

Abstract
Background
Haem oxygenase-1 (HO-1) catabolizes haem and has both cytotoxic and cytoprotective effects. Polymorphisms in the promoter of the Haem oxygenase-1 (HMOX1) gene encoding HO-1 have been associated with several diseases including severe malaria. The objective of this study was to determine the allele and genotype frequencies of two single nucleotide polymorphisms; A(−413)T and G(−1135)A, and a (GT)n repeat length polymorphism in the HMOX1 promoter in paediatric malaria patients and controls to determine possible associations with malaria disease severity.
Methods
Study participants were Ghanaian children (n=296) admitted to the emergency room at the Department of Child Health, Korle-Bu Teaching Hospital, Accra, Ghana during the malaria season from June to August in 1995, 1996 and 1997, classified as having uncomplicated malaria (n=101) or severe malaria (n=195; defined as severe anaemia (n=63) or cerebral malaria (n=132)). Furthermore, 287 individuals without a detectable Plasmodium infection or asymptomatic carriers of the parasite were enrolled as controls. Blood samples from participants were extracted for DNA and allele and genotype frequencies were determined with allele-specific PCR, restriction fragment length analysis and microsatellite analysis.
Results
The number of (GT)n repeats in the study participants varied between 21 and 46 with the majority of alleles having lengths of 26 (8.1%), 29/30 (13.2/17.9%) and 39/40 (8.0/13.8%) repeats, and was categorized into short, medium and long repeats. The (−413)T allele was very common (69.8%), while the (−1135)A allele was present in only 17.4% of the Ghanaian population. The G(−1135)A locus was excluded from further analysis after failing the Hardy-Weinberg equilibrium test. No significant differences in allele or genotype distribution of the A(−413)T and (GT)n repeat polymorphisms were found between the controls and the malaria patients, or between the disease groups, for any of the analysed polymorphisms and no associations with malaria severity were found.
Conclusion
These results contribute to the understanding of the role of HMOX1/HO-1. This current study did not find any evidence of association between HMOX1 promoter polymorphisms and malaria susceptibility or severe malaria and hence contradicts previous findings. Further studies are needed to fully elucidate the relationship between HMOX1 polymorphisms and malarial disease.
Keywords: HMOX1, Falciparum malaria, Polymorphisms, Severe malaria
Background
In malaria patients, a large number of infected erythrocytes rupture in the bloodstream, releasing considerable amounts of erythrocyte haemoglobin [1], which is oxidized and releases its haem moiety [2]. This results in large quantities of free haem, which can be highly cytotoxic to both host cells and parasites [1-3]. Survival of the host relies in part on ability to prevent the cytotoxic and inflammatory effects of the free haem. Free haem is only found under pathological conditions because excess haem is usually removed via the microsomal haem degradation (MHD) pathway. However, this pathway may become saturated in situations with large amounts of free haem [3,4]. The rate-limiting enzyme of the MHD pathway is Haem oxygenase (HO) [5]. Two isoforms (HO-1 and HO-2) have been characterized and are expressed in humans. HO-1 is the inducible isoform, whereas HO-2 is the constitutive isoform [6]. HO-1 is a highly inducible 32 kDa protein, with the highest activity in spleen, liver and bone marrow, where senescent erythrocytes are sequestered and degraded [7,8]. The HO-1-encoding gene HMOX1 is located on chromosome 22q12 [9], is approximately 14 kb long, and organized into 4 introns and 5 exons [7]. Transcriptional control of HMOX1 is governed by multiple regulatory elements localized in the promoter of the gene, as well as by enhancers, responsible for HMOX1 induction in response to increased haem concentration [10,11]. HO-1 catabolizes free haem into biliverdin (that is immediately converted to bilirubin), releasing ferrous iron (that is sequestered into the iron storage protein ferritin) and carbon monoxide (CO) [8]. All of these have been associated with the cytoprotective effects of HO-1. Indeed, bilirubin is a potent and abundant antioxidant in mammalian tissue [12,13] and ferritin is a cytoprotective molecule [14,15], whereas CO may affect the regulation of apoptosis [16-18], and inflammation, and has been suggested to mimic the cytoprotective effects of HO-1 [19]. However, high levels of HO-1 related products might also have damaging effects, resulting in an overall pro-oxidant effect, being cytotoxic and causing tissue damage [5,15,20-22]. Although both protective and damaging properties of HO-1 have been shown, HO-1 is essential and HO-1 deficiency leads to severe illness and death in both humans and mice [23-25].
Humans have been shown to differ quantitatively in their HMOX1/HO-1 activity due to polymorphisms in the HMOX1 promoter [26-29]. Two single-nucleotide polymorphisms (SNPs); T(−413)A and G(−1135)A, and a (GT)n repeat length polymorphism in the HMOX1 promoter have been described [7,27,28]. The T(−413)A SNP has been shown to influence promoter activity, with the A allele having a significantly higher activity in vitro compared to the T allele [27], while the functional importance of the G(−1135)A SNP is still unknown [28,30]. Finally, the (GT)n repeat length polymorphism has been described in several studies in various ethnic populations with repeat size varying from 13 to 45 repeats and main alleles at 23, 30 and 39 repeats [27,31-35]. This purine-pyrimidine alternating sequence can result in a Z-DNA conformation and negatively affect transcriptional activity [36,37]. (GT)n repeat length polymorphisms have been associated with many different diseases, including diabetes, cardiovascular, pulmonary, and neurological disease as reviewed by Exner et al.) and by Garcia-Santos & Chies, where long (GT)n repeats, associated with lower HO-1 expression, were identified as risk factors [38,39].
