons 12 and 14 but retaining the entire exon 15 and differing only for the spliced acceptor in exon 17, displayed the greatest difference between the premutation and control PMBCs, and to a lesser degree in the other two tissues, confirming the qRT-PCR data (Figure 5; Supplementary Figure 1).
Figure 4.

Histogram plots of exon start sites. A peak is representative of the start site for each exon. A low exon 12 peak is a direct representation of the magnitude of the different exon contributions as only very few isoforms include exon 12 within the pool examined (~10% in all three tissues) while in the majority, exon 12 is spliced out. On the contrary, exon 14 is spliced out in approximately 10-20% (in all three tissue). Of exons 9 through 17, only exons 15 (splice acceptors 1 and 2) and exon 17 (splice acceptor 1) exhibit alternative start sites. These are identified by the additional peak(s) in the respective histogram plots.
Figure 5.

Diagram representation of the 24 predicted isoforms. Exons can be distinguished in different colors and splicing acceptor sites are shown with number designations 1 or 2. The region between exon 1 and 10 is reduced to focus on the region known to be alternative spliced in the predicted isoforms. The relative percent of isoforms representation in PBMCs, brain and fibroblasts within each normal and premutation samples is shown for each corresponding isoform. The most abundant isoforms are Iso7 and Iso17 missing exon 12 and differing in exon 17, in both premutations and controls across the three tissues. A greater representation of isoforms with the full exon 15 but with missing exons 12 and 14 (Iso10 and Iso10b) was observed in the premutations compared to controls confirming qRT-PCR. Notably, Iso1, which contains the full FMR1 transcript, is present at low level in all three tissues examined.
These findings are intriguing in light of the fact that, compared to the other isoforms in group D (Figure 5) (Iso11, Iso11b, Iso12 and Iso20), Iso10 and Iso10b retain the full exon 15. The upstream sequence of exon 15 harbors functionally important phosphorylation sites for FMRP. However, although a frame-shift due to splicing of exon 14 eliminates the canonical phosphorylation sites known to be important for FMRP function (Figure 1A), it also generates potential new phosphorylation sites on Iso10 and Iso10b (Figure 1B). Interestingly, the putative Iso11 was only detected in trace amounts in the three tissues analyzed for either premutation or control alleles, under our filtering cutoff (at least 350 conforming reads). Verification by qRT-PCR revealed very low levels of this isoform, in agreement with a very small number of reads by sequencing, below our cutoff, and indicating the presence of Iso11 at <1% expression level in all 3 tissues.
In addition, we observed that isoforms containing all 17 exons, regardless of the splicing patterns at exons 15 and 17 (Iso1, Iso2, Iso3, Iso13, Iso14, Iso15) were few in number with no differences in proportion between premutations and controls, in any of the three tissues. Lastly, similar to Iso11, isoforms missing only exon 14 and varying in splicing of exons 15 and 17 (Iso4, Iso4b, Iso5, Iso5b, Iso6 and Iso16) were not detected under our cutoff conditions, yielding <0.5% abundance in both premutations and controls, in the three different tissues examined. In support of this observation is the qRT-PCR data showing low abundance of both Iso4 and Iso4b mRNAs (data not shown).
Alternative splicing of exon 3
The first half of the FMR1 transcript, spanning exons 1 through 9, was also investigated for the presence of splicing events using SMRT sequencing libraries constructed from PCR products generated using primers located at the 5'UTR of FMR1 and in exon 9. Splice variants lacking exon 3 were identified in approximately 1% of the transcript reads in both premutation and control samples. The presence of an FMR1 mRNA lacking exon 3 was confirmed by qRT-PCR and subsequent sequencing of the cDNA in both a normal and a premutation sample (Figure 6). qRT-PCR on total RNA derived from Cases 1 through 6 confirmed the existence of a splicing event involving exon 3 in both premutation and control samples. Interestingly, the expression levels of the isoforms missing exon 3 were the highest in brain tissue (normal =3.49 ± 0.23; premutation = 3.17 ± 0.26) compared to PBMCs (normal = 0.28 ± 0.09; premutation = 0.19 ± 0.03) with no statistical significant difference observed in a subgroup of premutation subjects (n=30) compared to control subjects (n=15).
Figure 6.
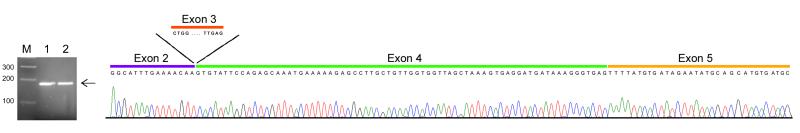
qRT-PCR shows the presence of an amplicon spanning from exon 2 to exon 5 but missing exon 3 confirming the presence of a novel alternative splicing event. Electrophoregram showing the amplicon’s sequence.
