

Abstract
Protein tyrosine phosphatases (PTPs) tightly regulate tyrosine phosphorylation essential for cell growth, adhesion, migration, and survival. We performed a mutational analysis of the PTP gene family in cutaneous metastatic melanoma and identified 23 phosphatase genes harboring somatic mutations. Among these, receptor-type tyrosine–protein phosphatase delta (PTPRD) was one of the most highly mutated genes, harboring 17 somatic mutations in 79 samples, a prevalence of 21.5%. Functional evaluation of six PTPRD mutations revealed enhanced anchorage-dependent and anchorage-independent growth. Interestingly, melanoma cells expressing mutant PTPRD were significantly more migratory than cells expressing wild-type PTPRD or vector alone, indicating a novel gain-of-function associated with mutant PTPRD. To understand the molecular mechanisms of PTPRD mutations, we searched for its binding partners by converting the active PTPRD enzyme into a “substrate trap” form. Using mass spectrometry and coimmunoprecipitation, we report desmoplakin, a desmosomal protein that is implicated in cell–cell adhesion, as a novel PTPRD substrate. Further analysis showed reduced phosphatase activity of mutant PTPRD against desmoplakin. Our findings identify an essential signaling cascade that is disrupted in melanoma. Moreover, because PTPRD is also mutated in glioblastomas and adenocarcinoma of the colon and lung, our data might be applicable to a large number of human cancers.
Keywords: PTPRD, desmoplakin, cell migration, somatic mutations, desmosomes
Introduction
Melanoma is one of the most fatal skin cancers. As melanoma cells have a high rate of somatic mutations, identification of driver mutations might lead to novel therapeutic targets. Tyrosine phosphorylation is one of the major protein modifications that regulate cellular processes such as growth, survival, and migration. The degree of protein tyrosine phosphorylation is tightly regulated by tyrosine kinases and phosphatases, therefore mutations in these proteins can lead to deregulation of cellular processes and result in transformation. In this study, we performed a comprehensive mutational analysis of the tyrosine phosphatome in malignant melanoma and identified receptor-type tyrosine–protein phosphatase T (PTPRT) and receptor-type tyrosine–protein phosphatase delta (PTPRD; MIM #601598) as the most frequently mutated tyrosine phosphatase genes in melanoma.
We had previously identified PTPRD as a frequently mutated gene in melanomas (12%, 7/57) using a candidate gene sequencing approach [Solomon et al., 2008]. Aberrations in PTPRD have also been reported in several other cancers. The PTPRD locus is frequently deleted in glioblastomas [Solomon et al., 2008; Veeriah et al., 2009] and lung cancer [Weir et al., 2007; Veeriah et al., 2009; Kohno et al., 2010]. Multiple studies of the COSMIC database indicate that PTPRD is highly mutated in cancers of esophagus (12.1%, 21/173), lung (10.6%, 126/1193) [Ding et al., 2008], endometrium (9.3%, 26/281), large intestine (8.2%, 52/637) [Wang et al., 2004], skin (10.3%, 40/387), and stomach (4.3%, 2/47) (http://cancer.sanger.ac.uk/accessed April 27th, 2014). In this study, we describe the functional consequence of PTPRD mutations in melanoma.
PTPRD has recently been found to interact with several proteins such as STAT3 [Veeriah et al., 2009], aurora kinase [Meehan et al., 2012], polycystein [Boucher et al., 2011], and E-cadherin [Kosuke Funato et al., 2011] and was shown to regulate cell growth and migration in many cancers. Furthermore, knockdown of PTPRD induces cell migration, whereas low expression of PTPRD correlates with invasiveness in colon cancer [Kosuke Funato et al., 2011], indicating a role in cell adhesion. PTPRD is expressed in early development, when intracellular adhesion is required for synaptic connections in the neuroepithelium, inferring a role in cell–cell adhesion [Sommer et al., 1997]. Further identification of PTPRD partners is essential to elucidate its cell-signaling cascade and tumor-suppressive mechanism.
To identify new PTPRD interacting partners, we generated two doxycycline-inducible melanoma cell lines, one expressing the intracellular phosphatase domains of PTPRD and the other carrying a substrate-trapping PTPRD mutation (Asp1521Ala). These mutations were previously used by Xie et al. (2002) to detect PTP1B substrates and were suggested to be applicable for most of the Protein tyrosine phosphatases (PTPs). Furthermore, a similar approach was used to investigate PTPRT substrates in colon cancer [Zhang et al., 2007; Zhao et al., 2010].
Here, we describe the analysis and identification of novel somatic mutations in the tyrosine phosphatome of malignant melanoma. We show that PTPRT and PTPRD are the most frequently mutated genes in melanoma. Functional analysis of PTPRD mutations confirms its role as a tumor suppressor and inhibitor of melanoma cell migration. We further report desmoplakin (MIM #125647), an essential component of desmosomes that maintains cell–cell adhesion, as a novel interacting substrate of PTPRD. Desmosomes are highly organized structures containing three protein families: (1) plakins such as desmoplakin, (2) armadillo proteins such as plakoglobin and plakophilin, and (3) desmosomal cadherins such as desmoglein 1–4 and desmocollin 1–3 [Dusek and Attardi, 2011]. Desmosomes maintain cell–cell adhesion and cell differentiation; therefore, loss of desmosomes causes enhanced migratory and invasive phenotypes [Dusek and Attardi, 2011]. We show that mutant PTPRD is unable to dephosphorylate desmoplakin, suggesting a mechanism by which PTPRD controls melanoma cell migration.
Materials and Methods
Tumor Tissues
Melanoma cell lines and melanoma tissue used in this study were described previously [Palavalli et al., 2009; Prickett et al., 2009].
PCR, Sequencing, and Mutational Analysis
PCR and sequencing was done as described previously [Palavalli et al., 2009; Prickett et al., 2009]. Mutations in the phosphatase genes for primary and subsequent screens were analyzed using DNASTAR Lasergene software (DNASTAR Inc., Madison, WI).
Pooled Stable Expression
Full-length PTPRD constructs in pCDF1 backbone were cotransfected with pVSV-G and pFIV-34N helper plasmids using Lipofectamine 2000 into HEK 293T cells. Virus-containing medium was harvested 60 hr after transfection. Filtered virus medium was used to infect cells in antibody-free medium. Sk-Mel-28 and Sk-Mel-2 melanoma cells were infected with lentivirus for PTPRD (wildtype [WT], Gly446Glu, Glu1042Lys, Asp1248Asn, His1477Tyr, Pro1690Phe, and Gly1707Arg point mutants) and empty vector control as previously described [Solomon et al., 2008]. Cells were selected and maintained in 1.5 μg/ml of puromycin for further analysis. Stable expression of full-length WT PTPRD or mutant PTPRD was determined by quantitative TaqMan analysis or by SDS-PAGE analysis followed by immunoblotting with anti-PTPRD and anti-α-tubulin antibodies.
