

Abstract
When subjected to pressure overload, the ventricular myocardium shifts from fatty acids to glucose as its main source for energy provision and frequently increases its mass. Here, we review the evidence in support of the concept that metabolic remodeling, measured as increased myocardial glucose uptake using dynamic positron emission tomography (PET) with the glucose analogue 2-deoxy-2-[18F]-fluoro-D-glucose (FDG), precedes the onset of left ventricular hypertrophy (LVH) and heart failure. Consistent with this, early intervention with propranolol, which attenuates glucose uptake, prevents the maladaptive metabolic response and preserves cardiac function in vivo. We also review ex vivo studies suggesting a link between dysregulated myocardial glucose metabolism, intracellular accumulation of glucose 6-phosphate (G6P) and contractile dysfunction of the heart. G6P levels correlate with activation of mTOR (mechanistic target of rapamycin) and endoplasmic reticulum stress. This sequence of events could be prevented by pre-treatment with rapamycin (mTOR inhibition) or metformin (enzyme 5′-AMP-activated protein kinase activation ). In conclusion, we propose that metabolic imaging with FDG PET may provide a novel approach to guide the treatment of patients with hypertension-induced LVH.
Keywords: Left Ventricular Hypertrophy, 2-deoxy-2-[18F]-fluoro-D-glucose positron emission tomography, Glucose 6-Phosphate, mechanistic target of rapamycin Activation, Endoplasmic Reticulum Stress
1. Perspective
Systemic hypertension affects about 30% of the adult US population(1) and is a common cause of left ventricular hypertrophy (LVH) leading to heart failure (HF) (2). There is a lack of effective medical therapies to treat LVH, hence early detection and prevention are important. Much of the biology of LVH has been learned from small animal models of aortic constriction. We have previously provided evidence that an increase in workload prompts the heart muscle to switch from fatty acids to glucose as its main source for oxidative energy provision (3-6). This adaptive response is considered beneficial to cardiac function in an acute setting, but sustained pressure overload reactivates the fetal gene program and makes the metabolic change permanent (7). The switch to glucose metabolism, in turn, is associated with impaired metabolic flexibility and mitochondrial dysfunction, both of which characterize the failing heart (8-10). We have proposed earlier that alterations in glucose metabolism in the pressure-overloaded heart precede the development of LVH and impaired cardiac function (11;12) and, that, targeting these metabolic changes may thus prevent the development of LVH and HF in the pressure-overloaded heart. (13-15) Additionally, accurate metabolic imaging is able to identify changes in myocardial substrate metabolism before and during the development of LVH and may be used to monitor therapeutic interventions to prevent LVH (16-19). Specifically, 2-deoxy-2-[18F]-fluoro-D-glucose (FDG) is used to trace dysregulated glucose metabolism. This is analogous to cancer cell metabolism where increased FDG retention heralds cell growth and proliferation(20).
The aim of this review is to summarize the knowledge derived from our work (11;12) in support of the hypothesis that metabolic remodeling associated with increased growth signaling and functional changes precedes structural left ventricular remodeling induced by sustained pressure overload. We also discuss the role of pharmacological interventions targeting metabolic intermediates to prevent the progression to LVH and HF in the setting of pressure overload.
