

Abstract
FLT3 is a receptor-tyrosine-kinase that is expressed on leukemic cells of the myeloid and lymphoid lineage rather specifically. We here report on the construction and selection of bispecific FLT3 X CD3 antibodies in a new recombinant format, termed Fabsc, that resembles the normal antibody structure more closely than the well-established bispecific single chain (bssc)-format. Our preferred antibody, which emerged from an initial selection procedure utilizing different FLT3- and CD3-antibodies, contains the FLT3-antibody 4G8 and the CD3-antibody UCHT1. The 4G8 X UCHT1 Fabsc-antibody was found to be superior to a bssc-antibody with identical specificities with respect to (i) affinity to the target antigen FLT3, (ii) production yield by transfected cells, and (iii) the diminished formation of aggregates. T-cell activation in the presence and absence of cultured leukemic cells and killing of these cells was comparable for both molecules. In addition, the 4G8 X UCHT1 Fabsc-antibody was found to induce T-cell activation and efficient killing of leukemic blasts in primary peripheral blood mononuclear cell (PBMC) cultures of acute myeloid leukemia (AML) patients. In these experiments, the bispecific molecule was clearly superior to an Fc-optimized monospecific FLT3-antibody described previously, indicating that within PBMC of AML patients the recruitment of T cells is more effective than that of natural killer cells.
Introduction
One of the strategies used to improve on the therapeutic activity of monoclonal antitumor antibodies aims at enhancing the affinity of antibody Fc-parts by genetic engineering, e.g., by introducing defined amino acid modifications, such as the S239D and I332E exchanges (SDIE modification).1,2 This modification was shown to markedly enhance antibody-mediated cellular cytotoxicity (ADCC) by FcR-carrying immune effector cells. Several SDIE-modified antibodies, directed to the lymphoma associated antigens CD19 (ref. 3), CD30 (ref. 4), and CD40 (ref. 5) have been developed and are currently evaluated in early clinical trials. We have introduced the SDIE-modification into an antibody directed to FLT3, a receptor tyrosine kinase preferentially expressed on leukemic cells of the myeloid lineage. This antibody, termed 4G8SDIEM, exerted markedly improved ADCC activity against myeloid leukemia cells6 and its clinical evaluation at our institution is ongoing.
Conceptually, the recruitment of T cells appears to be more attractive than that of FcR-carrying effector cells. T cells are known to exert potent antitumor effects in numerous experimental settings and their proliferative capacity in vivo is superior. Curiously, antibodies capable of effectively recruiting T cells against tumor cells are available for quite some time: in the mid-eighties, it was demonstrated that bispecific antibodies directed to tumor-associated antigens (TAAs) and agonistic T-cell receptors, such as the TCR/CD3 complex and CD28, are capable of activating T cells, irrespective of their TCR specificity, resulting in specific lysis of cells carrying the respective TAAs.7,8,9 However, the clinical development of these attractive reagents has been severely hampered over the years by formidable scientific as well as technical hurdles: unspecific binding of bispecific TAA X CD3 antibodies either to a critical number of normal cells expressing the respective TAA or to Fc receptors may lead to excessive CD3 crosslinking and off-target T-cell activation. Because of the resulting cytokine release syndrome local rather than systemic application was preferred in most of the early10,11 and also in a few recent studies.12,13
In the early days of bispecific antibody development, we proposed the model of a tumor cell restricted T-cell activation using Fc-attenuated or fragmented antibodies.14,15 However, successful clinical development of such reagents, that is production in pharmaceutical quality and quantity had to await advanced genetic engineering techniques developed during the nineties. In 1995, Mack et al.16 reported a bispecific single chain (bssc)-antibody with EpCAM X CD3-specificity which could be easily produced in sufficient amounts after transfection of the respective gene construct in eukaryotic cells. This bssc-format, later designated bispecific T-cell engager (BiTE), has been used to construct, among others, a bispecific antibody with CD19 X CD3-specificity, termed Blinatumomab. Recently, it was demonstrated that this antibody exerts remarkable therapeutic activity in patients suffering from CD19-positive lymphoma or leukemia.17,18 However, as in earlier studies with different anti-CD3 containing bispecific antibodies,19,20 excessive T-cell activation and cytokine release induced upon in vivo application limits the safely applicable doses to less than 100 µg/day resulting in serum concentrations below 1 ng/ml.21 As far as Blinatumomab is concerned, we hypothesize that part of its toxicity is due to its binding to normal CD19 expressing B cells. In this respect, FLT3 appears to be a more favourable antigen because its expression on normal hematopoietic cells is more restricted.
Regardless of antibody specificity, the bssc-format as such, although being exceedingly efficient in mediating T-cell activation, suffers from weaknesses of the single chain format, such as its low serum half-life, a potential loss of affinity and the general tendency to form aggregates.22
We here report on the development of a new bispecific format, termed Fabsc, more closely resembling the physiological structure of an antibody molecule. It consists of a Fab part, a linker CH2 domain, containing modifications to attenuate FcR-binding, and a single chain antibody with a second specificity. At variance with the Fab containing format described by Schoonjans et al.,23 the Fabsc-format described here contains a CH2-domain separating the two specificities in order to reduce undesirable interactions between them.
