

Abstract
Action potential (AP) firing in mouse chromaffin cells (MCCs) is mainly sustained by Cav1.3 L-type channels that drive BK and SK currents and regulate the pacemaking cycle. As secretory units, CCs optimally recruit Ca2+ channels when stimulated, a process potentially dependent on the modulation of the AP waveform. Our previous work has shown that a critical determinant of AP shape is voltage-gated sodium channel (Nav) channel availability. Here, we studied the contribution of Nav channels to firing patterns and AP shapes at rest (−50 mV) and upon stimulation (−40 mV). Using quantitative RT-PCR and immunoblotting, we show that MCCs mainly express tetrodotoxin (TTX)-sensitive, fast-inactivating Nav1.3 and Nav1.7 channels that carry little or no Na+ current during slow ramp depolarizations. Time constants and the percentage of recovery from fast inactivation and slow entry into closed-state inactivation are similar to that of brain Nav1.3 and Nav1.7 channels. The fraction of available Nav channels is reduced by half after 10 mV depolarization from −50 to −40 mV. This leads to low amplitude spikes and a reduction in repolarizing K+ currents inverting the net current from outward to inward during the after-hyperpolarization. When Nav channel availability is reduced by up to 20% of total, either by TTX block or steady depolarization, a switch from tonic to burst firing is observed. The spontaneous occurrence of high frequency bursts is rare under control conditions (14% of cells) but leads to major Ca2+-entry and increased catecholamine release. Thus, Nav1.3/Nav1.7 channel availability sets the AP shape, burst-firing initiation and regulates catecholamine secretion in MCCs. Nav channel inactivation becomes important during periods of high activity, mimicking stress responses.
Key points
Mouse chromaffin cells (MCCs) of the adrenal medulla possess fast-inactivating Nav channels whose availability alters spontaneous action potential firing patterns and the Ca2+-dependent secretion of catecholamines.
Here, we report MCCs expressing large densities of neuronal fast-inactivating Nav1.3 and Nav1.7 channels that carry little or no subthreshold pacemaker currents and can be slowly inactivated by 50% upon slight membrane depolarization.
Reducing Nav1.3/Nav1.7 availability by tetrodotoxin or by sustained depolarization near rest leads to a switch from tonic to burst-firing patterns that give rise to elevated Ca2+-influx and increased catecholamine release.
Spontaneous burst firing is also evident in a small percentage of control MCCs.
Our results establish that burst firing comprises an intrinsic firing mode of MCCs that boosts their output. This occurs particularly when Nav channel availability is reduced by sustained splanchnic nerve stimulation or prolonged cell depolarizations induced by acidosis, hyperkalaemia and increased muscarine levels.
Introduction
Chromaffin cells (CCs) of the adrenal medulla represent the main hub of the sympathetic nervous system. Upon splanchnic nerve stimulation, they secrete catecholamines that are central players of the stress response (de Diego et al. 2008; Guérineau et al. 2012). To exert their function, CCs are endowed with a broad array of ion channels that support action potential (AP) generation in a spontaneous manner (Vandael et al. 2010). Upon splanchnic nerve discharge, acetylcholine leads to sustained cell depolarizations, which increase the firing frequency, Ca2+-entry and exocytosis of catecholamine-containing vesicles (Garcia et al. 2006).