Proliferation and Growth Inhibition Assays
To analyze the cell proliferation potential, Sk-Mel-28 cells stably infected with vector and PTPRD proteins (WT and mutants) were seeded in 96-well plates at 500 or 1,000 cells per well and incubated for 9–15 days. Cell count was estimated every 48 hr by lysing cells in 50 μl of 0.2% SDS per well followed by incubation at 37°C for 2 hr before addition of 150 μl SYBR Green I solution (1:750 v/v, SYBR Green I [Invitrogen-Molecular Probes, Grand Island, NY] in deionized H2O) per well. Sk-Mel-2 cells were trypsinized and counted using hemocytometer.
Soft Agar Assay
Single cell suspension of Sk-Mel-28 pooled clones expressing PTPRD proteins (WT and mutants) were seeded in duplicate at 1,000 cells and 500 cells per well in sterile 0.33% Bacto-Agar (catalog number 214010; BD, Franklin Lakes, NJ) and 10% fetal bovine serum (FBS) (catalog number SH30109.03; HyClone, Logan, UT) in 1× RPMI in a 24-well plate (top plugs). Sk-Mel-2 were seeded in duplicate at 2,500 cells per well. The lower plug contained sterile 0.5% Bacto-Agar and 10% FBS in 1X RPMI. The colonies were stained with crystal violet, photographed, and counted after 14 days. A student’s t-test was performed to determine significance of the results.
Foci Formation Assay
Single cell suspension of Sk-Mel-28 and Sk-Mel-2 pooled clones expressing PTPRD proteins (WT and mutants) were seeded in triplicates at 1,000 cells and 500 cells per T-25 flask in 10% serum containing medium. Colonies formed by single cells after 14 days were fixed with methanol, stained with crystal violet, photographed, and counted. A student’s t-test was performed to determine significance of the results.
Migration Assay
Single cell suspension of Sk-Mel-28 pooled clones expressing PTPRD proteins (WT and mutants) were seeded in 5% serum-containing medium in licate at 50,000 cells per well in Boyden Chamber (Catalog number 354578; Corning, Tewksbury, MA). Bottom well was filled with 10% serum-containing medium. Number of cells migrated after 24 hr were fixed with methanol, stained with 0.01% crystal violet, photographed, and counted using NIH Image J software. Cells were seeded in parallel to measure the difference in growth for the indicated time. A Student’s t-test, ANOVA, and Bonferroni’s multiple comparisons test were performed to determine the significance of the results.
Construction of Intracellular PTPRD Expression Vector
Both phosphatase domains in the human PTPRD (NM002839.2) clone were PCR amplified using forward primer (5′ aaaaaccggtATGAAAAGCAGCATACCGAACAATAAGGAGATCCC) that adds Age1 restriction enzyme site and ATG start codon, and with reverse primer that adds 3X-FLAG tag to the C-terminal domain, followed by STOP codon and Mlu1 restriction enzyme site (5′ aaaacgcgtTTAttcgtcatcgtcatccttgtaatcgatatcatgatctttataatcaccgtcatggtctttgtagtccgttgcatagtggtcaaagctgcccaggtactc). PCR-amplified insert cut with Age1 and Mlu1 was inserted in the Age1/Mlu1 cut tet-regulated TRIPZ lenti-viral plasmid. Catalytic mutation bearing insert was cloned using the PTPRD Mut Asp1521Ala + (5′CCGCCTGGCCTGCTCATGGTGTTCC 3′) and PTPRD Mut Asp1521Ala − (5′GGAACACCATGAGCAGGCCAGGCGG3′) primers by Phusion PCR reaction. The constructs were sequenced and tested for tetracycline-regulated expression by transfection in 293T cells followed by Western blotting for FLAG-tag.
Cell Culture and Intracellular PTPRD Expression
Sk-Mel-28, Sk-Mel-2 melanoma cell lines, and HEK 293T were purchased from ATCC, Manassas, VA and maintained in complete DMEM supplemented with 10% FBS, 1× nonessential amino acids, 2 mM L-glutamine, and 0.75% sodium bicarbonate. HEK 293T cells were cotransfected with intracellular PTPRD (icPTPRD) constructs and helper plasmids pVSV-G and p8.74 using FuGENE HD transfection reagent (Roche, Indianapolis, IN) at a 6:1 μl–μg ratio. TRIPZ-virus supernatant was obtained as described previously [Walia et al., 2009]. Cells infected with icPTPRD-3X-FLAG lentiviruses were selected and maintained in 3 μg/ml of puromycin for further analysis. Stable expression of icPTPRD proteins (WT and catalytic mutants) in Sk-Mel-28 cells was determined by SDS-PAGE analysis followed by immunoblotting with anti-FLAG, PTPRD, and anti-α-tubulin to show equivalent expression among pools.
Immunoprecipitation and Mass Spectrometry
Cell lines stably expressing FLAG-tagged icPTPRD, cPTPRD–Asp1521Ala mutant, and vector alone were lysed in a nondetergent lysis buffer as previously described [Kim et al., 2011]. icPTPRD protein complex bound to the FLAG beads were eluted using FLAG peptide and concentrated by trichloroacetic acid precipitation as described [Kim et al., 2011]. The gel bands stained with Coomassie Blue on SDS-PAGE were excised and in-gel trypsin digestion was performed to extract the peptides. Each sample was loaded on an Agilent 1100 nanocapillary HPLC system (Agilent Technologies, Santa Clara, CA) with a 10-cm integrated μRPLC-electrospray ionization emitter columns (made in-house), coupled online with a LTQ XP mass spectrometer (Thermo Fisher Scientific, Waltham, MA) for mRPLC–MS/MS analysis. Peptides were eluted using a linear gradient of 2% mobile phase B (acetonitrile with 0.1% formic acid) to 42% mobile phase B within 40 min at a constant flow rate of 0.25 mL/min. The seven most intense molecular ions in the MS scan were sequentially selected for MS/MS by collision-induced dissociation using normalized collision energy of 35%. The mass spectra were acquired at the mass range of m/z 350–1800. The dynamic exclusion was set at 60 sec to reduce the redundancy of peptide selection for MS/MS. The ion source capillary voltage and temperature were set at 1.7 kV and 200°C, respectively. The MS/MS data were searched against UniProt Homo Sapiens database from the European Bioinformatics Institute (http://www.ebi.ac.uk/integr8) using BioWorks 3.3.1 SP1 interfaced SEQUEST (Thermo Fisher Scientific) operating on a Beowulf parallel virtual machine computer cluster (Dell, Inc., Round Rock, Texas). Up to two tryptic missed cleavage sites were allowed and methionine oxidation (15.99492) was selected as a differential modification during the database search. The cut-off for legitimate identifications were: charge state-dependent cross-correlation (Xcorr) ≥ 2.0 for [M+H]1+, ≥2.5 for [M+2H]2+, and ≥3.0 for [M+3H]3+ with delta correlation (ΔCn) ≥ 0.10.