Two different antibodies, 4G8 and BV10, originally developed at our institution24 and directed to different epitopes of FLT3 were used to construct Fabsc-antibodies with FLT3 X CD3-specificity. The activity of these molecules was evaluated in various experimental settings and compared to that of an analogous molecule in the bssc-format. Finally, we used peripheral blood mononuclear cells (PBMC) of leukemia patients containing “physiologic” amounts of blasts to evaluate the activity of our preferred bispecific antibody under realistic conditions and to compare it to the Fc-optimized antibody developed previously.6
Results
Construction and selection of an optimized bispecific FLT3 X CD3-antibody in the Fabsc-format
The first Fabsc-constructs produced, contained the FLT3 antibody 4G8 as an N-terminal targeting antibody and three different C-terminal effector antibodies directed to the TCR/CD3-complex: UCHT1, OKT3, and BMA031. We noticed that the antibody containing UCHT1 was superior with respect to lower aggregation tendency and preservation of binding affinity towards CD3-expressing Jurkat cells (data not shown). Thus, we used this antibody for construction of an additional Fabsc-antibody containing the FLT3 antibody BV10 (Figure 1). BV10 is directed to domain 2 of the FLT3 molecule whereas 4G8 binds to domain 4, more proximal to the cell membrane.6 Figure 2a shows that binding of both Fabsc to CD3 expressing Jurkat cells was comparable. However, binding to REH cells of the 4G8 containing bispecific antibody was clearly superior to that comprising BV10. In addition, the former antibody not only induces increased T-cell activation and cytokine release in the presence of REH cells (Figure 2d,f), it also mediates superior cytotoxicity of preactivated cytoloytic T cells against the target cells (Figure 2c). We therefore selected the 4G8 X UCHT1 Fabsc-molecule for direct comparison with an analogous molecule in the single chain format (Figure 1), constructed and produced as described in the Materials and Methods section.
Figure 1.

Schematic representation of the two bispecific antibody formats used in this paper. Antibody clones and specificities are indicated. Four antibodies (4G8 X UCHT1, 4G8 X OKT3, 4G8 X BMA031, BV10 X UCHT1) were constructed in the Fabsc-format (a), one (4G8 X UCHT1) in the bssc-format (b). In a, X and O indicate replacement of cysteins in the hinge region and of several amino acid residues in the CH2 domain (see Materials and Methods section) to prevent dimerization and binding to Fc receptors, respectively. In b, L1 stands for the glycine-serine linker (G4S)3 and L2 for a sequence derived from the elbow fragment of human Igγ1.
Figure 2.
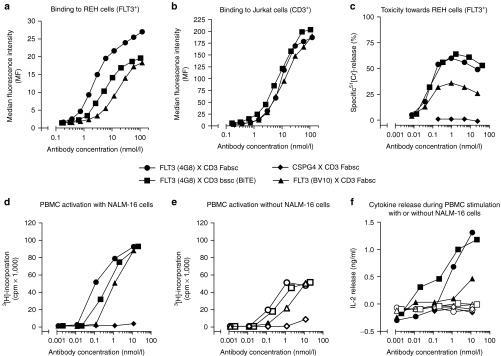
Functional characterizations of three different bispecific antibodies with FLT3 X CD3-specificity. Symbols (•, ▪, ▴) were assigned as indicated. In c–f, a Fabsc-antibody with CSPG4 X CD3 specificity (♦) was used as a negative control. In d–f, empty and filled symbols refer to the absence and presence, respectively of FLT3+ NALM-16 target cells. The binding experiment depicted in a and b was repeated once with almost identical results. In c–f, representative results from one out of three experiments with peripheral blood mononuclear cells from different healthy donors are shown.
Comparison of the bispecific Fabsc- and bssc (BiTE)-format
First, we noted that production of the antibody in the Fabsc-format by transfected Sp2/0 cells (2–3 µg/ml) was better than that in the bssc-format (~0.5 µg/ml). This could be due to mere chance during selection of clones. However, the numbers of screened clones were comparable, and in two other cases of bssc-antibodies, 9.2.27 X CD28 (ref. 25) and 2H7 X CD28 (ref. 26), we were not able to obtain clones producing more than 1 µg/ml, whereas concentrations measured in the supernatants of transfected Sp2/0 cells for 16 additional antibodies in the Fabsc-format ranged from 2 to 11 µg/ml (data not shown). This is somewhat less than the production rates obtained, when monospecific, Fc-optimized antibodies, e.g., directed to FLT3, CD19, or prostate-specific membrane antigen (PSMA) were produced in Sp2/0 cells (~5–15 µg/ml, data not shown). If compared to bssc-molecules, however, it appears that production yields are generally higher for antibodies expressed in the Fabsc-format.