Several studies have investigated a possible association between HO-1 and Plasmodium falciparum infections and have demonstrated both increased expression of HO-1 during malaria infection [40-42] and associations between HMOX1 promoter polymorphisms and malaria disease severity [32-35,43,44]. In mice, an up-regulation of HO-1 was associated protection against cerebral malaria, whereas associations between the short, highly inductive (GT)n repeat alleles and risk of severe malaria have been shown in human studies in both The Gambia, Myanmar, and Angola [32-34]. A lack of association between malaria severity and length of GT repeats has been documented in Thailand [35]. Still, the role of HO-1 during malaria remains unclear [44].
In the present study, the presence of the two single-nucleotide polymorphisms (T(−413)A and G(−1135)A) and the length of the (GT)n repeat were assessed in 583 Ghanaian children with malaria from 0–15 years of age to search for possible associations with malaria disease susceptibility and severity.
Methods
Study population
The study population consisted of patients admitted to the Department of Child Health, Korle-Bu Teaching Hospital, Accra, Ghana during the malaria season (June to August) in 1995, 1996 and 1997, as described in detail in earlier publications [45,46]. All patients were children between 0 and 14 years of age who fulfilled the general inclusion criteria of an asexual P. falciparum parasitaemia of more than 10,000 parasites/μl, and an axillary temperature of more than 37.5°C. A total of 296 malaria patients were enrolled; 101 with uncomplicated malaria and 195 with severe malaria (defined as severe anaemia (n=63) or cerebral malaria (n=132)). Patients with a positive sickling test or any other disease than malaria were excluded as a criterion used in the study the samples were originally collected for. Blood samples from healthy sickle cell-negative children between 0 and 15 years of age were collected as control samples from Dodowa, a nearby community (n=287). Both patients and controls were included after signed consent from patients or guardians after receiving standardized information in local language. Both patient and control population is a mixture of several ethnic groups, possibly with a slightly more uniform population in Dodowa. However, Ga-Adangme is the dominating ethnic group in both populations. The study population is well described with very thorough patient characterization and has been studied extensively, among other things, with regards to mannose-binding lectin genotypes [45] and complement receptor 1 [47]. The study was approved by the ethics and protocol review committee at the University of Ghana Medical School and the Ministry of Health, Ghana.
DNA extraction and whole genome amplification
Upon admission, venous blood samples were collected in EDTA-containing test tubes. Within two hours after collection, plasma was separated, and pellets frozen at −20°C. Genomic DNA was extracted as described previously [48]. Whole genome amplification of the extracted products was performed with Repli-g Mini Kits (Qiagen, Copenhagen, Denmark).
Determining SNPs in the HMOX1 promoter
Two SNPs were analysed in the HMOX1 promoter; the T(−413)A and G(−1135)A. A simple allele-specific PCR was developed to detect the T(−413)A SNP. Two primer-pairs were designed to amplify a 307-bp target sequence based on the nucleotide sequence of Genbank S58267, sharing the same forward primer, and only differing in the 3’-nucleotide end of the reverse primers, making them allele specific. Furthermore, to increase the specificity of the allele-specific primers, a mismatch near the 3’ends were introduced (Table 1). The PCR products were subsequently analysed by 1.5% agarose gel electrophoresis.
Table 1.
Primer sequences and conditions for the polymerase chain reactions (PCRs)
| Allele specific PCRs | Sequences |
|---|---|
| Fw | 5′-ACTGGCACTCTGCTTTATGTGTGA-3′ |
| Rw A(−413) | 5′-GGAGGCAGCGCTGCTCAGAGTAAT-3′ |
| Rw (−413)T | 5′-GGAGGCAGCGCTGCTCAGAGTAAA-3′ |
| Conditions | 95°C 15 min, 35 cycles: (94°C 30 sec, 60°C 30 sec, 72°C 30 sec), 72° 10 min |
| Restriction fragment length polymorphism | |
| Fw | 5′-TTATTTTATATTTTGTAGAGCC-3′ |
| Rw | 5′-AGATGATTCATACAGTCCTTTC-3′ |
| Conditions | 94°C 15 min, 45 cycles: (94°C 30 sec, 49°C 30 sec, 72°C 3 min), 72° 10 min |
| (GT) n repeat length polymorphism | |
| Fw (5’fam) | 5′-AGAGCCTGCAGCTTCTCAGA-3′ |
| Rw | 5′-ACAAAGTCTGGCCATAGGAC-3′ |
| Conditions | 95°C 15 min, 30 cycles: (95°C 30 sec, 64°C 30 sec, 72°C 30 sec), 72° 10 min |
Primer sequences and conditions for the PCR reactions used to determine the polymorphisms in the HMOX1 promoter. The primer pairs for the allele specific PCRs and the restriction fragment length PCR contains a mismatch (Italic). The forward primer for the (GT)n repeat length PCR is fluorescein-conjugated (highlighted). Primers for the allele specific PCRs were designed based on the HMOX1 nucleotide sequence (Genbank S58267). The primers for the restriction fragment length PCR and (GT)n repeat length PCR were designed by He et al. [30] and Takeda et al. [33], respectively.
A restriction fragment length polymorphism (RFLP) analysis was used to detect the G(−1135)A SNP. Primers, as described elsewhere [30], contained a mismatch (see Table 1), which creates a restriction site in the amplified product if the G allele is present. The PCR products were then digested overnight with the restriction enzyme HpaII at 37°C and visualized on a 1.5% agarose gel, showing one band of 225 bp (homozygote for the A allele), two bands of 23 and 202 bp (homozygote for the G allele) or all three bands (heterozygote).
For both PCR protocols, one μl DNA extract was amplified in a 20 μl reaction mix consisting of 4.0 μl H2O, 5.0 μl 2.0 μM primermix and 10.0 μl TEMPase Hot Start (Ampliqon, Odense, Denmark). The reactions were performed in a 96-well PCR plate (Starlab GmbH, Hamburg, Germany) in a VWRi Duo Cycler (VWR/Bie&Berntsen, Radnor, PA, USA). Conditions for amplification are provided in Table 1. Selected samples were sequenced by Sanger to verify the genotyping of the two SNPs.