Immunoprecipitation and Protein Blotting
Parental Sk-Mel-28 and 36T cell lines, pooled clones of Sk-Mel-28 expressing vector, WT and mutated PTPRD were lysed using ice-cold 0.5% Triton-X lysis buffer (1× Tris buffer saline, pH 7.4) for 20 min. Lysed cells were collected into a 1.5-ml microcentrifuge tube, lysed end over end for 20 min, and centrifuged for 10 min at 20,000g at 4°C. Five-hundred microgram to 2 mg of protein was immunoprecipitated overnight using 1 μg of PTPRD and desmoplakin antibody, respectively. Immunoprecipitates were washed and blotted for protein as previously described [Prickett et al., 2009]. Primary antibodies used in our analysis were anti-PTPRD (catalog number sc1118; Santa Cruz, Dallas, Texas), anti-Desmoplakin (catalog number ab71690; Abcam, Cambridge, MA), anti-PY20 (Zymed-Invitrogen, Grand Island, NY) and anti-α-tubulin (Calbiochem–EMD Biosciences, Billerica, MA). Desmoplakin constructs were a kind gift from Kathy Green (Northwestern University, IL).
Immunoblot Quantification Analysis
ImageJ (National Institutes of Health, Bethesda, MD) was used to analyze the protein blot films. Intensities of individual bands were noted and exported to Microsoft Excel for further analysis to determine intensity of phosphorylated desmoplakin.
Immunocytochemistry
Parental Sk-Mel-28, 36T, 21T, HEK cells transiently expressing desmoplakin-FLAG or desmoplakin–GFP, and Sk-Mel-28 cells overexpressing WT or mutant PTPRD were seeded in fibronectin-coated glass chamber slides (catalog number 154534; Nunc™ Lab-Tek™ II Chamber Slide™ System, Thermo Fisher Scientific) and allowed to grow for 48 hr. Cells were fixed in 4% paraformaldehyde in PBS for 10 min and then incubated in 1% BSA, 10% normal goat serum, 0.3 M glycine, 0.1% Tween containing PBS for 1 hr to permeabilize cells, and block nonspecific protein-antibody interactions. Cells were then incubated overnight with anti-FLAG (M2) antibody (Rabbit, Sigma F1804, St. Louis, MO) at 1:2,000 dilution, desmoplakin antibody (DP1, Rabbit antibody, Ab71690, 1 μg/μl; Abcam) at 1:500 dilution, and PTPRD antibody (PTPRD, Goat antibody, sc1118, 0.2 μg/μl; Santa Cruz) at 1:50 dilution in 2.5% BSA containing PBS. After three washings, secondary antibody TRITC (anti-Rabbit) was added at 1:1,500 and FITC (anti-Goat) at 1: 1,000 dilution for 2 hr. Chambers were washed three times with PBS containing 0.5% Tween-20 for 10 min at room temperature and fixed by adding 5 μl of glycerol containing DAPI (1.43 μM) for imaging.
Statistical Analysis
The significance of the ratio of nonsynonymous to synonymous mutations was determined by exact binomial test using an expected ratio of 2.5:1. To determine all other statistical calculations, the R statistical environment was used (http://www.r-project.org) [Sjoblom et al., 2006].
Results
Somatic Mutations in PTPs in Malignant Melanomas
We sequenced the coding exons of all 57 members of the tyrosine phosphatome in 24 melanomas (Supp. Tables S1 and S2). A total of 515 exons were sequenced with dye-terminator chemistry using 590 primer pairs (Supp. Table S3). Tumor-specific or somatic mutations were determined by comparing the sequence of the gene in the tumor sample with that of matched normal tissue and “c.” a coding DNA reference sequenced is used in this analysis. Sequencing of ~6.6 Mb of genome identified 23 tyrosine phosphatase genes containing a total of 165 somatic mutations, including both synonymous and nonsynonymous mutations (Supp. Table S4). We further extended our analysis by sequencing all coding exons of these 23 genes for somatic mutations in a total of 79 melanoma samples and identified 170 somatic mutations (Supp. Table S1; Supp. Fig. S1). The clinical profile of patients from whom melanoma samples were obtained is included in Supp. Table S5.
The identified somatic mutations could be “drivers” that functionally promote tumorigenesis or “passengers” representing nonfunctional changes. We found 120 nonsynonymous and 45 synonymous mutations in the 23 mutated genes (Supp. Table S4). The ratio of nonsynonymous to synonymous changes was 2.66:1, which is higher than the value of 2:1 predicted for passenger mutations [Sjoblom et al., 2006], suggesting that the majority of the mutations that we identified may promote melanoma progression. We further looked for the UV-specific mutation signatures typical of melanomas [Greenman et al., 2007]. We found that C>T mutations were significantly more frequent than other nucleotide substitutions in melanomas than lung or esophageal cancers, with nearly 14-fold increased incidence of C:G > T:A transitions (P < 1 × 10−4) (Supp. Fig. S2).
PTPRT and PTPRD were the most highly mutated genes identified in our study (Supp. Table S1; Supp. Fig. S1). Cancer cells often acquire loss of heterozygosity (LOH) mutations in tumor suppressors to inactivate its function. As PTPRD harbored seven LOH mutations compared with only three in PTPRT, we focused on PTPRD and investigated the functional effects of its mutations in melanoma.
