

Abstract
We report the engineering and characterization of paraoxonase-3 knockout mice (Pon3KO). The mice were generally healthy but exhibited quantitative alterations in bile acid metabolism and a 37% increased body weight compared to the wild-type mice on a high fat diet. PON3 was enriched in the mitochondria-associated membrane fraction of hepatocytes. PON3 deficiency resulted in impaired mitochondrial respiration, increased mitochondrial superoxide levels, and increased hepatic expression of inflammatory genes. PON3 deficiency did not influence atherosclerosis development on an apolipoprotein E null hyperlipidemic background, but it did lead to a significant 60% increase in atherosclerotic lesion size in Pon3KO mice on the C57BL/6J background when fed a cholate-cholesterol diet. On the diet, the Pon3KO had significantly increased plasma intermediate-density lipoprotein/LDL cholesterol and bile acid levels. They also exhibited significantly elevated levels of hepatotoxicity markers in circulation, a 58% increase in gallstone weight, a 40% increase in hepatic cholesterol level, and increased mortality. Furthermore, Pon3KO mice exhibited decreased hepatic bile acid synthesis and decreased bile acid levels in the small intestine compared with wild-type mice. Our study suggests a role for PON3 in the metabolism of lipid and bile acid as well as protection against atherosclerosis, gallstone disease, and obesity.—Shih, D. M., Yu, J. M., Vergnes, L., Dali-Youcef, N., Champion, M. D., Devarajan, A., Zhang, P., Castellani, L. W., Brindley, D. N., Jamey, C. Auwerx, J., Reddy, S. T., Ford, D. A., Reue, K., Lusis, A. J. PON3 knockout mice are susceptible to obesity, gallstone formation, and atherosclerosis.
Keywords: bile acid, cholesterol, hyperlipidemia, mitochondria, mitochondria-associated membrane
The paraoxonase gene family contains 3 members, PON1, PON2, and PON3, located as a cluster on mouse chromosome 6 or human chromosome 7. PON1 is expressed primarily in the liver and is associated with HDL particles in the blood (1, 2). PON2 is ubiquitously expressed and associated with endoplasmic reticulum (ER), nuclear membrane, and mitochondria of the cell (3–7). PON3 is expressed primarily in the liver, although lower expression levels are also observed in a variety of other tissues (8, 9). In humans and rabbits, low levels of PON3 are present on HDL (10, 11). However, murine PON3 is absent from the HDL and circulation, and it is entirely cell associated (12). Within the cells, PON3 was reported to be localized to ER and mitochondria (13), although we show here that in liver it is primarily in the mitochondria-associated membrane (MAM). All 3 PON members exhibit quorum-quenching lactonase activities (14, 15). PON1 also exhibits the capacity to hydrolyze organophosphates such as organophosphorus insecticides (16). Studies with genetically engineered mice and human epidemiologic studies suggest that the PON family members can inhibit oxidative stress, suppress inflammation, and protect against atherosclerosis (7, 17–24). While PON1 and PON2 have been quite extensively characterized, the function of PON3 is less clear. Transgenic mice overexpressing PON3 were shown to be protected against atherosclerosis and obesity (12), but the mechanism involved is unknown. Recently, PON3 was shown to be up-regulated in cancer tissues and protect against mitochondrial superoxide-mediated cell death (13).
In the present report, we engineered mice that lack PON3 and used these mice to examine the functions of PON3 under a variety of conditions. Although the mice were generally healthy, they showed quantitative alterations in mitochondrial functions, bile acid metabolism, and body fat. When stressed with a cholate-cholesterol (CC) diet, they exhibited substantial variation in cholesterol metabolism and increased atherosclerosis.
MATERIALS AND METHODS
Generation of Pon3KO mice, animal breeding, feeding, and atherosclerosis lesion development
A Pon3 targeting vector was constructed by subcloning a 3.6 kb ClaI fragment containing exon 3 upstream and a 2.8 kb BamHI fragment containing exon 5 downstream of the neomycin-resistance gene expression cassette in the pMC1TKpA vector (Supplemental Fig. S1A). The gene targeting vector was linearized and electroporated into RW-4 embryonic stem cells derived from mouse strain 129X1/SvJ. G418/gancyclovir-resistant clones were screened by Southern blot analysis, and embryonic stem cells carrying the disrupted allele were microinjected into blastocysts of mouse strain C57BL/6J to produce chimeric mice. Chimeric mice were crossed with C57BL/6J mice to produce F1 mice heterozygous for the PON3 null mutation. F1 PON3 heterozygous mice were then backcrossed with C57BL/6J mice for 9 more generations before intercrossing to produce mice homozygous for the Pon3 mutation. To introduce the PON3 null mutation onto the apolipoprotein E (apoE) knockout (KO) background, N5 mice carrying the PON3 null mutation were crossed with apoE KO mice. The offspring mice heterozygous for both the PON3 and apoE null mutations were then intercrossed to generate apoE KO mice and Pon3KO/apoE KO mice. Only female mice were included in the studies. ApoE KO and Pon3KO/apoE KO mice were maintained on chow diet for atherosclerosis assessment. Diet induced atherosclerosis was assessed using Pon3KO and wild-type (WT) mice on the C57BL/6J background using a CC diet containing 15.8% fat, 1.25% cholesterol, and 0.5% sodium cholate (TD.90221; Harlan Laboratories, Indianapolis, IN, USA). Atherosclerotic lesion size at the aortic root region was determined as previously described (25). For obesity study, Pon3KO and WT mice were fed a high-fat Western diet (TD.88137; Harlan Laboratories) for 10 wk. All animal studies were approved by the UCLA Animal Research Committee.
Lipid and bile acid levels, serum chemistry, liver lipid extraction and analysis by mass spectrometry, and lovastatinase activity assay
For plasma lipid and lipoprotein level determinations, mice were fasted for 16 h before bleeding. Total cholesterol, HDL cholesterol, unesterified/free cholesterol, triglycerides, and free fatty acid levels were determined by enzymatic colorimetric assays (25). Phosphatidylcholine levels were assayed using an enzymatic colorimetric assay from Wako (Richmond, VA, USA). Serum chemistry tests were performed by Pathology and Laboratory Medicine Services of the Department of Laboratory Animal Management at UCLA. Total bile acids levels were assayed using a kit from Diazyme Laboratories (Poway, CA, USA) according to the manufacturer’s protocol. This kit measures the 3α-hydroxyl group of bile acids. For lipid extraction, 50 mg of liver were homogenized in PBS. The lipids in the homogenate were then extracted using the Folch method (26). The extracted lipids were dried down and resuspended in 1% Triton X-100 before lipid assays were performed as described above. Tissue homogenates made from the livers of Pon3KO and WT mice were used in lovastatinase activity assay as previously described (12). In other analyses, mass spectrometry was performed using lipid extracts prepared by Bligh-Dyer extraction (27) with either water or water enriched with 1% acetic acid (for phosphatidic acid analyses) and using lipid class specific internal standards (1,2,3-triheptadecenoyl-sn-glycerol, heptadecanoyl cholesteryl ester, 1,2-diarachidoyl-sn-glycero-3-phosphocholine, 1,2-dimyristoyl-sn-glycerol, and 1,2-dimyristoyl-sn-glycero-3-phosphate). Electrospray ionization mass spectrometry (Quantum Ultra; Thermo-Fisher, Waltham, MA, USA) with lipid class (or molecular species)–specific tandem mass spectrometric analysis was utilized to quantify liver triglyceride [neutral loss scanning (NLS) of fatty acid losses with fingerprinting] (28), cholesteryl ester (NLS 368.5) (29), phosphatidylcholine (NLS 59.1) (30), diacylglycerol (selected reaction monitoring) (29), and phosphatidic acid (PA) (product ion scanning m/z = 183) (30) levels.