2. In vivo characterization of glucose metabolism changes in the pressure-overloaded heart using serial FDG Positron Emission Tomography imaging
2.1. FDG Positron Emission Tomography imaging of the mouse heart during induction of pressure overload
Non-invasive positron emission tomography (PET) imaging is used to identify metabolic substrate utilization (19). However, myocardial glucose uptake rate estimates have been inaccurate in mouse hearts due to partial volume effects and spillover of radioactivity from the myocardium into the LV blood pool and vice versa. In our novel quantitative method, blood input function with spillover and partial volume coefficients and metabolic rate constants in a compartment model are simultaneously estimated from attenuation-corrected, gated, and dynamic high resolution FDG PET images of mouse heart in vivo (11;21). In conjunction with magnetic resonance imaging (MRI), used for measuring cardiac structure and function, we tested the hypothesis in vivo, that early metabolic remodeling, recognized by increased myocardial FDG uptake, precedes structural left ventricular remodeling and decrease in cardiac function, in a mouse model of transverse aortic constriction (TAC) induced pressure overload LVH (11;12). In mice subjected to TAC surgery, there is a continuous increase in FDG uptake from the first day up to 4 weeks after surgery, whereas sham-operated animals show no change in FDG uptake over the same period (Figures 1A-B). However, 1 day after TAC, when myocardial FDG uptake was significantly increased, there were no appreciable changes in the ratio of heart weight to body weight and in end-diastolic wall thickness, as measured using MRI. Significant decreases in ejection fraction assessed by MRI, however, followed the metabolic changes (12). In addition, there was a significant increase in glucose 6-phosphate levels 1 day after TAC in harvested hearts, and the increase was more pronounced 2 weeks after TAC (Figure 1C). In order to unravel the molecular mechanisms leading to the structural changes observed in TAC mice, we investigated the participation of the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) in the model of TAC induced LVH (22). mTORC1 is a major regulator of myocardial protein synthesis located downstream of the phosphatidylinositol-3 kinase signaling pathway and is a major driver of cardiac hypertrophy (23). Already on day 1 after TAC the mTORC1 targets, p70S6 kinase and 4E-binding protein 1 were activated and remained so for 2 weeks (Figure 1D). Thus, significant metabolic and cell signaling changes are present 1 day after TAC surgery and precede significant structural changes.
Figure 1.

Metabolic remodeling and mechanistic target of rapamyicn (mTOR) activation precede structural remodeling in hearts subjected to high workload in vivo. BSL=Baseline. (A) Representative serial transverse, end-diastolic PET slices for TAC and sham-operated mice 1 day, 2 weeks, and 4 weeks after surgery. (B) Quantification of the rate of cardiac FDG uptake (Ki) in PET images. #p<0.05: 2 weeks versus BSL or day 1; **p< 0.05: 4 weeks versus BSL, day 1 or 2 weeks; *p<0.001. Comparisons between TAC and sham groups at the same points; ^p<0.05. (C) Tissue glucose 6-phosphate levels in hearts after TAC or sham operation at baseline and after 1 day and 2 weeks. (D) Western blot analysis illustrating mTOR activation at 1 day and 2 weeks after TAC compared to shams. Modified and reproduced with permission from the Journal of American Heart Association (12).
2.2. In vivo FDG PET during pharmacologic intervention
Metabolic imaging of the heart has been used to evaluate the response to therapeutic interventions. Studies using 31P Magnetic Resonance Spectroscopy (MRS) show that decreased cardiac phosphocreatine/adenosine triphosphate ratios in failing hearts of patients with dilated cardiomyopathy increase and normalize in response to β-blocker therapy (24;25). Metabolic changes are also accompanied by improved cardiac efficiency in patients with left ventricular dysfunction (26). Measurements, such as percent glucose uptake with FDG PET, have also been used to evaluate the beneficial effect of β-blocker treatment in patients with dilated cardiomyopathy (27). Using a 3-compartment kinetic model we measured rates of myocardial FDG uptake (Ki) and rate of myocardial glucose utilization (rMGU) in TAC mice treated with propranolol. Myocardial FDG uptake rates increased over a period of 7 days in TAC mice together with a reduction in cardiac function as compared to shams. Treatment with propranolol attenuated myocardial FDG uptake and simultaneously improved cardiac function (Figures 2A-B). Since rMGU in TAC mice does not increase linearly with FDG uptake rates, but rather increases at a much slower rate, a mismatch of 29% between FDG uptake and rMGU results (11). Note that this mismatch correlates with significant myocardial G6P accumulation observed as early as 1 day after TAC (Figure 1C). Early treatment with propranolol prevents the drastic increase in myocardial FDG uptake in the hearts of TAC mice, reduces the mismatch between FDG uptake and rMGU and preserves the left ventricular ejection fraction (Figures 2A-B) (11). Thus in vivo FDG PET studies are able to identify changes in myocardial glucose metabolism as a possible readout for cardiac efficiency.
Figure 2.

Rate of myocardial FDG uptake and cardiac function in mouse heart in vivo subjected to TAC surgery without and with propranolol treatment over a period of 7 days. *p<0.05 versus baseline, TAC treated with propranolol and sham groups. Top End-diastolic transverse PET images for sham, TAC, and TAC mice treated with propranolol at baseline and at day 1 and day 7 after surgery. (A) Measured Ki (ml/g/min). (B) Ejection fraction measured using high resolution gated PET images. Modified and reproduced with permission from the Journal of Nuclear Medicine (11).