Figure 2 shows that, whereas binding to CD3-expressing Jurkat cells is comparable for all antibodies tested, the affinity of the bssc-antibody towards FLT3 is moderately reduced, if compared to the Fabsc-antibody with identical specificities (Figure 2a,b). This is probably due to the fact that the N-terminal FLT3 antibody in this molecule is expressed in the more physiologic Fab- rather than the single chain format. Other functional parameters of the two bispecific molecules, that is T-cell activation in the absence and presence of target cells (Figure 2d,e) and T-cell–mediated killing of target cells (Figure 2c) were comparable. That off-target activation of PBMC in the absence of leukemic target cells is caused by specific binding of the antibodies to monocytes expressing trace amounts of FLT3, is supported by the following findings: (i) detection of small amounts of FLT3 (300–600 molecules per cell) on normal CD14+ monocytes within bone marrow cultures (Supplementary Figure S1), specific blockade of PBMC activation after depletion of monocytes with magnetic beads carrying CD14 and CD33 (Supplementary Figure S2) or by an excess of parental FLT3 antibody (data not shown). In addition, no proliferation was induced by the 9.2.27 X UCHT1 Fabsc-antibody, that binds to an unrelated chondroitin sulfate proteoglycan (CSPG4) preferentially expressed on melanoma cells (Figure 2e). The latter finding confirms that Fc attenuation within the CH2 domain has been successful. Otherwise binding of the construct to FcR-expressing cells in the PBMC preparations would have resulted in CD3-mediated T-cell activation. A significant difference between the antibodies was again noted, if the release of the index cytokine IL-2 during the activation of PBMC cultures was measured. Cytokine release could be detected only in the presence of FLT3-expressing leukemia target cells and was more pronounced in the case of the bssc-antibody with FLT3 X CD3-specificity (Figure 2f).
Differences were also observed, when we analyzed the two proteins by size exclusion chromatography. The amount of both, dimers as well as aggregates, although detectable, were reproducibly low in the case of the Fabsc-molecule (three batches) and >90% of the material was migrating in a peak corresponding to the expected molecular weight of 87 kDa (Figure 3a). In contrast, four different batches of the bssc-material yielded ~70, 35, 50, and 70%, respectively, of multimers and aggregates (Figure 3b). In the case of the bssc-molecule, we therefore used monomeric material, purified by size exclusion chromatography, in all experiments comparing the activity of the two proteins.
Figure 3.

Size exclusion chromatography of bispecific antibodies in two different formats. Two FLT3 X CD3-antibodies with identical specificities (4G8 X UCHT1) in the Fabsc- (a) and bssc-format (b) were analyzed on a Superdex 200 PC3.2/30 column. Approximately 70% of the bssc-material consists of dimers and higher molecular weight aggregates. This percentage was found to be approximately 35, 50, and 70% respectively, in three additional, independently produced batches.
Figure 4 shows that the serum elimination of both antibodies does obviously not follow a first order kinetic. Elimination of the bssc-molecule within the first 2 hours is ~50% faster than that of the Fabsc-molecule. Half lifes between hours 1 and 2 were 40 and 60 minutes, respectively. At later time points, that is up to 20 hours, when the detection limit of 1 ng/ml was reached, no significant difference between the two molecules was observed and their half life increased to ~3 hours.
Figure 4.

Serum clearance of the Fabsc- and the bssc-antibody in mice. C57BL/6 mice were injected i.v. with 25 µg of 4G8 X UCHT1 antibodies in the bssc-(O)- and Fabsc-format (•). Serum concentrations of the antibodies were determined using an ADCC reporter bioassy as described in the Material and Methods section. Values obtained by bleeding three mice per time point are indicated. Serum concentrations at the 20 hours time point (2–3 ng/ml) were significantly above background, but below that of the standard sample with the lowest concentration. Those values were calculated by extrapolating the calibration curve.
Further characterization of the 4G8 X UCHT1 Fabsc-antibody
Having established that the bispecific 4G8 X UCHT1 antibody in the new Fabsc-format is a stable, monomeric molecule capable of inducing target cell restricted T-cell activation and tumor cell killing in well-established in vitro assays, we next wanted to evaluate the therapeutic activity as well as the potential toxicity of the new molecule under conditions as realistic as possible. In addition, we were interested to compare its activity to that of a monospecific Fc-optimized FLT3 antibody developed previously. Currently, this antibody is clinically used at our institution on a compassionate need basis and a phase 1/2 study is in preparation.
To mimic conditions of clinical application as closely as possible, we incubated PBMC of 10 AML patients with antibodies and determined the percentage of remaining blasts and of activated T cells after 3 days by flow cytometry. The expression of FLT3 on the leukemic cells was low to moderate ranging from 400 to 3,300 molecules per cell. In one sample (#6), it was below the detection threshold. The ratio of CD4+ and CD8+ cells to leukemic blasts was far below 1 in most of the samples (columns B and C in Table 1). After 3 days of incubation with the FLT3 X CD3 Fabsc antibody, T cells were activated (as determined by the expression of CD69 or CD25), and had proliferated in all samples (except that of patient #6 without detectable FLT3 expression). Leukemic cells were markedly reduced in all FLT3 expressing samples with the notable exception of patient#5. In this sample, an unexpected increase in blast counts was noted despite pronounced T-cell activation and proliferation. In Figure 5, representative results for one of the eight responding patients (#8) are shown. Maximal activity with respect to both, T-cell activation and blast reduction requires an antibody concentration of ~0.1 µg/ml and the activity of the Fabsc- and the bssc (BiTE)-molecule was almost identical. In two additional samples (#9 and #10), the activities of the two molecules were compared and were found to be very similar. Notably, the monospecific 4G8SDIEM antibody was effective in only 1 of the 10 samples (#7, column G in Table 1). This indicates that natural killer (NK)-cell–mediated killing is generally less effectivewithin PBMC preparations of AML patients.
Table 1. Killing of leucemic blasts within PBMC of AML patients.
