

Abstract
We recently reported the use of desorption electrospray ionization (DESI) as a novel interface to couple high-performance liquid chromatography (HPLC) with mass spectrometry (MS) (Chem. Commun. 2011, 47, 4171). One of the benefits of such an interface is that post-column derivatization of separated analytes can be integrated with ionization via a “reactive” DESI approach in which a derivatizing reagent is doped into the spray solvent. The reactive DESI interface allows analyte desorption/ionization from the end of the chromatographic column with prompt MS detection; a short time delay of ~20 ms was demonstrated. In this study, we extended this application by “supercharging” proteins following HPLC separation using a DESI spray solvent containing supercharging reagents, m-nitrobenzyl alcohol (m-NBA) or sulfolane. Proteins (insulin, ubiquitin, lysozyme and α-lactalbumin) eluted out of the LC column can be supercharged with the protein charge state distributions (CSDs) significantly increased (to higher charge), which would be advantageous for subsequent top-down MS analysis of proteins. Interestingly, supercharging combined with reactive DESI enhances tolerance towards trifluoroacetic acid (TFA), which is known to be a superior additive in the mobile phase for premium peptide/protein chromatographic separation but has severe signal suppression effects for conventional electrospray ionization (ESI). In comparison to electrosonic spray ionization (ESSI), a variant form of ESI, the sensitivity of protein analysis using LC/DESI-MS with the mobile phase containing TFA can be improved by up to 70-fold for lysozyme and α-lactalbumin by including m-NBA in the DESI spray solvent. Presumably, by reducing TFA dissociation in the droplet, supercharging agents lower trifluoroacetate anion concentrations and concomitantly reduce ion pairing to analyte cationic sites. The reduced ion pairing therefore decreases the TFA signal suppression effect. The supercharging capability and the reduction of TFA signal suppression suggest that LC/DESI-MS is a valuable method for protein analysis.
Keywords: Liquid chromatography/mass spectrometry, reactive DESI, protein supercharging, TFA ion suppression
INTRODUCTION
Electrospray ionization mass spectrometry (ESI-MS)[1] coupled with reversed-phase high-performance liquid chromatography (RP-HPLC) has become one of the most powerful techniques for the analysis of biomolecules and pharmaceuticals. Recently, with the advent of ambient ionization techniques, such as desorption electrospray ionization (DESI)[2] and direct analysis in real time (DART)[3], the coupling techniques with HPLC to MS have been more diversified with increased capability of LC/MS. For instance, the capability of directly ionizing eluents containing phosphate buffer (which otherwise suppresses ESI signals) has been demonstrated by the coupling of DART with HPLC[4]. In our laboratory, a DESI interface has been used to couple with HPLC, in which the eluent with a high flow rate up to 1.8 mL/min can be directly sampled and ionized by DESI-MS[5]. Taking advantage of the freedom to choose different DESI spray solvents, reactive DESI has been used in the coupling of LC-DESI for selective analyte derivatization. In addition, the integration of an electrochemical cell with LC/MS is made possible and such a LC/EC/DESI-MS system allows separated protein digests to undergo redox conversion prior to MS detection [5].
For protein and peptide analysis by MS, there are practical benefits to enhance analyte charge[6], and the ability to increase analyte charge during LC/MS analysis of proteins would be equally beneficial. The advantages of increasing analyte charge are several fold: (a) charge enhancement for proteins and peptides can be of great value to further structural analysis via top-down approaches using electron-based tandem MS techniques, such as electron capture dissociation (ECD)[7] or electron transfer dissociation (ETD)[8]; (b) ion signal-to-noise (S/N) ratio, mass resolution, and mass accuracy can also benefit from increased charges when high resolution mass analyzers such as orbitrap[9] and ion cyclotron resonance[10, 11] are used, as enhancing analyte charge moves the effective mass-to-charge (m/z) ratio to lower ranges where most analyzers have better performance. Different methods to increase the protein/peptide charge states have been proposed, including modifying the solvent system, adjustment of solution pH, and the reduction of disulfide bonds of proteins (if present). Another approach is to use the supercharging reagents such as m-nitrobenzyl alcohol (m-NBA) which was first reported by Williams’ group[12–15]. We have studied the supercharging effect on proteins and protein complexes using m-NBA and its analogues, as well as newly identified supercharging reagents such as sulfolane under the native solution environment of ammonium acetate or denaturing solution conditions[16–18]. Jensen and coworkers first added m-NBA as a supercharging modifier to the HPLC mobile phase for the analysis of tryptic peptides to improve the ETD MS/MS efficiency for peptide sequencing and identification[19]. However, in this study, the addition of m-NBA in mobile phase caused LC peak broadening and consequently decreased the analyte S/N ratio, which would reduce the sensitivity of the analysis. Marshall and coworkers incorporated different supercharging reagents (DMF, DMSO, etc) in the LC mobile phase; some of the supercharging modifiers such as DMSO appears to improve sensitivity, charging and chromatographic resolution for intact proteins up to 78 kDa [6]. Zaia’s group added sulfolane to a make-up flow of an HPLC chip interface to MS[20].