DISCUSSION
The full-length FMRP, the Iso1 protein, harbors RNA-binding domains (two KH domains and an RGG box),[25 ,26], nuclear export and localization signals,[7 ,26], and phosphorylation and methylation sites within exon 15,[7 ,27]. As the function of any given protein is dictated by the properties of its domains, the splicing/removal of exon sequences that encode functional motifs in the FMR1 gene, will likely impact the functional properties of FMRP. The consequent variations in expression levels and/or in ratios between the expressed isoforms could potentially contribute to the FMR1-associated disorders observed in premutations, including FXTAS. Splicing enhancers and silencers are keys to this process, usually accomplished by RNA binding proteins, such as Sam68, whose role in alternative splicing has been demonstrated for a number of genes including CD44,[28 ,29], Bcl-X,[30] and SMN2,[31]. In this regard, we have recently demonstrated that Sam68 is a specific and early component of CGG inclusions in FXTAS, and is depleted as a consequence of its sequestration by the expanded CGG repeats, leading in turn to an altered splicing-regulatory function,[32].
FMR1 isoforms contain different discrete exonic combinations due to alternative splicing at exons 12, 14, 15 and 17. Exon 12 or both exons 12 and 14 are entirely removed in certain isoforms and exons 15 and 17 contain two and one alternative splicing site acceptors, respectively. Earlier studies have identified the existence of these isoforms in humans,[7 ,33] and more recently in mouse,[10 ,11]. Importantly, a number of FMR1 alternative spliced isoforms were shown to co-sediment with polyribosomes in sucrose gradient ultracentrifugation experiments performed in adult mouse brain lysates, suggesting that those isoforms can be properly translated,[10]. These findings, including our current observations, highlight the importance of understanding the expression of multiple FMR1 isoforms and their functional properties. To our knowledge this is the first study that has used (SMRT) sequencing to identify which FMR1 isoforms are generated from splicing of the FMR1 transcript and to determine their relative abundance in human controls and premutations. As noted, several studies using a qRT-PCR approach have demonstrated the presence of a number of different FMR1 isoforms,[7 ,10 ,11 ,33]; however PCR-based methodologies only allow the individual splice sites to be analyzed separately and therefore fail to provide the combinations of different splice sites within the same RNA molecule. Our approach, which has utilized the single-molecule long-read sequencing technology, has allowed us to obtain a transcript map of all of the splice combinations within a single FMR1 transcript, and more importantly in both premutation and control individuals. As premutation carriers express elevated levels of FMR1 transcripts, changes in expression of specific isoforms could be playing a relevant role in the pathogenesis of the premutation associated disorders.
In the current study we have determined the existence of at least 16 out of 24 predicted alternative spliced FMR1 isoforms (nucleotide and polypeptide alignments are shown in Supplementary Figure 2 and 3) in PBMCs, brain tissue, and fibroblasts; and we have determined the differences in level between premutations and controls in these tissues.
Our qRT-PCR data and sequencing analysis are consistent with previous findings that the most common spliced isoforms are those missing exon 12,[10 ,11], indicating that these splice variants are likely the ones that have a more critical gene function,[34], or that inclusion of exon 12 might have a negative impact on one or more of those functions. In this study we have been able to assess the contribution to FMRP expression of all the single isoforms lacking exon 12, including exon 14 but differing in the other alternative splicing sites. All the isoforms tested and belonging to group A, B and C, although expressed at different levels were increased in premutations. Thus, the relative ratio of the most abundant variant isoforms (Iso7, Iso17, Iso8, Iso18, Iso9 and Iso19) was significantly higher in premutation samples compared to controls, as expected based on the increased bulk expression of FMR1 mRNA in the premutation range. Detailed examination of spliced variants within this group, spliced at exon 12, revealed that Iso7 and Iso17 are the two most highly expressed isoforms in both the premutation and the control groups, also showing increased levels in the premutation range. The importance of this splicing events is not completely clear, as exon 12 is part of an approximately 76 amino acid long unstructured variable loop encoded from the end of exon 10 through exon 12. This loop localizes between β sheets, β2 and β' in the eukaryotic FMRP KH2 domain, sitting outside of the Type I KH domain minimal motif,[15]. It is not known whether alternative splicing resulting in the removal of exon 12 and therefore in the shortening of the variable loop by 21 amino acids (encoded by exon 12) would change the arrangement of the β strands or modify the orientation of the KH2 domain affecting its ligand binding properties; although the crystal structure of FMRP KH1-KH2 domains with a variable loop truncated to 10 amino acids does not demonstrate a compromised folding of the domain,[15]. However, shortening of the loop may have some effect on KH2 RNA binding properties, as the direct comparison of in vitro binding of KC1 (kissing complex 1) RNA with FMR1 isoforms including or excluding exon 12 showed greater affinities in the absence of exon 12. No differences were observed with binding of poly(rG) sequences,[11]. Interestingly, the same authors also reported that isoforms including exon 12 are enriched in P7 and P15 mice synaptoneurosomes, suggesting that these isoforms may play a role in nerve terminals of neuronal processes,[11]. Finally, it has been shown that although the isoform excluding exon 12 is predominant during mouse development, the expression of isoforms including exon 12 increases from E7 to E17 indicating its developmental regulation,[11].
In those isoforms simultaneously spliced at exons 12 and 14 in addition to introducing a shorter variable loop in the KH2 domain, the splicing event also eliminates a nuclear export signal identified in exon 14 and introduces a frame shift that creates a novel C-terminus,[33], with a nuclear localization of the truncated protein isoforms as likely consequence,[7 ,10]. Strikingly, our data shows that two isoforms in this group, namely Iso10 and Iso10b, which differ only in their splicing pattern in exon 17, show the highest overexpression in the premutation samples as suggested by their greater representation relative to controls by sequencing analysis and by the relative expression levels measured by qRT-PCR. (Figure 2d, 2h and Figure 5).
