

Abstract
Cell penetrating peptides (CPPs) and their synthetic mimics (CPPMs) represent a class of molecules that facilitate the intracellular delivery of various cargo. Previous studies indicated that the presence of aromatic functionalities improved CPPM activity. Given that aromatic functionalities play prominent roles in membrane biology and participate in various π-interactions, we explored whether these interactions could be optimized for improved CPPM activity. CPPMs were synthesized by ring-opening metathesis polymerization using monomers that contained aromatic rings substituted with electron donating and electron withdrawing groups and covered an electrostatic potential range from -29.69 to +15.57 kcal/mol. These groups altered the quadrupole moments of the aromatic systems and were used to test if such structural modifications changed CPPM activity. CPPMs were added to dye-loaded vesicles and the release of carboxyfluorescein was monitored as a function of polymer concentration. Changes in the effective polymer concentration to release 50% of the dye (EC50) were monitored. Results from this assay showed that the strength of the electron donating and electron withdrawing groups incorporated in the CPPMs did not alter polymer EC50 values or activity. This suggests that other design parameters may have a stronger impact on CPPM activity. In addition, these results indicate that a wide range of aromatic groups can be incorporated without negatively impacting polymer activity.
Introduction
Cell penetrating peptides (CPPs) and their synthetic mimics (CPPMs) represent a unique class of molecules that is capable of crossing biological membranes.[1] The peptides are generally short, cationic sequences rich in arginine and/or lysine residues, with some containing hydrophobic residues such as leucine, phenylalanine, or tryptophan.[1b, 1g, 1i, j] They derive inspiration from proteins with translocation abilities, such as HIV-1 Tat and Antennapedia Homeodomain protein.[2] It has been shown that the cation-rich domains of these proteins, referred to as protein transduction domains (PTDs), are primarly responsibile for their uptake abilities.[2a, 3] Many studies have highlighted the ability of CPP(M)s to facilitate the intracellular delivery of various cargo, including, but not limited to, small molecules, siRNA, pDNA, and proteins via covalent or non-covalent interactions.[1b, c, 1e, 1g-j, 4] Although their mechanism of uptake is debated in the literature, various forms of endocytosis, macropinocytosis, protein-dependent translocation, and energy-independent translocation are involved in the internalization process.[5]
In efforts to elucidate the mechanisms of CPP(M) uptake and assess the structural components of CPP(M)s necessary for uptake, model vesicle membrane studies have frequently been used.[6] Vesicle experiments represent a simpler system for evaluating energy-independent methods of transduction than using cells, where it is difficult to decouple various methods of cellular uptake. Previously, Matile and coworkers have used model vesicle systems to show that polyarginine, a widely used CPP, requires hydrophobic counterions to efficiently cross lipid membranes.[6b, 7] For these studies, lipids were swollen in a solution of carboxyfluorescein, which is a hydrophilic, anionic dye that self-quenches at high concentrations, and dye release was monitored as a function of peptide concentration. Changes in peptide activity were assessed by calculating the effective concentrations to release 50% of the dye (EC50). Similar assays have also been used by Almeida and coworkers to explore CPP internalization mechanisms.[8] The hydrophobic counterions selected for Matile and coworkers' studies were said to help mask the overall cationic charge of the peptides to aid in transduction, a process referred to as activation.[6b, 7] Although these studies showed that bulky aromatic activators, such as pyrene butyrate, outperformed aliphatic activators, the roles of hydrophobicity and aromaticity were not fully understood.