Determining the HMOX1 promoter (GT)n repeat length polymorphism
A PCR product containing the (GT)n repeat was amplified with a fluorescein-conjugated forward primer and an unlabelled reverse primer designed by Takeda et al. [33] One μl DNA was amplified in a 10 μl reaction consisting of 1.5 μl H2O, 2.5 μl 1.0 μM primermix and 5.0 μl TEMPase Hot Start DNA Polymerase (Ampliqon, Odense, Denmark). The reactions were performed in a 96-well PCR plate (Starlab GmbH, Hamburg, Germany) in a VWRi Duo Cycler (VWR/Bie&Berntsen, Radnor, PA, USA). Primer sequences and amplification conditions are provided in Table 1. One μl PCR product was added to a 10.5 μl reaction containing 9.25 μl HiDi formamideTM (Applied Biosystems, Foster City, CA, USA) and 0.25 μl GeneScanTM 500 LIZTM Dye Size Standard (Applied Biosystems, Foster City, CA, USA) in a 96 well MicroAmp® Optical Reaction Plate (Applied Biosystems, Foster City, CA, USA) and denatured for 3 minutes at 95°C before analysis with an ABI 3730 XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Subsequent allele scoring of the microsatellites was performed using GeneMapper version 4.1 (Applied Biosystems, Foster City, CA, USA). Alleles were divided into short repeats “S” (<27 repeats), medium “M” (27–32 repeats) and long “L” (>32 repeats) based on earlier classifications [26,34,49]. Selected samples were sequenced to verify the determination of repeats.
Statistical analysis
Deviations from the Hardy-Weinberg equilibrium at the two loci; T(−413)A and G(−1135)A were tested using The Court Lab Calculator [50]. Cut-off was set to p < 0.05. Allele and genotype frequencies were compared between the disease groups with Chi-square or Fisher’s exact test (SigmaPlot 12.3 SPSS Inc., USA) for the SNPs and the repeat length polymorphism. Associations between alleles, genotypes, or haplotypes, and disease groups were investigated with logistic regression models to determine odds ratio and p values, with disease group as outcome variable (defined as controls, uncomplicated malaria patients, and severe malaria patients (collectively and divided into severe anaemia and cerebral malaria). Age and gender were included as covariates; p-values < 0.05 were considered significant. Calculations were performed using SAS ver. 9.2, (2002–2008, SAS Institute Inc., Cary, NC, USA). Linkage disequilibrium was calculated using Arlequin [51].
Results
Population demographics
In total, blood samples were collected from 287 controls and 296 patients; 101 with uncomplicated malaria, and 195 with severe malaria (defined as severe anaemia (n=63) or cerebral malaria (n=132)). All patients were children 0–15 years of age (see Table 2).
Table 2.
Demographics of the study population
| Controls (n=287) | Uncomplicated malaria (n=101) | Severe anaemia (n=63) | Cerebral malaria (n=132) | |
|---|---|---|---|---|
| Age (years) | ||||
| Mean ± SD | 8.01 ± 3.97 | 5.51 ± 3.32 | 2.92 ± 2.62 | 4.81 ± 2.77 |
| Minimum/Maximum | 0-15 | 0.5-14 | 0-12 | 0.5-13 |
| Sex (n) | ||||
| Male | 145 (50.52%) | 55 (54.46%) | 40 (63.49%) | 69 (52.27%) |
| Female | 142 (49.48%) | 46 (45.54%) | 23 (36.51%) | 63 (47.72%) |
| Parasitaemia (p/ul) | ||||
| Median | - | 52.000 | 50.265 | 97.157 |
| Percentiles 25 and 75 | - | 24.941-121.912 | 17.730-114.295 | 33.788-212.900 |
| Haemoglobin (g/dl) | ||||
| Mean ± SD | - | 10.49 ± 1.89 | 4.10 ± 1.17 | 7.56 ± 2.26 |
| Minimum/Maximum | - | 6.80-17.5 | 1.80-11.20 | 0.80-13.40 |
Demographic data for the Ghanaian control, uncomplicated malaria, severe anaemia and cerebral malaria groups.
Allele and genotype frequencies of the SNPs in the HMOX1 promoter
The allele-specific PCR and RFLP analysis successfully determined the allele distribution of the T(−413)A SNP in 556 of 583 samples (95.4%) and the G(−1135)A SNP in 478 of 583 samples (82.0%), respectively. Results were confirmed by sequencing of selected samples representing the six possible genotypes. As can be seen in Table 3, the T(−413) allele was common in the study population (69.8%), reflected in high frequencies of both the heterozygote A/T (41.0%) and homozygote T/T (49.3%) genotype. The G(−1135) allele was common (82.6%) with a frequency of 75.1% of the homozygote genotype G/G and 15.1% of the heterozygote G/A. The T(−413)A genotype distribution was found to be in Hardy-Weinberg equilibrium (Χ2=0.42, p=0.51), whereas, the G(−1135)A genotype distribution was not in equilibrium, (Χ2=107.9, p < 0.0001) and this SNP was therefore excluded from further analysis. Analysis of the T(−413)A alleles and genotype frequencies showed no significant differences between the control, uncomplicated or severe malaria (cerebral malaria, severe anaemia and total) groups (p > 0.3 in all cases). Furthermore, logistic regression, adjusted for age and sex, showed no significant association between the T(−413)A alleles or genotypes and severity of malaria (p > 0.4 in all cases).
Table 3.