Recent studies have shown the importance of Cav1.3 L-type Ca2+ channels (LTCCs) in driving spontaneous firing in mouse chromaffin cells (MCCs) (Marcantoni et al. 2007; Marcantoni et al. 2010). Nevertheless, reports suggesting a partial contribution of voltage-gated Na+ channels (Nav) to CC excitability can be found. Early studies showed the existence of a background sodium conductance that could drive Na+-influx at rest in rat and gerbil CCs (Biales et al. 1976; Brandt et al. 1976). Tetrodotoxin (TTX) application or Na+ removal from the extracellular medium reduces but does not block rat CCs firing (Kidokoro & Ritchie, 1980). In MCCs, Nav channel blockade by TTX preserves AP firing (Nassar-Gentina et al. 1988), suggesting that Nav channels play a different role in excitability than LTCCs (Vandael et al. 2010). Furthermore, Nav channels in MCCs activate at 24 mV more positive potentials than Cav1.3, suggesting a minor contribution to subthreshold pacemaker currents (Mahapatra et al. 2011). As in most neurons, Nav channels are shown to be critical for shaping the AP waveform in MCCs (Vandael et al. 2012). Subtle changes in spike shape can lead to drastic changes in Ca2+ channel recruitment and in AP-induced Ca2+ transients (Li et al. 2007). AP broadening is indeed a common way to optimize presynaptic Ca2+ influx at central presynaptic terminals (Engel & Jonas, 2005).
At present, Nav1.7 is considered to be the main voltage-gated Nav channel expressed in bovine and human adrenal tissues (Klugbauer et al. 1995; Sangameswaran et al. 1997). In the rat adrenal gland, mRNA encoding Nav1.7 is expressed only in the medulla (Morinville et al. 2007), with a plausible localization in CCs, as also reported in PC12 cells (Toledo-Aral et al. 1997). The few functional studies performed with CC Nav channels show that they are TTX-sensitive, fast-inactivating and rather insensitive to closed-state inactivation (Fenwick et al. 1982; Lou et al. 2003; Wada et al. 2008; Mahapatra et al. 2011). The contribution of Nav channels to CC spiking at rest and upon stimulation remains controversial and studies that have considered Nav properties and availability at physiologically relevant potentials (−55 to −40 mV) are lacking.
In the present study, using quantitative RT-PCR and western-blot analysis, we show that, besides Nav1.7, Nav1.3 is highly expressed together with β1–3 subunits. The two principal MCC Nav channels possess rather similar activation–inactivation properties (Catterall et al. 2005) and contribute to spike generation but not to the pacemaker current. This is evident when both channels are blocked by TTX or when their availability is strongly reduced by steady inactivation during slight membrane depolarizations. Block or reduced Nav1.3/Nav1.7 availability induces a switch of firing from tonic single APs to doublet and triplet bursts of APs, which cause a paradoxical increase of Ca2+-entry and catecholamine secretion. Nav channel inactivation thus appears to comprise an important phenomenon that boosts MCC output during strong splanchnic nerve discharges as occurs during the stress response (Garcia et al. 2006) or during prolonged depolarizations near rest induced by acidosis, hyperkalaemia or increased muscarine levels.
Methods
Ethical approval
Ethical approval for all experimental protocols was obtained from the University of Torino Animal Care and Use Committee, Torino, Italy. All experiments were conducted in accordance with the National Guide for the Care and Use of Laboratory Animals adopted by the Italian Ministry of Health. Every effort was made to minimize animal suffering and the number of animals used. For removal of tissues, animals were deeply anaesthetized with CO2 inhalation and rapidly killed by cervical dislocation.
Cell culture
Chromaffin cells were obtained from male C57BL/6 J mice (Harlan, Correzzano, Italy) aged 3 months. Animals were killed by cervical dislocation. All procedures were performed in accordance with the guidelines established by the National Council on Animal Care and were approved by the local Animal Care committee of the University of Turin. Under sterile conditions, the abdomen was opened, and then the adrenal glands were isolated and transferred to an ice-cold Ca2+ and Mg2+ free Locke's buffer containing (in mm) 154 NaCl, 3.6 KCl, 5.6 NaHCO3, 5.6 glucose and 10 Hepes (pH 7.4) (Marcantoni et al. 2009; Vandael et al. 2012). Under a dissecting microscope, the adrenal glands were decapsulated and subsequently subjected to an enzymatic dissociation with 20–25 units ml–1 papain (Worthington Biochemical Corporation, Segrate, Italy) dissolved in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Invitrogen Life Technologies, Monza, Italy) supplemented with 1.5 mm of l-cysteine, 1 mm of CaCl2 and 0.5 mm of EDTA (Sigma-Aldrich, Munich, Germany) for 25 −30 min at 37°C in a water saturated atmosphere with 5% CO2. Afterwards, two washing steps were performed with DMEM supplemented with 1 mm CaCl2 and 10 mg ml–1 of BSA (Sigma-Aldrich). Adrenal medulla's were re-suspended in DMEM containing 1% penicillin/streptomycin and 15% fetal bovine serum (both from Sigma-Aldrich) and were mechanically dissociated with a fire polished Pasteur pipette. A drop (100 μl) of this concentrated cell suspension was plated on poly-ornithine (1 mg ml–1) and laminin (5 μg ml–1) coated Petri dishes and, subsequently (30 min later), 1.9 ml of DMEM containing 1% penicillin/streptomycin and 15% fetal bovine serum (all from Sigma-Aldrich) was added. The primary CC culture was maintained in an incubator at 37°C at water saturated atmosphere with 5% CO2. Measurements were performed on cultured MCCs 2–5 days after plating.