Motivated by these studies, our lab previously developed a series of oxanorbornene imide-based CPPMs to assess the effect of hydrophobicity on CPPM activity.[6d, e] Instead of using external activators, the hydrophobic components were chemically incorporated into the polymeric structures to yield self-activating polymers.[6d, e] These polymers were correctly predicted to outperform their counterparts that only contained cationic residues.[6a, 6d, e] Initially, various aliphatic chains were incorporated into the CPPMs to assess the effect of chain length on activity.[6e] These results were evaulated by assessing differences in reported EC50 values from vesicle dye release assays.[7b] Although polymer activity improved by increasing the alkyl chain lengths from one carbon to four carbons, longer alkyl chains were less water soluble and thus led to poorer performance.[6e]
Another series of polymers was designed to evaluate the impact of various aromatic, cyclic non-aromatic, and alkyl hydrophobic moieties of similar hydrophobicity on polymer activity.[6d] This was done to gain a better understanding of the interplay between hydrophobicity and aromaticity. Aromaticity was the cornerstone of that report because of the significant role it plays in protein-membrane interactions. The aromatic amino acids tyrosine and tryptophan are present as part of aromatic belts that flank either end of transmembrane proteins.[9] These residues sit at the interface between the hydrophobic core and the more hydrophilic external environment to enhance stability at those regions.[9] Although not typically present in aromatic belts, phenylalanine has also been shown to aid in anchoring proteins in the membrane.[10] All three of these aromatic amino acids have been shown to provide favorable energies of insertion into membranes.[10] It was further reasoned that aromatic moieties are ideal for incorporation into CPPMs because such residues are found in many CPPs such as Penetratin, Pep-1, and MPG and have been shown in some cases to be critical for uptake.[1e, 11] Experiments in which aromatic residues in Tat and Penetratin were replaced with non-aromatic hydrophobic residues led to a reduction in cellular interalization efficiencies.[1e, 11]
Using HPLC retention times to assess relative hydrophobicity of the polymer side chains[12], these values were compared to the polymer EC50 values to illustrate that the effects of hydrophobicity and aromaticity could be distinguished. Through these studies, it was suggested that aromatic hydrophobic moieties were superior activators.[6d] Similar results were obtained by Matile and coworkers when they monitored dye release of polyarginine with various external activators.[7d]
Given these observations and the different electronic properties of tyrosine, phenylalanine, and tryptophan (Figure S1, Table S1), the role of aromaticity in CPP(M) activity was studied by exploring the effect that changes in quadrupole moments have on these systems. The flat, planar structures of these aromatic rings and their associated quadrupole moments are thought to enable various π-interactions, such as π-π, π-cation, π-anion, and π-polar interactions within the cellular environment that can aid in membrane interactions.[10a, 13] Since the quadrupole moment collects the electron density on the face of these planar, aromatic rings, it was hypothesized that by strengthening or weakening this phenomenon, the corresponding π-membrane interactions would provide additional handles for tuning of CPPM activity. Specifically, this was attempted by incorporating electron donating and electron withdrawing groups into the aromatic systems as a way to alter the electron density of the ring system. Although nature offers an electrostatic potential range for its aromatic amino acids between -31.41 kcal/mol (Trp) and -23.48 (Tyr) kcal/mol, by using synthetic systems, it was possible to examine a much wider electrostatic potential window of -29.69 kcal/mol (Scheme 1, R= b = CH3) to +15.57 kcal/mol (Scheme 2, R′= c = NO2). All values are summarized in Table S1-2.
Scheme 1.
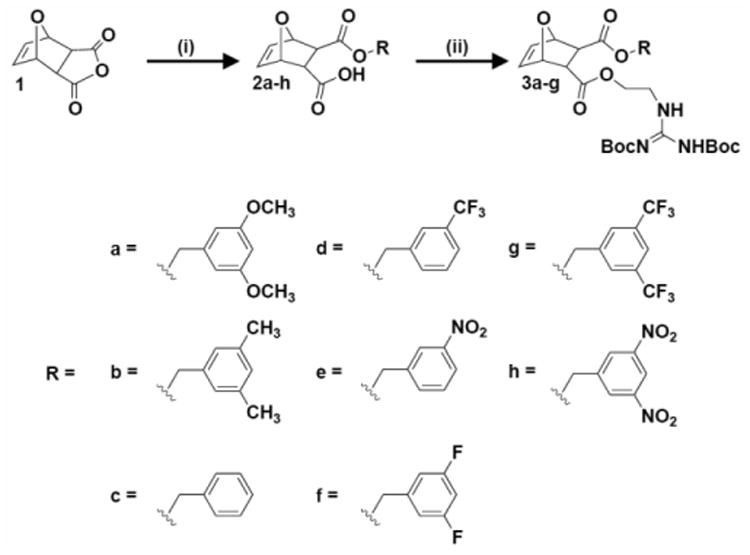
Synthesis of diester monomers containing π-rich and π-poor aromatic rings. i)R-OH, DMAP, CH2Cl2, RT, overnight; ii) 1,3-di-boc-2-(2-hydroxyethyl)guanidine, EDC, DMAP, CH2Cl2, 0°C to RT, overnight.
Scheme 2.
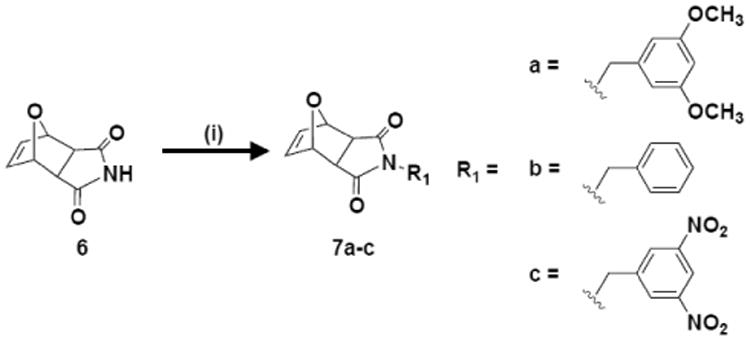
Synthesis of imide monomers containing π-rich and π-poor aromatic rings. i) R1-OH, PPh3, DIAD, THF, RT, 18 hr.