PTPRD Mutations Enhance Growth and Migration of Melanoma Cells
To investigate the biological effects of PTPRD (NM 002839.2) mutations in melanoma, we functionally analyzed six mutations: c.1337G>A(p.Gly446Glu), c.3124G>A(p.Glu1042Lys), c.3742G> A(p.Asp1248Asn), c.4429C>T(p.His1477Tyr), c.[5068C>T; 5069C> T](p.Pro1690Phe), and c.5119G>A(p.Gly1707Arg). Two of these mutations, Gly446Glu and Asp1248Asn, were previously reported to inhibit the induction of apoptosis by PTPRD [Solomon et al., 2008]. We generated Sk-Mel-28 melanoma cells stably expressing WT PTPRD, mutant PTPRD, or vector control. Western blot analysis showed similar expression levels of PTPRD in all of the pooled clones (Fig. 1A).
Figure 1.
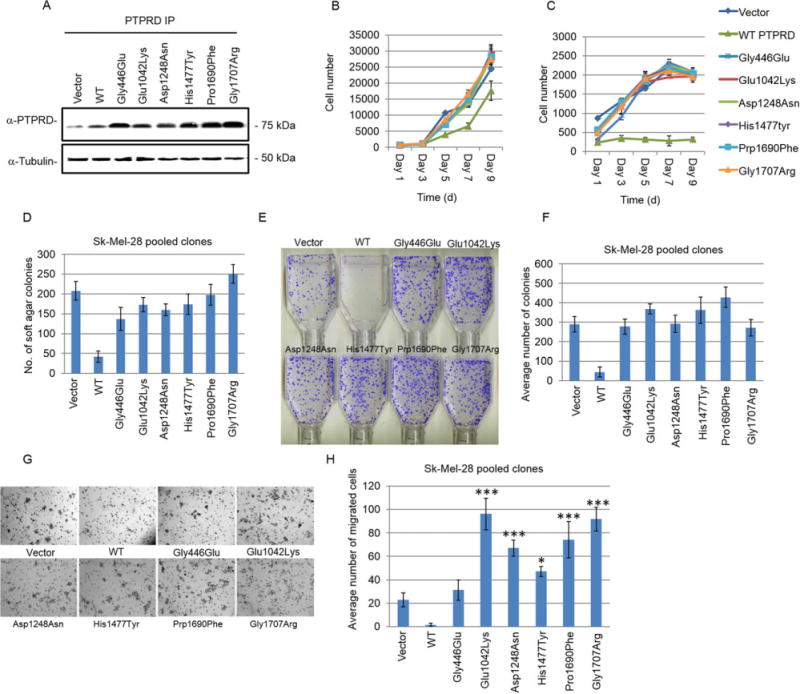
Somatic mutations of PTPRD promote melanoma cell survival. A: Western blot analysis for the expression of indicated vector (control) and PTPRD constructs in Sk-Mel-28 pooled clones. Cell lysates were immunoprecipitated and detected with anti-PTPRD antibody. Loading control: anti-α-tubulin. B: Cellular proliferation of Sk-Mel-28 pooled clones transduced with an empty vector, WT PTPRD, and indicated mutants was measured in the presence of 10% serum for 9 days. Average cell number was determined by assessing the DNA content in four replicate wells using SYBR green. C: Cellular proliferation assay was performed as described in (B) in the presence of 1% serum. D: Anchorage-independent proliferation of Sk-Mel-28 cell clones expressing indicated constructs was analyzed by counting the number of colonies formed in soft agar. The graph indicates average number of colonies formed after 14 days of growth. Mutants were compared with WT using t-test, P < 0.0055. E and F: Foci formation by Sk-Mel-28 pooled clones expressing the indicated empty vector (control), WT, or mutant PTPRD constructs seeded at 1,000 cells per T25 flask in 10% serum. The graph indicates average number of colonies formed after 14 days of growth. Mutants were compared with WT using t-test, P < 0.0015. E: Representative images of the colonies formed in (F). G and H: Sk-Mel-28 cells expressing the indicated constructs were seeded in Boyden chambers and assessed for their migratory ability. Average number of cells that migrated post 24 hr seeding was plotted. Top well contained 5% serum and bottom well contained 10% serum containing medium. G: Representative images of histogram shown in (H) of the migrated cells fixed with methanol and stained with crystal violet in one focal plane. Mutants were compared with WT PTPRD expressing cells using ANOVA and Bonferroni’s multiple comparisons test. Glu1042Lys (P < 0.0001***), Asp1248Asn (P < 0.0001), Pro1690Phe (P < 0.0001), and Gly1707Arg (P < 0.0001) mutants showed highly significant increase in migration. Migration induced by His1477Tyr was low but still significant (P < 0.0065)*, whereas Gly446Glu did not significantly induce migration. Comparison between vector alone versus mutants also showed increase in migration by PTPRD mutants such as Glu1042Lys (P < 0.0001), His1477Tyr (P < 0.009), Pro1690Phe (P < 0.0005), and Gly1707Arg (P < 0.0001).
To examine the effects of these mutations on cell growth, we assayed growth rate on plastic in the presence of 1% and 10% serum. Cells expressing WT PTPRD had a slower growth rate in 10% serum than cells expressing vector control or mutant protein (Fig. 1B). Moreover, reduction of serum concentration to 1% further promoted growth inhibition of WT PTPRD overexpressing cells compared with a modest decrease in cells expressing vector or mutant protein (Fig. 1C). These results demonstrate that expression of mutant PTPRD had little to no effect on inhibiting the growth rate of cells, suggesting that PTPRD mutations may have a loss of function effect.
Next, we used a soft agar assay to determine whether the expression of PTPRD (WT or mutants) affected anchorage-independent growth of cells stably expressing the proteins. Expression of WT PTPRD reduced Sk-Mel-28 melanoma cell growth when compared with vector or mutant PTPRD expressing cells in a colony formation assay in soft agar (Fig. 1D). In parallel, we created another melanoma cell line, Sk-Mel-2, stably expressing WT PTPRD, mutant PTPRD, or vector control (Supp. Fig. S3a and b) and obtained comparable induction in the growth of mutant PTPRD-expressing cells (Supp. Fig. S3C and D). Thesedata suggest that mutant PTPRD can support growth in anchorage-dependent and -independent conditions.
Melanoma cells with invasive and migratory abilities can penetrate the basement membrane and enter into the circulatory system to metastasize and form colonies in distant organs. We observed enhanced ability of mutant PTPRD to form foci at low cell density in the presence of 10% serum than cells expressing WT PTPRD (Fig. 1E and F; Supp. Fig. S3E). We further tested whether PTPRD mutations affect the migratory ability of melanoma cells and found that cells expressing mutant PTPRD were significantly more migratory than those expressing WT protein or vector alone (Fig. 1G and H). Notably, mutations close to the transmembrane segment, such as Glu1042Lys (4.5-fold) and Asp1248Asn (threefold), and in the second phosphatase domains of PTPRD, such as Pro1690Phe (3.5-fold) and Gly1707Arg (3.9-fold), had the most significant impact on the migratory ability of Sk-Mel-28 cells compared with vector control (Fig. 1H). No significant differences were found in the growth rate of the cells at 24 hr, indicating that the difference in migration rates exclusively reflected the mutant phenotypes. These data indicate that PTPRD mutations increase the migratory ability of melanoma cells.