Prevalence of the T(−413)A and G(−1135)A alleles and genotypes
| T(−413)A Alleles and genotypes N (%) | Controls (n=281) | Uncomplicated malaria (n=95) | Severe anaemia (n=61) | Cerebral malaria (n=119) | |
|---|---|---|---|---|---|
| Alleles | A | 175 (31.1) | 56 (29.5) | 35 (28.7) | 70 (29.4) |
| T | 387 (68.9) | 134 (70.5) | 87 (71.3) | 168 (70.6) | |
| Genotypes | A/A | 26 (9.3) | 11 (11.6) | 5 (8.2) | 12 (10.1) |
| A/T | 123 (43.8) | 34 (35.8) | 25 (41.0) | 46 (38.7) | |
| T/T | 132 (47.0) | 50 (52.6) | 31 (50.8) | 61 (51.3) | |
| G(−1135)A Alleles and genotypes N (%) | Controls (n=238) | Uncomplicated malaria (n=78) | Severe anaemia (n=55) | Cerebral malaria (n=107) | |
| Alleles | G | 397 (83.4) | 123 (78.9) | 91 (82.7) | 179 (83.6) |
| A | 79 (16.6) | 33 (21.2) | 19 (17.3) | 35 (16.4) | |
| Genotypes | G/G | 179 (75.2) | 57 (73.1) | 42 (76.4) | 81 (75.7) |
| G/A | 39 (16.4) | 9 (11.5) | 7 (12.7) | 17 (15.9) | |
| A/A | 20 (8.4) | 12 (15.4) | 6 (10.9) | 9 (8.4) | |
Prevalence of alleles and genotypes in controls, uncomplicated malaria, severe anaemia and cerebral malaria groups. No significant differences in the allele or genotype distribution were found between any of the groups (p > 0.05. G(−1135)A was excluded from further analysis since the control group failed the Hardy-Weinberg equilibrium test.
Allele and genotype frequencies of the (GT)n repeat length polymorphism in the HMOX1 promoter
The (GT)n repeat length polymorphism were successfully genotyped in 572 of 583 samples (98.1%). Sequencing of selected samples was performed to define the size of the repeats (data not shown). The distributions in the control, uncomplicated malaria, severe anaemia and cerebral malaria groups are shown in Figure 1. In total, twenty-six (GT)n alleles were identified, ranging from 21 to 46 repeats. The majority of alleles had lengths of 26 (8.1%), 29/30 (13.2/17.0%) or 39/40 (8.0/13.8%) GT repeats. The alleles were categorized into: short “S” (<27 repeats), medium “M” (27–32 repeats), or long “L” (>32 repeats). Based on these three categories, the patients were classified as having a S/S, S/M, S/L, M/M, M/L or L/L genotype. The allele and genotype frequencies are shown in Table 4. The long repeat alleles (L) were the most prevalent in all four study groups ranging from 42.6 to 45.7%, with high frequencies of the M/L (23.2-41.4%) and L/L (19.5-22.7%) genotypes, whereas the short repeat alleles (S) were found with the lowest frequencies, ranging from 19.1 to 21.6%, and genotype frequencies of 4.3-6.3% (S/S) and 12.6-18.0% (S/M). The distributions of alleles and genotypes were compared between the groups; controls, uncomplicated malaria patients, severe anaemia and cerebral malaria patients (and as a combined severe malaria patient group of severe anaemia and cerebral malaria). No significant differences were found between any of the groups (p > 0.5 in all cases). Logistic regression models were used to test for an association between the alleles or genotypes and severity of malaria. All models were adjusted for age and sex. No significant associations were found (p > 0.7 in all cases). Furthermore, the data was analysed comparing genotypes containing at least one “L” allele (S/L, M/L, L/L) against non-L carriers (S/S, S/M, M/M) and no significant differences were found (p > 0.9).
Figure 1.
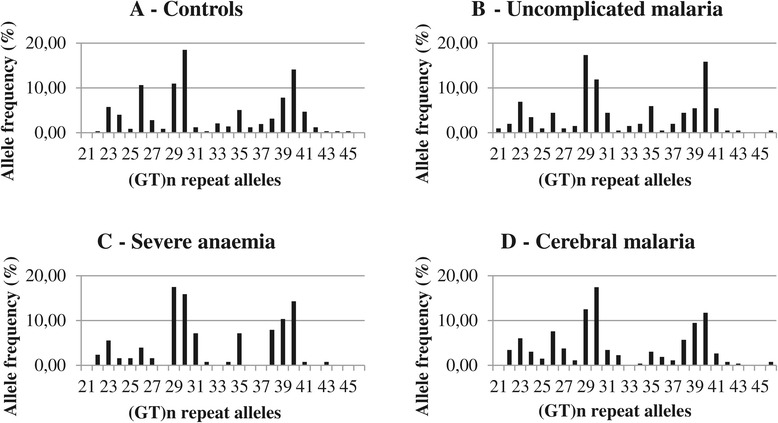
Frequency distribution of the (GT)n repeats in the study groups. Frequency distribution of the (GT)n repeat alleles in the four study groups.1A: The control group. 1B: The uncomplicated malaria group. 1C: The severe anaemia group. 1D: The cerebral malaria group.
Table 4.
Prevalence of the categorized (GT) n repeat alleles and genotypes
| Microsatellites Alleles & genotypes N (%) | Controls (n=564) | Uncomplicated malaria (n=95) | Severe anaemia (n=58) | Cerebral malaria (n=128) | |
|---|---|---|---|---|---|
| Alleles | S | 114 (20.2) | 41 (21.6) | 23 (19.8) | 48 (18.8) |
| M | 210 (37.2) | 68 (35.8) | 40 (37.5) | 100 (39.1) | |
| L | 240 (42.6) | 81 (42.6) | 53 (45.7) | 108 (42.2) | |
| Genotypes | S/S | 12 (4.3) | 6 (6.3) | 3 (5.2) | 6 (4.7) |
| S/M | 41 (14.5) | 12 (12.6) | 6 (10.3) | 23 (18.0) | |
| S/L | 49 (17.4) | 17 (17.9) | 11 (19.0) | 13 (10.2) | |
| M/M | 44 (15.6) | 17 (17.9) | 5 (8.6) | 20 (15.6) | |
| M/L | 81 (28.7) | 22 (23.2) | 24 (41.4) | 37 (28.9) | |
| L/L | 55 (19.5) | 21 (22.1) | 9 (15.5) | 29 (22.7) | |
Frequencies of the alleles and genotypes of the categorized (GT)n repeat alleles in Short “S” (<27), Medium “M” (27–32) and Long “L” (>32). No significant differences in the allele or genotype distribution were found between any of the groups (p > 0.05).