As part of this study, π-rich and π-poor CPPMs were designed and synthesized based on both the diester and imide ROMP scaffolds. CPPMs based on the diester system were synthesized since the dual-functional monomers offer greater potential for polymeric structure variations. Also, CPPMs based on the imide scaffold were synthesized as a direct comparison to polymers from previous hydrophobicity structure activity relationships (SARs) with model membranes.[6d, e] These CPPMs were designed to contain π-rich and π-poor aromatic functionalities in order to assess the role of π-interactions in tuning membrane activity.
Results and Discussion
Monomer Synthesis
Diester monomers were synthesized using a two-step process, as depicted in Scheme 1. These procedures were adapted from previously described methods with modifications.[4a, 14] In brief, oxanorbornene anhydride (1) was ring-opened using various aromatic alcohols (a-h) and DMAP to yield the half-ester intermediates.
Half-esters 2a-f were then further reacted with 1,3-di-boc-2-(2-hydroxyethyl)guanidine using EDC coupling conditions to yield monomers 3a-f. Half-esters 2g-h were not used for monomer synthesis because they proved to be unstable in solution at room temperature. As shown in Figure 1A, half-esters 2g-h underwent a spontaneous retro-Diels-Alder reaction to yield 4g-h and furan (5). This was demonstrated by isolating the retro-Diels-Alder product, 4g, using column chromatography and verifying its chemical composition using 1H NMR, 13C NMR, and mass spectrometry (MS). Retro-Diels-Alder product 4h proved more difficult to isolate because of additional nitro-based impurities.
Figure 1.
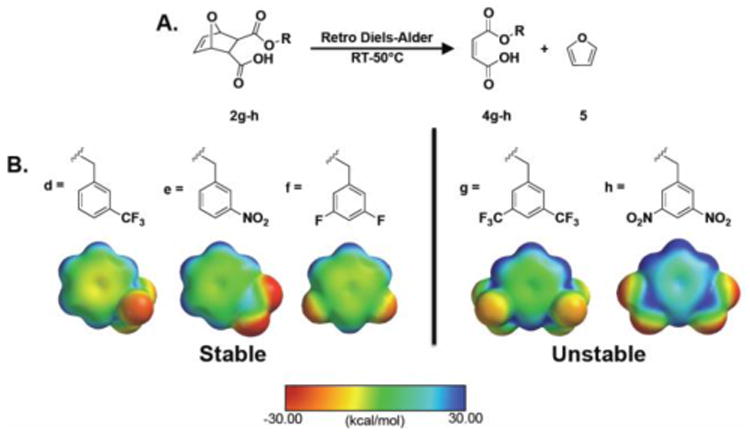
Stability of π-poor monomers. A) Retro-Diels-Alder reaction that occurs for monomers 4g-h. B) Stable and unstable π-poor monomer aromatic groups with their corresponding electrostatic potential maps. The range for electrostatic potential was set between -30.00 and 30.00 kcal/mol. The color scale bar reflects this range with red representing electron rich surfaces and blue representing electron poor surfaces. Surfaces were calculated at the HF level using the 3-21G* basis set.
However, peaks for the retro-Diels-Alder product were observed in the 1H NMR of 2h (see SI). Since 2a-f did not appear to undergo the retro-Diels-Alder reaction, it was hypothesized that this reaction was related to the electron withdrawing substituents attached to the aromatic rings. To investigate this, all π-poor aromatic rings were modeled using Spartan molecular modeling software as shown in Figure 1B. The most electron poor (most blue in color) aromatic rings were the rings associated with the unstable half esters. From this and a study by Nanjappan and Czarnik, it was concluded that electron withdrawing groups destabilized Diels-Alder adducts and accelerated the retro-Diels-Alder reaction.[15]
Since 2a-f did not appear to undergo the retro-Diels-Alder reaction, it was hypothesized that this reaction was related to the electron withdrawing substituents attached to the aromatic rings. To investigate this, all π-poor aromatic rings were modeled using Spartan molecular modeling software as shown in Figure 1B. The most electron poor (most blue in color) aromatic rings were the rings associated with the unstable half esters. From this and a study by Nanjappan and Czarnik, it was concluded that electron withdrawing groups destabilized Diels-Alder adducts and accelerated the retro-Diels-Alder reaction.[15] 2g-h were not pursued for monomer formation because the retro-Diels-Alder impurities 4g-h have the same reactive functional groups (-COOH, C=C) as 2a-f. All stable half-esters and monomers were characterized by 1H NMR, 13C NMR, and MS. In terms of electrostatic potential values, the six stable monomers covered an electrostatic potential range from -4.66 kcal/mol (Scheme 1, R= e= NO2) to +29.69 kcal/mol (Scheme 1, R= b= CH3). Electrostatic potential values are summarized in Table S2. All characterization data is provided in the supporting information.
Imide monomers were synthesized using a one-step process adapted from Som et.al., as illustrated in Scheme 2.[6d, e] Unlike the diester system, there were no issues with stability for the imide system and no retro-Diels-Alder products were observed. All monomers were characterized by 1H NMR, 13C NMR, and MS. In terms of electrostatic potential values for the aromatic groups incorporated, all stable monomers covered an electrostatic potential range from -18.10 kcal/mol (Scheme 2, R1 = a = OCH3) to 15.57 kcal/mol (Scheme 2, R1 = c = NO2). These monomers expand the negative end of the electrostatic potential range so that in total the monomer design spans -29.69 to +15.57 kcal/mol as summarized in Table S2.