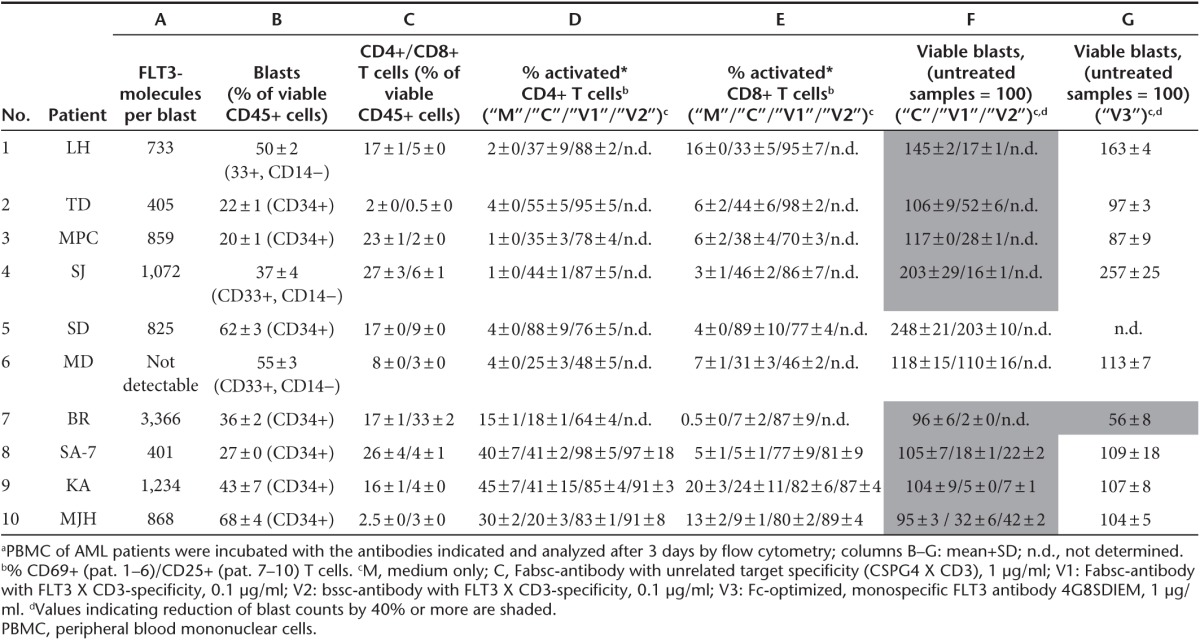
Figure 5.

T-cell activation and reduction of leukemic blasts in primary peripheral blood mononuclear cells (PBMC) cultures of a patient with acute myeloid leukemia. PBMC of patient #8 were incubated for 3 days with the indicated concentrations of the 4G8 X UCHT1 Fabsc- (○, •) or bssc (□, ▪) antibody and reduction of blasts (a) and T-cell activation (b) were determined as described in the Methods section. Empty and filled symbols in b refer to CD4+ and CD8+ T cells, respectively. A bispecific Fabsc-antibody targeting the melanoma associated CSPG4-antigen (9.2.27 X UCHT1) was used as a negative control at 1 µg/ml (bars on the right). Corresponding data for additional patient samples are summarized in Table 1.
Given the well-established expression of FLT3 on normal hematopoietic precursor cells, we finally wanted to address the issue of stem cell toxicity exerted by the 4G8 X UCHT1 Fabsc-antibody. Figure 6a shows that no significant reduction of colony forming units was observed after incubation of bone marrow cells of three different healthy donors with up to 0.1 µg/ml of the antibody. In two of the donors, however, a moderate reduction was observed at a concentration of 1 µg/ml. In contrast, if the antibody was incubated with bone marrow cells of three normal donors for 24 hours, significant activation of CD4+ and CD8+ T cells (Figure 6b) and moderate reduction of CD34+ precursor cells (Figure 6c) was observed at an antibody concentration of 0.1 µg/ml. When we extended the duration of these experiments to 3 days in the presence of 10% human serum, again a moderate reduction of CD34+ precursor cells by 10–20% and of CD14+ monocytes by 20–40% was noted (data not shown).
Figure 6.
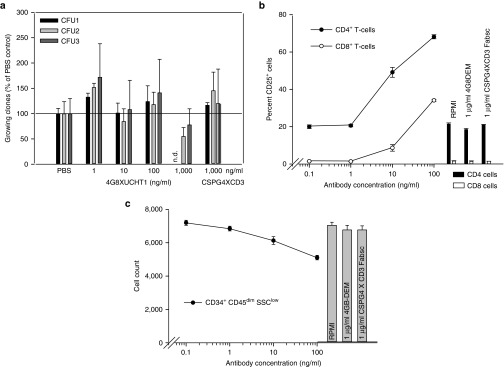
Effects of the 4G8 X UCHT1 Fabsc antibody on normal bone marrow cells. Normal bone marrow cells were incubated with bispecific antibodies for 24 hours. Colony forming unit formation (a), T-cell activation (b), and the number of CD34+ hematopoetic precursor cells (c) were determined as described in the Materials and Methods section. In a, results with bone marrow cells of three donors are depicted. The results shown in b and c are representative for experiments with cells from three normal donors.