Thus, whereas there is a general increase in the expression of all the detected FMR1 isoform mRNAs consistent with the elevated FMR1 mRNA levels detected in premutation carriers,[20], which is thought to lead to RNA toxicity and ultimately to FMR1 associated disorders,[1], it also appears that not all FMRP isoforms are equally represented in premutation carriers. How altered abundances of the FMR1 isoforms, particularly of Iso10 and Iso10b, may impact FMRP function in the premutations is not currently known. One possibility would involve a gain of function mechanism in which an increased relative abundance of truncated isoforms lacking the function of the C-terminal RGG box and NES can effectively execute specific interactions mediated by the N-terminus (containing NES, Tudor and KH domains) that over time may not only deplete a significant pool of FMRP binding factors but reduce functions mediated by the cross-talk with the missing C-terminus. The most recent model of FMRP function in translational repression, derived from studies in D. melanogaster, implicates the direct binding of FMRP direct binding to ribosomes through the KH domains,[35]. However, repression of translation was reduced by either an I244N mutation in the KH1 RNA binding domain or a deletion of the RGG box domain, suggesting that the joint activity of the FMRP N- and C-terminus may be essential for FMRP function in the translational regulation of its targets.
Although both Iso10 and Iso10b isoforms retain the full exon 15 sequence, the frame-shift resulting from exon 14 splicing eliminates the canonical sites for phosphorylation implicated in FMRP function as a translational repressor,[7] and introduces novel putative phosphorylation sites prior to the early stop codon,[33]. One important interaction eliminated by the deletion of the RGG box is that of the microtubule-associated protein 1B (MAP1B) that is involved in the development of the nervous system and normal physiology in adult brain,[36]. Indeed, it is intriguing that heterozygous MAP1B KO mice exhibit motor system abnormalities including cerebellar ataxia and tremors,[37], both phenotypes of FXTAS,[38]. Using the most stringent settings of the group-based prediction system (GPS), ver 2.0,[39] Ser460 or Thre455 and Thre456 were identified as potential sites of phosphorylation by the Ser/Thre kinase casein kinase 1 (CK1). CK1 phosphorylation has been associated with nuclear localization of its targets,[40] and is required for activation of cdk5 under the regulation of metabotropic glutamate receptors (mGluRs),[41 ,42]. Whether FMRP is a target of CK1 and the potential biological implications has not been contemplated thus far. Interestingly, a recent report implicates FMRP in the regulation of neurotransmitter release via synaptic vesicle exocytosis by modulating the density of N-type calcium channels through direct interaction with FMRP’s C-terminus,[43]. N-type calcium channels are coincidentally regulated by cdk5, which impacts neurotransmitter release at presynaptic terminals,[44]. The absence of the C-terminus in a larger pool of truncated isoforms may create an imbalance affecting N-type calcium channel densities and therefore, neurotransmitter release. Therefore, the question of whether the overabundance of these truncated FMRP isoforms may degrade FMRP function and contribute to pathology is of great interest.
Additionally, the significance of splicing events at exon 17 is not presently understood. It has been speculated that the absence of the extended loop removed by this splicing event could provide FMRP specificity reducing the repertoire of mRNA interactions perhaps serving as a mechanism for selection, or discrimination, of targets for FMRP mediated translational repression,[10]. However, direct evidence for this mechanism of action has yet to be reported. As the biological function of these mRNA isoforms and the proteins they encode remain unknown, elucidating the function of the truncated FMRP will be crucial for determining their biological role in premutation disorders. Presumably any of the splicing events could lead to altered FMRP function due to changes in thermodynamic and isoelectric properties affecting FMRP binding activity. Indeed, fluorescence spectroscopy RNA binding experiments using recombinant FMRP produced from constructs containing spliced Iso1 (containing all exonic regions), spliced Iso2 (all exonic regions and exon 15 spliced at the first acceptor site) and Iso3 (all exonic regions and exon 15 spliced at the second acceptor site) demonstrated that each splicing event in exon 15 leads to a significant increase in RNA binding affinity of the RGG box,[45]. The authors suggested that either conformational changes reducing the distance of the two RNA binding regions that form the RGG box, or perhaps the removal of negatively charged amino acids, could be responsible for these affinity changes. It is interesting to note that all isoforms containing the entire exon 15, regardless their splicing at exon 12, 14 and 17, were the most highly represented compared to those isoforms spliced at the first or at the second acceptor site in exon 15 (Figure 5).