Polymers were synthesized using ROMP with Grubbs 3rd generation catalyst, as illustrated in Schemes 3-5.
Scheme 3.
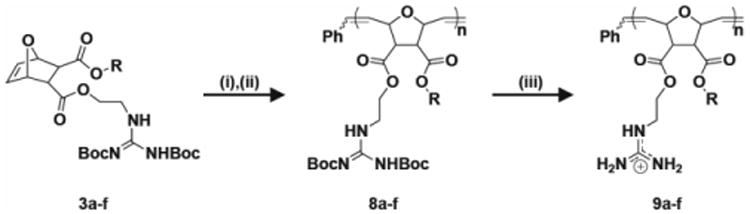
Synthesis of diester homopolymers containing π-rich and π-poor aromatic rings. i) Dichloro-di(3-bromopyridino)-N,N′-Dimesitylenoimidazolino-Ru=CHPh (G3) catalyst, CH2Cl2, RT, 45 min; ii) Ethyl vinyl ether, RT, overnight; iii) TFA/CH2Cl2 (1:1), RT, overnight. Products 9a-f further purified by dialysis with molecular weight cut-off : 2,000 g/mol. All polymers were synthesized with n=20. R was defined in Scheme 1.
Scheme 5.
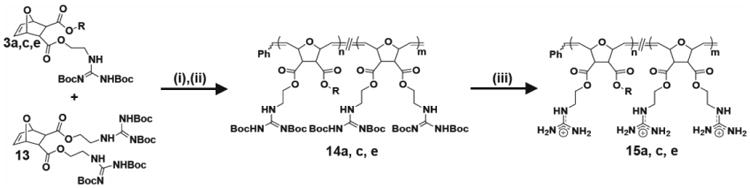
Synthesis of diester random copolymers containing π-rich and π-poor aromatic rings. i) Dichloro-di(3-bromopyridino)-N,N′-Dimesitylenoimidazolino- Ru=CHPh (G3) catalyst, CH2Cl2, RT, 45 min; ii) Ethyl vinyl ether, RT, overnight; iii) TFA/CH2Cl2 (1:1), RT, overnight. Products 15a,c,e further purified by dialysis with molecular weigth cut-off: 1,000 g/mol. All polymers were synthesized with n=8 and m=12. R was defined in Scheme 1.
All boc-protected polymers (8a-f, 10a-c, 13a,c,e) were characterized by 1H NMR to assess chemical composition and gel permeation chromatography (GPC) to assess relative molecular weights. GPC data is summarized in Table 1.
Table 1.
A. Molecular characteristics of π-rich and π-poor CPPMs.
| Diester Homopolymers | ||
|---|---|---|
|
| ||
| CPPM | Mna | Đa |
| 8a | 11,600 | 1.05 |
| 8b | 11,200 | 1.05 |
| 8c | 11,300 | 1.05 |
| 8d | 12,600 | 1.05 |
| 8e | 11,400 | 1.05 |
| 8f | 11,500 | 1.05 |
| B. | |||
|---|---|---|---|
|
| |||
| Imide and Diester Random Copolymers | |||
|
| |||
| CPPM | n:mb | Mna | Đa |
| 11a | 56:44 | 16,200 | 1.06 |
| 11b | 55:45 | 13,700 | 1.07 |
| 11c | 58:42 | 17,000 | 1.06 |
| 14a | 38:62 | 10,600 | 1.10 |
| 14c | 40:60 | 12,100 | 1.08 |
| 14e | 39:61 | 10,700 | 1.14 |
Number average molecular weight (Mn) and polydispersity indices (Đ=Mw/Mn) determined by GPC using polymethyl methacrylate (PMMA) standards for diester polymers and polystyrene standards for the imide polymers using tetrahydrofuran (THF) as the eluent and toluene as the flow marker.
Ratio of residues, where n represents the percentage of hydrophobic residues and m represents the percentage of guanidine-containing residues.
Polymers were subsequently deprotected using trifluoroacetic acid (TFA) and CH2Cl2 (1:1) overnight.[4a, 6d, e, 14] TFA was removed by azeotropic distillation with methanol. Diester polymers were dialyzed for three days in water using membranes with a molecular weight cut-off of 2,000 g/mol for homopolymers and 1,000 g/mol for random copolymers. All polymers were then dissolved in water and lyophilized to yield dry 9a-f, 12a-c, and 15a,c,e.
All polymers were tested using a vesicle dye release assay to assess relative polymer activity using a fluorescence plate reader.[6a, 6c-e] This high throughput screening method enabled the testing of all samples in a 12-well plate at the same time. Carboxyfluorescein (CF) filled phosphatidylcholine (PC) or PC/phosphatidylserine (PS) vesicles were prepared as described in the supporting information and then used for these experiments.