Discussion
Recruitment of T cells against tumor cells with bispecific antibodies directed to TAAs and activating T-cell receptors is a strategy, which has been developed almost three decades ago. Given the recent clinical success of Blinatumomab, a bssc-antibody with CD19 X CD3-specificity, this attractive concept might therefore be considered a late bloomer in experimental tumor immunotherapy. This is, because demanding design and production procedures for bispecific antibodies add to the already cumbersome translational process for therapeutic antibodies: the choice of the targeting antibody, for example, is particularly critical for bispecific reagents, since binding to normal cells expressing the target antigen may contribute to “off- target” T-cell activation and dose-limiting cytokine release. The safely applicable daily doses in earlier clinical studies with different bispecific antibodies19,20 as well as in the more recent Blinatumomab trials17,18 were in the order of 50 µg per day. This is several orders of magnitude lower than the doses applied in studies with conventional monospecific antitumor antibodies and certainly not sufficient to achieve maximal biologic activity in vivo. In the case of Blinatumomab, off-target cytokine release is most likely due to the expression of the target antigen CD19 on normal B cells present in peripheral blood, bone marrow, and lymphatic tissue. The recently described bssc-antibodies with CD33 X CD3-specificity27,28 might face a similar problem, since CD33 is known to be expressed on most normal cells of the myeloid lineage. Compared to CD19, the expression of FLT3 on normal blood cells is rather restricted but not completely absent: some off-target T-cell activation was observed with all bispecific reagents targeting FLT3, due to the expression of small amounts of this antigen on normal monocytes.
Besides the choice of the antigen, robust production of bispecific antibodies has been a major issue and the debate, which of the numerous recombinant formats is best suited, is ongoing. While single-chain antibodies are an essential component in almost all recombinant bispecific formats suggested so far, it is clear that they come with several drawbacks: (i) molecular weight, (ii) affinity, and (iii) production rates are reduced and thus compare unfavorably with conventional antibodies. In addition, a marked tendency to form multimers and/or aggregates may further aggravate production problems.22 With the bispecific Fabsc-format introduced by this paper, we intended to address these disadvantages. In fact, comparison with a corresponding bispecific single chain antibody with identical FLT3 X CD3 specificities, confirmed that the Fabsc format was superior with respect to better production rates and markedly lower aggregation tendency. However, we observed only minor differences in serum elimination: for both molecules serum half life in normal mice increased from ~40–60 minutes in the first 2 hours to ~3 hours after 1 day. This is more than 20 times lower than the serum half life of intact, monospecific antibodies and most likely due to the, desired, lack of Fc-binding domains in both molecules.
As expected, binding of the FLT3 X CD3 bssc-molecule is moderately impaired if compared to its Fabsc counterpart. This is consistent with the notion that the single-chain format is more vulnerable to loss of affinity than the more physiologic Fab-format. In fact, with single-chain antibodies directed to CD3, CD16, and CD95, we have observed a loss of affinity that was much more pronounced than that described here for the FLT3 antibody 4G8 (data not shown).
Apart from binding affinity toward FLT3, other activities evaluated, that is the capability to mediate target cell restricted T-cell activation and killing of FLT3+ target cells by activated CD8+ T cells, were comparable for the Fabsc- and bssc-format. What was more pronounced in the case of the bssc-antibody, albeit to a moderate extent, was the secretion of the index cytokine IL-2. It is open to doubt, however, whether this is really advantageous, since cytokine release is the single most important dose-limiting parameter for T-cell activating bispecific antibodies. In any case, it was not self-evident, that the bispecific FLT3 X CD3 antibodies tested here are capable of mediating effective target cell dependent T-cell activation and killing of leukemia cells, since FLT3 expression on such cells is rather low and varies from 500–4,000 molecules per cell, whereas the expression of, e.g., CD19 on leukemic cell lines and blasts is usually several fold higher.29
For the evaluation of the antileukemic activity of our preferred FLT3 X CD3 Fabsc antibody, we paid special attention to “treat” AML cells with antibodies under realistic conditions, that is, by using PBMC preparations of AML patients that contain leukemic blasts and autologous T cells at “physiological” ratios. When we incubated those PBMC for 3 days with the bispecific FLT3 X CD3 Fabsc antibody, we detected marked T-cell activation, T-cell proliferation, and reduction of the number of leukemic cells in all but one of nine FLT3 expressing samples. In a recent paper of Aigner et al.,27 the authors report a marked reduction of AML blasts in a similar setting using a bssc-antibody with CD33 X CD3-specificity. However, killing of the blasts in 16 out of 20 of their samples required an incubation time of 5 days and was only marginal at day 3 possibly due to the low antibody concentration used (1 ng/ml). In our experiments, at least 10 ng/ml were required to achieve significant reduction of leukemic blasts at day 3.
When we used the previously described monospecific, Fc-optimized FLT3 antibody rather than the bispecific reagent, reduction of blasts was observed in only one of the nine FLT3+ samples (#7). Since this was the one with the highest FLT3 expression, it is tentative to speculate that the level of the target antigen might be more critical for mono- as compared to bispecific antibodies. In any case, it is obvious that under the stringent conditions used here, that is effector:target ratios <<1 and low expression level of the target antigen, T-cell–mediated killing is more effective than that exerted by NK cells, even if an Fc-optimized antibody with a strongly enhanced capability to mediate ADCC is used. Whether this is due to differences in the number or the activity of the respective effector cell populations remains an open question.