Identification of PTPRD-Binding Partners and Substrates
To identify the underlying signaling cascade of mutant and WT PTPRD, we used a comprehensive proteomics approach to screen for PTPRD substrates by substrate-trapping immunoprecipitation and mass spectroscopy. We created two lentiviral vectors expressing FLAG-tagged WT and substrate-trapping Asp1521Ala mutant icPTPRD under a tetracycline-regulated promoter (Fig. 2A). This mutation was engineered based on its homology to the well-characterized substrate-trapping D1074A mutant of PTPRT (ENSP00000362294) and D181A mutant of PTP1B (ENSP00000360683) (Supp. Fig. S4), which have impaired phosphatase activity and higher substrate affinity [Flint et al., 1997; Xie et al., 2002; Zhang et al., 2007]. We created two doxycycline-inducible Sk-Mel-28 melanoma cell lines expressing the FLAG-tagged WT icPTPRD or the substrate trapping Asp1521Ala mutant of PTPRD (icPTPRD–Asp1521Ala). The expression of the respective proteins could be induced by doxycycline in a dose-dependent manner (Fig. 2B).
Figure 2.
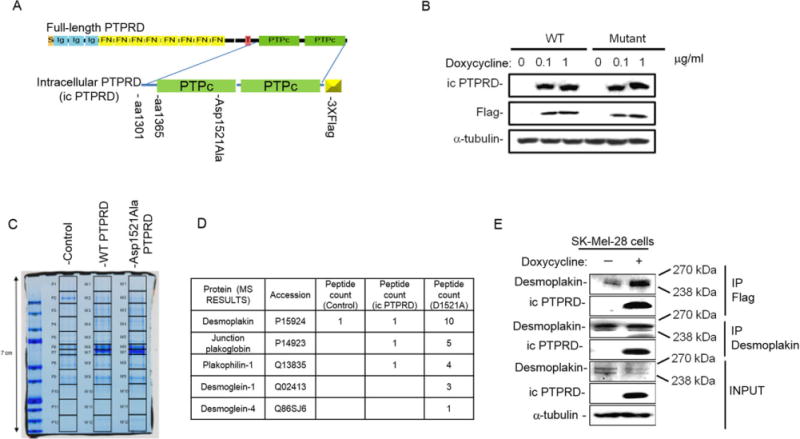
Mass spectrometry analysis of PTPRD interactome revealed its interaction with desmosomal proteins. A: icPTPRD represents PTPRD truncated at the transmembrane domain that includes two phosphatase domains of PTPRD tagged with 3× FLAG and cloned into tet-inducible modified TRIPZ lentivirus vector. Asp1521Ala mutation represents putative substrate-trapping mutation of PTPRD. Boxes represent functional domains: S, signal sequence; Ig, immunoglobulin-like domain; FN, fibronectin domain; T, transmembrane segment; PTPc, PTP domain I and II. B: Immunoblots of WT icPTPRD and icPTPRD–Asp1521Ala showing induction by indicated doses of doxycycline by 24 hr and detected by anti-PTPRD antibody and FLAG antibody. C: Coomassie-stained gel showing PTPRD-interacting proteins pulled down from Sk-Mel-28 cells post 20 hr induction by doxycycline. Lanes were cut and peptides were analyzed using mass spectrometry. D: Table showing number of peptides of desmosomal proteins pulled down by WT icPTPRD and icPTPRD–Asp1521Ala. E: Immunoblots representing coimmunoprecipitation of desmoplakin and WT icPTPRD by FLAG antibody and desmoplakin antibody. Lysates containing similar amount of total protein was determined by immunoblotting with anti-α-tubulin.
To identify novel PTPRD substrates, we performed an immunoprecipitation experiment with anti-FLAG antibody-conjugated beads, concentrated the immunoprecipitates, and separated the proteins by SDS-PAGE. The proteins were then extracted and analyzed by mass spectrometry [Kim et al., 2011] (Fig. 2C). A total of 1,541 nonredundant protein peptides, 581 from WT icPT-PRD and 868 from icPTPRD–Asp1521Ala, were pulled down and identified (Supp. Table S6). A control melanoma cell line with no PTPRD-FLAG bait showed background binding of keratin, immunoglobulin, and actin. Proteins for which at least two peptides were detected by mass spectroscopy in PTPRD–FLAG expressing cells upon normalization to control cells are shown in Supp. Table S7. We found that WT icPTPRD interacted with 107 proteins and icPTPRD–Asp1521Ala interacted with 162 peptides, whereas 59 protein peptides were common to both. These data represent an enriched pool of potential novel PTPRD-interacting partners and substrates (Supp. Fig. S5).
Among the PTPRD Asp1521Ala mutant-binding partners identified by mass spectroscopy (Supp. Table S7), we identified various desmosomal proteins, namely, desmoplakin, junction plakoglobin, plakophilin-1, desmoglein-1, and desmoglein-4 (Fig. 2D). Desmosome formation is a characteristic of cell differentiation and intercellular adhesion, whereas desmosomal loss drives tumorigenesis and early migration of tumor cells [Lorch, 2004 and Dusek and Attardi, 2011]. Because the loss of desmoplakin promotes migration by disrupting desmosomes and PTPRD mutation was shown to significantly accelerate cell migration (Fig. 1G and H), we validated desmoplakin interaction with icPTPRD by coimmunoprecipitation. Immunoprecipitation of Sk-Mel-28 cell lysates expressing WT icPTPRD with FLAG beads and immunoblotting for desmoplakin confirmed that desmoplakin directly interacted with icPTPRD. Conversely, immunoprecipitation of desmoplakin with antidesmoplakin antibody also pulled down icPTPRD (Fig. 2E). These data suggest that desmoplakin is a novel binding partner of PTPRD.