Traditionally, in the separation of complex protein mixtures by RP-HPLC, trifluoroacetic acid (TFA) is often used as a mobile phase modifier to enhance chromatographic performance as TFA not only serves to adjust pH but also acts as an ion-pairing agent[21]. However, TFA is not typically suitable for ESI-MS analysis due to its significant signal suppression effect[22]. TFA anions can neutralize or decrease the charges of the protonated analytes by forming strong ion pairs that are not dissociated during the ionization process. Alternatively, the pseudo neutral solvated ion pairs may prevent ion emission by “masking” charge from the electric field. For this reason, ESI-based detection sensitivity is typically reduced by a factor of 10 compared to that using other acid additives such as acetic acid or formic acid[21]. Although formic acid can provide good sensitivity for LC/MS, the use of TFA can produce superior chromatographic performances with the ideal peak shapes which is not exceeded by the use of other additives[23].
Another earlier method termed “TFA-fix” aimed to solve the deleterious effect of TFA[24] by using a large excess of a weaker acid (e.g., propionic acid) that is introduced using a post-column Tee to force the equilibrium with TFA back to its free acid form so that fewer TFA anions are available to ion pair with protonated proteins/peptides during ESI. However, this method requires additional pumps and involves the dilution of the eluate[22]. Recently, a novel technique was reported to overcome the TFA ion suppression issue using a fabricated post-column device for electrophoretic mobility control (EMC) of the ions[22]. The concept of EMC is to apply different voltages to the eluent out of the HPLC column to dissociate the ion pairs between TFA anions and the protonated analyte ions so that the TFA anions had a net mobility in the direction to the junction reservoir (anode) while the positively charged analytes would migrate to the ESI sprayer (cathode)[22].
It is clear that both increasing protein charge and increasing analyte ion signal with a mobile phase containing TFA would be desirable for LC/MS analysis of proteins. In this study, we demonstrate a solution for both desired features by combining supercharging reagents with reactive DESI-MS and LC/MS. Proteins of interest in the HPLC eluent containing TFA were directly sampled and ionized by DESI using a spray solvent doped with supercharging reagents (m-NBA or sulfolane). Experimentally, it is advantageous to employ reactive DESI rather than ESI to enable derivatization such as supercharging as there is no need to use a post-column Tee to introduce derivatizing reagents for DESI and therefore fast MS detection is possible. Remarkably, both signal enhancement and protein charge enhancement were achieved simultaneously in such an LC/reactive DESI-MS experiment. This study suggests a novel and valuable LC/MS method for protein analysis, as separation of proteins using TFA does not compromise MS-based detection and increased protein charge is available for subsequent top-down MS/MS.
EXPERIMENTAL SECTION
Materials
Insulin (from bovine pancreas, HPLC grade), ubiquitin (from bovine erythrocytes), lysozyme (from chicken egg white, ≥90%), α-lactalbumin (from bovine milk), trifluoroacetic acid (reagent plus, 99%), 3-nitrobenzyl alcohol (m-NBA, for mass spectroscopy, ≥99.5%) and sulfolane (99%) were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. Acetic acid was purchased from Fisher Chemicals (Pittsburgh, PA). HPLC-grade methanol was purchased from GFS Chemicals (Columbus, OH). HPLC-grade acetonitrile was purchased from EMD Chemicals Inc. (Billerica, MA). The de-ionized water used for sample preparation was obtained using a Nano-pure Diamond Barnstead purification system (Barnstead International, Dubuque, IA).