Phosphatidic acid phosphatase activity assay
Phosphatidic acid phosphatase (PAP) activity was measured on liver tissue extracts as described previously (31, 32). Briefly, fresh mouse tissues were directly homogenized in lysis buffer [250 mM sucrose, 2 mM DTT, protein phosphatase inhibitor mixtures 2 and 3 (Sigma-Aldrich, St. Louis, MO, USA), protease inhibitor mixture (Roche Diagnostics, Basel, Switzerland), and 0.15% Tween-20]. PAP-1 activity was measured in a final volume of 0.1 ml containing 100 mM Tris maleate buffer, pH 7.5, 5 mM Mg2+, and 0.6 mM PA labeled with [3H]palmitate (about 75 Ci/mol) dispersed in 0.4 mM phophatidylcholine and 1 mM EDTA, and the reactions were incubated at 37°C. Chloroform containing 0.08% olive oil was added to stop the reaction. Then basic alumina was added to absorb the PA and any [3H] palmitate formed by phospholipase A–type activities. The [3H] diacylglycerol product was then isolated and quantified by scintillation counting. Three different protein concentrations were analyzed for each sample to ensure the proportionality of the assay. Parallel analyses were done in the presence of excess N-ethylmaleimide (5 mM) to assess the contribution of lipid phosphate phosphatase activity, and this latter activity was subtracted from the total activity to yield true PAP activity values.
RNA isolation and quantitative RT-PCR analyses
Total RNA samples from tissues were isolated using Trizol reagent (Life Technologies) according to the manufacturer’s protocol. The cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was performed using gene-specific primers (Supplemental Table S1) and the Roche SYBR green master mix in a Roche Lightcycler 480 system. The mRNA levels of specific genes were normalized to the mRNA levels of the housekeeping gene, Rpl13a, of the same sample.
Quantification of gallstones, collection of gallbladder and hepatic bile, bile lipid analysis, and bile acid composition analysis by mass spectrometry
After cholecystectomy, the gallbladder was cut at the fundus to collect bile and gallstones. After drying overnight at room temperature, stones were weighed. Hepatic bile was collected for 30 min as described (33). Bile samples were diluted with water containing 1% Triton X-100, followed by determination of cholesterol, phospholipid, and total bile acid concentrations using colorimetric assays as described above. The bile acid composition of plasma, liver, gallbladder bile, and small intestine samples was examined and quantified using mass spectrometry as previously described (34).
Determination of average adipocyte size
Hematoxylin and eosin–stained histologic sections of fat pads were used for determination of adipocyte size as previously described (35).
Isolation of organelles and immunoblotting
Four mice from each group were fasted overnight before killing and collection of livers. The pooled liver samples were then used for isolation of organelles by ultracentrifugation according to the protocol of Wieckowski et al. (36). For immunoblotting, equal amounts of purified organelles or liver lysates were fractionated by SDS-PAGE, transferred onto a nylon membrane, incubated with various primary antibodies, washed, incubated with a secondary antibody, and detected using electrochemiluminescence (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The primary antibodies against PON3 and calnexin were purchased from R&D Systems (Minneapolis, MN, USA) and Santa Cruz Technology (Santa Cruz, CA, USA), respectively. Primary antibodies against cytochrome c and voltage-dependent anion channel (VDAC) were purchased from Cell Signaling Technology (Danvers, MA). Primary antibody against Cyp7a1 (cytochrome P450, family 7, subfamily A, polypeptide 1) was a gift from Simon Hui of UCLA. Lipin-1 antibody was a gift from Maroun Bou Kahlil (University of Ottawa, Ottawa, ON, Canada). Lipin-2 antibody was a gift from Brian Finck (Washington University, St. Louis, MO, USA).
Mitochondrial functional assays and TUNEL assay
Mitochondria were isolated from mouse liver as described by Rogers et al. (37). The mitochondrial function of isolated mitochondria (5 μg per well) was determined using an XF24-3 Extracellular Flux Analyzer (Seahorse Bioscience) as described (37). Succinate (10 mM), rotenone (2 μM), ADP (2 mM), oligomycin (25 µM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 40 µM), and antimycin A (15 µg/ml) were used to assay complex II dependent respiration. Oxygen consumption rate (OCR) in fresh tissues were measured with the XF24 Analyzer as described (38). Briefly, freshly isolated tissues were minced and rinsed in PBS, placed in a Seahorse Islet Plate (3–5 mg per well), and incubated with 625 μl of unbuffered DMEM (containing 25 mM glucose) 1 h prior to measurements. Oxygen consumption was measured after 200 µM palmitate or 0.75 µM FCCP injection and expressed as percentage OCR of baseline. For OCR in primary white adipocyte, stromal vascular cells were collected, cultured, and differentiated as previously described (39). OCR was measured at baseline and after the sequential injection of 1 µM oligomycin, 0.75 µM FCCP and 1 µM rotenone/myxothiazol. The TUNEL assay was performed on frozen sections of liver samples, using the In situ Cell Death Detection kit purchased from Roche, according to the manufacturer’s manual.
Statistical analyses
Log-rank (Mantel-Cox) test was used for comparison of survival curves shown in Fig. 1B. For the rest of the study, Student’s t test was used for comparison of means.
Figure 1.
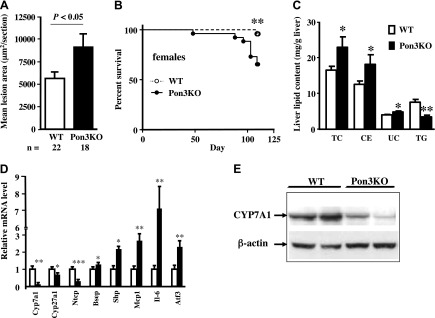
Pon3KO mice exhibit increased atherosclerosis, premature death, increased hepatic cholesterol accumulation, and altered hepatic gene expression when fed a CC diet. Three-month-old WT and Pon3KO mice were maintained on a CC diet for 16 wk before (A) atherosclerotic lesion at the aortic root region, (B) survival, (C) liver lipid content, (D) hepatic gene expression, and (E) hepatic Cyp7A1 protein levels were determined. CE, cholesterol ester; TC, total cholesterol; TG, triglyceride; UC, unesterified cholesterol. *P < 0.05; **P < 0.01; ***P < 0.001 between the 2 genotype groups.
RESULTS
General characteristics of mice deficient in PON3
We constructed mice targeted for Pon3 using classic gene targeting approaches (Supplemental Fig. S1). The strategy is shown in Supplemental Fig. S1A. Supplemental Figure S1B shows the expected Southern blot pattern following cleavage with PstI in heterozygous targeted mice. The homozygous Pon3KO mice lacked PON3 mRNA (Supplemental Fig. S1C), and protein (Supplemental Fig. S1D) in liver. We previously showed that PON3 is able to hydrolyze lovastatin (12). The Pon3KO mice had no lovastatinase activity (Supplemental Fig. S1E) in liver, indicating that PON3 is entirely responsible for lovastatin hydrolysis. The data indicate that the targeting creates a PON3 null mutation.
We generated the Pon3KO on the background of strain 129X1/SvJ. Since strain 129 mice are difficult to breed, we transferred the null mutation onto the genetic background of C57BL/6J through a series of backcrosses for 10 generations. The Pon3KO mice did not exhibit any discernible effects on appearance, fertility, or life span. When maintained on a chow diet, there was no evidence of liver pathology. However, small but significant differences in plasma triglycerides and free fatty acid levels were observed between Pon3KO and WT mice (Supplemental Table S2).
PON3 deficiency does not influence atherosclerosis in a hyperlipidemic apoE null background
Previously, we had shown that transgenic mice overexpressing PON3 in liver exhibited reduced atherosclerosis on a LDL receptor null background (12). To test whether the Pon3KO mice would exhibit increased atherosclerosis, we bred PON3 KO mice onto the background of apoE null mice and examined the double KO mice for effects on atherosclerotic lesion development. Surprisingly, there were no discernible differences effects on atherosclerosis (Supplemental Fig. S1F). Plasma lipoprotein and lipid levels were similar between the double KO and apoE KO mice, except small but significant decreases of HDL and free fatty acid levels in the double KO mice compared to the apoE KO (Supplemental Table S3).