Analysis of the combination of the A(−413)T and (GT)n repeat length polymorphisms
The T(−413)A and (GT)n repeat alleles were next considered together, and the frequencies are shown in Figure 2. (−413)A/(GT)29, (−413)A/(GT)30 and T(−413)/(GT)40 were the most common combinations with frequencies of 22.4%, 30.4% and 19.1%, respectively. It seems that the longer repeats are more often present with a T(−413) allele than the shorter repeats (p < 0.001), however, no linkage disequilibrium (LD) was found (data not shown). In Table 5, the combinations of the (GT)n repeat genotypes and the T(−413)A genotypes are shown. The most prevalent combinations were ML/TA (16.7%), LL/TT (14.9%), SL/TT (12.1%), ML/TT (10.6%) and SM/TA (10.0%), whereas the combination SS/AA was not found. A hypothesis based on earlier human studies was that the combination LL/TT should confer protection against severe malaria. However, analysis with the logistic regression model showed that no combination were more likely to develop severe malaria compared to LL/TT (p > 0.05).
Figure 2.
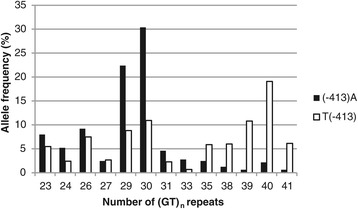
Allele frequencies of the T(−413)A single nucleotide polymorphism for each (GT)n repeat length polymorphism. Frequency of the T(−413)A single nucleotide polymorphism for each (GT)n repeat length. The A(−413) allele is shown in closed bars, the (−413)T in open bars. (GT)n repeats with allele frequencies of less than 2% of both alleles of the A(−413)T single nucleotide polymorphism are not shown.
Table 5.
Combinations of the (GT)n repeat and A(−413)T genotypes
| (GT)n | T(−413)A | N (%) | OR (95% CI) | P value |
|---|---|---|---|---|
| SS | AA | 0 (0.0) | - | - |
| SS | AT | 3 (0.6) | 1.77 (0.15-21.34) | 0.65 |
| SS | TT | 23 (4.3) | 0.63 (0.22-1.84) | 0.40 |
| SM | AA | 3 (0.6) | <0.001 (<0.001- > 999.99) | 1.0 |
| SM | AT | 54 (10.0) | 1.01 (0.46-2.22) | 1.0 |
| SM | TT | 22 (4.1) | 1.12 (0.42-3.37) | 0.75 |
| SL | AA | 2 (0.4) | <0.001 (<0.001- > 999.99) | 1.0 |
| SL | AT | 21 (3.9) | 0.39 (0.09-1.58) | 0.19 |
| SL | TT | 65 (12.1) | 0.70 (0.33-1.50) | 0.36 |
| MM | AA | 37 (6.9) | 0.66 (0.27-1.58) | 0.35 |
| MM | AT | 27 (5.0) | 0.70 (0.24-2.04) | 0.52 |
| MM | TT | 18 (3.4) | 0.59 (0.14-2.45) | 0.47 |
| ML | AA | 9 (1.7) | 1.43 (0.28-7.21) | 0.66 |
| ML | AT | 90 (16.7) | 1.30 (0.66-2.57) | 0.45 |
| ML | TT | 57 (10.6) | 0.71 (0.31-1.62) | 0.42 |
| LL | AA | 2 (0.4) | <0.001 (<0.001- > 999.99) | 0.99 |
| LL | AT | 25 (4.7) | 0.13 (0.02-1.08) | 0.06 |
| LL | TT | 80 (14.9) | Reference | - |
Frequencies of the T(−413)A genotypes combined with the genotypes of the (GT)n repeats. The combinations ML/AT, LL/TT, SL/TT, ML/TT, SM/AT were the most prevalent. No significant association with severity of malaria was found by analysis with logistic regression models, adjusted for age and sex. Significance level p < 0.05.
Discussion
This study investigated the possible association between HMOX1 polymorphisms and severity of malaria. Based on earlier studies, it was hypothesized that certain polymorphisms in the promoter of the HMOX1 gene encoding HO-1 could confer protection against severe malaria. However, this study could not provide evidence for such an association.
The (GT)n repeat length polymorphism have been studied extensively in the past decade, relating short or long repeat alleles to the risk of many different diseases [38,39]. In this current study, the alleles ranged from 21 to 46 repeats, which is similar to findings in studies in Angola and The Gambia [32,34]. However, there were exceptions; short alleles down to 13 repeats were found in The Gambia [34] and repeats > 41 found in the present study were not found in Angola [32]. The majority of the alleles had lengths of 26, 29/30 and 39/40 repeats, consistent with the study in The Gambia [34]. In Angola, the distribution was slightly different, with the most frequent alleles being 23, 29 and 38 repeats. The differences in the alleles around 29/30 and 38–40 repeats found in the present study as compared to the two previous studies, could be due to small discrepancies in the analysis of the (GT)n nucleotide repeats. However, this did not affect the length-category analysis since both the 29 and 30 repeat alleles were categorized as medium-sized and 38–40 as large. Studies outside Africa have found much lower frequencies of the 39-repeat allele than the current study of less than 3% in Japan [28], Thailand [35] and Greece [31], and absent in a study in Myanmar [33]). In contrast, the allele with 23 repeats was more prevalent in the studies outside Africa with frequencies up to 30% [28,31,33,35] compared to the 6% found here. In a Brazilian study, the frequencies of both the 23- and 39-repeat alleles were low (<2%), whereas alleles with 28–30 repeats all had high frequencies (>65%) [44].