Electrophysiology
Macroscopic whole-cell currents and APs were recorded in perforated-patch conditions using a multiclamp 700-B amplifier and pClamp 10.0 software (Molecular Devices, Sunnyvale, CA, USA) (Marcantoni et al. 2010; Vandael et al. 2012). Traces were sampled at 10 kHz using a digidata 1440 A acquisition interface (Molecular Devices) and filtered using a low-pass Bessel filter set at 1–2 kHz. Borosilicate glass pipettes (Kimble Chase Life Science, Vineland, NJ, USA) with a resistance of 2–3 MΩ were dipped in an Eppendorf tube containing intracellular solution before being back filled with the same solution containing 500 μg ml–1 of amphotericin B (Sigma-Aldrich), dissolved in dimethyl sulphoxide (Sigma-Aldrich) (Cesetti et al. 2003). Recordings were initiated after amphotericin B lowered the access resistance below 15 MΩ (5–10 min). Series resistance was compensated by 80% and monitored throughout the experiment. Fast capacitive transients during stepwise depolarizations (in voltage-clamp mode) were minimized online by the use of patch clamp analogue compensation. Uncompensated capacitive currents were further reduced by subtracting the averaged currents in response to P/4 hyperpolarizing pulses. To eliminate any additional leakage current, the recorded ion current during step depolarizations was subtracted from traces measured in the presence of saturating concentrations of TTX (300 nM) (Carbone & Lux, 1986). The same was applied for the AP-clamp experiments in which the cell was clamped to a train of previously recorded APs. L-type Ca2+ currents were isolated by subtracting the non-L-type current (in the presence of nifedipine, 3 μm) from the whole-cell Ca2+ current.
Single channel recordings were performed on perfor-ated microvesicles that were pulled out of the cell after amphotericin B lowered the access resistance below 15 MΩ (Levitan & Kramer, 1990). Glass pipettes for this purpose were pulled and fired polished to obtain electrode resistances of ∼7 MΩ (Carabelli et al. 2001). Single channel recordings were sampled at 20 kHz and low-pass filtered at 2 kHz. Blockade by TTX at the end of the recording gave rise to single channel-free baseline traces that were averaged and used to correct for the capacitive artefacts. Further baseline correction was performed manually using Clampfit software (Molecular Devices). Mean open and closed times were analysed using pClamp software (Molecular Devices). The criteria for selecting the detection levels of channel opening and closing were similar to those described previously (Carbone & Lux, 1987; Carabelli et al. 2001). The effective minimal duration of detectable opening and closing was set to 150 μs (Carbone & Lux, 1986)
Carbon fibres (tip diameter of 5 μm) were purchased from Ala Scientific Instruments Inc. (Westbury, NY, USA). Electrochemical recordings were performed using a HEKA EPC-10 amplifier (HEKA Elektronik Dr Schulze GmbH, Lambrecht/Pfalz, Germany). Amperometric measurements were performed by positioning the carbon fibre microelectrode (polarized at +800 mV) adjacent to the cell membrane (Carabelli et al. 2007b; Marcantoni et al. 2009). MCCs were current clamped and spontaneous firing was suppressed by the injection of −5 pA. Subsequently, inhibition was relieved and cells were allowed to fire spontaneously for 40 s. Amperometric currents were sampled at 4 kHz and low-pass filtered at 1 kHz. Data were analysed by IGOR macros (WaveMetrics, Lake Oswego, OR, USA) as described previously (Segura et al. 2000). The analysis of individual exocytotic events was carried out by measuring the parameters: maximum oxidation current (Imax), spike width at half-height (t1/2), total charge of the spike (Q), ascending slope of the spike (m) and time to reach the spike (tp). All experiments were performed at room temperature.