Finally, the presence of alterative splicing in the first half of the FMR1 gene has not previously been reported. We have detected the absence of exon 3 in approximately 1% of the sequenced transcripts. This finding is especially interesting given the presence of two Tudor (Agenet) motifs,[33] implicated in the recently proposed chromatin binding-dependent DNA damage response activity of FMRP during development,[46]. The tandem Tudor domains are highly conserved in the FMRP paralogs FXR1P and FXR2P,[47] and are implicated in RNA metabolism and specific protein-protein interactions,[48]. One of such interaction is the binding of FMRP to brain cytoplasmic RNA (BC1), a small non-coding RNA involved in the translation of certain targets of FMRP. Recently, it was shown that unmethylated BC1, found at synapses, has a higher affinity for FMRP, within the second FMRP Tudor domain,[49]. Additionally, a KH domain RNA binding protein Src-associated in mitosis, 68 kDa (Sam68) has been shown to potentially regulate the alternative splicing of FMR1 at exon 3,[50]. This regulation suggests that the observed sequestration of Sam68 by the FMR1 CGG repeat locus,[32] may result in increased FMR1 exon 3 splicing. It is unknown which FMR1 mRNA isoforms harboring splicing events between exons 12 through 17 also become spliced at exon 3. However, the low expression of splicing in exon 3 suggests that possibly only those isoforms present in low abundance may carry this event. It is also possible that relative representation and expression levels of the various isoforms missing exon 3 and alternatively spliced in other exonic regions of the FMR1 gene could be different during development or in different tissues. Thus, further studies to understand the implications of exon 3 splicing events and to verify the presence of the corresponding translated products will be necessary to define its biological significance which may involve altered mRNA translational activity at the synapses.
In conclusion, the characterization of the expression levels of the FMR1 isoforms is fundamental for understanding regulation of the expression of the FMR1 gene as well as to elucidate the mechanism by which “toxic gain of function” of the FMR1 mRNA and intranuclear inclusion formation may take place in FXTAS and/or in the other FMR1-associated conditions. These findings also suggest that in addition to the elevated levels of FMR1 isoforms, the altered abundance/ratio of the corresponding FMRP isomers may affect the overall function of FMRP in premutations. Thus, future research should address these questions and to whether or not the proteins for which the splicing of exon 14 creates a frame shift that leads to the loss of the canonical phosphorylation sites are translated, and, determine the impact of FMRP variants and the biogenesis of specific FMR1 isoforms in the neurological and neurodevelopmental clinical presentation in premutation carriers, particularly of FXTAS.
MATERIALS AND METHODS
Study Subjects
For qRT-PCR analysis, total RNA was isolated from PBMCs from subjects with a normal allele [n=40, (mean±StdDev), CGG repeats = 30.9 ± 5.4; range 19-47 CGG, age range= 8-73 years) and from premutation carriers (n=70; mean CGG repeats = 100 ± 25; range 58-180 CGG, age range= 17-74 years). Among the premutation carriers, 60 had FXTAS stage 3-5 as defined by ,[51] and post-mortem brain tissue from premutation carriers with FXTAS (n=22, mean CGG= 87 ± 16.4; range 57-118 CGG; age range= 66-87 years) and controls (n=7; mean CGG= 30 ± 7.7; age range 53-88 years).
For sequencing analysis, PBMCs were collected from two different male subjects, one carrying a normal and one a premutation FMR1 allele (Case 1, CGG=30, age=35 years; Case 2, CGG=170, age= 19 years). Primary fibroblast cultures were established from two different male subjects one carrying a normal and one a premutation FMR1 allele (Case 3, CGG= 20; Case 4, CGG=107). Post-mortem brain tissue from a control brain tissue (Case 5, CGG = 30, age=69 years) was obtained from the Harvard Brain Bank and a premutation carrier with FXTAS (Case 6, CGG= 92, age= 81 years) was obtained from the UC Davis Brain Tissue Repository.
All samples were collected under approved UC Davis Institutional Review Board (IRB) protocols.
RNA isolation
Total RNA was isolated from 3ml of blood collected in Tempus tubes according to manufacture instructions (Applied Biosystems, Foster City, CA). Total RNA from post-mortem cerebellar tissues or from 1x 106 primary fibroblast cells were isolated using Trizol (Life Technologies, Carlsbad, CA) from samples stored at −80°C and pulverized in liquid nitrogen. All RNA isolations were performed in a clean RNA designated area. TotRNA quantification and quality control were carried out using the Agilent 2100 Bioanalyzer system.
mRNA expression levels
qRT-PCR was performed using custom designed TaqMan primers and probe assays (Applied Biosystems, Foster City, CA). Custom designed probes were used to detect both the full-length FMR1 gene and the β-glucuronidase (GUS) gene that was used for normalization; details are as previously described ,[20]. Probe and primer assays were also designed to quantify: total FMR1 mRNA (which included the transcripts derived for all possible isoform combinations; forward primer 5’-TGG CTT CAT CAG TTG TAG CAG G-3’, reverse primer 5’-TCT CTC CAA ACG CAA CTG GTC-3’), FMR1 isoforms in Group A (forward primer 5’ TGGCTTCATCAGTTGTAGCAGG 3’; reverse primer 5’ TCTCTCCAAACGCAACTGGTC 3’); FMR1 isoforms within Group B, specifically Iso4 and Iso4b (forward primer 5’ TGGCTTCATCAGTTGTAGCAGG 3’, reverse primer 5’ CAGAATTAGTTCCTTTAAATAGTTCAGG 3’); FMR1 isoforms in Group C (forward primer 5’ TCCAGAGGGGTATGGTACCATT 3’, reverse primer 5’ TCCAAACGCAACTGGTCTACTTC 3’); FMR1 Iso7 and Iso17 (forward primer 5’ TCCAGAGGGGTATGGTACCATT 3’, reverse primer 5’ GCTTCAGAATTAGTTCCTGAAGTATATCC 3’), FMR1 Iso10 and Iso10b isoforms (forward primer 5’ TCCAGAGGGGTATGGTACCATT 3’, reverse primer 5’ CAGAATTAGTTCCTTTAAATAGTTCAGG 3’). Two sets of primers were also designed to verify the alternative splicing at exon 3 (forward1 primer 5'-GCA TTT GAA AAC AAG TGT ATT CCA G 3', reverse1 primer 5' TGG CAG GTT TGT TGG GAT TAA CAG A-3', forward2 primer-2 5’-AGG CAT TTG TAA AGG ATG TTC ATG-3’ and reverse2 primer-2 5’-GCA AGG CTC TTT TTC ATT TGC T-3’). mRNA expression levels for each isoform were compared between premutation and control samples using two-sample t-tests conducted on log transformed data.