2.3. In vivo FDG PET in other animal models of LVH
The TAC mouse model lacks key features of the human disease, most importantly the slow progressive development of pressure overload. This makes it impossible to identify a window for aggressive therapeutic strategies that could be readily translated to clinical practice. The spontaneously hypertensive rat and the Dahl salt-sensitive rat are widely used models to study the transition from stable compensated LVH to systolic HF (28-31). Recent PET imaging studies in vivo by Hernandez et al. in the spontaneously hypertensive rat heart over a period of 20 months observed significant increases in myocardial glucose and fatty acid utilization at both early and late stages, and these precede mechanical changes during LVH progression (32). Studies in the Dahl salt-sensitive rat model ex vivo,(31) also revealed increased glucose uptake early in the progression from LVH to heart failure, but in contrast to Hernandez et al,(32) a late decrease in fatty acid metabolism was observed. Although evidence supporting an increase in glucose metabolism preceeding LVH and HF is observed in both genetic and TAC induced LVH animal models, there is no clear consensus yet on the changes and/or role of fatty acid metabolism in the stressed heart in vivo. This is mainly due to the lack of adequate compartment models to non-invasively quantify myocardial fatty acid metabolism that complements the conventional graphical Patlak analysis(32).
3. Probing for mechanisms ex vivo
3.1 G6P and mTOR activation
We have previously shown that the uptake of FDG by the isolated working rat heart is linear with alterations in substrate supply and workload (33). The in vivo FDG PET studies and the metabolic analysis on the harvested TAC hearts indicate a possible role of glucose-mediated mTOR activation in the pressure overload mouse heart. To further elucidate the underlying mechanism, rates of glucose uptake and oxidation were simultaneously quantitated in ex vivo glucose-perfused isolated working rat hearts by Sen et al (19). At normal workload, rates of glucose uptake matched rates of glucose oxidation. However, when the workload was increased, rates of glucose exceeded rates of glucose oxidation, and G6P accumulated. In hearts from rats pretreated with rapamycin, the mismatch between glucose uptake and oxidation disappeared, as did the accumulation of G6P (Figures 3A-B) (12). Because mTOR activation was associated with a mismatch between glucose uptake and oxidation, we further investigated whether G6P is a signal for mTOR activation when the heart workload is increased. For this purpose, two groups of hearts were perfused with either one of two glucose analogues, 3-O-methylglucose or 2-deoxyglucose. 3-O-methylglucose is transported into the cell cytoplasm but not phosphorylated, whereas 2-deoxyglucose is phosphorylated to 2-deoxyglucose 6-phosphate, but not metabolized further (with the exception of cycling in and out of glycogen). Of the two analogues, only 2-deoxyglucose increased phosphorylation of mTOR and its downstream targets in hearts perfused at a high workload (Figure 3C). In vitro studies in cardiomyocytes suggest that hexokinase-II binds and inhibits mTORC1 during glucose deprivation. An increase in intracellular glucose concentration results in hexokinase-II mTORC1 unbinding and subsequent activation of mTORC1(34). Collectively, these experiments provide strong evidence that intracellular G6P levels are associated with mTOR activation in hearts subjected to increased workload. This mechanism of glucotoxic remodeling in pressure-overload induced cardiac dysfunction differs from previously described glucoprotective remodeling in the hibernating myocardium(35) and probably relates to enhanced glycogen metabolism in the latter(36-39).
Figure 3.

In response to a high workload, rates of glucose uptake exceed rates of glucose oxidation, glucose 6-phosphate accumulates, and mTOR is activated. (A) Rates of glucose uptake and oxidation in glucose-perfused rat hearts at high workload pretreated with vehicle (left) and rapamycin (right). Data shown are means ± SEM; n=5-6 per group, n.s.=Not significant. *p=0.07 with the Mann-Whitney rank sum test. (B) Glucose 6-phosphate (G6P) levels in freeze-clamped rat hearts pretreated with vehicle or rapamycin. (C) Representative Western blots of mTOR activation in rat hearts perfused with glucose or glucose analogues 2-deoxyglucose (2DG), or 3-O-methylglucose (3OMG) at normal and high work load. Modified and reproduced with permission from the Journal of American Heart Association (12).