In both the Myanmar study and the two African studies in Angola and The Gambia, short repeat alleles were positively correlated with severity of malaria. Studies in Thailand and Brazil have shown conflicting results [35,44]. In the Brazilian study, long (GT)n repeats were associated with symptomatic malaria, however, the patients were mainly infected with P. vivax and only five severe cases were included [44]. In Thailand, no association between the (GT)n repeats and severity of malaria was found, however, limited sample size in some groups might have influenced results and furthermore, the study group consisted of both P. falciparum and P. vivax infected patients [35]. Although an association between the (GT)n repeat polymorphism and severity of malaria has been shown in two other African populations, this study could not confirm such association. In The Gambia, the allelic and genotypic distributions were different from this current study Thus, the severe malaria patients with short repeat alleles were more prevalent in The Gambia than in Ghana (50% vs 19%), and the long repeat alleles more prevalent in Ghana (43% vs 26%) [34]. This was also reflected in the genotypes, with frequencies of 28% (S/S) and 8% (L/L) in the severe malaria patients in The Gambia compared to frequencies of 5% and 20% in Ghana, respectively. The patient sample size was equivalent to the Gambian and Angolan studies; however, differences in study population, study design, age, or malaria transmission might influence results.
As with pro-inflammatory cytokines for example, excessive levels are cytotoxic [34] and the optimal levels of HO-1 might be a balancing act since both low and very high levels of the enzyme are associated with cytotoxic effects [21,22,52,53]. HMOX1 has been associated with several diseases [38,39], which may have blurred a possible selective force of malaria on HMOX1.
Two single nucleotide polymorphisms (SNPs) in HMOX1 were also determined. No analysis was done regarding the G(−1135)A SNP, since none of the groups were in Hardy-Weinberg equilibrium. The T(−413)A SNP has been associated with differences in promoter activity, however, it has not been studied as extensively as the (GT)n repeat length polymorphism. The frequency distribution of the T(−413)A SNP reported here is similar to previous findings in a Chinese population [30] and a study based on North Americans and Europeans [54]. However, it differed significantly from a Japanese population where the AA genotype was more prevalent than in the Ghanaian population (26.9 vs 9.7%) whereas the TT genotype was more prevalent than in the Japanese population (49.3% vs 24.5%) [27]. In contrary to our initial hypothesis, no association between genotypes and malaria severity was found.
When alleles of the T(−413)A SNP and (GT)n repeat alleles were considered together, long repeats were mostly found with the T(−413) allele, which is consistent with the findings of the Japanese study [27]. However, there was no linkage disequilibrium between the alleles. Furthermore, the T(−413)A and (GT)n repeat category genotypes were combined; no association with severity of malaria was found. Thus in summary, based on the findings of this study together with the fact that previously found associations between malaria and HMOX1 have shown effects in opposing directions suggest that malaria does not seem to be a major selective force on the polymorphisms of the HMOX1 promoter.
Conclusion
The GT)n repeat allele frequencies found in this study are similar to those of other African studies. However, the association between the (GT)n repeat alleles and severity of malaria was not confirmed in this well-characterized Ghanaian population [45]. Furthermore, the A(−413)T SNP showed no association with severity of malaria alone or in combination with the (GT)n repeat alleles. In this population, the polymorphisms of the HMOX1 promoter are not associated with severity of malaria, and another selective force may be influencing these alleles.
Acknowledgements
We are grateful to all study participants, parents and guardians and staff at the Department of Child Health, Korle-Bu Teaching Hospital, Accra, Ghana. Sylvia Mathiasen and Ulla Abildtrup, University of Copenhagen, are thanked for technical assistance and the staff at The Section of Biostatistics, University of Copenhagen is thanked for statistical support.
Abbreviations
- CO
Carbon Monoxide
- HO
Haem Oxygenase
- HMOX1
Haem Oxygenase 1
- LD
Linkage Disequilibrium
- L
Long
- MHD
Microsomal Haem Degradation
- M
Medium
- PCR
Polymerase Chain Reaction
- RFLP
Restriction Fragment Length Polymorphism
- S
Short
- SNP
Single Nucleotide Polymorphism
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BQG, FNN, OPR and JALK designed the clinical study. BQG and OPR conducted the clinical study. JALK did the preparatory laboratory work and handled data management. HHH, CB and EL carried out the laboratory work. HHH analysed the data and wrote the manuscript. MA, JALK, ICB, MLS, CH and LM contributed to study design and interpretation of data and revised the manuscript for important intellectual content. BQG, FNN, and OPR edited and revised the manuscript. All authors contributed to the manuscript and read and approved the final version.
Contributor Information
Helle H Hansson, Email: hellehan@sund.ku.dk.
Lasse Maretty, Email: lassemaretty@binf.ku.dk.
Christina Balle, Email: frk.balle@gmail.com.
Bamenla Q Goka, Email: bamenla@yahoo.co.uk.
Elisa Luzon, Email: luzonelisa@gmail.com.
Francis N Nkrumah, Email: fnkrumah@noguchi.ug.edu.gh.
Mette L Schousboe, Email: mettelettes@gmail.com.
Onike P Rodrigues, Email: onikerodrigues@yahoo.co.uk.
Ib Christian Bygbjerg, Email: Iby@sund.ku.dk.