WT PTPRD Interacts with Desmoplakin and Localizes at Cell–Cell Border
To confirm this interaction, we performed an immunocytochemical labeling of overexpressed desmoplakin and WT PTPRD, and found colocalization at the cell–cell boundaries (Fig. 3A). Immunoprecipitation of ectopically expressed WT PTPRD using PTPRD antibody pull down desmoplakin in Sk-Mel-28 cells and HEK-293T. Conversely, immunoprecipitation of endogenous desmoplakin using desmoplakin antibody coimmunoprecipitated WT PTPRD in these cells (Supp. Fig. S6A and B). Immunocytochemistry of endogenous desmoplakin and endogenous WT PTPRD in Sk-Mel-28 cells showed strong colocalization at the cell–cell border (Fig. 3B, top). The Gly1707Arg PTPRD mutant in 36T melanoma cells and the Asp1248Asn PTPRD mutant in 21T melanoma cells showed intracellular colocalization with desmoplakin, and the absence on the cell border (Fig. 3B, middle and bottom). Notably, 21T cells had low desmoplakin expression. Quantification of desmoplakin at the cell border in Sk-Mel-28, 32T, and 21T cells showed that desmoplakin localizes with WT PTPRD at cell–cell borders (Fig. 3C). Similar results were obtained when we overexpressed WT and mutant PTPRD in Sk-Mel-28 cells. WT PTPRD-expressing cells showed enhanced desmoplakin localization at cell borders and mutant PTPRD showed a diffused signal (Fig. 3D; Supp. Fig. S7). Furthermore, co-immunoprecipitation of endogenous desmoplakin and WT PTPRD in Sk-Mel-28 cells confirmed that PTPRD and desmoplakin were in complexes (Fig. 3E). Moreover, in 36T melanoma cells, Gly1707Arg PTPRD mutant could also form a complex with desmoplakin (Fig. 3F). The coimmunoprecipitation experiments were not possible in 21T cells as they have low desmoplakin expression. Together, these results indicate that both WT and mutant PTPRD bind desmoplakin and this may regulate desmoplakin function.
Figure 3.
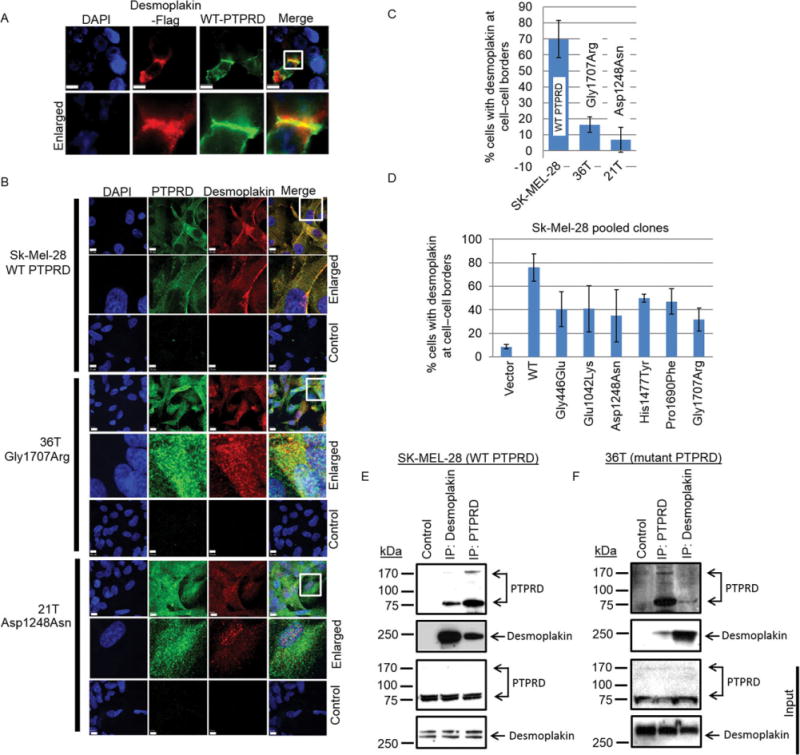
WT PTPRD interacts with desmoplakin and localizes at cell–cell border. A: Immunocytochemistry representing colocalization of desmoplakin-FLAG and WT PTPRD (scale 10u). Bottom panel represents an enlarged frame from the top panel. B: Immunocytochemistry of endogenous desmoplakin (red) with WT PTPRD (green) in Sk-Mel-28 cells (top panel), Gly1707Arg mutant (green) in 36T melanoma cells (middle panel), and Asp1248Asn mutant (green) in 21T cells (bottom panel) is shown. The control lane represents staining with secondary antibodies in the respected cell lines (scale 10u). Images were acquired using an oil objective of 63× magnification. C: Borders of desmoplakin from the immunocytochemistry experiments were counted and quantified. Two-thirds staining along the length of the cell border is counted as a single unit. Quantification of desmoplakin at cell border in Sk-Mel-28, 32T, and 21T is shown. D: Sk-Mel-28 overexpressing WT PTPRD showed most significant colocalization along with desmoplakin around the cell border. Individual mutants were compared with WT using t-test, P < 0.003. E: Coimmunoprecipitation of endogenous desmoplakin with WT PTPRD in Sk-Mel-28 cells, and Gly1707Arg mutant in 36T melanoma cells (F) is shown. Full-length PTPRD (~175 kDa) and one of the cleaved fragments (~75 kDa), and desmoplakin (~250 kDa) are shown.
PTPRD Regulates Tyrosine Phosphorylation Status on Desmoplakin
Recently, Moritz et al. (2010) identified more than 300 substrates of the oncogenic receptor tyrosine kinase family in cancer cell lines. They identified phosphorylation events on serine, threonine, and tyrosine residues of downstream signaling proteins upon activation of c-Met, epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR)-alpha signaling pathways. Interestingly, desmoplakin was one of 31 substrates that were phosphorylated at tyrosine and serine/threonine residues. Although serine/threonine phosphorylation has been previously described for desmoplakin [Amar et al., 1999], the role of tyrosine phosphorylation on desmoplakin remains unclear. Therefore, we investigated whether PTPRD regulates desmoplakin phosphorylation.