3.2 G6P mediated mTOR activation and 5′AMP-activated protein kinase downregulation
The enzyme 5′AMP-activated protein kinase (AMPK) regulates substrate metabolism in the heart. Saha et al.(40) observed glucose-induced protein synthesis with downregulation of AMPK in the rat skeletal muscle. Similarly, we observed that rat hearts perfused with glucose at a high workload showed a decrease in phosphorylation of AMPK and its downstream targets acetyl-CoA carboxylase and tuberin. These changes were prevented by pretreatment with rapamycin (12) (Figure 4A). To determine the functional consequences of AMPK activation, hearts from rats pretreated with metformin in vivo for 7 days were also perfused with glucose at a high workload. Metformin pretreatment resulted in tight coupling of glucose uptake and oxidation, reduced G6P levels and increased cardiac power (Figure 4B-D).
Figure 4.

G6P-dependent mTOR activation is associated with downregulation of AMPK. (A) Representative Western blots in hearts perfused with glucose at normal and high workload demonstrate downregulation of AMPK and its downstream targets. (B) Contractile performance in the working heart perfused with glucose in the presence and absence of metformin. Data shown are means ± SEM; n=5-7 for each group. Metformin improves cardiac power at high workload. *p <0.05, #p=0.08, using the Mann-Whitney rank sum test. (C) Rates of glucose uptake and oxidation by hearts from rats receiving vehicle or metformin pretreatment for 7 days. ‡p=0.021 using the Mann-Whitney rank sum test; ns= not significant. (D) G6P levels in the same hearts (C) at normal and high workloads. Modified and reproduced with permission from the Journal of American Heart Association (12).
3.3 mTOR Activation and Endoplasmic Reticulum Stress
Sustained mTOR activation results in protein synthesis unmatched by the folding capacity of the endoplasmic reticulum (ER) and induces ER stress (41). Sen et al examined the possibility that sustained mTOR activation by G6P results in the induction of ER stress which may be responsible for the decline in cardiac power in hearts perfused at high workload. Protein levels of GRP78, an ER chaperone protein, increased in hearts perfused with glucose at high workload. The increase in GRP78, protein levels was not observed in hearts from animals pretreated with rapamycin or metformin (Figure 5A). Also, an ER “stress relieving” agent, the histone deacetylase inhibitor 4-phenylbutyrate (PBA), reduced the protein level of GRP78 (Figure 5A) and improved cardiac power (Figure 5B).
Figure 5.
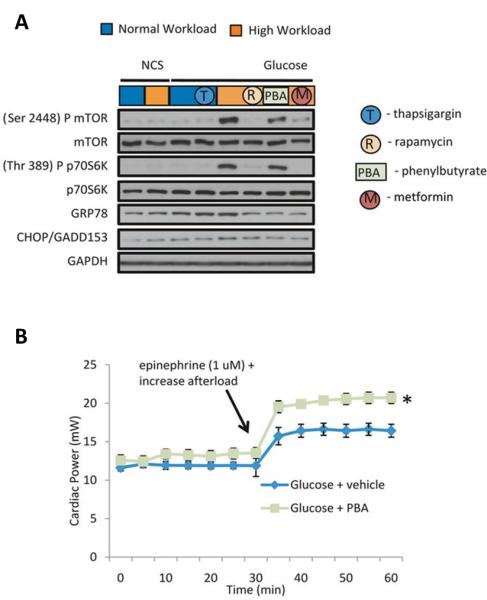
Relief of ER stress improves contractile function in hearts perfused with glucose at high workload. (A) Representative Western blots of mTOR, p70S6K, GRP78, and CHOP. Note, rapamycin, metformin and Phenylbutyrate (PBA) decrease levels of GRP78. However, PBA achieves this independently of changes in mTOR signaling. NCS: Noncarbohydrate substrate (B) Effect of the addition of PBA (10 mmol/L) to the perfusate on contractile performance of hearts perfused with glucose. Data shown are means ± SEM (n=5-8 for each group). PBA improved cardiac power at high workload. *p<0.004 using the Mann-Whitney rank sum test. Modified and reproduced with permission from the Journal of American Heart Association (12).