For this dye release assay, the baseline fluorescence (F0) of Tris saline buffer with carboxyfluorescein filled vesicles was determined. Then, polymer solutions (in DMSO) of varying concentrations were added to the vesicle-containing solutions. After 10 minutes, the fluorescence intensity (Ff,0) was measured again. Triton X-100 (5% in DMSO) was then added to each experimental solution to lyse the vesicles and release all of the dye. The final fluorescence measurement (Ft) was taken after five minutes. Complete experimental details for these experiments can be found in the supporting information (Figure S18). The results were normalized according to the baseline and Triton controls to yield fractional dye release (If) according to Equation 1.
| Equation 1 |
For Hill analysis, If was plotted against polymer concentration, c, and fit to the Hill equation, Equation 2, to give the EC50, where If,0 and If, max are the minimum and maximum value of If obtained for each well, respectively.
| Equation 2 |
The first set of polymers tested were 9a-f and 12a-c, since both sets of polymers had comparable hydrophobic and hydrophilic contents (roughly 1:1) but different backbone compositions. A summary of the EC50, If,max, and n values obtained from testing 9a-f and 12a-c with PC vesicles are displayed in Table 2.
Table 2.
EC50, Ymax, and Hill coefficient n values for diester homopolymer and imide random copolymer-based CPPM activity using 100 nm PC large unilamellar vesicles swelled with carboxyfluorescein.
| CPPM | EC50a (nM) | Ymaxb | nc |
|---|---|---|---|
| 9a | 8.27 ± 0.84 | 0.89 ± 0.00 | 2.01 ± 0.04 |
| 9b | 7.36 ± 1.87 | 0.92 ± 0.02 | 1.59 ± 0.04 |
| 9c | 9.07 ± 0.79 | 0.90 ± 0.01 | 1.93 ± 0.02 |
| 9d | 7.48 ± 0.50 | 0.91 ± 0.01 | 1.43 ± 0.01 |
| 9e | 9.93 ± 0.50 | 0.92 ± 0.02 | 1.50 ± 0.06 |
| 9f | 8.63 ± 0.52 | 0.85 ± 0.02 | 1.79 ± 0.01 |
| 12a | 10.09 ± 1.96 | 0.87 ± 0.01 | 1.59 ± 0.07 |
| 12b | 11.85 ± 0.19 | 0.84 ± 0.05 | 1.63 ± 0.14 |
| 12c | 10.64 ± 0.21 | 0.72 ± 0.02 | 1.93 ± 0.15 |
Effective concentrations (EC50) needed to reach Ymax/2,
Maximum fraction of carboxyfluorescein released compared to total dye released upon addition of Triton-X 100.
n is the Hill coefficient. Standard deviation from three independent experiments is reported.
In addition, representative overlays of π-rich and π-poor polymers from both sets of polymers are shown in Figure 2. All EC50 values were similar (7-12 nM) and, with the exception of the lower If,max for 12c, the Hill plots were also almost identical. Only EC50 values that differ over several orders of magnitiude represent significant changes, as observed in previous studies where the aliphatic hydrophobic group incorporated was changed from a methyl group (EC50 = 6.4 μM) to a butyl group (EC50 = 0.003 μM).[6e]
Figure 2.

Diester vs. imide Hill plots for π-rich CPPMs 9a and 12a (red) and π-poor CPPMs 9e and 12c (blue) using 100 nm PC large unilamellar vesicles swelled with carboxyfluorescein. Data was fit to the Hill Equation and If represents the fraction of dye released. Solid lines represent diester-based CPPMs and dashed lines represent imide random copolymer-based CPPMs.
Within each series, it was determined that the nature of the π-rich or π-poor aromatic rings incorporated did not have a significant impact on CPPM activity. By comparing CPPMs from both series, it was determined that the nature of the CPPM backbone also had a negligible effect on CPPM activity. Although many of our previous SAR studies were based on the imide system, in this paper all further testing was conducted with the diester system since backbone architecture had little impact on results and this system offers more options for structural tuning.
To further probe the effect of π-electronics on CPPM activity, dye-swelled vesicles were prepared by adding negatively charged PS lipids (20 mol%) to PC lipids. This lipid composition was selected in order to exploit potentially favorable π-anion interactions that can occur between anionic lipids and electron deficient aromatic systems while also capitalizing on electron repulsions between anionic lipids and electron-rich aromatic systems.[16] Based on the nature of π-interactions, it was anticipated that CPPM activity would trend based on π-electron density, with π-poor CPPMs exhibiting better activity due to favorable π-anion interactions. In contrast, π-rich polymers were expected to have weaker activity due to electron repulsion between the anionic lipids and the electron-rich aromatic rings. For these studies, polymers 9a, c, and e were tested and the results were compared to those obtained for PC vesicles. A summary of the EC50, If,max, and n values can be found in Table S3. In addition, representative overlays of π-rich and π-poor polymers tested with PC and PC/PS vesicles can be found in Figure 3. EC50 values for polymers 9a, c, and e tested with PC/PS vesicles were similar to those obtained from studies with PC vesicles. There was also little difference in the Hill Plots for these polymers, regardless of the nature of the π-rich or π-poor aromatic ring incorporated or type of vesicles used for the study.