Sample Preparation
For preparing protein mixture samples, insulin was dissolved in TFA/H2O (0.1/99.9 by volume) and ubiquitin, lysozyme and α-lactalbumin were dissolved in pure water and stored as stock solutions. m-NBA and sulfolane were dissolved in water and then diluted with CH3OH/H2O/HOAc (50/50/1 by volume). For the online LC/DESI-MS experiments, a 10 μL of 125 μM protein mixture sample in ACN/H2O containing 0.1% TFA was injected into the HPLC while either 50 mM m-NBA or 200 mM sulfolane in CH3OH/H2O/HOAc (50/50/1 by volume) was used as the DESI spray solvent for supercharging.
Apparatus
Figure 1 shows the apparatus for coupling a commercial Perkin Elmer HPLC system (Perkin Elmer, Shelton, CT) and a Thermo Finnigan LCQ DECA ion trap mass spectrometer (San Jose, CA) with a home-built liquid DESI source. A gradient program from 25% B to 45% B in 30 minutes was used to separate protein mixtures via a Waters XB Bridge TM 300 C4 column (3.5 μm, 4.6×150mm) with the mobile phase composition A, 0.1% TFA in H2O and B, 0.085% TFA in 95% ACN. The UV detector was set at the absorbance wavelength of 220 nm. PEEK tubing was connected to the UV detector outlet and a piece of silica capillary (i.d. 100 μm, length 4.2 cm) was joined to the end of the PEEK tubing. A fast flying liquid jet containing protein eluent flowing out of the silica capillary with an elution rate of 1.0 mL/min was directly sampled/ionized by DESI using a spray solvent containing CH3OH/H2O/HOAc or supercharging reagents (m-NBA or sulfolane) in CH3OH/H2O/HOAc. In the comparison experiments, the eluent was split at a ratio of 100:1 and then ionized by electrosonic spray ionization (ESSI) [25, 26], a variant form of ESI.
Figure 1.
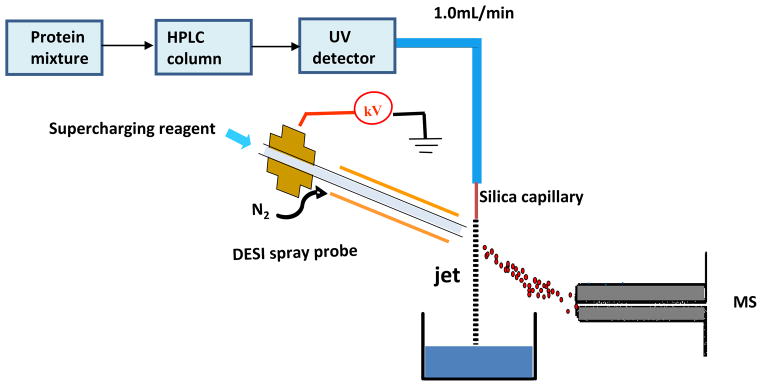
Scheme showing the online coupling of HPLC with the DESI-MS apparatus. A DESI spray probe was used to sample/ionize the analytes of interests in the eluent directly from the fast flow jet.
For the online coupling LC with DESI-MS apparatus, an alternative configuration was adopted as shown in Figure S-5a (Supplemental Materials). In this experimental setup, the PEEK tubing and UV detector was removed. A silica capillary was connected to the LC column outlet to form a jet that was then ionized by DESI.
RESULTS AND DISCUSSION
Direct infusion for the supercharging reactive DESI analysis
Previous studies have shown that DESI can be used to increase the charge state of large proteins and protein complexes simply by doping supercharging reagents into the DESI spray solvent[16]. For example, by spraying 40 mM m-NBA from the DESI solvent, the most abundant charge state for enolase (93 kDa) increases from +20 to +25 and the maximum charge state from to +22 to +29 [16]. In this study, we first tested if the “supercharging” reactive DESI can be applicable to proteins in TFA solution.
Figure 2a shows the DESI-MS spectrum of 14 kDa lysozyme in methanol/water (50:50 by volume) containing 0.1% TFA. A charge state distribution (CSD) ranging from +8 to +12 was measured with the highest intensity at 2.05E3 (arbitrary units). It is most likely that the low intensity of the lysozyme ions is in agreement with previous reports that TFA causes significant signal suppression and decreases the net average charge of peptide and protein ions observed in ESI-MS analysis [24, 27, 28]. In contrast, when 50 mM m-NBA was doped into the DESI spray, the CSD shifted dramatically to higher charge number, shifting the maximum charge state from +12 to +17 and the average charge state (qaverage)[12] from +10.1 to +11.6 with the intensity increasing by approximately 50-fold (Figure 2b). A similar phenomenon was observed in the case of 8.6 kDa ubiquitin. Compared with the regular DESI analysis for 10 μM ubiquitin in 0.1% TFA (Figure 2c), when spraying 50 mM m-NBA supercharging reagent, qaverage increases from +9.1 to +10.7 with an increase in ion signal by 7-fold (Figure 2d).