4. General considerations
Based on the literature discussed, we propose that dysregulation of glucose metabolism is a prominent feature of the maladapted failing heart. Furthermore, we propose that metabolic changes precede and trigger structural and functional remodeling of the pressure overloaded heart (Figure 6). The glucometabolic footprint of the stressed heart manifests as follows:
4.1. During acute pressure overload and inotropic stimulation (6), rates of glucose uptake and oxidation are increased. It is not yet completely clear how this relates to left ventricular function during pressure overload LVH. Studies using Single Photon Emission Computed Tomography imaging and PET have already demonstrated that the metabolic switch can be measured both in small animals and in human beings (42). However, serial studies on the temporal relationship between glucose uptake and cardiac structure and function in mice in vivo using PET have thus far never been done. As demonstrated earlier (21), a model corrected blood input function applied to an image-derived blood input function obtained from high-resolution gated images (43) with less severe spill-over and partial volume effects is quantitative and also repetitive as compared to that obtained from low resolution ungated images (44). Applying improved quantitative methods, we found that an increase in glucose metabolism in the form of increased FDG uptake precedes and possibly triggers the functional and structural remodeling of the heart in LVH in mice in vivo (12). We also observed that early treatment with propranolol attenuated myocardial glucose uptake and rescued cardiac function in vivo (11). The attenuation of increased glucose uptake and the improved contractile function early in the course of treatment with propranolol suggests that an early detection of metabolic remodeling may offer a therapeutic opportunity to prevent the onset of severe contractile dysfunction. Thus, metabolic imaging with FDG PET may guide treatment of patients with hypertension-induced LVH and improve clinical outcomes. This conclusion differs from FDG PET studies of patients with advanced cardiomyopathy(45;46), possibly for the following reasons: the subjects already had contractile dysfuntion, hence beyond the mere correlation, it was impossible to establish a timeline or causality as we have with our studies. Furthermore, Taylor et al. (46) based the expected FDG uptake on findings observed by other investigators. Therefore, the difference observed between healthy individuals and the study group may lie in the protocol used, since graphical Patlak analysis without considering the confounding effects of partial volume and spillover and variability in the lumped constant used for the fatty acid and glucose tracers may limit the accuracy of this method. Furthermore, most of the patients included in their study had ischemic cardiomyopathy; hence, measurements in areas of non-viable myocardium could falsely lower FDG uptake readings. Also, the patients were treated with angiotensin-converting enzyme inhibitors and digoxin, which may have further compromised myocardial metabolism.
4.2. Although it is known that mTOR signaling is responsible for cardiac growth in hypertrophy (23), the role of G6P-mediated mTOR activation in cardiac dysfunction is a recent observation (47). The findings in the working rat heart ex vivo demonstrate that acute increases in workload require the presence of glucose to activate mTOR, suggesting that ‘load-induced’ hypertrophic signaling through mTOR is substrate-specific. More specifically, mTOR is activated by the metabolic signal G6P, which may induce HK-II release from mTORC1.
4.3. G6P accumulation, most likely via sustained mTOR activation, induces ER stress, elicits the unfolded protein response (UPR) and impairs contractile function. Pretreatment with rapamycin, metformin or PBA relieve ER stress, thereby rescuing cardiac function in glucose-perfused rat hearts under a high workload ex vivo.
4.4. Pharmacological interventions that prevent G6P accumulation and mTORC1 activation result in restoration of cardiac power and decrease the progression of LVH. Propranolol treatment improves cardiac function in mice subjected to TAC, most likely by attenuating glucose uptake and preventing G6P accumulation. Treatment with metformin couples the rate of glucose uptake and oxidation and restores cardiac power in pressure overloaded hearts. It is tempting to speculate that metformin could be a novel attractive adjunct treatment in patients with hypertension-induced LVH for the prevention of contractile dysfunction. Evidence for the beneficial use of metformin to treat diabetic patients with cardiac failure has recently been reviewed elsewhere (48).
Figure 6.