Generation of PCR amplicons for SMRT libraries
Two PCR products, covering the entire FMR1 coding region, were amplified. One amplicon (plus and minus exon 3), ~847 bp in length was amplified using primers located in the 5’UTR (5’GCA GGG CTG AAG AGA AGA TG 3’) and a reverse primer located at the exon 8/9 boundary (5’ CAC TGC ATC CTG ATC CTC TC 3’). The second amplicon (including al the expressed isoforms) of ~1119 bp in length was obtained using a forward primer located at exon 8/9 boundary (5’ GAG AGG ATC AGG ATG CAG TG 3’) and a reverse primer located in the 3’UTR of the FMR1 gene (5’ CCT GTG CCA TCT TGC CTA C 3’). cDNAs were made as described in Tassone et al. (2000) and were derived from total RNA isolated from normal and premutation PBMCs Case 1 and 2), from normal and premutation primary fibroblasts (Case 3 and 4) and from normal and premutation cerebellum tissue (Case 5 and 6). cDNA synthesis was performed as previously described in,[19] with minor modifications; briefly the annealing temperature in the RT reaction was 55°C for 40 min. PCR reactions were performed in 50-ul aliquots containing 1 X High Fidelity Buffer, 0.2 mM each dNTP, primers each at 0.2uM, 1.25U Platinum Taq DNA Polymerase high fidelity, 2 mM MgSO4 (Life Technology-Invitrogen, Grand Island, NY).
Library preparation and polymerase binding
SMRT bell sequencing libraries were constructed for each PCR amplicon for the six cases, using the DNA Template Prep Kit 1.0 (Pacific Biosciences, Menlo Park, CA), followed by primer annealing and polymerase binding using the DNA Polymerase Binding Kit (Pacific Biosciences, Menlo Park, CA) as described in,[52].
SMRT sequencing
Sequencing was performed on the RS I version 1.3.3.1.116585 software and C2 chemistry. Each run consisted of a 45’ movie that interrogated 75,000 zero mode waveguides (ZMW). Two to three movies were acquired per sample, using diffusion loading yielding an average of 13,463 reads per sample (Table 1).
Table 1.
Filtering summary of sequencing reads by condition and tissue type
| Category | Tissue | Initial reads | Exon 9 included | Exon 17 included | Exons 9 and 17 included | Percent agreement* | Percent of discrete sequences** |
Final number of reads*** |
|---|---|---|---|---|---|---|---|---|
| Normal | Brain | 16158 | 92.2% | 99.4% | 91.6% | 98.8% | 98.7% | 14732 (91.2%) |
| Premutation | Brain | 15824 | 90.2% | 98.8% | 89.1% | 99.1% | 99.0% | 13973 (88.3%) |
| Normal | PBMC | 18047 | 77.3% | 99.7% | 76.9% | 98.7% | 98.5% | 13840 (76.7%) |
| Premutation | PBMC | 10771 | 86.8% | 98.8% | 85.6% | 99.3% | 99.1% | 9188 (85.3%) |
| Normal | Fibroblasts | 10846 | 75.4% | 99.5% | 75.0% | 95.1% | 94.7% | 8025 (74%%) |
| Premutation | Fibroblasts | 22556 | 93.4% | 99.4% | 93.3% | 99.6% | 99.7% | 21024 (93.2%) |
Percent of agreement with reference sequence (NM_002024.5).
Percent of discrete sequences indicates the percent of reads containing the correct exon sequence for each isoform
Final number of reads indicate the number of reads after the filtering steps. In parenthesis is the percent.
Full length transcript analysis
We used intramolecular circular consensus sequence (CCS) alignment to generate high accuracy individual sequence reads,[53]. The reads were aligned to our reference sequence, Iso1 by using MUMmer (NUCmer (NUCleotide MUMmer) version 3.07),[54]. A cut-off match alignment of >97% was used to keep high quality agreements. The data were then collated on a per-molecule basis to construct the full transcript using R (http://www.r-project.org). Only molecules that contained both ends of the cDNA strand amplified by RT-PCR (exons 1 and 9 or exons 9 and 17 for the amplicons covering the first and the second half of the FMR1 gene) were selected for further analyses. All samples had rates of 90% or greater for inclusion of exon 17, but varied for inclusion of exon 9 from 75% to 93%. (Table 1) shows the number of sequences that were filtered and total number of reads). Subsequent filtering criteria included strand agreement to the mapped reference piece.