Pon3KO mice exhibit significant alterations in lipid metabolism and atherosclerosis when fed a diet containing cholic acid and cholesterol
We had previously shown that transgenic PON3 mice fed a CC diet, exhibited significantly reduced atherosclerosis compared to control mice (12). We, therefore, tested whether the Pon3KO mice would also exhibit altered atherosclerosis when fed the CC diet. When fed the diet for 16 wk, Pon3KO mice exhibited a significant 60% increase in atherosclerotic lesion size compared to WT mice (Fig. 1A). During the course of these studies we noticed that while 96% (23 out of 24) of the WT mice survived, the Pon3KO mice exhibited significant mortality with only 65% (17 out of 26) survival (Fig. 1B). Furthermore, there were significant increases in liver total cholesterol (40%), cholesterol ester (44%), and unesterified cholesterol (22%) levels, and a decrease in liver triglyceride (56%) level in the Pon3KO mice (Fig. 1C).
Because of the mortality and the altered liver lipids, we carried out more detailed metabolic studies of the mice on the CC diet. When maintained on the CC diet for 16 wk, PON3 mice exhibited significant alterations in plasma lipids and bile acid levels. The Pon3KO mice exhibited significant increases in total cholesterol (320%), very low-density lipoprotein (VLDL)/intermediate-density lipoprotein (IDL)/LDL cholesterol (466%), unesterified cholesterol (896%), triglycerides (267%), and bile acids (273%) in the plasma (Table 1). These lipid effects could contribute to the increased atherosclerosis in Pon3KO mice compared to WT mice.
TABLE 1.
Plasma lipid, glucose, and bile acid levels of Pon3KO and WT mice on a CC diet for 16 wk
| Genotype | n | Triglycerides | Total cholesterol | HDL cholesterol | VLDL/IDL/LDL cholesterol | Unesterified cholesterol | Free fatty acids | Glucose | Bile acids |
|---|---|---|---|---|---|---|---|---|---|
| WT | 16 | 6 (0.5) | 250 (13) | 74 (5) | 176 (9) | 68 (6) | 29 (1) | 128 (6) | 73 (10) |
| Pon3KO | 6 | 22 (9.8)* | 1051 (335)* | 55 (16) | 996 (349)* | 677 (280)* | 29 (6) | 194 (42) | 272 (90)* |
All values shown are means (se) in mg/dl except bile acids values, which are shown in μM.
P < 0.05, Pon3KO vs. WT, using log2 transformed data.
Given that the Pon3KO mice exhibited significantly increased mortality, we also examined evidence of hepatic toxicity using plasma levels of alanine aminotransferase, aspartate aminotransferase, and direct bilirubin (Table 2). Compared to WT mice, the Pon3KO mice exhibited a dramatic increase in all 3 parameters, consistent with increased hepatoxicity in response to the CC diet compared to WT mice.
TABLE 2.
Plasma ALT, AST, and direct bilirubin levels of Pon3KO and WT mice on a CC diet for 16 wk
| Genotype | n | ALT (U/L) | AST (U/L) | Direct bilirubin (mg/dl) |
|---|---|---|---|---|
| WT | 11 | 262 (37) | 216 (17) | 0.19 (0.02) |
| Pon3KO | 6 | 853 (281)* | 935 (312)* | 8.65 (3.99)* |
All values shown are means (SE). ALT, alanine aminotransferase; AST, aspartate aminotransferase.
P < 0.05, Pon3KO vs. WT, using log2 transformed data.
PON3 deficiency alters lipid and bile acid metabolism
To determine the basis of the differences in liver lipids observed in the atherosclerosis studies, we examined the gene expression profiles in liver of several genes involved in either bile acid metabolism or inflammation. As shown in Fig. 1D, in the livers of Pon3KO maintained on the CC diet for 16 wk, the levels of Cyp7a1 mRNA were decreased by about 90% compared to WT mice. The Pon3KO mice also exhibited a substantial decrease in the levels of CYP7A1 protein (Fig. 1E), the rate limiting enzyme in bile acid synthesis. The mRNA levels of the alternative bile acid synthesis pathway mediated by Cyp27a1 (cytochrome P450, family 27, subfamily A, polypeptide 1) were also significantly reduced, as was the bile acid transporter Ntcp (Na+-taurocholate cotransporting polypeptide) (Fig. 1D). On the other hand, the transcriptional repressor of bile acid synthesis Shp (small heterodimer partner), exhibited significantly increased expression (Fig. 1D). Thus, the increased cholesterol in livers of Pon3KO mice fed the CC diet likely results in part from reduced bile acid synthesis.
Atherosclerosis and inflammation in Pon3KO mice
Atherosclerosis is known to be enhanced by local or systemic inflammation (40). To test whether hepatic inflammation might contribute to the increased lesion development that we observed in Pon3KO mice, we examined the expression of Mcp-1, a chemokine, and Il-6, an inflammatory cytokine. Both were substantially increased in Pon3KO mice (Fig. 1D). We also showed that Atf3 (activating transcription factor 3), a member of the unfolded protein response, was increased (Fig. 1D).
Lipid and lipoprotein metabolism in Pon3KO mice
We conducted another 10 wk CC diet feeding study to avoid mortality in the Pon3KO mice and further examine lipoprotein and lipid metabolism. The Pon3KO mice exhibited significantly increased total, VLDL/IDL/LDL cholesterol, and total bile acid levels, and decreased HDL cholesterol levels compared to WT mice (Table 3). The plasma lipoprotein profile was further examined by FPLC. As shown in Fig. 2A, the Pon3KO exhibited higher IDL/LDL and lower HDL cholesterol levels compared to WT mice. Mass spectrometry analysis of liver lipid extracts showed decreased triglycerides and diacylglycerol, but increased phosphatidate levels in the Pon3KO liver compared to those of the controls (Fig. 2B), suggesting lower PAP activity in the Pon3KO liver. We determined that recombinant PON3 does not exhibit PAP activity (data not shown), suggesting that the observed phosphatidate accumulation may be related to altered activity of the lipin PAP enzymes. Lipins 1, 2, and 3 are known to exhibit PAP activity that converts PA to diacylglycerol, the precursor of triglyceride and phospholipid biosynthesis (41). We could not detect Lipin3 mRNA in any liver samples. Quantitative PCR and Western blot analyses revealed no differences in hepatic Lipin1 or Lipin2 mRNA levels (Fig. 2C), or Lipin1 protein levels (data not shown) between Pon3KO and controls. However, Lipin2 protein levels were elevated in the Pon3KO liver (Fig. 2D). Liver PAP activity, on the other hand, was similar between Pon3KO and WT mice (Fig. 2E).
TABLE 3.
Plasma lipid, glucose, and bile acid levels of Pon3KO and WT mice on a CC diet for 10 wk
| Genotype | n | Triglycerides | Total cholesterol | HDL | VLDL/IDL/LDL | Free fatty acids | Bile acids |
|---|---|---|---|---|---|---|---|
| WT | 11 | 9.9 (1.9) | 193 (11) | 62 (3) | 131 (10) | 31 (2) | 48 (5) |
| Pon3KO | 12 | 7.6 (1.3) | 240 (11)* | 50 (3)* | 190 (10)* | 29 (2) | 74 (9)* |
All values shown are means (se) in mg/dl, except bile acids values are shown in μM.
P < 0.01, Pon3KO vs. WT.
Figure 2.

Altered plasma lipoprotein and liver lipid levels in Pon3KO mice fed the CC diet for 10 wk. Pon3KO and WT mice were maintained on the CC diet for 10 wk before tissue/plasma collection. A) Plasma lipoprotein profiles of Pon3KO and WT mice as fractionated by FPLC. B) Liver lipid concentration as determined by mass spectrometry. C) Liver gene expression as determined by qPCR. D) Immunoblotting of mouse lipin-2 protein in liver protein lysate. E) Liver PAP activity was determined as described in Materials and Methods. #P = 0.09, *P < 0.05, **P < 0.01, ***P < 0.001 between the 2 genotype groups.