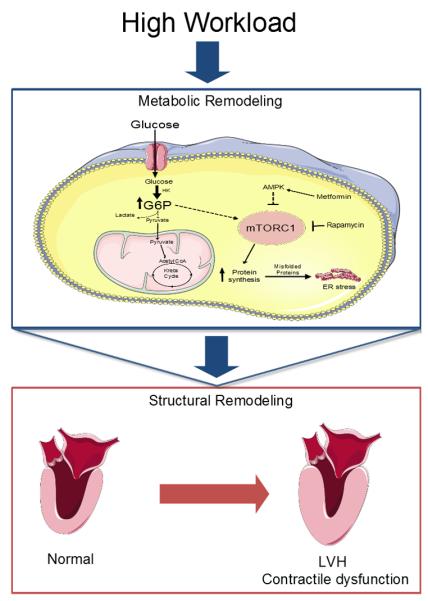
Proposed mechanism by which metabolic remodeling precedes and triggers structural and functional remodeling in the heart. Sustained pressure overload leads to increased glucose metabolism. However, a mismatch between glucose uptake and oxidation leads to accumulation of glucose 6-phosphate (G6P). This metabolite activates the mechanistic target of rapamycin complex 1 (mTORC1) by unknown mechanisms, resulting in increased protein synthesis and endoplasmic reticulum (ER) stress. As a consequence, the heart hypertrophies (increased protein synthesis) and fails (contractile dysfunction induced by ER Stress). Metformin and rapamycin decrease mTOR activity and may thus prevent LVH and HF. AMPK: 5′AMP-activated protein kinase. HK: Hexokinase-II. LVH: Left ventricular hypertrophy.
In conclusion, we have developed a new application of FDG PET as a tool to identify myocardial metabolic changes that precede irreversible structural changes of the left ventricle. The method will be of use to stratify the risk of hypertensive patients for developing heart failure.
Acknowledgements
We thank Mrs. Roxy A. Tate for expert editorial assistance and Rebecca Salazar B.S. for assistance with images. Figure 6 was produced using Servier Medical Art (http://www.servier.com).
Grant Support : US Public Health Service HL R21-102627 (to BKK) and HL R01-61483 (to HT)
Reference List
- 1.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katz AM. Cardiomyopathy of Overload - A Major Determinant of Prognosis in Congestive Heart-Failure. N Engl J Med. 1990;322:100–110. doi: 10.1056/NEJM199001113220206. [DOI] [PubMed] [Google Scholar]
- 3.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 4.Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem.J. 1980;186:701–711. doi: 10.1042/bj1860701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allard MF, Schonekess BO, Henning SL, et al. Contribution of Oxidative-Metabolism and Glycolysis to Atp Production in Hypertrophied Hearts. Am J Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 6.Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol.Chem. 1998;273:29530–29539. doi: 10.1074/jbc.273.45.29530. [DOI] [PubMed] [Google Scholar]
- 7.Young ME, Yan J, Razeghi P, et al. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul.Syst.Bio. 2007;1:251–262. doi: 10.4137/grsb.s222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taegtmeyer H, Golfman L, Sharma S, et al. Linking gene expression to function: Metabolic flexibility in the normal and diseased heart. Ann.N.Y.Acad Sci. 2004;1015:202–213. doi: 10.1196/annals.1302.017. [DOI] [PubMed] [Google Scholar]
- 9.de las Fuentes L, Soto PF, Cupps BP, et al. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J.Nucl.Cardiol. 2006;13:369–377. doi: 10.1016/j.nuclcard.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Doenst T, Pytel G, Schrepper A, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc.Res. 2010;86:461–470. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- 11.Zhong M, Alonso CE, Taegtmeyer H, et al. Quantitative PET Imaging Detects Early Metabolic Remodeling in a Mouse Model of Pressure-Overload Left Ventricular Hypertrophy In Vivo. J Nucl.Med. 2013;54:609–615. doi: 10.2967/jnumed.112.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen S, Kundu BK, Wu HC, et al. Glucose Regulation of Load-Induced mTOR Signaling and ER Stress in Mammalian Heart. J Am.Heart Assoc. 2013;2:e004796. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taegtmeyer H. Cardiac Metabolism as a Target for the Treatment of Heart Failure. Circulation. 2004;110:894–896. doi: 10.1161/01.CIR.0000139340.88769.D5. [DOI] [PubMed] [Google Scholar]
- 14.Lopaschuk GD. Optimizing cardiac fatty acid and glucose metabolism as an approach to treating heart failure. Semin.Cardiothorac.Vasc.Anesth. 2006;10:228–230. doi: 10.1177/1089253206291150. [DOI] [PubMed] [Google Scholar]