The specificity of FLT3 for malignant cells compares favorably to that of CD19 or CD33, since the latter two antigens are abundantly expressed on normal B cells and myeloid cells, respectively. On the other hand, FLT3 is expressed at low levels on normal monocytes and hematopoietic precursor cells. Whereas a transient reduction of monocytes appears to be tolerable, significant depletion of hematopoietic precursor cells is a matter of concern. In fact, incubation of the FLT3 X CD3 Fabsc-antibody with normal bone marrow cells resulted in a reduction of CD34-positive precursor cells after 24 hours of incubation. Like most drugs used within conventional antileukemic treatment schemes, the bispecific antibody described here might therefore induce some stem cell toxicity. However, since FLT3 expression on normal hematopoietic cell is significantly lower than that detected on most leukemic cells,6 we expect this toxicity, if it occurs, to be tolerable and outweighed by therapeutic efficiency.
Materials and Methods
Production and purification of recombinant antibodies. The variable domains of the antibodies 9.2.27 (anti-chondroitin sulphate proteoglycan, CSPG4), 4G8 (anti-FLT3), BV10 (anti-FLT3), OKT3 (anti-CD3), and UCHT1 (anti-CD3) were cloned from the respective hybridoma cells as previously described.6,25 ScFv fragments derived from the variable domains of the TCR-antibody BMA031 (VH-VL)30 and of a humanized version of UCHT1 (VL-VH) were synthesized de novo at Geneart (Regensburg, Germany).
At the C-terminus of the Fab fragment of antibodies 9.2.27, BV10, and 4G8, a modified CH2 domain of human Igγ1 and the respective scFv-fragments of OKT3, BMA031, and UCHT1 were added resulting in a format, termed Fabsc. Modification of the CH2 domain of human Igγ1 consists of the amino acid substitutions and deletions C226S; C229S; E233P; L234V; L235A; ΔG236; D265G; N297Q; A327Q; A330S (EU-index) which abrogate FcR binding and complement fixation and prevent glycosylation as well as disulfide mediated homotypic dimerization.
Bispecific Fabsc- and bssc-antibodies were purified from culture supernatants of transfected Sp2/0 cells by affinity chromatography on KappaSelect- (GE Healthcare, Munich, Germany) and protein L-columns (Thermo Fisher Scientific, Rockford, IL), respectively, and elution at pH 2.3. The antibodies were subjected to analytical and preparative size exclusion chromatography on Superdex 200 (GE Healthcare) using PC3.2/30 and HiLoad 16/60 columns, respectively.
Cells and media. PBMCs, isolated by density gradient centrifugation (LSM 1077, Lonza, Basel, Switzerland), hybridoma cells, NALM-16 cells (ACC 680, DSMZ, Braunschweig, Germany), and REH cells (ATCC, Manassas, VA) were kept in RPMI 1640, mouse Sp2/0-Ag14 cells (ATCC) in Iscove's modified Dulbecco's medium (IMDM; Lonza). All media were supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 1 mmol/l sodium-pyruvate, nonessential amino-acids, 2 mmol/l L-glutamine, and 50 µmol/l β-mercaptoethanol.
Antibodies and flow cytometry. CD33-APC (clone WM53), CD34-APC/Cy7 (clone 581), CD4-PaBlue (clone OKT4), CD8a-FITC (clone HIT8a), CD25-PE (clone BC96), CD14-PE/Cy7 (clone HCD14), CD69-PE (clone FN50), and corresponding isotype control antibodies were purchased from BioLegend (San Diego, CA), CD45-AmCyan (clone 2D1) from BD Biosciences (Heidelberg, Germany). All antibodies were incubated with cells for 30 minutes at 4 °C unless otherwise stated. For indirect immunofluorescence, PE- or APC-conjugated goat-anti-mouse F(ab)2-fragments and goat-anti-human F(ab)2-fragments were used (Jackson ImmunoResearch, West Grove, PA). Cells were analyzed on a FACSCanto II or a FACSCalibur (BD Biosciences). Beads for the quantitative analysis of indirect immunofluorescence (QIFIKIT) were purchased from Dako (Hamburg, Germany) and used according to the protocol of the manufacturer.
To analyze binding affinity, different FLT3 X CD3 antibodies were incubated with REH cells (FLT3+) and Jurkat cells (CD3+) in 96-well plates (1.5 × 105/well) for 60 minutes at 4 °C and washed. The cells were then stained with a protein L-Biotin-conjugate (Thermo Scientific, Waltham, MA) followed by incubation with a streptavidin-APC-complex (Life Technologies, Paisley, UK).
To determine T-cell–mediated killing of AML blasts, PBMC of AML-patients were incubated in 96-well plates (5 × 105 cells/well) with different concentrations of bispecific 4G8 X UCHT1 antibodies or controls in triplicates. After incubation for 72 hours, the cells were stained with antibodies and finally resuspended in FACS-buffer containing 7-AAD (BioLegend) and negative control compensation particles (BD Biosciences). Malignant cells were defined as CD34+CD45dim, or CD33+CD45dimCD14-. T cells as CD45+CD4+ or CD45+CD8+ and activated T cells within these populations as CD25+ or in some experiments as CD69+. By the acquisition of equal numbers of compensation particles in each sample, absolute cell counts could be determined and used to calculate the amount of target cell lysis.