In mice that were fed the CC diet for 10 wk, we also observed decreased expression of Cyp7a1, Cyp8b1 (cytochrome P450, family 8, subfamily B, polypeptide 1), and Ntcp and increased expression of Shp in the livers of Pon3KO mice compared to those of the WT mice (Fig. 2C). Expression of these genes is known to be regulated by farnesoid X receptor (FXR). In addition, the Pon3KO mice exhibited decreased expression of hepatic enzymes involved in lipogenesis, including Acc, Me1, Scd1 (stearoyl-CoA desaturase-1), and Agpat6 (1-acylglycerol-3-phosphate O-acyltransferase 6) (Fig. 2C). Furthermore, the expression of genes involved in cholesterol homeostasis, including Ldlr and Hmgcr (3-hydroxy-3-methylglutaryl-CoA reductase), was significantly decreased in the Pon3KO livers (Fig. 2C).
Altered bile composition and increased gallstone formation in Pon3KO mice
Gallbladder bile composition was analyzed in Pon3KO and WT mice on the CC diet for 10 wk. The decreased synthesis of bile acids in the liver (Fig. 2C) was accompanied by increased cholesterol and phospholipid levels in the bile of Pon3KO mice (Fig. 3A). In addition, we observed a significant 39% increase in cholesterol levels in the hepatic bile samples collected from the Pon3KO mice (Fig. 3B). As shown in Fig. 3C, Pon3KO mice fed the CC diet exhibit a 58% increase in gallstone weight. Presumably, the decreased bile acid synthesis and increased cholesterol excretion in the Pon3KO mice resulted in an increased ratio of cholesterol to bile acids in the gall bladder, favoring the formation of gallstones.
Figure 3.
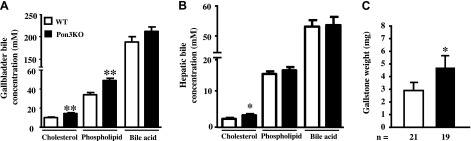
Gallstone formation is increased in Pon3KO mice fed the CC diet. WT and Pon3KO mice were maintained on the CC diet for 10 wk (A, C) or 2 wk (B) before (A) gallbladder bile composition (8 WT and 10 Pon3KO mice), (B) hepatic bile composition (6 WT and 6 Pon3KO mice), and (C) gallstone weight were determined. Symbols as in Fig. 1.
We performed mass spectrometry analysis to examine bile acid composition of plasma, liver, bile, and small intestine samples of Pon3KO and WT mice on the CC diet (Fig. 4). Since the CC diet contains 0.5% cholic acid (CA), the major bile acid found in the samples of these mice was CA (Fig. 4). The rest of the bile acids in these samples are deoxycholic acid, the secondary bile acid generated by the gut bacteria using CA as the substrate, and other endogenously synthesized bile acids including muricholic acid (MCA), chenodeoxycholic acid (CDCA), and ursodeoxycholic acid (UDCA) (Fig. 4). In contrast to humans, UDCA is a primary bile acid in rodents (42). In the plasma, there are significantly increased levels of all of the bile acid species in the Pon3KO mice compared to those of the WT mice (Fig. 4A). In the liver, the total bile acid content in the Pon3KO mice was not different from the WT mice (Fig. 4B). However, the levels of several unconjugated bile acids, including β-MCA, α-MCA, CDCA, and UDCA, were significantly decreased in the Pon3KO compared to those of the WT mice (Fig. 4C). This might have been caused by the decreased expression of bile acid synthesis genes observed in the Pon3KO liver (Fig. 1D), or by decreased uptake of bile acid returning from the small intestine as a result of decreased expression of the bile acid transporter, Ntcp. In the bile samples we observed a significant decrease in deoxycholic acid levels and a trend of decreased levels of other bile acids in the Pon3KO compared to WT mice (Fig. 4D). Finally, in the small intestine, while the levels of CA and deoxycholic acid, presumably derived from the diet, were not different between the 2 groups of mice, the levels of endogenously synthesized bile acids, MCA, CDCA, and UDCA, were significantly decreased in the Pon3KO mice compared to those of the WT mice (Fig. 4E).
Figure 4.

Bile acid composition analysis. WT and Pon3KO mice were maintained on the CC diet for 16 wk before various tissues were collected for bile acid composition analysis. Shown are data from (A) plasma, (B, C) liver, (D) bile, and (E) small intestine. Data are the sum of both conjugated and unconjugated bile acids, except for (C), where only levels of unconjugated bile acids are shown. Symbols as in Fig. 1.
Pon3KO mice exhibit altered gene expression in small intestine and kidney when maintained on the CC diet
The mRNA levels of FXR target genes, Mrp2 (multidrug resistance protein 2) and Abcg8 (ATP-binding cassette subfamily G member 8) were significantly decreased in the ileum samples of Pon3KO mice fed the CC diet compared to those of the WT mice (Supplemental Fig. S2A). On the other hand, the expression of other FXR-regulated genes including Shp, Ostα and Ostβ (organic solute transporter α and β), I-babp (ileal bile acid-binding protein), Fgf15 (fibroblast growth factor 15), Abcg5 (ATP-binding cassette subfamily G member 5), and Asbt (apical sodium-dependent bile salt transporter) was not significantly different between these 2 groups of mice (Supplemental Fig. S2A). In kidney, the expression of FXR target genes, Shp and Mrp2, was significantly increased in the Pon3KO compared to WT mice (Supplemental Fig. S2B), probably resulted from the elevated circulating bile acid levels found in the Pon3KO mice (Fig. 4A). However, the expression of other FXR target genes, including Ostα, Ostβ, and Asbt was not different between the 2 genotype groups (Supplemental Fig. S2B).
Pon3KO mice exhibit elevated plasma total bile acid levels when maintained on a Western diet
We also determined plasma total bile acid levels of Pon3KO and WT mice maintained on either low fat chow diet or a Western diet. While there was no significant difference in plasma total bile acid levels between Pon3KO and WT mice on chow diet (Supplemental Fig. S3A), Pon3KO exhibited significantly elevated levels of plasma total bile acid compared to WT mice when fed the Western diet (Supplemental Fig. S3A). Hepatic expression levels of genes involved in bile acid homeostasis, including Bsep (bile salt export pump), Cyp7a1, and Ntcp, were not different between Pon3KO and WT mice, whereas Shp mRNA level was significantly elevated in the Pon3KO mice when maintained on the Western diet (Supplemental Fig. S3B).
PON3 is localized in MAM and influences mitochondrial respiration and hepatic inflammation
Previous studies had suggested that PON2 may influence mitochondrial functions (6, 43). Therefore, we sought to test whether the effects of PON3 deficiency on bile acid metabolism might involve various mitochondrial functions. We began by examining the subcellular localization of PON3. WT and Pon3KO livers were subjected to subcellular fractionation on density gradients to produce fractions enriched in ER, crude mitochondria (CM), highly purified mitochondria (PM), and MAM. We used Western blot analysis to examine the presence in these fractions of PON3, calnexin (MAM and ER marker), VDAC, and cytochrome c (mitochondrial markers). As can be seen, PON3 is present in crude mitochondrial fractions but not in highly purified fractions, and it colocalizes with calnexin in ER and MAM, with higher abundance of PON3 in MAM than ER (Fig. 5A). The PON3 antibody we used exhibits no reactivity with any of the fractions in Pon3KO mice, indicating its specificity (Fig. 5A).
Figure 5.