Assignment of isoform constraints
FMR1 isoforms contained different discrete exonic combinations due to alternative splicing at exons 12, 14, 15 and 17. Exon 12 or both exons 12 and 14 are entirely removed in certain isoforms and exons 15 and 17 contain two and one alternative splicing start sites, respectively (Figure 1). Nomenclature of the identified isoforms is as illustrated in Table 2. Limited by the size of transcripts sequenced (from the 5’UTR to exon 9), transcripts with exon 3 spliced out were not assigned isoform designations as downstream splicing events in exons 12, 14, 15, and 17 were undetermined.
Table 2.
Isoform nomenclature used in this study
| Nomenclature | Isoform structure |
|---|---|
| Iso1 | it has everything +12, +14, +full 15, +full 17 |
| Iso13 | +12, +14, +full 15, +alt1 17 |
| Iso2* | +12, +14, +alt 1 15, +full 17 |
| Iso14 | +12, +14, +alt 1 15, +alt 1 17 |
| Iso3 | +12, +14, +alt 2 15, +full 17 |
| Iso15 | +12, +14, +alt 2 15, +alt1 17 |
| Iso4 * | +12, −14, +full 15, +full 17 |
| Iso4b * | +12, −14, +full 15, +alt1 17 |
| Iso5 * | +12, −14, +alt 1 15, +full 17 |
| Iso5b * | +12, −14, +alt 1 15, +alt 1 17 |
| Iso6 * | +12, −14, +alt 2 15, +full 17 |
| Iso16 * | +12, −14, +alt 2 15, +alt 1 17 |
| Iso7 | −12, +14, +full 15, +full 17 |
| Iso17 | −12, +14, +full 15, +alt 1 17 |
| Iso8 | −12, +14, +alt 1 15, +full 17 |
| Iso18 | −12, +14, +alt 1 15, +alt 1 17 |
| Iso9 | −12, +14, +alt 2 15, +full 17 |
| Iso19 | −12, +14, +alt 2 15, +alt 1 17 |
| Iso10 | −12, −14, +full 15, +full 17 |
| Iso10b | −12, −14, +full 15, +alt 1 17 |
| Iso11* | −12, −14, +alt 1 15, +full 17 |
| Iso11b | −12, −14, +alt 1 15, +alt 1 17 |
| Iso12 | −12, −14, +alt 2 15, +full 17 |
| Iso20 | −12, −14, +alt 2 15, +alt 1 17 |
Isoform nomenclature is as described in Sittler et al. 1996. Isoforms named (ISO4), (ISO 5), (ISO10) and (ISO11) by Sittler et al. 1996 have been renamed Iso4b, Iso5b, Iso10b and Iso11b, respectively.
These isoforms were not detected in any of the three tissues or groups by (SMRT®).
Supplementary Figure 1. Relative isoform proportions observed in PBMCs, brain and fibroblasts from premutation and control samples. A larger proportion of Iso10 and Iso10b is observed in the premutation, in particular in PBMCs and brain tissue (cerebellum).
Supplementary Material
Supplementary Text 1. Nucleotide alignment (generated using expasy, SIB Bioinformatics, CLUSTAL2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) of the 16 isoforms identified in this study. Nomenclature is as reported in Table 1.
Supplementary Text 2. Polypeptide alignment (generated using expasy, SIB Bioinformatics, CLUSTAL2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) of the 16 isoforms identified in this study. Nomenclature is as reported in Table 1.
ACKNOWLEDGEMENTS
This work is dedicated to the memory of Matteo. We thank Elizabeth Berry-Kravis for providing one fibroblast cell line.
FUNDING
This work was supported by the National Institutes of Health (NIH) through the research award HD02274 and HD040661 and the National Center for Advancing Translational Sciences research grant UL1 TR000002. Brain tissue samples were obtained from the UC Davis Brain Repository, from the Harvard Brain Tissue Resource Center, which was supported in part by PHS grant number R24MH 068855; the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, Maryland supported by NICHD contract # NO 1-HD-4-3368 and NO1-HD-4-3383.
Footnotes
COMPETING INTEREST
FT has received funds from Roche and had consulted with Novartis and Genentech. FT and PJH hold patents for sizing of the CGG repeat and for quantification of FMRP. PJH and FT are collaborators with Pacific Biosciences on an NIH STTR grant. The other authors declare no conflicts of interest.