For assessment of stem cell toxicity, bone marrow cells of normal donors were purified by density gradient centrifugation, incubated with different concentrations of bispecific antibodies in triplicates for 24 hours in medium containing 10% FCS and processed and analyzed as described above. In some experiments, the incubation period was extended to 3 days and FCS was replaced by human AB-serum. Stem cells and progenitor cells were defined as CD45dimCD34+ with lymphocyte scatter-characteristics according to the ISHAGE protocol.
T-cell activation. PBMC (105/well) were seeded in triplicates in 96-well plates and incubated with various concentrations of bispecific antibodies with or without irradiated (100 Gy) FLT3 expressing NALM-16 cells (5 × 104/well). After 48 hours, cells were pulsed for another 20 hours with 3H-methyl-thymidine (0.5 µCi/well) and harvested on filtermats. Incorporated radioactivity was determined by liquid scintillation counting in a 2450 Microplate counter (Perkin Elmer, Waltham, MA). In some experiments, 50 µl of supernatant were removed from the wells after 24 hours and IL-2 content was determined by enzyme-linked immunosorbent assay. To this end high absorbance plates (Thermo Fisher Scientific, Waltham, MA) were coated with capture antibody directed to IL-2 (BD Biosciences), washed, incubated with Biotin conjugated detection antibody (BD Biosciences), washed again, incubated with HRP-conjugated streptavidin (Immunotools, Friesoythe, Germany) and finally stained using a peroxidase containing substrate kit (KPL, Gaithersburg, MD).
51[Cr]-release assays. REH cells were used as targets and activated CD8+ T cells as effector cells. For the preactivation of these cells, PBMC were stimulated with 20 ng/ml of intact UCHT1 antibody for 3 days and washed. CD8+ cells were then isolated by positive selection with antibody coated magnetic beads using LS columns according to the manufacturer's instructions (Miltenyi, Bergisch Gladbach, Germany). Chromium release assays were performed as previously described.6 Briefly, labeled target cells and activated CD8+ T cells were incubated at 37 °C for 4 or 8 hours in 96-well flat bottom plates in the presence of various concentrations of antibodies at an effector:target ratio of 10:1. Percentage of specific 51[Cr]-release was calculated according to the formula: ((cpm (test)−cpm (spontaneous))/((cpm (triton lysis))–cpm (spontaneous)) × 100).
Colony forming unit assay. Bone marrow cells were obtained by lavage of the femoral head of patients undergoing hip surgery. The cells were purified by density gradient centrifugation and seeded at 6.7 × 106/ml in IMDM medium containing antibodies as indicated. After 24 hours of incubation, cells were transfered into antibody containing methylcellulose medium (1.3 × 104 cells/ml, MethoCult H4434 classic, Stemcell Technologies, Grenoble, France). The assay was performed in triplicates. After 12 days, colonies were counted and classified. The use of human material for cytotoxicity- and CFU-assays has been approved by the ethics commitee of the University of Tübingen.
Serum half-life in mice. Experiments were carried out in accordance with the German animal protection law. C57BL/6 mice (three animals per time point) were injected i.v. with 25 µg of the respective antibodies. Mice were bled after 0.5, 1, 2, 4, 8, and 20 hours and serum concentration of the bispecific antibody was determined using the ADCC reporter bioassay (Promega, Mannheim, Germany) that uses gene modified Jurkat cells as “reporter” effector cells. Serum samples at 1:10 and 1:1,000 dilutions were incubated with NALM-16 target cells and the Jurkat cells for 6 hours. Luminescence induced after activation of the Jurkat cells by bispecific antibodies bound to the NALM-16 cells was measured in a SpectraMax L luminometer (Molecular Devices, Biberach, Germany). Serum concentrations were calculated by comparison with a standard curve obtained by dilution of purified antibodies in pooled sera of C57BL/6 mice.
SUPPLEMENTARY MATERIAL Figure S1. FACS analysis of FLT3 expression on different subpopulations of normal bone marrow cultures. Figure S2. Activation of normal PBMC and PBMC subpopulations by the 4G8 X UCHT1 Fabsc-antibody.
Acknowledgments
The authors thank Sandra Drescher, Sabrina Grimm, and Beate Pömmerl for expert technical assistance. The work described was supported by the GO-Bio programme of the Federal Ministry of Education and Research (BMBF), Germany (FZK 0315096/0316070). This programme was implemented to support the foundation of spin off companies from academic institutions in the field of Biotechnology. G.J., L.G.H., and H.G.R. are cofounders of the Synimmune GmbH, Tübingen, a company that emerged from the GO-Bio programme. M.D., M.H., L.G.H., and G.J. receive compensations from the company. The remaining authors declare no competing financial interests.