We transfected HEK293T cells with desmoplakin tagged with GFP or FLAG and PTPRD (WT, Gly446Glu, Glu1042Lys, Asp1248Asn, Gly1707Arg, or empty vector control) and performed immunoprecipitation with anti-GFP or anti-PTPRD antibodies to show reciprocal binding between the two proteins. We found that desmoplakin bound all forms of PTPRD (WT, Gly446Glu, Glu1042Lys, Asp1248Asn, and Gly1707Arg) (Fig. 4A; Supp. Fig. S8). Similarly, reciprocal experiments using anti-PTPRD antibody revealed no difference in complex formation between WT or mutant forms of PTPRD and desmoplakin (Supp. Fig. S8). Immunocytochemistry and immunoprecipitation analysis of endogenous PTPRD or mutant PTPRD from parental cell lines further confirmed that both WT and mutant PTPRD complexes with desmoplakin (Fig. 3A, B, E, and F). To test the phosphatase activity of the PTPRD mutants against desmoplakin, we measured phospho-tyrosine levels on immunoprecipitated desmoplakin–GFP in the presence of PTPRD (WT, Gly446Glu, Glu1042Lys, Asp1248Asn, Gly1707Arg, or empty vector control). Figure 4B shows that the phospho-tyrosine (pY) levels of desmoplakin were higher in the presence of mutant forms of PTPRD (two to three fold) compared with WT PTPRD, indicating that mutant forms of PTPRD may have reduced phosphatase activity. Therefore, these results demonstrate that both WT and mutant forms of PTPRD bind desmoplakin but mutant PTPRD has reduced phosphatase activity toward desmoplakin, suggesting that desmoplakin is a novel PTPRD substrate.
Figure 4.
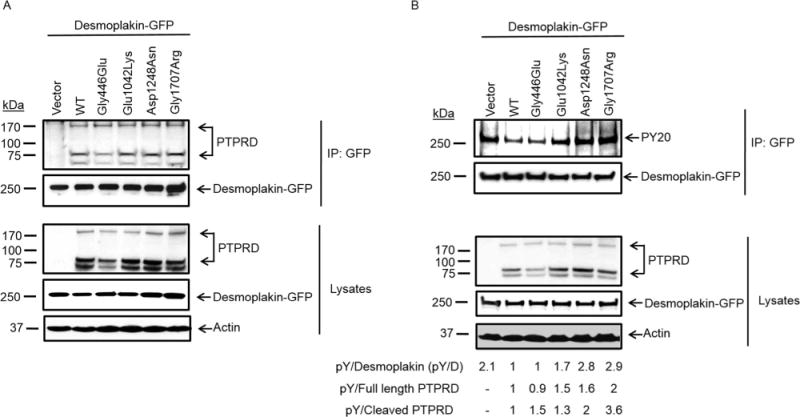
PTPRD regulates desmoplakin phosphorylation. A: Immunoblots representing coimmunoprecipitation of WT and mutant PTPRD with desmoplakin–GFP. B: Immunoblots showing tyrosine-phosphorylation status of desmoplakin in mutant and WT PTPRD expressing cells. Quantification of immunoblots from PY-20 antibody upon normalization with total desmoplakin shows that desmoplakin remains phosphorylated at tyrosine residues in mutant PTPRD cells but not in WT PTPRD expressing cells. Normalization by total desmoplakin shows two to three fold increase, by full-length PTPRD shows up to two fold increase and by cleaved PTPRD shows up to four fold increase in phosphorylation.
Discussion
The activation of tyrosine kinases as a result of mutations is a well-established phenomenon in cancer in general and specifically in melanomas [Prickett et al., 2009]. In contrast, the role of mutations in tyrosine phosphatases is poorly understood. Here, we analyzed the spectrum of somatic mutations in a complete family of tyrosine phosphatase genes in melanoma and found that PTPRT and PTPRD are the most frequently mutated genes. In particular, PTPRD bears the highest number of LOH mutations relative to its size among all tyrosine phosphatases. Reconstitution of PTPRD mutations in melanoma cells caused increased growth in the presence and absence of growth factors and promoted anchorage-independent growth. Furthermore, PTPRD mutations promoted melanoma cell migration.
We used a tetracycline-inducible icPTPRD-bearing WT or mutant catalytic domains with a 3× FLAG tag at the C-terminal end as a novel tool for the identification of interacting partners of PTPRD. Because constitutive ectopic expression of PTPRD suppresses growth, we induced cells for 20 hr with doxycycline to identify the most significant immediate changes. Similar approaches have been described for the identification of PTPRT- and PTP1B-interacting genes [Xie et al., 2002; Zhang et al., 2007; Zhao et al., 2010]. Although we identified many binding partners for PTPRD, we were unable to detect the Stat-3–PTPRD complex that was previously described for glioblastomas in our mass spectrometry screen. This may reflect the distinct proteomic signature of melanoma compared with glioblastoma.
Our finding that PTPRD directly interacts with desmoplakin suggested its involvement in the intercellular adhesion pathway. Desmoplakin functions in the formation of desmosomal junctions that are essential for maintaining cell–cell adhesion [Dusek and Attardi, 2011]. Loss of desmoplakin has been associated with invasiveness and a migratory phenotype of several cancers [Davies et al., 1999; Green and Simpson, 2007; Papagerakis et al., 2009; Chun and Hanahan, 2010]. Whether PTPRD-mediated dephosphorylation of desmoplakin at tyrosine residues is essential for desmosome formation and intercellular adhesion needs to be further evaluated. It is possible that PTPRD regulates phosphorylation of other desmosomal proteins that were identified in our screen such as plakoglobin, plakophilin, desmoglein-1, and desmoglein-4; or of adherens junction proteins such as E-cadherin. Our study has established a novel link between PTPRD and desmoplakin and identifies a phosphatase implicated in desmosome formation (Fig. 5).
Figure 5.
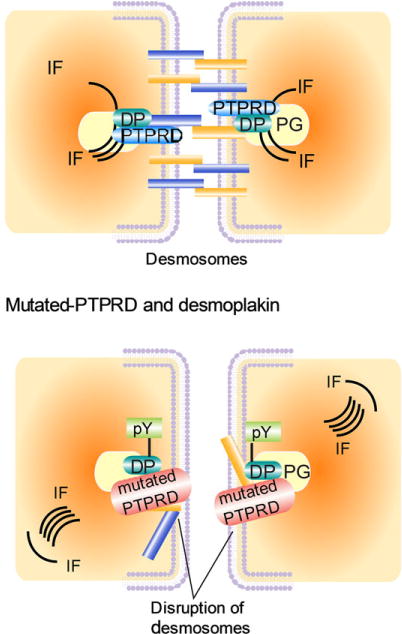
Model representing PTPRD regulation of desmosomes. (Top) WT PTPRD maintains desmoplakin in dephosphorylated state that promotes desmosome assembly. (Bottom) Inactivating mutations in PTPRD uncheck its control on phosphorylation of tyrosine residues in desmoplakin, thereby promoting loss of cell–cell contact and enhanced migratory abilities. DP, desmoplakin; pY, tyrosine phosphorylation on desmoplakin; IF, intermediate filaments; desmosomal plaque consist of plakoglobulin; desmocollin and desmoflein.