- 15.Kolwicz SC, Jr., Tian R. Metabolic therapy at the crossroad: how to optimize myocardial substrate utilization? Trends Cardiovasc.Med. 2009;19:201–207. doi: 10.1016/j.tcm.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gropler RJ, Beanlands RS, Dilsizian V, et al. Imaging myocardial metabolic remodeling. J Nucl.Med. 2010;51(Suppl 1):88S–101S. doi: 10.2967/jnumed.109.068197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taegtmeyer H. Tracing cardiac metabolism in vivo: one substrate at a time. J.Nucl.Med. 2010;51(Suppl 1):80S–87S. doi: 10.2967/jnumed.109.068205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osterholt M, Sen S, Dilsizian V, et al. Targeted metabolic imaging to improve the management of heart disease. JACC.Cardiovasc.Imaging. 2012;5:214–226. doi: 10.1016/j.jcmg.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gropler RJ. Recent advances in metabolic imaging. J Nucl.Cardiol. 2013;20:1147–1172. doi: 10.1007/s12350-013-9786-z. [DOI] [PubMed] [Google Scholar]
- 20.Gallamini A, Zwarthoed C, Borra A. Positron Emission Tomography (PET) in Oncology. Cancers.(Basel) 2014;6:1821–1889. doi: 10.3390/cancers6041821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong M, Kundu BK. Optimization of a Model Corrected Blood Input Function From Dynamic FDG-PET Images of Small Animal Heart In Vivo. IEEE Trans Nucl Sci. 2013;60:3417–3422. doi: 10.1109/TNS.2013.2269032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma S, Guthrie PH, Chan SS, et al. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc.Res. 2007;76:71–80. doi: 10.1016/j.cardiores.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proud CG. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc.Res. 2004;63:403–413. doi: 10.1016/j.cardiores.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Neubauer S, Krahe T, Schindler R, et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation. 1992;86:1810–1818. doi: 10.1161/01.cir.86.6.1810. [DOI] [PubMed] [Google Scholar]
- 25.Neubauer S, Horn M, Pabst T, et al. Contributions of 31P-magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur.Heart J. 1995;16(Suppl O):115–118. doi: 10.1093/eurheartj/16.suppl_o.115. [DOI] [PubMed] [Google Scholar]
- 26.Beanlands RS, Nahmias C, Gordon E, et al. The effects of beta(1)-blockade on oxidative metabolism and the metabolic cost of ventricular work in patients with left ventricular dysfunction: A double-blind, placebo-controlled, positron-emission tomography study. Circulation. 2000;102:2070–2075. doi: 10.1161/01.cir.102.17.2070. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa S, Kusuoka H, Maruyama K, et al. Myocardial positron emission computed tomographic images obtained with fluorine-18 fluoro-2-deoxyglucose predict the response of idiopathic dilated cardiomyopathy patients to beta-blockers. J Am Coll.Cardiol. 2004;43:224–233. doi: 10.1016/j.jacc.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Brooks WW, Shen SS, Conrad CH, et al. Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: Structure, function, and transcript analysis. Genomics. 2010;95:84–92. doi: 10.1016/j.ygeno.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 29.Bing OH, Brooks WW, Robinson KG, et al. The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. J Mol.Cell Cardiol. 1995;27:383–396. doi: 10.1016/s0022-2828(08)80035-1. [DOI] [PubMed] [Google Scholar]
- 30.Brooks WW, Conrad CH, Robinson KG, et al. L-arginine fails to prevent ventricular remodeling and heart failure in the spontaneously hypertensive rat. Am.J Hypertens. 2009;22:228–234. doi: 10.1038/ajh.2008.334. [DOI] [PubMed] [Google Scholar]