PON3 is localized in MAM and influences mitochondrial function. A) Distribution of PON3 protein in various organelles isolated from the WT and Pon3KO livers. CM, crude mitochondria; ER, endoplasmic reticulum; MAM, mitochondria-associated membrane; PM, pure mitochondria; T, total lysate. Distribution patterns of calnexin, VDAC, and cytochrome c are also shown. B, C) Complex II–IV-dependent respiration of mitochondria isolated from the liver of WT and Pon3KO mice fed a chow diet. D–F) Mice were maintained on the CC diet for 10 wk before liver samples were collected for determination of (D) mitochondrial superoxide levels, (E) mitochondrial complex II + III activity, and (F) extent of apoptosis as measured by TUNEL assay. *P < 0.05, ***P < 0.001 between the 2 genotype groups.
To examine whether the Pon3KO mice exhibited alterations in mitochondrial functions, we used a Seahorse Bioscience (Lowell, MA, USA) XF24-3 instrument to measure OCR, an indicator of mitochondrial respiration. In the presence of succinate and rotenone (for measurement of respiration driven by complex II–IV activity), Pon3KO mitochondria exhibited significantly lower OCR in response to ADP (state 3) and FCCP (state 3u), indicating decreased mitochondrial respiration (Fig. 5B, C).
Mitochondria are a major source of superoxide (44), and we investigated whether superoxide might contribute to the increased inflammation observed in mice fed the CC diet. As shown in Fig. 5D, Pon3KO mice exhibited about twice the level of superoxide compared to WT mice. Complexes II–III are essential parts of mitochondrial electron transport chain. As shown in Fig. 5E, Pon3KO mice exhibited a significantly decreased complex II–III activity compared to WT mice when fed the CC diet, suggesting impaired mitochondrial respiration. Because the Pon3KO mice on the CC diet exhibited increased superoxide and inflammatory gene expression, we wondered whether there might be an increase in apoptosis. As shown in Fig. 5F, Pon3KO mice exhibited a 66% increase in TUNEL-positive cells compared to WT mice.
Increased obesity in Pon3KO mice due to impaired mitochondrial respiration and decreased fatty acid oxidation
After a 10 wk feeding of a Western diet, the Pon3KO mice exhibited a significant 37% increase in body weight compared to WT mice (Fig. 6A), despite similar daily food consumption between the 2 groups (data not shown). Furthermore, the weights of gonadal, retroperitoneal, and subcutaneous fat pads of the Pon3KO mice were significantly increased compared to those of the WT mice (Fig. 6B). The average size of adipocytes of the gonadal fat pad was also significantly increased by 25% in Pon3KO compared to WT mice (Fig. 6C). Thus, PON3 deficiency results in increased obesity.
Figure 6.
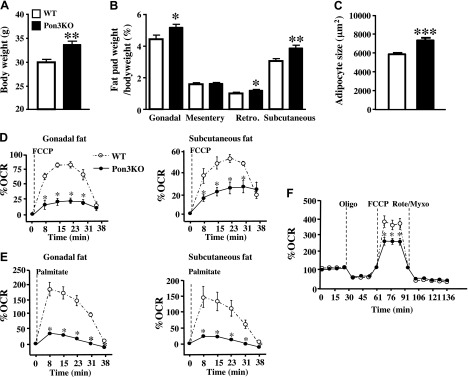
Increased obesity and impaired mitochondrial function in the white adipose tissues of Pon3KO mice. The 5.5-mo-old Pon3KO and WT mice were fed a Western diet for 10 wk before (A) body weight and (B) fat pad weight (expressed as percentage of body weight) were determined (n = 13 for each genotype). Retro, retroperitoneal. C) The average size of adipocytes of the gonadal fat pad is shown. Gonadal fat pads from 6 mice of each group were examined. The oxygen consumption rate of gonadal and subcutaneous fat pads was examined in the presence of (D) FCCP and (E) palmitate. F) OCR of differentiated white adipocytes derived from the Pon3KO and WT mice was examined at baseline and after the sequential injection of oligomycin, FCCP, and rotenone/myxothiazol. Symbols as in Fig. 1.
We then examined mitochondrial respiration of fat pads isolated from the 2 groups of mice. Tissues from the gonadal and subcutaneous fat pads were assessed for their oxygen consumption rate after injection of the uncoupler FCCP. By revealing the maximal respiration capacity, FCCP can be used to uncover disrupted electron transport chain activity. Pon3KO mice showed decreased OCR in response to FCCP in the fat depots compared to those of the WT mice (Fig. 6D). The fact that the Pon3KO tissues cannot increase respiration under stress to the same degree as WT mice suggests that the Pon3KO mice have impaired mitochondrial respiration. This defect was confirmed when tissues were challenged with palmitate to induce fatty acid oxidation. The OCR response to palmitate was substantially decreased in the fat depots from Pon3KO mice (Fig. 6E).
To confirm the mitochondrial respiration defect in the Pon3KO fat pads, we isolated stromal vascular cells from the subcutaneous fat pads. Cells were differentiated for 7 d and were found to harbor a white adipocyte phenotype with large lipid droplet–filled cells (data not shown). OCR was monitored during a mitochondrial assay where oligomycin, FCCP, and a mix of rotenone and myxothiazol were sequentially injected after the baseline measures. This allowed for an estimation of the contribution of ATP-linked mitochondrial oxygen consumption, nonmitochondrial respiration, and maximal mitochondrial respiratory capacity. OCR response from the Pon3KO adipocyte were not different from the WT cells after oligomycin or rotenone/myxotiazol (Fig. 6F), suggesting no difference in ATPase activity or nonmitochondrial respiration. In contrast, the response to FCCP was substantially lower in Pon3KO compared to WT cells (Fig. 6F), confirming the defect in mitochondrial respiration capacity.
DISCUSSION
We report the generation of Pon3KO mice and their characterization to further understand the functions of this enzyme. Several novel and unexpected findings emerged. First, PON3 deficiency clearly influences bile acid metabolism by an as yet unknown mechanism. The increased gallstone formation observed in the Pon3KO mice is probably caused by the decreased bile acid secretion and increased cholesterol excretion into bile. Second, PON3 is enriched in the mitochondria-associated membrane fraction of the hepatocytes, and PON3 deficiency has a clear impact on mitochondrial function and oxidative stress. The increased diet-induced obesity observed in the Pon3KO mice may be explained in part by impaired mitochondrial function and fatty acid oxidation observed in the fat pads isolated from the Pon3KO mice. Third, PON3 deficiency results in increased superoxide levels and apoptosis in liver as well as increased expression of inflammatory genes. PON3 deficiency also leads to a proatherogenic lipoprotein profile, including elevated IDL/LDL and decreased HDL cholesterol levels, when fed the CC diet. The proatherogenic lipoprotein profile and increased inflammation in the Pon3KO mice likely lead to the increased atherosclerotic lesion formation compared to WT mice. On the other hand, we cannot rule out the notion that PON3 deficiency in the artery wall may also contribute to atherogenesis because mouse Pon3 is expressed in the macrophages (45). However, there is no evidence that mouse Pon3 is expressed by endothelial or smooth muscle cells.
A recent publication described generation of another line of PON3 null mice independently; Kempster et al. (46) concluded that PON3 deficiency led to embryonic lethality in mice. In the present study, we showed a complete loss of PON3 at the mRNA, protein, and activity (based on lovastatinase activity) levels, yet we did not observe any embryonic lethality of Pon3KO mice (data not shown). In fact, PON3 null allele was inherited in a Mendelian fashion with a 1:2:1 genotype ratio of WT, heterozygous, and homozygous PON3 mutant mice being observed in the offspring of a cross between PON3 heterozygous mice (data not shown). There are 2 plausible explanations for the difference in lethality observed between these 2 lines of Pon3KO mice. First, different genetic backgrounds have been shown to influence the phenotype and lethality of mice harboring null mutations (47, 48). Our PON3 null mutation was generated using an embryonic stem cell line derived from the 129X1/SvJ background. The null mutation was then introduced onto the C57BL/6J genetic background by 10 generations of backcrossing. Kempster et al. (46) obtained their Pon3KO mice from Texas A&M Institute for Genomic Medicine (TIGM). According to information obtained from the TIGM website, their Pon3KO mice were on a mixed background of 129S5/SvEvBrd and C57BL/6. This difference in genetic background between the 2 Pon3KO mice may have caused difference in lethality, but the rather closely related backgrounds suggest that this is unlikely. Second, a spontaneous null mutation of a passenger gene near the Pon3 locus may have occurred during embryonic stem cell manipulation, and this mutation of the passenger gene could be the cause of embryonic lethality observed in Pon3KO mice obtained from the TIGM. An example of this has been previously reported (49), and this seems the more likely explanation. A detailed expression analysis of passenger genes near the Pon3 locus of the TIGM Pon3KO mice could help to determine whether these mice harbor additional null mutations that could be the cause of embryonic lethality.