Figure 2.

DESI mass spectra of 10 μM lysozyme in 0.1% TFA using a spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) (a) without m-NBA and (b) with 50 mM m-NBA; DESI mass spectra of 10 μM ubiquitin in 0.1% TFA using a spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) (c) without m-NBA and (d) with 50 mM m-NBA. The peaks of m/z 700~1100 in (a) were from background leftover.
The addition of the supercharging agent sulfolane (or m-NBA) has similar effects in the presence of TFA for direct infusion ESI-MS measurements, as the addition of 200 mM sulfolane (or 0.5% m-NBA) to a lysozyme solution (50/50/0.1 H2O/ACN/TFA) increases charging and signal intensity (Figure S-1, Supplemental Materials). These results show that supercharging is applicable to proteins in solution containing TFA, and the TFA suppression effect can be mitigated.
Online Coupling RP-HPLC with DESI-MS
Based on the direct infusion results (above), online coupling of HPLC with TFA-containing mobile phases and MS is demonstrated using the reactive DESI interface. The prototype LC/DESI-MS apparatus (Figure 1) allows the direct DESI-MS analysis of the eluent with a high flow rate without splitting. This characteristic of DESI has been noted in our previous study in coupling LC with MS [5], and in the development of sub-millisecond time-resolved mass spectrometry[29]. A 10 μL protein mixture (125 μM each) consisting of insulin, ubiquitin, lysozyme, and α-lactalbumin was injected to the HPLC column (Waters XB Bridge TM 300 C4 column) and subjected to gradient separation with 0.1% TFA in the mobile phase. The eluent with the flow rate of 1.0 mL/min flowing through the UV detector (detection wavelength was set at 220 nm) was subject to ionization by DESI. As shown in the UV chromatogram (Figure 3), four protein peaks were well separated, corresponding to insulin, ubiquitin, lysozyme, and α-lactalbumin, respectively. The separation order follows the size order of proteins, which is in line with previous reports[6]. The connection between the UV detector and DESI ion source was a piece of fused silica capillary joined to the end of PEEK tubing. As a result of the high eluent flow rate of 1.0 mL/min, a stable liquid jet from the outlet of capillary was produced. The extracted ion chromatogram (XIC) for each protein eluent agrees well with the UV chromatogram with the delay time of less than 5 s between the UV detection and DESI-MS detection. This suggests again that DESI is a feasible interface for LC/MS, even at a high flow rate which would suit the usage of an ordinary analytical separation column without splitting or fits the needs of preparative chromatography [5].
Figure 3.

UV chromatogram showing the HPLC separation of a protein mixture containing insulin, ubiquitin, lysozyme and α-lactalbumin.
As shown in Figure 4a, the direct infusion of lysozyme eluent by ESSI analysis, a variant form of ESI, after 100:1 splitting (10 μL/min) yielded the mass spectrum showing low intensity protein peaks (with the intensity of the +10 ion as 1.80E3) and a high level of noise. However, when lysozyme eluent coming out of the LC system without splitting was sampled/ionized directly by a DESI spray probe consisting CH3OH/H2O/HOAc (50:50:1 by volume), a mass spectrum with higher intensity of protein peaks (6.06E3 intensity for the +9 ion) was observed (Figure 4b). This further demonstrates the feasibility of DESI to ionize large flow rate of eluent, which could not be achieved easily by normal ESI, for such a high flow rate stream which otherwise overloads the ion source. Splitting causes sample loss, which might account for lower sensitivity of ESSI compared to DESI. When the DESI spray was doped with supercharging reagent, such as 50 mM m-NBA, the intensity of protein peaks (Figure 4c) was increased significantly. In comparison to the ESSI signal (Figure 4a), the intensity of protein ions was enhanced by 73-fold using LC/DESI-MS by including m-NBA in the DESI spray solvent (Figure 4c). Furthermore, the average charge state shifted from +9.4 to +11.6 and the maximum charge state from +11 to +17 compared to the ESSI data. Another supercharging reagent, sulfolane, was also doped into the DESI spray to test this concept. Using 200 mM sulfolane as part of the DESI spray, the intensity of the lysozyme peaks (Figure 4d) was increased by 57-fold and lysozyme average charging increased from +9.4 to +9.9 compared to the ESSI result. Under this specific experimental condition, besides both reagents being able to overcome TFA suppression, m-NBA has slightly better supercharging effect over sulfolane.