- 31.Kato T, Niizuma S, Inuzuka Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ.Heart Fail. 2010;3:420–430. doi: 10.1161/CIRCHEARTFAILURE.109.888479. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez AM, Huber JS, Murphy ST, et al. Longitudinal evaluation of left ventricular substrate metabolism, perfusion, and dysfunction in the spontaneously hypertensive rat model of hypertrophy using small-animal PET/CT imaging. J Nucl.Med. 2013;54:1938–1945. doi: 10.2967/jnumed.113.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen VT, Mossberg KA, Tewson TJ, et al. Temporal analysis of myocardial glucose metabolism by 2-[18F]fluoro-2-deoxy-D-glucose. Am.J.Physiol. 1990;259:H1022–H1031. doi: 10.1152/ajpheart.1990.259.4.H1022. [DOI] [PubMed] [Google Scholar]
- 34.Roberts DJ, Tan-Sah VP, Ding EY, et al. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol.Cell. 2014;53:521–533. doi: 10.1016/j.molcel.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunken R, Schwaiger M, Grover-McKay M, et al. Positron emission tomography detects tissue metabolic activity in myocardial segments with persistent thallium perfusion defects. J Am.Coll.Cardiol. 1987;10:557–567. doi: 10.1016/s0735-1097(87)80198-5. [DOI] [PubMed] [Google Scholar]
- 36.Depre C, Vanoverschelde JL, Melin JA, et al. Structural and metabolic correlates of the reversibility of chronic left ventricular ischemic dysfunction in humans. Am.J Physiol. 1995;268:H1265–H1275. doi: 10.1152/ajpheart.1995.268.3.H1265. [DOI] [PubMed] [Google Scholar]
- 37.Bolukoglu H, Goodwin GW, Guthrie PH, et al. Metabolic fate of glucose in reversible low-flow ischemia of the isolated working rat heart. Am.J Physiol. 1996;270:H817–H826. doi: 10.1152/ajpheart.1996.270.3.H817. [DOI] [PubMed] [Google Scholar]
- 38.Goodwin GW, Arteaga JR, Taegtmeyer H. Glycogen turnover in the isolated working rat heart. J Biol.Chem. 1995;270:9234–9240. doi: 10.1074/jbc.270.16.9234. [DOI] [PubMed] [Google Scholar]
- 39.Russell RR, III, Cline GW, Guthrie PH, et al. Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J Clin.Invest. 1997;100:2892–2899. doi: 10.1172/JCI119838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saha AK, Xu XJ, Lawson E, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59:2426–2434. doi: 10.2337/db09-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glembotski CC. The role of the unfolded protein response in the heart. J Mol.Cell Cardiol. 2008;44:453–459. doi: 10.1016/j.yjmcc.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mochizuki T, Tsukamoto E, Ono T, et al. Sequential change of BMIPP uptake with age in spontaneously hypertensive rat model. Ann.Nucl Med. 1997;11:299–306. doi: 10.1007/BF03165297. [DOI] [PubMed] [Google Scholar]
- 43.Locke LW, Berr SS, Kundu BK. Image-Derived Input Function from Cardiac Gated Maximum a Posteriori Reconstructed PET Images in Mice. Mol.Imaging Biol. 2011;13:342–347. doi: 10.1007/s11307-010-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang YH, Muzic RF., Jr. Spillover and Partial-Volume Correction for Image-Derived Input Functions for Small-Animal 18F-FDG PET Studies. J.Nucl.Med. 2008;49:606–614. doi: 10.2967/jnumed.107.047613. [DOI] [PubMed] [Google Scholar]
- 45.Wallhaus TR, Taylor M, Degrado TR, et al. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441. [DOI] [PubMed] [Google Scholar]
- 46.Taylor M, Wallhaus TR, Degrado TR, et al. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in Patients with Congestive Heart Failure. J Nucl.Med. 2001;42:55–62. [PubMed] [Google Scholar]
- 47.Ellis JM, Mentock SM, Depetrillo MA, et al. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol.Cell Biol. 2011;31:1252–1262. doi: 10.1128/MCB.01085-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ekeruo IA, Solhpour A, Taegtmeyer H. Metformin in Diabetic Patients with Heart Failure: Safe and Effective? Curr.Cardiovasc.Risk Rep. 2013;7:417–422. doi: 10.1007/s12170-013-0355-4. [DOI] [PMC free article] [PubMed] [Google Scholar]