Pon3KO mice exhibited increased plasma total bile acid levels when maintained on a CC diet containing cholesterol and cholic acid (Tables 1 and 3). This increase of plasma bile acids observed in the Pon3KO mice was likely due to the deceased Ntcp expression (Figs. 1D and 2C), a transporter that mediates bile acid portal uptake. Liver gene expression pattern showed increased activation of FXR in the Pon3KO mice, as evidenced by increased expression of Shp and Bsep and decreased expression of Cyp7a1, compared to WT mice when fed the CC diet (Fig. 1D). However, we failed to observe higher total concentrations of bile acids in the livers of Pon3KO mice compared to WT mice (Fig. 4B). This could be because we did not perfuse the livers before tissue collection, which could obscure the difference between the 2 groups of liver samples. The significantly decreased expression of Cyp7a in the livers of Pon3KO mice could be due to the actions of the FXR-SHP-dependent pathway (50) and of FXR-independent pathways such as the inhibitory effects of inflammatory cytokines on the expression of Cyp7a1 (51). Because there was increased expression of inflammatory genes in the livers of Pon3KO mice (Fig. 1D), this could contribute to further down-regulation of Cyp7a1 gene expression in these mice. The elevated plasma total bile acid levels and significantly increased hepatic expression of Shp in the Pon3KO mice fed the Western diet suggest that altered bile acid metabolism in these mice can occur without the feeding of cholic acid, a bile acid known to cause inflammation in the liver (52). The Western diet, which is high in fat and contains a moderate amount of cholesterol (0.15%), is known to induce hepatic inflammatory gene expression as well (53). However, we did not observe significant differences in the hepatic expression of inflammatory genes, including Mcp-1, between Pon3KO and WT mice fed the Western diet (data not shown), suggesting the preferentially increased inflammation in the livers of Pon3KO mice only occurs in the presence of cholic acid feeding.
We observed increased mortality in the Pon3KO mice when maintained on the CC diet for 16 wk (Fig. 1B). The cause of death in these mice was likely cholestasis caused by increased gallstone formation (Fig. 3C) that led to bile duct obstruction. In fact, we observed jaundice in critically ill Pon3KO mice (data not shown). Significantly elevated plasma bilirubin, total bile acids, and total cholesterol levels were also observed in the Pon3KO mice that survived the 16 wk feeding of the CC diet (Tables 1 and 2), consistent with the notion that these mice might have cholestasis.
Our study showed that Pon3KO mice have a proatherogenic lipoprotein profile with elevated IDL/LDL and decreased HDL levels when maintained on the CC diet (Fig. 2A). We also observed increased cholesterol and decreased triglyceride levels in the livers of the Pon3KO mice fed the CC diet (Fig. 1C). These changes are likely caused by the altered bile acid metabolism observed in the Pon3KO mice. Decreased Cyp7a1 protein levels observed in the livers of Pon3KO mice lead to decreased conversion of cholesterol to bile acid, causing increased cholesterol accumulation in the liver. Increased cholesterol accumulation in the livers of the Pon3KO mice may then result in decreased hepatic LDL receptor expression (Fig. 2C) that leads to decreased clearance of IDL/LDL from circulation. Bile acids are known to lower liver triglyceride accumulation through inhibition of expression of lipogenic genes by a pathway involving FXR, SHP, and SREBP-1c (54). Likewise, we observed decreased expression of lipogenic genes (Fig. 2C) and decreased triglyceride accumulation (Fig. 1C) in the livers of Pon3KO mice fed the CC diet.
To our knowledge, ours is the first report to localize PON3 to MAM (Fig. 5A). MAM is the physical association location between ER and mitochondria. The functions of MAM include phospholipid synthesis, lipid transport, calcium homeostasis, and control of apoptosis (55, 56). It remains unclear how PON3 influences MAM functions. However, we demonstrated that PON3 deficiency leads to impaired respiratory function and increased superoxide levels in the mitochondria isolated from the livers of Pon3KO mice (Fig. 5). Hydrophobic bile acids are known to induce reactive oxygen species generation, and release of cytochrome c and apoptosis-inducing factors by the mitochondria (57). Our previous findings demonstrated the role of PON3 in protecting against superoxide formation in mitochondria and superoxide-induced apoptosis in cultured cells (13). We reason that the increased hepatic bile acid levels caused by cholic acid feeding lead to the significantly increased mitochondrial superoxide levels (Fig. 5D) and increased apoptosis (Fig. 5F) observed in the Pon3KO mice. Furthermore, the accumulation of cholesterol has been shown to increase ER stress, inflammation, oxidative stress, and apoptosis in various cell types (58–64). The increased accumulation of cholesterol in the livers of CC diet–fed Pon3KO mice (Fig. 1C) could also contribute to increased oxidative stress, inflammation, and apoptosis observed in these mice.
PON3 deficiency also leads to impaired mitochondrial respiration and fatty acid oxidation in the fat pads and adipocyte cultures derived from the Pon3KO mice (Fig. 6). The mitochondrial dysfunction in the white adipose tissues of Pon3KO mice could in part contribute to increased adiposity observed in the Pon3KO mice compared to WT mice (Fig. 6). However, our study could not exclude the notion that lack of PON3 in other tissues might contribute to the increased adiposity observed in Pon3KO mice. In addition, inflammation is known to contribute to obesity as well. For example, mice that are deficient in TNF-α or protease-activated receptor 2, a substantial contributor to inflammation, are both protected against obesity (65, 66). Thus, the increased inflammation associated with PON3 deficiency could also contribute to obesity.
In summary, our data demonstrate that PON3 deficiency leads to impaired hepatic mitochondrial function, increased oxidative stress, inflammation, and cell death when the Pon3KO mice were challenged with a CC diet. In addition, our results suggest a role for PON3 in bile acid metabolism. However, because Pon3KO mice on a low-fat chow diet did not exhibit altered bile acid levels, it is also possible that the PON3 effect on bile acid metabolism is the result of liver injury caused by the CC diet. This study demonstrates a protective role of PON3 against atherosclerosis, gallstone disease, and obesity.
Supplementary Material
Acknowledgments
The authors thank Yu-Rong Xia, Yi-Shou Shi, Yonghong Meng, Judy Wu, Xu-Ping Wang, Zhiqiang Zhou, and Sarada Charugundla for excellent technical support. This study was supported by the following grants: U.S. National Institutes of Health (NIH) HL030568-26A1 (to A.J.L. and D.M.S.), and R01 HL074214 and RR019232 (to D.A.F.); National Center for Research Resources S10RR026744 and NIH P01 HL028481 (to K.R.), and 2 RO1 HL71776 (to S.T.R. and D.M.S.).