REFERENCES
- 1.Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet neurology. 2013;12(8):786–98. doi: 10.1016/S1474-4422(13)70125-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–30. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 3.Maenner MJ, Baker MW, Broman KW, Tian J, Barnes JK, Atkins A, McPherson E, Hong J, Brilliant MH, Mailick MR. FMR1 CGG expansions: prevalence and sex ratios. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2013;162B(5):466–73. doi: 10.1002/ajmg.b.32176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, Nguyen D, Mu LY, Laffin J, Bailey DB, Hagerman RJ. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome medicine. 2012;4(12):100. doi: 10.1186/gm401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashley CT, Jr., Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science (New York, NY) 1993;262(5133):563–6. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 6.Huang T, Li LY, Shen Y, Qin XB, Pang ZL, Wu GY. Alternative splicing of the FMR1 gene in human fetal brain neurons. American journal of medical genetics. 1996;64(2):252–5. doi: 10.1002/(SICI)1096-8628(19960809)64:2<252::AID-AJMG3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Sittler A, Devys, D, Weber, C, Mandel JL. Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of fmr1 protein isoforms. Human Molecular Genetics. 1996;5(1):95–102. doi: 10.1093/hmg/5.1.95. [DOI] [PubMed] [Google Scholar]
- 8.Verheij C, Bakker, CE, de Graaff E, Keulemans, J, Willemsen, R, Verkerk, AJ, Galjaard, H, Reuser, AJ, Hoogeveen, AT, Oostra BA. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363(6431):722–4. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- 9.Verkerk AJ, de Graaff E, De Boulle K, Eichler, EE, Konecki, DS, Reyniers, E, Manca, A, Poustka, A, Willems, PJ, Nelson, DL, Oostra BA. Alternative splicing in the fragile X gene FMR1. Hum Mol Genet. 1993;2(8):1348. doi: 10.1093/hmg/2.8.1348. [DOI] [PubMed] [Google Scholar]
- 10.Brackett DM, Qing F, Amieux PS, Sellers DL, Horner PJ, Morris DR. FMR1 transcript isoforms: association with polyribosomes; regional and developmental expression in mouse brain. PloS one. 2013;8(3):e58296. doi: 10.1371/journal.pone.0058296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie W, Dolzhanskaya N, LaFauci G, Dobkin C, Denman RB. Tissue and developmental regulation of fragile X mental retardation 1 exon 12 and 15 isoforms. Neurobiology of disease. 2009;35(1):52–62. doi: 10.1016/j.nbd.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Dolzhanskaya N, Merz G, Denman RB. Alternative splicing modulates protein arginine methyltransferase-dependent methylation of fragile X syndrome mental retardation protein. Biochemistry. 2006;45(34):10385–93. doi: 10.1021/bi0525019. [DOI] [PubMed] [Google Scholar]
- 13.Evans TL, Blice-Baum AC, Mihailescu MR. Analysis of the Fragile X mental retardation protein isoforms 1, 2 and 3 interactions with the G-quadruplex forming semaphorin 3F mRNA. Molecular bioSystems. 2012;8(2):642–9. doi: 10.1039/c1mb05322a. [DOI] [PubMed] [Google Scholar]
- 14.Valverde R, Edwards L, Regan L. Structure and function of KH domains. The FEBS journal. 2008;275(11):2712–26. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- 15.Valverde R, Pozdnyakova I, Kajander T, Venkatraman J, Regan L. Fragile X mental retardation syndrome: structure of the KH1-KH2 domains of fragile X mental retardation protein. Structure (London, England : 1993) 2007;15(9):1090–8. doi: 10.1016/j.str.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Dury AY, El Fatimy R, Tremblay S, Rose TM, Cote J, De Koninck P, Khandjian EW. Nuclear Fragile X Mental Retardation Protein is localized to Cajal bodies. PLoS genetics. 2013;9(10):e1003890. doi: 10.1371/journal.pgen.1003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10(14):1449–54. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- 18.Peprah E, He W, Allen E, Oliver T, Boyne A, Sherman SL. Examination of FMR1 transcript and protein levels among 74 premutation carriers. Journal of human genetics. 2010;55(1):66–8. doi: 10.1038/jhg.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. American journal of medical genetics. 2000;94(3):232–6. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 20.Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66(1):6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004;114(5):439–47. doi: 10.1007/s00439-004-1086-x. [DOI] [PubMed] [Google Scholar]
- 22.Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. Journal of medical genetics. 2004;41(4):e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pretto DI, Kumar M, Cao Z, Cunningham CL, Durbin-Johnson B, Qi L, Berman R, Noctor SC, Hagerman RJ, Pessah IN, Tassone F. Reduced excitatory amino acid transporter 1 and metabotropic glutamate receptor 5 expression in the cerebellum of fragile X mental retardation gene 1 premutation carriers with fragile X-associated tremor/ataxia syndrome. Neurobiology of aging. 2014;35(5):1189–97. doi: 10.1016/j.neurobiolaging.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Real-time DNA sequencing from single polymerase molecules. Science (New York, NY) 2009;323(5910):133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 25.Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74(2):291–8. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 26.Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5(8):1083–91. doi: 10.1093/hmg/5.8.1083. [DOI] [PubMed] [Google Scholar]
- 27.Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12(24):3295–305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- 28.Cheng C, Sharp PA. Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Molecular and cellular biology. 2006;26(1):362–70. doi: 10.1128/MCB.26.1.362-370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420(6916):691–5. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 30.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. The Journal of cell biology. 2007;176(7):929–39. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedrotti S, Sette C. Spinal muscular atrophy: a new player joins the battle for SMN2 exon 7 splicing. Cell cycle (Georgetown, Tex) 2010;9(19):3874–9. doi: 10.4161/cc.9.19.13153. [DOI] [PubMed] [Google Scholar]