Figure 4.

Mass spectra of lysozyme following the HPLC separation obtained by (a) ESSI analysis after splitting the eluent (split ratio: 100:1), (b) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume), (c) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) containing 50 mM m-NBA, and (d) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) containing 200 mM sulfolane. The asterisk labeled peaks in (c) probably correspond to the protein adduct ions with m-NBA.
Similar phenomena were observed for the protein α-lactalbumin (Figure 5). Direct DESI analysis of LC eluent without splitting produced cleaner spectra than that obtained from LC-splitting-ESSI analysis in which, again, the TFA suppression effect is apparent. When the DESI spray was doped with supercharging reagent, m-NBA or sulfolane, the TFA suppression issue can be overcome with the increased abundance for protein peaks. Compared with ESSI mass spectrum shown in Figure 5a, supercharging DESI with 50 mM m-NBA doped in the spray (Figure 5c) reduced TFA signal suppression, increased sensitivity by 72 times, and shifted the maximum charge state from +10 to +16 and the average charge state from +8.7 to +11.7. When 200 mM sulfolane was doped in the DESI spray instead of m-NBA (Figure 5d), sensitivity of protein peaks increased by 106-fold, the average charge state increased from +8.7 to +9.2, and the maximum charge state shifted from +10 to +12.
Figure 5.

Mass spectra of α-lactalbumin following the HPLC separation obtained by (a) ESSI analysis after splitting the eluent (split ratio: 100:1), (b) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume), (c) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) containing 50 mM m-NBA, and (d) DESI analysis of the jet eluent using the spray solvent of CH3OH/H2O/HOAc (50:50:1 by volume) containing 200 mM sulfolane. The asterisk labeled peaks in (c) probably correspond to the protein adduct ions with m-NBA.
This enhancement effect on protein signal and charge was also observed in the analysis of insulin and ubiquitin (Figures S-2 and S-3, Supplemental Materials). However, the signal enhancement effect by m-NBA is modest (by 2-fold increment for insulin and by 5-fold increment for ubiquitin). This is probably due to the fact that there is less TFA suppression involved in the ESSI of these two smaller proteins.
Rationale of supercharging and reduction of TFA suppression effect
It has been suggested that the primary cause of ion suppression is strong ion pairing between the trifluoroacetate anion and the protonated sample cation[21]. Ion-pairing “masks” the protonated sample cations from the electric fields by rendering them “neutral”, thus inhibiting ion emission. An explanation for the effect of supercharging reagents to overcome TFA-induced signal suppression is that they reduce trifluoroacetic acid dissociation within the droplet[30]. The corresponding reduction in the concentration of trifluoroacetate anions decreases the extent of ion pairing to analyte cationic sites.
By spraying a mixture of TFA and m-NBA, a [NBA+TFA-H]− (m/z 266) ion was observed (Figure S-4a, Supplemental Materials). This ion dissociates into [TFA-H]− (m/z 113) upon collision-induced dissociation (CID), confirming its structure (Figure S-4b, Supporting Information). This result suggests that m-NBA does have some interaction with TFA anions by forming adducts. More studies are needed to fully elucidate the mechanism of the supercharging and signal enhancement in our reactive DESI experiment.
Online RP-HPLC Coupling with DESI-MS with Small Delay Time
In order to shorten the delay time after HPLC separation of protein mixtures for MS analysis, an alternative apparatus shown in Figure S-5a (Supplemental Materials) was used by removing the UV detector between the LC column and mass spectrometer, and by assembling a short piece of fused silica capillary (length: 4.2 cm) to the LC column. Based on the flow rate of the eluent as well as the dimension of the capillary, a delay time of about 20 ms was achieved. The DESI apparatus shown in Figure 1 was used to test this interface. Similar results were obtained, in which by spraying the supercharging reagents, TFA ion suppression issue could be minimized and the protein CSD shifted to higher charge state, as exemplified by α-lactalbumin (Figure S-5b, Supplemental Materials).