Glossary
- apoE
apolipoprotein E
- CA
cholic acid
- CC
cholate-cholesterol
- CDCA
chenodeoxycholic acid
- Cyp7a1
cytochrome P450, family 7, subfamily A, polypeptide 1
- ER
endoplasmic reticulum
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- FXR
farnesoid X receptor
- IDL
intermediate-density lipoprotein
- KO
knockout
- MAM
mitochondria-associated membrane
- MCA
muricholic acid
- NLS
neutral loss scanning
- Ntcp
Na+-taurocholate cotransporting polypeptide
- OCR
oxygen consumption rate
- Ost
organic solute transporter
- PA
phosphatidic acid
- PAP
phosphatidic acid phosphatase
- Pon3
paraoxonase-3
- Shp
small heterodimer partner
- TIGM
Texas A&M Institute for Genomic Medicine
- UDCA
ursodeoxycholic acid
- VDAC
voltage-dependent anion channel
- VLDL
very low-density lipoprotein
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Hassett C., Richter R. J., Humbert R., Chapline C., Crabb J. W., Omiecinski C. J., Furlong C. E. (1991) Characterization of cDNA clones encoding rabbit and human serum paraoxonase: the mature protein retains its signal sequence. Biochemistry 30, 10141–10149 [DOI] [PubMed] [Google Scholar]
- 2.Mackness M. I., Walker C. H. (1988) Multiple forms of sheep serum A–esterase activity associated with the high-density lipoprotein. Biochem. J. 250, 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothem L., Hartman C., Dahan A., Lachter J., Eliakim R., Shamir R. (2007) Paraoxonases are associated with intestinal inflammatory diseases and intracellularly localized to the endoplasmic reticulum. Free Radic. Biol. Med. 43, 730–739 [DOI] [PubMed] [Google Scholar]
- 4.Stoltz D. A., Ozer E. A., Recker T. J., Estin M., Yang X., Shih D. M., Lusis A. J., Zabner J. (2009) A common mutation in paraoxonase-2 results in impaired lactonase activity. J. Biol. Chem. 284, 35564–35571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horke S., Witte I., Wilgenbus P., Krüger M., Strand D., Förstermann U. (2007) Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation 115, 2055–2064 [DOI] [PubMed] [Google Scholar]
- 6.Devarajan A., Bourquard N., Hama S., Navab M., Grijalva V. R., Morvardi S., Clarke C. F., Vergnes L., Reue K., Teiber J. F., Reddy S. T. (2011) Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 14, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng C. J., Wadleigh D. J., Gangopadhyay A., Hama S., Grijalva V. R., Navab M., Fogelman A. M., Reddy S. T. (2001) Paraoxonase-2 is a ubiquitously expressed protein with antioxidant properties and is capable of preventing cell-mediated oxidative modification of low density lipoprotein. J. Biol. Chem. 276, 44444–44449 [DOI] [PubMed] [Google Scholar]
- 8.Shih D. M., Xia Y. R., Yu J. M., Lusis A. J. (2010) Temporal and tissue-specific patterns of Pon3 expression in mouse: in situ hybridization analysis. Adv. Exp. Med. Biol. 660, 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Précourt L. P., Amre D., Denis M. C., Lavoie J. C., Delvin E., Seidman E., Levy E. (2011) The three-gene paraoxonase family: physiologic roles, actions and regulation. Atherosclerosis 214, 20–36 [DOI] [PubMed] [Google Scholar]
- 10.Draganov D. I., Stetson P. L., Watson C. E., Billecke S. S., La Du B. N. (2000) Rabbit serum paraoxonase 3 (PON3) is a high density lipoprotein–associated lactonase and protects low density lipoprotein against oxidation. J. Biol. Chem. 275, 33435–33442 [DOI] [PubMed] [Google Scholar]
- 11.Reddy S. T., Wadleigh D. J., Grijalva V., Ng C., Hama S., Gangopadhyay A., Shih D. M., Lusis A. J., Navab M., Fogelman A. M. (2001) Human paraoxonase-3 is an HDL-associated enzyme with biological activity similar to paraoxonase-1 protein but is not regulated by oxidized lipids. Arterioscler. Thromb. Vasc. Biol. 21, 542–547 [DOI] [PubMed] [Google Scholar]
- 12.Shih D. M., Xia Y. R., Wang X. P., Wang S. S., Bourquard N., Fogelman A. M., Lusis A. J., Reddy S. T. (2007) Decreased obesity and atherosclerosis in human paraoxonase 3 transgenic mice. Circ. Res. 100, 1200–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schweikert E. M., Devarajan A., Witte I., Wilgenbus P., Amort J., Förstermann U., Shabazian A., Grijalva V., Shih D. M., Farias-Eisner R., Teiber J. F., Reddy S. T., Horke S. (2012) PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell Death Differ. 19, 1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozer E. A., Pezzulo A., Shih D. M., Chun C., Furlong C., Lusis A. J., Greenberg E. P., Zabner J. (2005) Human and murine paraoxonase 1 are host modulators of Pseudomonas aeruginosa quorum-sensing. FEMS Microbiol. Lett. 253, 29–37 [DOI] [PubMed] [Google Scholar]
- 15.Teiber J. F., Horke S., Haines D. C., Chowdhary P. K., Xiao J., Kramer G. L., Haley R. W., Draganov D. I. (2008) Dominant role of paraoxonases in inactivation of the Pseudomonas aeruginosa quorum-sensing signal N-(3-oxododecanoyl)-l-homoserine lactone. Infect. Immun. 76, 2512–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa L. G., Li W. F., Richter R. J., Shih D. M., Lusis A., Furlong C. E. (1999) The role of paraoxonase (PON1) in the detoxication of organophosphates and its human polymorphism. Chem. Biol. Interact. 119-120, 429–438 [DOI] [PubMed] [Google Scholar]
- 17.Mackness B., Durrington P., McElduff P., Yarnell J., Azam N., Watt M., Mackness M. (2003) Low paraoxonase activity predicts coronary events in the Caerphilly Prospective Study. Circulation 107, 2775–2779 [DOI] [PubMed] [Google Scholar]
- 18.Shih D. M., Gu L., Xia Y. R., Navab M., Li W. F., Hama S., Castellani L. W., Furlong C. E., Costa L. G., Fogelman A. M., Lusis A. J. (1998) Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 394, 284–287 [DOI] [PubMed] [Google Scholar]
- 19.Shih D. M., Xia Y. R., Wang X. P., Miller E., Castellani L. W., Subbanagounder G., Cheroutre H., Faull K. F., Berliner J. A., Witztum J. L., Lusis A. J. (2000) Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 275, 17527–17535 [DOI] [PubMed] [Google Scholar]
- 20.Tward A., Xia Y. R., Wang X. P., Shi Y. S., Park C., Castellani L. W., Lusis A. J., Shih D. M. (2002) Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 106, 484–490 [DOI] [PubMed] [Google Scholar]
- 21.Ng C. J., Bourquard N., Grijalva V., Hama S., Shih D. M., Navab M., Fogelman A. M., Lusis A. J., Young S., Reddy S. T. (2006) Paraoxonase-2 deficiency aggravates atherosclerosis in mice despite lower apolipoprotein-B-containing lipoproteins: anti-atherogenic role for paraoxonase-2. J. Biol. Chem. 281, 29491–29500 [DOI] [PubMed] [Google Scholar]
- 22.Bhattacharyya T., Nicholls S. J., Topol E. J., Zhang R., Yang X., Schmitt D., Fu X., Shao M., Brennan D. M., Ellis S. G., Brennan M. L., Allayee H., Lusis A. J., Hazen S. L. (2008) Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 299, 1265–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarvik G. P., Rozek L. S., Brophy V. H., Hatsukami T. S., Richter R. J., Schellenberg G. D., Furlong C. E. (2000) Paraoxonase (PON1) phenotype is a better predictor of vascular disease than is PON1(192) or PON1(55) genotype. Arterioscler. Thromb. Vasc. Biol. 20, 2441–2447 [DOI] [PubMed] [Google Scholar]
- 24.Rozenberg O., Rosenblat M., Coleman R., Shih D. M., Aviram M. (2003) Paraoxonase (PON1) deficiency is associated with increased macrophage oxidative stress: studies in PON1-knockout mice. Free Radic. Biol. Med. 34, 774–784 [DOI] [PubMed] [Google Scholar]
- 25.Mehrabian M., Qiao J. H., Hyman R., Ruddle D., Laughton C., Lusis A. J. (1993) Influence of the apoA-II gene locus on HDL levels and fatty streak development in mice. Arterioscler. Thromb. 13, 1–10 [DOI] [PubMed] [Google Scholar]
- 26.Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 27.Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 28.Han X., Gross R. W. (2001) Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal. Biochem. 295, 88–100 [DOI] [PubMed] [Google Scholar]