Jørgen AL Kurtzhals, Email: Joergen.Kurtzhals@regionh.dk.
Michael Alifrangis, Email: micali@sund.ku.dk.
Casper Hempel, Email: casperhempel@gmail.com.
References
- 1.Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 2.Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Eaton JW, et al. Haem, haem oxygenase and ferritin in vascular endothelial cell injury. Mol Nutr Food Res. 2005;49:1030–43. doi: 10.1002/mnfr.200500076. [DOI] [PubMed] [Google Scholar]
- 3.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell haem uptake from haem proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90:9285–9. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, et al. Different faces of the haem-haem oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–71. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 5.Dong Z, Lavrovsky Y, Venkatachalam MA, Roy AK. Haem oxygenase-1 in tissue pathology: the Yin and Yang. Am J Pathol. 2000;156:1485–8. doi: 10.1016/S0002-9440(10)65019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maines MD. Haem oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–68. [PubMed] [Google Scholar]
- 7.Shibahara S, Sato M, Muller RM, Yoshida T. Structural organization of the human haem oxygenase gene and the function of its promoter. Eur J Biochem. 1989;179:557–63. doi: 10.1111/j.1432-1033.1989.tb14583.x. [DOI] [PubMed] [Google Scholar]
- 8.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of haem to bilirubin by microsomal haem oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–55. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kutty RK, Kutty G, Rodriguez IR, Chader GJ, Wiggert B. Chromosomal localization of the human haem oxygenase genes: haem oxygenase-1 (HMOX1) maps to chromosome 22q12 and haem oxygenase-2 (HMOX2) maps to chromosome 16p13.3. Genomics. 1994;20:513–16. doi: 10.1006/geno.1994.1213. [DOI] [PubMed] [Google Scholar]
- 10.Immenschuh S, Ramadori G. Gene regulation of haem oxygenase-1 as a therapeutic target. Biochem Pharmacol. 2000;60:1121–8. doi: 10.1016/S0006-2952(00)00443-3. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi K, Sun J. The haem-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. 2006;8:107–18. doi: 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 12.Gopinathan V, Miller NJ, Milner AD, Rice-Evans CA. Bilirubin and ascorbate antioxidant activity in neonatal plasma. FEBS Lett. 1994;349:197–200. doi: 10.1016/0014-5793(94)00666-0. [DOI] [PubMed] [Google Scholar]
- 13.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 14.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, et al. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–53. [PubMed] [Google Scholar]
- 15.Ryter SW, Tyrrell RM. The haem synthesis and degradation pathways: role in oxidant sensitivity. Haem oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/S0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Kim SH, Yoon HJ, Shin DH, Park SS, Kim YS, et al. NAT2, CYP2C9, CYP2C19, and CYP2E1 genetic polymorphisms in anti-TB drug-induced maculopapular eruption. Eur J Clin Pharmacol. 2011;67:121–7. doi: 10.1007/s00228-010-0912-4. [DOI] [PubMed] [Google Scholar]
- 17.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by haem oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000;192:1015–26. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2007;282:1718–26. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 19.Otterbein LE, Bach FH, Alam J, Soares M, Tao LH, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–8. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 20.Galbraith R. Haem oxygenase: who needs it? Proc Soc Exp Biol Med. 1999;222:299–305. doi: 10.1046/j.1525-1373.1999.d01-147.x. [DOI] [PubMed] [Google Scholar]
- 21.Suttner DM, Dennery PA. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999;13:1800–9. doi: 10.1096/fasebj.13.13.1800. [DOI] [PubMed] [Google Scholar]
- 22.Lamb NJ, Quinlan GJ, Mumby S, Evans TW, Gutteridge JM. Haem oxygenase shows pro-oxidant activity in microsomal and cellular systems: implications for the release of low-molecular-mass iron. Biochem J. 1999;344(Pt 1):153–8. doi: 10.1042/0264-6021:3440153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human haem oxygenase-1 deficiency. J Clin Invest. 1999;103:129–35. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poss KD, Tonegawa S. Reduced stress defense in haem oxygenase 1-deficient cells. Proc Natl Acad Sci U S A. 1997;94:10925–30. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poss KD, Tonegawa S. Haem oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A. 1997;94:10919–24. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, Suzuki S, Shibahara S, Sasaki H. Microsatellite polymorphism in haem oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood. 2003;102:1619–21. doi: 10.1182/blood-2002-12-3733. [DOI] [PubMed] [Google Scholar]
- 27.Ono K, Mannami T, Iwai N. Association of a promoter variant of the haeme oxygenase-1 gene with hypertension in women. J Hypertens. 2003;21:1497–503. doi: 10.1097/00004872-200308000-00013. [DOI] [PubMed] [Google Scholar]
- 28.Ono K, Goto Y, Takagi S, Baba S, Tago N, Nonogi H, Iwai N. A promoter variant of the haem oxygenase-1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis. 2004;173:315–9. doi: 10.1016/j.atherosclerosis.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, et al. Microsatellite polymorphism in the haem oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet. 2000;66:187–95. doi: 10.1086/302729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Z, Hu Y, Feng L, Lu Y, Bao D, Xi Y, et al. Association between HMOX-1 genotype and cardiac function during exercise. Appl Physiol Nutr Metab. 2008;33:450–60. doi: 10.1139/H08-016. [DOI] [PubMed] [Google Scholar]