Figure 3.

Anionic vs. zwitterionic vesicle Hill plots for π-rich polymer 9a (red) and π-poor polymer 9e (blue) using two types of 100 nm large unilamellar vesicles swelled with carboxyfluorescein: PC (solid lines) and PC/PS (80/20, dashed lines). Data was fit to the Hill Equation. If represents the fraction of dye released.
Based on these results, a set of diester random copolymers was designed that had a more dilute hydrophobic content to be sure that the results observed were not due to the CPPM hydrophobic content being too high. The CPPMs designed are shown in Scheme 4. A summary of the EC50, If,max, and n values can be found in Table S4 for PC and Table S5 for PC/PS vesicles. In addition, representative overlays for the diester random copolymers as they compare to their corresponding diester homopolymers for PC and PC/PS vesicles can be observed in Figures 4 and 5, respectively.
Scheme 4.
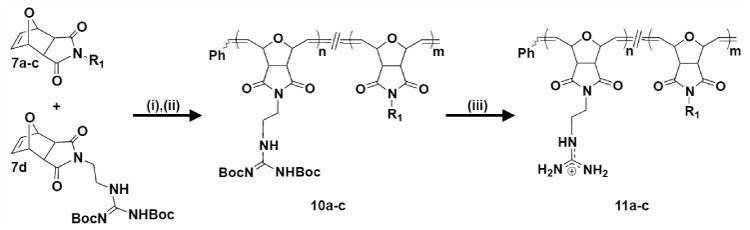
Synthesis of imide random copolymers containing π-rich and π-poor aromatic rings. i) Dichloro-di(3-bromopyridino)-N,N′-Dimesitylenoimidazolino- Ru=CHPh (G3) catalyst, CH2Cl2, RT, 45 min; ii) Ethyl vinyl ether, RT, overnight; iii) TFA/CH2Cl2 (1:1), RT, overnight. All polymers were synthesized with n=20 and m=20. R1 was defined in Scheme 2.
Figure 4.

Homopolymer vs. random copolymer Hill plots for π-rich polymers 9a and 15a (red) and π-poor polymers 9e and 15e (blue) using 100 nm PC large unilamellar vesicles swelled with carboxyfluorescein. Data was fit to the Hill Equation and If represents the fraction of dye released. Solid lines represent diester homopolymers and dashed lines represent diester random copolymers.
Figure 5.

Homopolymer vs. random copolymer Hill plots for π-rich polymers 9a and 15a (red) and π-poor polymers 9e and 15e (blue) using 100 nm PC/PS (80/20) large unilamellar vesicles swelled with carboxyfluorescein. Data was fit to the Hill Equation and If represents the fraction of dye released. Solid lines represent diester homopolymers and dashed lines represent random copolymers.
EC50 values for 14a, c, and e were similar to those obtained for 9a, c, and e when tested with PC vesicles and the overlays of the Hill plots in Figure 4 further suggest that there was little difference between the activity of the diester homopolymers and the less hydrophobic random copolymers. When tested with PC/PS vesicles, there was a slight increase in EC50 values for the random copolymers as compared to the homopolymers and there was a noticeable shift in the Hill plots in Figure 5. However, the shift was about the same for all diester random copolymers and thus attributed to the lower hydrophobic content and not due to the π-electronics of the system. Since no trend was observed, it was concluded that π-electronics do not play a major role in CPPM activity. Alternatively, it is possible that the assay used here does not have the fidelity to distinguish the subtleties of π –interactions despite the fact that these same assays previously illustrated that adding hydrophobicity improves activity, with aromatic groups outperforming aliphatic groups.[6d, e] Even though we were able to synthesize a series of polymers that contained aromatic groups with an electrostatic potential range of -29.69 to 15.57 kcal/mol, it is also possible that the structural modifications made to the polymers may not have been significant enough to impact CPPM activity. While it is likely that overall CPPM hydrophobicity and cationic charge are more influential design parameters than π-electronics, these results indicate that a wide range of aromatic groups can be incorporated into the polymer structures with limited impact on CPPM activity. From a design standpoint, this opens up additional ways in which CPPMs can be modified without inhibiting their performance.
Conclusion
For this study, aromatic groups containing electron donating and electron withdrawing groups were chemically incorporated into CPPM structures as a way to tune π-interactions. It was hypothesized that tuning the quadrupole moments of the aromatic rings would provide additional control over CPPM activity. When synthesizing the monomers for these studies, it was established through small molecule synthesis that highly electron-withdrawing groups could not be chemically incorporated into the diester versions of our oxanorbornene monomers because it spontaneously induced a retro-Diels-Alder reaction.