- 29.Bowden J. A., Albert C. J., Barnaby O. S., Ford D. A. (2011) Analysis of cholesteryl esters and diacylglycerols using lithiated adducts and electrospray ionization–tandem mass spectrometry. Anal. Biochem. 417, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han X., Gross R. W. (2005) Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 24, 367–412 [DOI] [PubMed] [Google Scholar]
- 31.Dwyer J. R., Donkor J., Zhang P., Csaki L. S., Vergnes L., Lee J. M., Dewald J., Brindley D. N., Atti E., Tetradis S., Yoshinaga Y., De Jong P. J., Fong L. G., Young S. G., Reue K. (2012) Mouse lipin-1 and lipin-2 cooperate to maintain glycerolipid homeostasis in liver and aging cerebellum. Proc. Natl. Acad. Sci. USA 109, E2486–E2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang P., O’Loughlin L., Brindley D. N., Reue K. (2008) Regulation of lipin-1 gene expression by glucocorticoids during adipogenesis. J. Lipid Res. 49, 1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mardones P., Quiñones V., Amigo L., Moreno M., Miquel J. F., Schwarz M., Miettinen H. E., Trigatti B., Krieger M., VanPatten S., Cohen D. E., Rigotti A. (2001) Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I–deficient mice. J. Lipid Res. 42, 170–180 [PubMed] [Google Scholar]
- 34.Argmann, C. A., Houten, S. M., Champy, M. F., and Auwerx, J. (2006) Lipid and bile acid analysis. In Current Protocols in Molecular Biology (Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K., eds.), pp 29B.2.1–29B.2.24, J. Wiley, New York [DOI] [PubMed] [Google Scholar]
- 35.Vergnes L., Beigneux A. P., Davis R., Watkins S. M., Young S. G., Reue K. (2006) Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 47, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P. (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 37.Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N. (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 6, e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin L. J., Lau E., Singh H., Vergnes L., Tarling E. J., Mehrabian M., Mungrue I., Xiao S., Shih D., Castellani L., Ping P., Reue K., Stefani E., Drake T. A., Bostrom K., Lusis A. J. (2012) ABCC6 localizes to the mitochondria-associated membrane. Circ. Res. 111, 516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang, K., ed. (2008) Adipose Tissue Protocols, 2nd ed., Humana Press, New York [Google Scholar]
- 40.Libby P. (2012) Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Csaki L. S., Dwyer J. R., Fong L. G., Tontonoz P., Young S. G., Reue K. (2013) Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog. Lipid Res. 52, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayin S. I., Wahlström A., Felin J., Jäntti S., Marschall H. U., Bamberg K., Angelin B., Hyötyläinen T., Orešič M., Bäckhed F. (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235 [DOI] [PubMed] [Google Scholar]
- 43.Altenhöfer S., Witte I., Teiber J. F., Wilgenbus P., Pautz A., Li H., Daiber A., Witan H., Clement A. M., Förstermann U., Horke S. (2010) One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J. Biol. Chem. 285, 24398–24403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenblat M., Draganov D., Watson C. E., Bisgaier C. L., La Du B. N., Aviram M. (2003) Mouse macrophage paraoxonase 2 activity is increased whereas cellular paraoxonase 3 activity is decreased under oxidative stress. Arterioscler. Thromb. Vasc. Biol. 23, 468–474 [DOI] [PubMed] [Google Scholar]
- 46.Kempster S. L., Belteki G., Licence D., Charnock-Jones D. S., Smith G. C. (2012) Disruption of paraoxonase 3 impairs proliferation and antioxidant defenses in human A549 cells and causes embryonic lethality in mice. Am. J. Physiol. Endocrinol. Metab. 302, E103–E107 [DOI] [PubMed] [Google Scholar]
- 47.Akahoshi N., Kobayashi C., Ishizaki Y., Izumi T., Himi T., Suematsu M., Ishii I. (2008) Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum. Mol. Genet. 17, 1994–2005 [DOI] [PubMed] [Google Scholar]
- 48.Lu S. Y., Jin Y., Li X., Sheppard P., Bock M. E., Sheikh F., Duckworth M. L., Cattini P. A. (2010) Embryonic survival and severity of cardiac and craniofacial defects are affected by genetic background in fibroblast growth factor-16 null mice. DNA Cell Biol. 29, 407–415 [DOI] [PubMed] [Google Scholar]
- 49.Westrick R. J., Mohlke K. L., Korepta L. M., Yang A. Y., Zhu G., Manning S. L., Winn M. E., Dougherty K. M., Ginsburg D. (2010) Spontaneous Irs1 passenger mutation linked to a gene-targeted SerpinB2 allele. Proc. Natl. Acad. Sci. USA 107, 16904–16909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. (2008) Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 [DOI] [PubMed] [Google Scholar]
- 51.Davis R. A., Miyake J. H., Hui T. Y., Spann N. J. (2002) Regulation of cholesterol-7alpha-hydroxylase: BAREly missing a SHP. J. Lipid Res. 43, 533–543 [PubMed] [Google Scholar]
- 52.Vergnes L., Phan J., Strauss M., Tafuri S., Reue K. (2003) Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J. Biol. Chem. 278, 42774–42784 [DOI] [PubMed] [Google Scholar]
- 53.Kassel K. M., Owens A. P. III, Rockwell C. E., Sullivan B. P., Wang R., Tawfik O., Li G., Guo G. L., Mackman N., Luyendyk J. P. (2011) Protease-activated receptor 1 and hematopoietic cell tissue factor are required for hepatic steatosis in mice fed a Western diet. Am. J. Pathol. 179, 2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayashi T., Rizzuto R., Hajnoczky G., Su T. P. (2009) MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Brito O. M., Scorrano L. (2010) An intimate liaison: spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 29, 2715–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sokol R. J., Dahl R., Devereaux M. W., Yerushalmi B., Kobak G. E., Gumpricht E. (2005) Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J. Pediatr. Gastroenterol. Nutr. 41, 235–243 [DOI] [PubMed] [Google Scholar]
- 58.Yao P. M., Tabas I. (2000) Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J. Biol. Chem. 275, 23807–23813 [DOI] [PubMed] [Google Scholar]
- 59.Yao P. M., Tabas I. (2001) Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J. Biol. Chem. 276, 42468–42476 [DOI] [PubMed] [Google Scholar]
- 60.Devries-Seimon T., Li Y., Yao P. M., Stone E., Wang Y., Davis R. J., Flavell R., Tabas I. (2005) Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 171, 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hung Y. C., Hong M. Y., Huang G. S. (2006) Cholesterol loading augments oxidative stress in macrophages. FEBS Lett. 580, 849–861 [DOI] [PubMed] [Google Scholar]
- 62.Wang N., Tabas I., Winchester R., Ravalli S., Rabbani L. E., Tall A. (1996) Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J. Biol. Chem. 271, 8837–8842 [DOI] [PubMed] [Google Scholar]
- 63.Lu X., Liu J., Hou F., Liu Z., Cao X., Seo H., Gao B. (2011) Cholesterol induces pancreatic β cell apoptosis through oxidative stress pathway. Cell Stress Chaperones 16, 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Subramanian S., Goodspeed L., Wang S., Kim J., Zeng L., Ioannou G. N., Haigh W. G., Yeh M. M., Kowdley K. V., O’Brien K. D., Pennathur S., Chait A. (2011) Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor–deficient mice. J. Lipid Res. 52, 1626–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim J., Iyer A., Liu L., Suen J. Y., Lohman R. J., Seow V., Yau M. K., Brown L., Fairlie D. P. (2013) Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism. FASEB J. 27, 4757–4767 [DOI] [PubMed] [Google Scholar]
- 66.Ventre J., Doebber T., Wu M., MacNaul K., Stevens K., Pasparakis M., Kollias G., Moller D. E. (1997) Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes 46, 1526–1531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.