Moritz et al. (2010) identified phosphorylated tyrosine residues at positions Y-28 and Y-56 on desmoplakin. Treatment with SU11274, a Met-kinase inhibitor, reduced phosphorylation at Y-56 (16.8-fold) and at Y-28 (2.9-fold), whereas gefitinib, an EGFR inhibitor, inhibited phosphorylation at Y-28 (2.9-fold) [Moritz et al., 2010]. Interestingly, both SU11274 and gefitinib also reduced serine/threonine phosphorylation on desmoplakin. Gleevec, a PDGFR-alpha inhibitor, had relatively little effect on phosphorylation of serine, threonine, and tyrosine residues of desmoplakin [Moritz et al., 2010], suggesting that Met signaling may regulate desmoplakin phosphorylation. Rapid tyrosine phosphorylation of desmoplakin could also be accompanied by serine/threonine phosphorylation to modulate desmoplakin function and this needs to be investigated in future studies.
We have described a novel link between the PTPRD and desmoplakin and speculate that PTPRD may inhibit tyrosine phosphorylation of desmoplakin and thus modulate cell–cell adhesion. A previous report of tyrosine phosphorylation of desmoplakin by activation of the HGF/c-Met pathway [Moritz et al., 2010] suggests that PTPRD may regulate c-Met substrates. As PTPRD is mutated in multiple cancers, PTPRD-regulated desmosomal proteins may be novel therapeutic targets in tumors bearing genetic mutations in PTPRD.
Supplementary Material
Acknowledgments
We thank Allison Burrell, Sean Bhalla, and Catherine Cheng for bioinformatics help.
Contract grant sponsors: 5R01CA115699; Intramural Research Programs of the National Human Genome Research Institute; National Cancer Institute; National Institutes of Health; The Harry J. Lloyd Charitable Trust; Gideon Hamburger; Israel Science Foundation grant numbers 877/13; ERC (St. G-335377).
Footnotes
Additional Supporting Information may be found in the online version of this article.
Data Access
Somatic variants are listed in Supp. Table S8, and are deposited to ClinVar. http://www.ncbi.nlm.nih.gov/clinvar/
References
- Amar LS, Shabana AH, Oboeuf M, Martin N, Forest N. Involvement of desmoplakin phosphorylation in the regulation of desmosomes by protein kinase C, in HeLa cells. Cell Adhes Commun. 1999;7:125–138. doi: 10.3109/15419069909034396. [DOI] [PubMed] [Google Scholar]
- Boucher CA, Ward HH, Case RL, Thurston KS, Li X, Needham A, Romero E, Hyink D, Qamar S, Roitbak T, Powell S, Ward C, et al. Receptor protein tyrosine phosphatases are novel components of a polycystin complex. Biochim Biophys Acta. 2011;1812:1225–1238. doi: 10.1016/j.bbadis.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun MG, Hanahan D. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010;6:e1001120. doi: 10.1371/journal.pgen.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies EL, Gee JM, Cochrane RA, Jiang WG, Sharma AK, Nicholson RI, Mansel RE. The immunohistochemical expression of desmoplakin and its role in vivo in the progression and metastasis of breast cancer. Eur J Cancer. 1999;35:902–907. doi: 10.1016/s0959-8049(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–323. doi: 10.1038/nrc3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci USA. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KJ, Simpson CL. Desmosomes: new perspectives on a classic. J Invest Dermatol. 2007;127:2499–2515. doi: 10.1038/sj.jid.5701015. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Xu X, Li H, Solomon D, Lane WS, Jin T, Waldman T. Mechanistic analysis of a DNA damage-induced, PTEN-dependent size checkpoint in human cells. Mol Cell Biol. 2011;31:2756–2771. doi: 10.1128/MCB.01323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Otsuka A, Girard L, Sato M, Iwakawa R, Ogiwara H, Sanchez-Cespedes M, Minna JD, Yokota J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer. 2010;49:342–352. doi: 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuke Funato Y, Takeaki Y, Oda T, Akiyama T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp Ther Med. 2011;2:457–463. doi: 10.3892/etm.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, Stack MS, Green KJ. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem. 2004;279:37191–37200. doi: 10.1074/jbc.M405123200. [DOI] [PubMed] [Google Scholar]
- Meehan M, Parthasarathi L, Moran N, Jefferies CA, Foley N, Lazzari E, Murphy D, Ryan J, Ortiz B, Fabius AW, Chan TA, Stallings RL. Protein tyrosine phosphatase receptor delta acts as a neuroblastoma tumor suppressor by destabilizing the aurora kinase A oncogene. Mol Cancer. 2012;11:6. doi: 10.1186/1476-4598-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palavalli LH, Prickett TD, Wunderlich JR, Wei X, Burrell AS, Porter-Gill P, Davis S, Wang C, Cronin JC, Agrawal NS, Lin JC, Westbroek W, et al. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet. 2009;41:518–520. doi: 10.1038/ng.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagerakis S, Shabana AH, Pollock BH, Papagerakis P, Depondt J, Berdal A. Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum Pathol. 2009;40:1320–1329. doi: 10.1016/j.humpath.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, Ressom H, Jean W, Bigner D, Yan H, Samuels Y, Waldman T. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68:10300–10306. doi: 10.1158/0008-5472.CAN-08-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer L, Rao M, Anderson DJ. RPTP delta and the novel protein tyrosine phosphatase RPTP psi are expressed in restricted regions of the developing central nervous system. Dev Dyn. 1997;208:48–61. doi: 10.1002/(SICI)1097-0177(199701)208:1<48::AID-AJA5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci USA. 2009;106:9435–9440. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia V, Ding M, Kumar S, Nie D, Premkumar LS, Elble RC. hCLCA2 is a p53-inducible inhibitor of breast cancer cell proliferation. Cancer Res. 2009;69:6624–6632. doi: 10.1158/0008-5472.CAN-08-4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, Van Der Heijden MS, Parmigiani G, Yan H, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Zhang YL, Zhang ZY. Design and characterization of an improved protein tyrosine phosphatase substrate-trapping mutant. Biochemistry. 2002;41:4032–4039. doi: 10.1021/bi015904r. [DOI] [PubMed] [Google Scholar]
- Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, Wang ZJ. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci USA. 2007;104:4060–4064. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang X, Guda K, Lawrence E, Sun Q, Watanabe T, Iwakura Y, Asano M, Wei L, Yang Z, Zheng W, Dawson D, et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci USA. 2010;107:2592–2597. doi: 10.1073/pnas.0914884107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.