Supplementary Material
References
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp Cell Res. 2011;317:1278–1285. doi: 10.1016/j.yexcr.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008;68:8049–8057. doi: 10.1158/0008-5472.CAN-08-2268. [DOI] [PubMed] [Google Scholar]
- Foyil KV, Bartlett NL. Anti-CD30 Antibodies for Hodgkin lymphoma. Curr Hematol Malig Rep. 2010;5:140–147. doi: 10.1007/s11899-010-0053-y. [DOI] [PubMed] [Google Scholar]
- Horton HM, Bernett MJ, Peipp M, Pong E, Karki S, Chu SY, et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood. 2010;116:3004–3012. doi: 10.1182/blood-2010-01-265280. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Große-Hovest L, Nübling T, Pyż E, Bamberg ML, Aulwurm S, et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia. 2012;26:1228–1237. doi: 10.1038/leu.2011.372. [DOI] [PubMed] [Google Scholar]
- Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314:628–631. doi: 10.1038/314628a0. [DOI] [PubMed] [Google Scholar]
- Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316:354–356. doi: 10.1038/316354a0. [DOI] [PubMed] [Google Scholar]
- Jung G, Ledbetter JA, Müller-Eberhard HJ. Induction of cytotoxicity in resting human T lymphocytes bound to tumor cells by antibody heteroconjugates. Proc Natl Acad Sci USA. 1987;84:4611–4615. doi: 10.1073/pnas.84.13.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canevari S, Stoter G, Arienti F, Bolis G, Colnaghi MI, Di Re EM, et al. Regression of advanced ovarian carcinoma by intraperitoneal treatment with autologous T lymphocytes retargeted by a bispecific monoclonal antibody. J Natl Cancer Inst. 1995;87:1463–1469. doi: 10.1093/jnci/87.19.1463. [DOI] [PubMed] [Google Scholar]
- Nitta T, Sato K, Yagita H, Okumura K, Ishii S. Preliminary trial of specific targeting therapy against malignant glioma. Lancet. 1990;335:368–371. doi: 10.1016/0140-6736(90)90205-j. [DOI] [PubMed] [Google Scholar]
- Marmé A, Strauss G, Bastert G, Grischke EM, Moldenhauer G. Intraperitoneal bispecific antibody (HEA125xOKT3) therapy inhibits malignant ascites production in advanced ovarian carcinoma. Int J Cancer. 2002;101:183–189. doi: 10.1002/ijc.10562. [DOI] [PubMed] [Google Scholar]
- Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127:2209–2221. doi: 10.1002/ijc.25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Eberhard HJ. An in-vitro model for tumor immunotherapy with antibody heteroconjugates. Immunol Today. 1988;9:257–260. doi: 10.1016/0167-5699(88)91304-7. [DOI] [PubMed] [Google Scholar]
- Jung G, Freimann U, Von Marschall Z, Reisfeld RA, Wilmanns W. Target cell-induced T cell activation with bi- and trispecific antibody fragments. Eur J Immunol. 1991;21:2431–2435. doi: 10.1002/eji.1830211020. [DOI] [PubMed] [Google Scholar]
- Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA. 1995;92:7021–7025. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29:2493–2498. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- Kroesen BJ, Buter J, Sleijfer DT, Janssen RA, van der Graaf WT, The TH, et al. Phase I study of intravenously applied bispecific antibody in renal cell cancer patients receiving subcutaneous interleukin 2. Br J Cancer. 1994;70:652–661. doi: 10.1038/bjc.1994.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibben JG, Boerman OC, Massuger LF, Schijf CP, Claessens RA, Corstens FH. Pharmacokinetics, biodistribution and biological effects of intravenously administered bispecific monoclonal antibody OC/TR F(ab')2 in ovarian carcinoma patients. Int J Cancer. 1996;66:477–483. doi: 10.1002/(SICI)1097-0215(19960516)66:4<477::AID-IJC11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- Schoonjans R, Willems A, Schoonooghe S, Fiers W, Grooten J, Mertens N. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J Immunol. 2000;165:7050–7057. doi: 10.4049/jimmunol.165.12.7050. [DOI] [PubMed] [Google Scholar]
- Rappold I, Ziegler BL, Köhler I, Marchetto S, Rosnet O, Birnbaum D, et al. Functional and phenotypic characterization of cord blood and bone marrow subsets expressing FLT3 (CD135) receptor tyrosine kinase. Blood. 1997;90:111–125. [PubMed] [Google Scholar]
- Grosse-Hovest L, Hartlapp I, Marwan W, Brem G, Rammensee HG, Jung G. A recombinant bispecific single-chain antibody induces targeted, supra-agonistic CD28-stimulation and tumor cell killing. Eur J Immunol. 2003;33:1334–1340. doi: 10.1002/eji.200323322. [DOI] [PubMed] [Google Scholar]
- Otz T, Grosse-Hovest L, Hofmann M, Rammensee HG, Jung G. A bispecific single-chain antibody that mediates target cell-restricted, supra-agonistic CD28 stimulation and killing of lymphoma cells. Leukemia. 2009;23:71–77. doi: 10.1038/leu.2008.271. [DOI] [PubMed] [Google Scholar]
- Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27:1107–1115. doi: 10.1038/leu.2012.341. [DOI] [PubMed] [Google Scholar]
- Arndt C, von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I, et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia. 2013;27:964–967. doi: 10.1038/leu.2013.18. [DOI] [PubMed] [Google Scholar]
- Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Catovsky D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol. 1998;51:364–369. doi: 10.1136/jcp.51.5.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman CW, Pollock D, White G, Hehir K, Moore GP, Kanzy EJ, et al. Construction, expression and characterization of humanized antibodies directed against the human alpha/beta T cell receptor. J Immunol. 1991;147:4366–4373. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.