- 31.Katana EP, Skoura LG, Scouras ZG, Daniilidis MA. Genotype and allele frequencies of haem oxygenase-1 promoter region in a Greek cohort. Chin Med J (Engl ) 2011;124:3408–11. [PubMed] [Google Scholar]
- 32.Sambo MR, Trovoada MJ, Benchimol C, Quinhentos V, Goncalves L, Velosa R, et al. Transforming growth factor beta 2 and haem oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS One. 2010;5:e11141. doi: 10.1371/journal.pone.0011141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda M, Kikuchi M, Ubalee R, Na-Bangchang K, Ruangweerayut R, Shibahara S, et al. Microsatellite polymorphism in the haem oxygenase-1 gene promoter is associated with susceptibility to cerebral malaria in Myanmar. Jpn J Infect Dis. 2005;58:268–71. [PubMed] [Google Scholar]
- 34.Walther M, De CA, Aka P, Njie M, Amambua-Ngwa A, Walther B, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012;8:e1002579. doi: 10.1371/journal.ppat.1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuesap J, Hirayama K, Kikuchi M, Ruangweerayut R, Na-Bangchang K. Study on association between genetic polymorphisms of haem oxygenase-1, tumour necrosis factor, cadmium exposure and malaria pathogenicity and severity. Malar J. 2010;9:260. doi: 10.1186/1475-2875-9-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naylor LH, Clark EM. d(TG)n.d(CA)n sequences upstream of the rat prolactin gene form Z-DNA and inhibit gene transcription. Nucleic Acids Res. 1990;18:1595–601. doi: 10.1093/nar/18.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delic J, Onclercq R, Moisan-Coppey M. Inhibition and enhancement of eukaryotic gene expression by potential non-B DNA sequences. Biochem Biophys Res Commun. 1991;181:818–26. doi: 10.1016/0006-291X(91)91263-C. [DOI] [PubMed] [Google Scholar]
- 38.Exner M, Minar E, Wagner O, Schillinger M. The role of haem oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med. 2004;37:1097–104. doi: 10.1016/j.freeradbiomed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Santos D, Chies JA. HO-1 polymorphism as a genetic determinant behind the malaria resistance afforded by haemolytic disorders. Med Hypotheses. 2010;74:807–13. doi: 10.1016/j.mehy.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Medana IM, Mai NT, Day NP, Hien TT, Bethell D, Phu NH, et al. Cellular stress and injury responses in the brains of adult Vietnamese patients with fatal Plasmodium falciparum malaria. Neuropathol Appl Neurobiol. 2001;27:421–33. doi: 10.1046/j.0305-1846.2001.00360.x. [DOI] [PubMed] [Google Scholar]
- 41.Schluesener HJ, Kremsner PG, Meyermann R. Haem oxygenase-1 in lesions of human cerebral malaria. Acta Neuropathol. 2001;101:65–68. doi: 10.1007/s004010000250. [DOI] [PubMed] [Google Scholar]
- 42.Clark IA, Awburn MM, Harper CG, Liomba NG, Molyneux ME. Induction of HO-1 in tissue macrophages and monocytes in fatal falciparum malaria and sepsis. Malar J. 2003;2:41. doi: 10.1186/1475-2875-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunnington AJ, Kendrick SF, Wamola B, Lowe B, Newton CR. Carboxyhemoglobin levels in Kenyan children with Plasmodium falciparum malaria. Am J Trop Med Hyg. 2004;71:43–47. [PubMed] [Google Scholar]
- 44.Mendonca VR, Luz NF, Santos NJ, Borges VM, Goncalves MS, Andrade BB, et al. Association between the haptoglobin and haem oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect Immun. 2012;80:1445–54. doi: 10.1128/IAI.05933-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garred P, Nielsen MA, Kurtzhals JA, Malhotra R, Madsen HO, Goka BQ, et al. Mannose-binding lectin is a disease modifier in clinical malaria and may function as opsonin for Plasmodium falciparum-infected erythrocytes. Infect Immun. 2003;71:5245–53. doi: 10.1128/IAI.71.9.5245-5253.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtzhals JA, Adabayeri V, Goka BQ, Akanmori BD, Oliver-Commey JO, Nkrumah FK, et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet. 1998;351:1768–72. doi: 10.1016/S0140-6736(97)09439-7. [DOI] [PubMed] [Google Scholar]
- 47.Hansson HH, Kurtzhals JA, Goka BQ, Rodriques OP, Nkrumah FN, Theander TG, et al. Human genetic polymorphisms in the Knops blood group are not associated with a protective advantage against Plasmodium falciparum malaria in Southern Ghana. Malar J. 2013;12:400. doi: 10.1186/1475-2875-12-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikuchi A, Yamaya M, Suzuki S, Yasuda H, Kubo H, Nakayama K, et al. Association of susceptibility to the development of lung adenocarcinoma with the haem oxygenase-1 gene promoter polymorphism. Hum Genet. 2005;116:354–60. doi: 10.1007/s00439-004-1162-2. [DOI] [PubMed] [Google Scholar]
- 50.Court M. Court Lab Calculator [http://www.scribd.com/doc/246388807/Court-Lab-HW-Calculator#scribd]. 2008.
- 51.Excoffier L, Laval LG, Scheinder S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 52.Abraham NG, Lavrovsky Y, Schwartzman ML, Stoltz RA, Levere RD, Gerritsen ME, et al. Transfection of the human haem oxygenase gene into rabbit coronary microvessel endothelial cells: protective effect against haem and hemoglobin toxicity. Proc Natl Acad Sci U S A. 1995;92:6798–802. doi: 10.1073/pnas.92.15.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dennery PA, Sridhar KJ, Lee CS, Wong HE, Shokoohi V, Rodgers PA, et al. Haem oxygenase-mediated resistance to oxygen toxicity in hamster fibroblasts. J Biol Chem. 1997;272:14937–42. doi: 10.1074/jbc.272.23.14937. [DOI] [PubMed] [Google Scholar]
- 54.Bean CJ, Boulet SL, Ellingsen D, Pyle ME, Barron-Casella EA, Casella JF, et al. Haem oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. 2012;120:3822–28. doi: 10.1182/blood-2011-06-361642. [DOI] [PMC free article] [PubMed] [Google Scholar]