Within the synthetically accessible series, vesicle dye release experiments were performed as a way to determine the CPPMs' EC50 values and assess their relative activities. It was shown using PC vesicles that polymer backbone did not impact activity for the π-rich/π-poor CPPMs and that the electron donating or electron withdrawing groups as well as the relative hydrophobic content did not impact activity either. Diester CPPMs were also tested with PC vesicles containing 20% PS anionic lipids in efforts to more thoroughly understand π-membrane interactions.
However, it would seem that only overall hydrophobicity dictated polymer activity and not the incorporated electron donating or electron withdrawing groups. Although it is possible that the assay used could not distinguish the subtleties of π-interactions, it is also likely that the structural modifications made to the polymers were not significant enough to impact CPPM activity, despite the fact that molecules containing aromatic rings with an electrostatic potential range of -29.69 kcal/mol to 15.57 +kcal/mol were explored. This suggests that other design parameters, such as overall hydrophobicity and cationic charge, have a greater impact on CPPM activity. The results also indicate that a wide range of aromatic groups can be incorporated into the polymer structures with limited impact on CPPM activity. This is encouraging from a design standpoint as it expands potential functionality without impacting activity. Understanding these design principles will help guide the development of future CPPMs.
Experimental Section
All experimental details are provided in the supporting information.
Supplementary Material
Acknowledgments
The work was funded by the NIH (T32 GMO8515) and NSF (CHE-0910963). The authors would like to thank Dr. Hitesh D. Thaker, Dr. Jing Jiang, Prof. Ke Zhang, and Dr. Michael Lis for invaluable scientific discussions and help with half-ester and monomer purifications. The authors would also like to thank Ms. Catherine N. Walker, Mr. Ilker Ozay, and Mr. Nicholas Posey for their feedback on early drafts of this manuscript. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Facility, which is supported in part by NSF.
Footnotes
Supporting Information: Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.a) Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J Am Chem Soc. 2009;131:16401–+. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]; c) Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Proc Natl Acad Sci U S A. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lindgren M, Langel U. Cell-Penetrating Peptides: Methods and Protocols. 2011;683:3–19. doi: 10.1007/978-1-60761-919-2_1. [DOI] [PubMed] [Google Scholar]; e) Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]; f) Opalinska JB, Gewirtz AM. Nat Rev Drug Discovery. 2002;1:503–514. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]; g) Patel LN, Zaro JL, Shen WC. Pharm Res. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]; h) Sgolastra F, deRonde BM, Sarapas JM, Som A, Tew GN. Acc Chem Res. 2013;46:2977–2987. doi: 10.1021/ar400066v. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Bechara C, Sagan S. FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 2.a) Joliot A, Pernelle C, Deagostinibazin H, Prochiantz A. Proc Natl Acad Sci U S A. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Green M, Loewenstein PM. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]; c) Frankel AD, Pabo CO. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 3.a) Derossi D, Joliot AH, Chassaing G, Prochiantz A. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]; b) Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Proc Natl Acad Sci U S A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vives E, Brodin P, Lebleu B. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 4.a) Tezgel AO, Telfer JC, Tew GN. Biomacromolecules. 2011;12:3078–3083. doi: 10.1021/bm200694u. [DOI] [PubMed] [Google Scholar]; b) Yin LC, Tang HY, Kim KH, Zheng N, Song ZY, Gabrielson NP, Lu H, Cheng JJ. Angew Chem, Int Ed Engl. 2013;52:9182–9186. doi: 10.1002/anie.201302820. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gabrielson NP, Lu H, Yin LC, Kim KH, Cheng JJ. Mol Ther. 2012;20:1599–1609. doi: 10.1038/mt.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tang HY, Yin LC, Kim KH, Cheng JJ. Chem Sci. 2013;4:3839–3844. doi: 10.1039/C3SC51328A. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Stanzl EG, Trantow BM, Vargas JR, Wender PA. Acc Chem Res. 2013;46:2944–2954. doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tezgel AO, Gonzalez-Perez G, Telfer JC, Osborne BA, Minter LM, Tew GN. Mol Ther. 2013;21:201–209. doi: 10.1038/mt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Futaki S. Adv Drug Deliv Rev. 2005;57:547–558. doi: 10.1016/j.addr.2004.10.009. [DOI] [PubMed] [Google Scholar]; b) Mishra A, Lai GH, Schmidt NW, Sun VZ, Rodriguez AR, Tong R, Tang L, Cheng J, Deming TJ, Kamei DT, Wong GC. Proc Natl Acad Sci U S A. 2011;108:16883–16888. doi: 10.1073/pnas.1108795108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zaro JL, Shen WC. Exp Cell Res. 2005;307:164–173. doi: 10.1016/j.yexcr.2005.02.024. [DOI] [PubMed] [Google Scholar]; d) Thoren PE, Persson D, Isakson P, Goksor M, Onfelt A, Norden B. Biochem Biophys Res Commun. 2003;307:100–107. doi: 10.1016/s0006-291x(03)01135-5. [DOI] [PubMed] [Google Scholar]; e) Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]; f) Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 6.a) Hennig A, Gabriel GJ, Tew GN, Matile S. J Am Chem Soc. 2008;130:10338–10344. doi: 10.1021/ja802587j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sakai N, Matile S. J Am Chem Soc. 2003;125:14348–14356. doi: 10.1021/ja037601l. [DOI] [PubMed] [Google Scholar]; c) Schmidt NW, Lis M, Zhao K, Lai GH, Alexandrova AN, Tew GN, Wong GC. J Am Chem Soc. 2012;134:19207–19216. doi: 10.1021/ja308459j. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Som A, Reuter A, Tew GN. Angew Chem, Int Ed Engl. 2012;51:980–983. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]; e) Som A, Tezgel AO, Gabriel GJ, Tew GN. Angew Chem, Int Ed Engl. 2011;50:6147–6150. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 7.a) Perret F, Nishihara M, Takeuchi T, Futaki S, Lazar AN, Coleman AW, Sakai N, Matile S. J Am Chem Soc. 2005;127:1114–1115. doi: 10.1021/ja043633c. [DOI] [PubMed] [Google Scholar]; b) Nishihara M, Perret F, Takeuchi T, Futaki S, Lazar AN, Coleman AW, Sakai N, Matile S. Org Biomol Chem. 2005;3:1659–1669. doi: 10.1039/b501472g. [DOI] [PubMed] [Google Scholar]; c) Sakai N, Futaki S, Matile S. Soft Matter. 2006;2:636–641. doi: 10.1039/b606955j. [DOI] [PubMed] [Google Scholar]; d) Sakai N, Takeuchi T, Futaki S, Matile S. Chembiochem. 2005;6:114–122. doi: 10.1002/cbic.200400256. [DOI] [PubMed] [Google Scholar]
- 8.a) Almeida PF, Pokorny A. Biochemistry. 2009;48:8083–8093. doi: 10.1021/bi900914g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Almeida PF, Pokorny A. Methods Mol Biol. 2010;618:155–169. doi: 10.1007/978-1-60761-594-1_11. [DOI] [PubMed] [Google Scholar]
- 9.a) Yau WM, Wimley WC, Gawrisch K, White SH. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]; b) Killian JA, von Heijne G. Trends Biochem Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 10.a) White SH, Wimley WC. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]; b) Wimley WC, White SH. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 11.a) Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morris MC, Deshayes S, Heitz F, Divita G. Biol Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]; c) Caesar CE, Esbjorner EK, Lincoln P, Norden B. Biochemistry. 2006;45:7682–7692. doi: 10.1021/bi052095t. [DOI] [PubMed] [Google Scholar]; d) Yezid H, Konate K, Debaisieux S, Bonhoure A, Beaumelle B. J Biol Chem. 2009;284:22736–22746. doi: 10.1074/jbc.M109.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Gabriel GJ, Pool JG, Som A, Dabkowski JM, Coughlin EB, Muthukumar M, Tew GN. Langmuir. 2008;24:12489–12495. doi: 10.1021/la802232p. [DOI] [PubMed] [Google Scholar]; b) Thaker HD, Sgolastra F, Clements D, Scott RW, Tew GN. J Med Chem. 2011;54:2241–2254. doi: 10.1021/jm101410t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Braun P, von Heijne G. Biochemistry. 1999;38:9778–9782. doi: 10.1021/bi990923a. [DOI] [PubMed] [Google Scholar]; b) Strandberg E, Morein S, Rijkers DT, Liskamp RM, van der Wel PC, Killian JA. Biochemistry. 2002;41:7190–7198. doi: 10.1021/bi012047i. [DOI] [PubMed] [Google Scholar]; c) John Haynes W, Zhou XL, Su ZW, Loukin SH, Saimi Y, Kung C. FEBS Lett. 2008;582:1514–1518. doi: 10.1016/j.febslet.2008.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gromiha MM, Suwa M. Int J Biol Macromol. 2005;35:55–62. doi: 10.1016/j.ijbiomac.2004.12.001. [DOI] [PubMed] [Google Scholar]; e) Gromiha MM. Biophys Chem. 2003;103:251–258. doi: 10.1016/s0301-4622(02)00318-6. [DOI] [PubMed] [Google Scholar]; f) Arbuzova A, Wang L, Wang J, Hangyas-Mihalyne G, Murray D, Honig B, McLaughlin S. Biochemistry. 2000;39:10330–10339. doi: 10.1021/bi001039j. [DOI] [PubMed] [Google Scholar]
- 14.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J Am Chem Soc. 2008;130:9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nanjappan P, Czarnik AW. J Org Chem. 1986;51:2851–2853. [Google Scholar]
- 16.Schottel BL, Chifotides HT, Dunbar KR. Chem Soc Rev. 2008;37:68–83. doi: 10.1039/b614208g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.