- 32.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Adams JS. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010 doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashley CT, Sutcliffe JS, Kunst CB, Leiner HA, Eichler EE, Nelson DL, Warren ST. Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nature genetics. 1993;4(3):244–51. doi: 10.1038/ng0793-244. [DOI] [PubMed] [Google Scholar]
- 34.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126(1):37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 35.Chen E, Sharma MR, Shi X, Agrawal RK, Joseph S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Molecular cell. 2014;54(3):407–17. doi: 10.1016/j.molcel.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villarroel-Campos D, Gonzalez-Billault C. The MAP1B case: An old MAP that is new again. Developmental neurobiology. 2014 doi: 10.1002/dneu.22178. [DOI] [PubMed] [Google Scholar]
- 37.Edelmann W, Zervas M, Costello P, Roback L, Fischer I, Hammarback JA, Cowan N, Davies P, Wainer B, Kucherlapati R. Neuronal abnormalities in microtubule-associated protein 1B mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(3):1270–5. doi: 10.1073/pnas.93.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brega AG, Reynolds A, Bennett RE, Leehey MA, Bounds LS, Cogswell JB, Hagerman RJ, Hagerman PJ, Grigsby J. Functional status of men with the fragile X premutation, with and without the tremor/ataxia syndrome (FXTAS) International journal of geriatric psychiatry. 2009;24(10):1101–9. doi: 10.1002/gps.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics : MCP. 2008;7(9):1598–608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Blume J, Knippschild U, Dequiedt F, Giamas G, Beck A, Auer A, Van Lint J, Adler G, Seufferlein T. Phosphorylation at Ser244 by CK1 determines nuclear localization and substrate targeting of PKD2. EMBO J. 2007;26(22):4619–33. doi: 10.1038/sj.emboj.7601891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Ma XH, Ule J, Bibb JA, Nishi A, DeMaggio AJ, Yan Z, Nairn AC, Greengard P. Regulation of cyclin-dependent kinase 5 and casein kinase 1 by metabotropic glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11062–8. doi: 10.1073/pnas.191353898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma P, Sharma M, Amin ND, Albers RW, Pant HC. Regulation of cyclin-dependent kinase 5 catalytic activity by phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11156–60. doi: 10.1073/pnas.96.20.11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nature communications. 2014;5:3628. doi: 10.1038/ncomms4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su SC, Seo J, Pan JQ, Samuels BA, Rudenko A, Ericsson M, Neve RL, Yue DT, Tsai LH. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron. 2012;75(4):675–87. doi: 10.1016/j.neuron.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans TL, Mihailescu MR. Recombinant bacterial expression and purification of human fragile X mental retardation protein isoform 1. Protein expression and purification. 2010;74(2):242–7. doi: 10.1016/j.pep.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stutzer A, Armache KJ, Simon MD, Xu C, Ali M, Murn J, Prisic S, Kutateladze TG, Vakoc CR, Min J, Kingston RE, Fischle W, Warren ST, Page DC, Shi Y. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell. 2014;157(4):869–81. doi: 10.1016/j.cell.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams-Cioaba MA, Guo Y, Bian C, Amaya MF, Lam R, Wasney GA, Vedadi M, Xu C, Min J. Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2. PloS one. 2010;5(11):e13559. doi: 10.1371/journal.pone.0013559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adinolfi S, Ramos A, Martin SR, Dal Piaz F, Pucci P, Bardoni B, Mandel JL, Pastore A. The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry. 2003;42(35):10437–44. doi: 10.1021/bi034909g. [DOI] [PubMed] [Google Scholar]
- 49.Lacoux C, Di Marino D, Boyl PP, Zalfa F, Yan B, Ciotti MT, Falconi M, Urlaub H, Achsel T, Mougin A, Caizergues-Ferrer M, Bagni C. BC1-FMRP interaction is modulated by 2'-O-methylation: RNA-binding activity of the tudor domain and translational regulation at synapses. Nucleic acids research. 2012;40(9):4086–96. doi: 10.1093/nar/gkr1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chawla G, Lin CH, Han A, Shiue L, Ares M, Jr., Black DL. Sam68 regulates a set of alternatively spliced exons during neurogenesis. Molecular and cellular biology. 2009;29(1):201–13. doi: 10.1128/MCB.01349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bacalman S, Farzin F, Bourgeois JA, Cogswell J, Goodlin-Jones BL, Gane LW, Grigsby J, Leehey MA, Tassone F, Hagerman RJ. Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: newly described fronto-subcortical dementia. The Journal of clinical psychiatry. 2006;67(1):87–94. doi: 10.4088/jcp.v67n0112. [DOI] [PubMed] [Google Scholar]
- 52.Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome research. 2013;23(1):121–8. doi: 10.1101/gr.141705.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic acids research. 2010;38(15):e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome biology. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text 1. Nucleotide alignment (generated using expasy, SIB Bioinformatics, CLUSTAL2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) of the 16 isoforms identified in this study. Nomenclature is as reported in Table 1.
Supplementary Text 2. Polypeptide alignment (generated using expasy, SIB Bioinformatics, CLUSTAL2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) of the 16 isoforms identified in this study. Nomenclature is as reported in Table 1.