CONCLUSIONS
A unique strategy was demonstrated to alleviate TFA ion suppression in LC/MS analysis and simultaneously increase charging of protein ions. Taking advantage of the freedom to choose DESI spray solvents, supercharging reagents m-NBA and sulfolane were doped into a DESI spray via reactive DESI to form the basis of a versatile interface to couple RP-HPLC with MS. Compared with the regular LC-ESSI analysis of protein eluent in TFA-containing mobile phases, supercharging reagents can overcome the TFA suppression issue by reducing the ion-pairs formed between TFA anion and protein/peptide. Moreover, m-NBA and sulfolane increase multiple charging for proteins, which can be significant for the further structure analysis by ECD and ETD. This concept of using supercharging reagents could provide a widely used solution for LC-MS applications using mobile phases containing TFA or some other MS non-compatible additives such as phosphate or inorganic salts, etc.
Supplementary Material
Acknowledgments
The financial support of this work by NSF (CHE-0911160) and NSF Career (CHE-1149367) to H.C. and by NIH (R01 RR020004, now reassigned as GM103479) to J.A.L. are gratefully acknowledged.
Footnotes
Supplemental Materials: Additional spectra and information are provided.
References
- 1.Whitehouse CM, Dreyer RN, Yamashita M, Fenn JB. Electrospray Interface for Liquid Chromatographs and Mass Spectrometers. Anal Chem. 1985;57:675–679. doi: 10.1021/ac00280a023. [DOI] [PubMed] [Google Scholar]
- 2.Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass Spectrometry Sampling under Ambient Conditions with Desorption Electrospray Ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 3.Cody RB, Laramee JA, Durst HD. Versatile New Ion Source for the Analysis of Materials in Open Air under Ambient Conditions. Anal Chem. 2005;77:2297–2302. doi: 10.1021/ac050162j. [DOI] [PubMed] [Google Scholar]
- 4.Eberherr W, Buchberger W, Hertsens R, Klampfl CW. Investigations on the Coupling of High-Performance Liquid Chromatography to Direct Analysis in Real Time Mass Spectrometry. Anal Chem. 2010;82:5792–5796. doi: 10.1021/ac1008496. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Yuan ZQ, Dewald HD, Chen H. Coupling of Liquid Chromatography with Mass Spectrometry by Desorption Electrospray Ionization (DESI) Chem Commun. 2011;47:4171–4173. doi: 10.1039/c0cc05736c. [DOI] [PubMed] [Google Scholar]
- 6.Valeja SG, Tipton JD, Emmett MR, Marshall AG. New Reagents for Enhanced Liquid Chromatographic Separation and Charging of Intact Protein Ions for Electrospray Ionization Mass Spectrometry. Anal Chem. 2010;82:7515–7519. doi: 10.1021/ac1016858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 8.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu QZ, Noll RJ, Li HY, Makarov A, Hardman M, Cooks RG. The Orbitrap: A New Mass Spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 10.Marshall AG, Hendrickson CL. Charge Reduction Lowers Mass Resolving Power for Isotopically Resolved Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectra. Rapid Commun Mass Spectrom. 2001;15:232–235. [Google Scholar]
- 11.Marshall AG, Hendrickson CL, Jackson GS. Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 12.Iavarone AT, Jurchen JC, Williams ER. Supercharged Protein and Peptide Lone Formed by Electrospray Ionization. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iavarone AT, Williams ER. Supercharging in Electrospray Ionization: Effects on Signal and Charge. Int J Mass Spectrom. 2002;219:63–72. [Google Scholar]
- 14.Iavarone AT, Williams ER. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J Am Chem Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iavarone AT, Williams ER. Collisionally Activated Dissociation of Supercharged Proteins Formed by Electrospray Ionization. Anal Chem. 2003;75:4525–4533. doi: 10.1021/ac034144i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson CN, Benchaar SA, Miao Z, Loo JA, Chen H. Direct Ionization of Large Proteins and Protein Complexes by Desorption Electrospray Ionization-Mass Spectrometry. Anal Chem. 2011;83:6468–6473. doi: 10.1021/ac201390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lomeli SH, Yin S, Loo RRO, Loo JA. Increasing: Charge While Preserving Noncovalent Protein Complexes for ESI-MS. J Am Soc Mass Spectrom. 2009;20:593–596. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lomeli SH, Peng IX, Yin S, Loo RRO, Loo JA. New Reagents for Increasing Esi Multiple Charging of Proteins and Protein Complexes. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kjeldsen F, Giessing AMB, Ingrell CR, Jensen ON. Peptide Sequencing and Characterization of Post-Translational Modifications by Enhanced Ion-Charging and Liquid Chromatography Electron-Transfer Dissociation Tandem Mass Spectrometry. Anal Chem. 2007;79:9243–9252. doi: 10.1021/ac701700g. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Shi XF, Yu X, Leymarie N, Staples GO, Yin HF, Killeen K, Zaia J. Improved Liquid Chromatography-MS/MS of Heparan Sulfate Oligosaccharides Via Chip-Based Pulsed Makeup Flow. Anal Chem. 2011;83:8222–8229. doi: 10.1021/ac201964n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhlmann FE, Apffel A, Fischer SM, Goldberg G, Goodley PC. Signal Enhancement for Gradient Reverse-Phase High-Performance Liquid Chromatography Electrospray Ionization Mass Spectrometry Analysis with Trifluoroacetic and Other Strong Acid Modifiers by Postcolumn Addition of Propionic Acid and Isopropanol. J Am Soc Mass Spectrom. 1995;6:1221–1225. doi: 10.1016/1044-0305(95)00571-4. [DOI] [PubMed] [Google Scholar]
- 22.Wang NH, Lee WL, Her GR. Signal Enhancement for Peptide Analysis in Liquid Chromatography-Electrospray Ionization Mass Spectrometry with Trifluoroacetic Acid Containing Mobile Phase by Postcolumn Electrophoretic Mobility Control. Anal Chem. 2011;83:6163–6168. doi: 10.1021/ac2003714. [DOI] [PubMed] [Google Scholar]
- 23.Temesi D, Law B. The Effect of LC Eluent Composition on MS Responses Using Electrospray Ionization. LC- GC N Am. 1999;17:626–632. [Google Scholar]
- 24.Apffel A, Fischer S, Goldberg G, Goodley PC, Kuhlmann FE. Enhanced Sensitivity for Peptide-Mapping with Electrospray Liquid-Chromatography Mass-Spectrometry in the Presence of Signal Suppression Due to Trifluoroacetic Acid-Containing Mobile Phases. J Chromatogr A. 1995;712:177–190. doi: 10.1016/0021-9673(95)00175-m. [DOI] [PubMed] [Google Scholar]
- 25.Takats Z, Wiseman JM, Gologan B, Cooks RG. Electrosonic Spray Ionization. A Gentle Technique for Generating Folded Proteins and Protein Complexes in the Gas Phase and for Studying Ion - Molecule Reactions at Atmospheric Pressure. Anal Chem. 2004;76:4050–4058. doi: 10.1021/ac049848m. [DOI] [PubMed] [Google Scholar]
- 26.Wiseman JM, Takats Z, Gologan B, Davisson VJ, Cooks RG. Direct Characterization of Enzyme-Substrate Complexes by Using Electrosonic Spray Ionization Mass Spectrometry. Angew Chem Int Ed. 2005;44:913–916. doi: 10.1002/anie.200461672. [DOI] [PubMed] [Google Scholar]
- 27.Mirza UA, Chait BT. Effects of Anions on the Positive-Ion Electrospray-Ionization Mass-Spectra of Peptides and Proteins. Anal Chem. 1994;66:2898–2904. doi: 10.1021/ac00090a017. [DOI] [PubMed] [Google Scholar]
- 28.Gustavsson SA, Samskog J, Markides KE, Langstrom B. Studies of Signal Suppression in Liquid Chromatography-Electrospray Ionization Mass Spectrometry Using Volatile Ion-Pairing Reagents. J Chromatogr A. 2001;937:41–47. doi: 10.1016/s0021-9673(01)01328-0. [DOI] [PubMed] [Google Scholar]
- 29.Miao Z, Chen H, Liu P, Liu Y. Development of Submillisecond Time-Resolved Mass Spectrometry Using Desorption Electrospray Ionization. Anal Chem. 2011;83:3994–3997. doi: 10.1021/ac200842e. [DOI] [PubMed] [Google Scholar]
- 30.Streuli CA. Titrations in Nonaqueous Solvents. Anal Chem. 1964;36:363R–369R. doi: 10.1021/ac60313a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.