Solutions
Intracellular solution for current-clamp and Na+ current/K+ current measurements in voltage-clamp or AP-clamp mode comprised (in mm): 135 KAsp, 8 NaCl, 20 Hepes, 2 MgCl2 and 5 EGTA (pH 7.4) (with NaOH; Sigma-Aldrich). For Ca2+ current recordings, the intracellular solution contained (in mm): 135 Cs-MeSO3, 8 NaCl, 2 MgCl2 and 20 Hepes (pH 7.4) (with CsOH; Sigma-Aldrich). The extracellular solution used for current-clamp measurements is based on a physiological Tyrode's solution containing (in mm): 130 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes and 10 glucose (pH 7.4) (with NaOH). The same solution was also used to measure K+ currents. KV currents were obtained by application of 200 μm Cd2+, whereas Ca2+-activated BK currents were estimated by subtracting KV from the total K+ currents. In rat chromaffin cell (RCCs) and MCCs, 500 μm Cd2+ are shown to be effective as 1 μm paxilline for blocking transient BK channels (Marcantoni et al. 2007; Marcantoni et al. 2010) and we have confirmed that, in 2 mm extracellular Ca2+, 200 μm Cd2+ is as effective as 500 μm Cd2+ for blocking Cav channels and Ca2+-activated BK channels (not shown). In addition, residual Cd2+-insensitive voltage-dependent BK currents contribute little (<5%) to the total BK currents at +20 to +30 mV (Berkefeld & Fakler, 2013). Thus, isolation of transient BK currents using 200 μm Cd2+ appears to be a justified protocol. The extracellular solution used for Na+ current measurements comprised (in mm): 104 NaCl, 30 TEACl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes and 10 glucose (pH 7.4) (with NaOH), plus 200 μM Cd2+. The extracellular solution used for Ca2+ current measurements in voltage- and AP-clamp configuration contained (in mm): 135 TEA, 2 CaCl, 2 MgCl2, 10 Hepes and 10 glucose (pH 7.4) (with TEA-OH; Sigma-Aldrich). TTX was purchased from Tocris Bioscience (Bristol, UK), dissolved in bi-distilled water and stored at −20°C until used. Solutions were applied using a gravity based perfusion system. Current-clamp data were not corrected for the liquid junction potential (15.4 mV at 22°C) (Marcantoni et al. 2010) because TTX (up to 1 μm) did not affect the liquid junction potential any further.
Real-time PCR and western blotting
Adrenal medullary tissue was prepared from six mice as described previously (Desarmenien et al. 2013). After removal, the glands were incubated in RNAlater RNA Stabilization Reagent (Sigma-Aldrich, Saint Quentin Fallavier, France) at 0–4°C for 24 h. Next, the glands were decapsulated and the cortical tissue was gently separated, under a microscope, from the medulla using micropincers and discarded. Both macrodissected adrenal medulla from each animal were pooled and used separately for RNA or protein extraction. Total RNA was extracted using the RNeasy micro kit (Qiagen, Courtaboeuf, France). For retrotranscription, total RNA (200 ng) from adrenal medulla of each animal was used and the reactions were performed using random hexamer primers and the QuantiTect Reverse Transcription kit (Qiagen). Real-time PCR assays were carried out on a LightCycler 480 Instrument II (Roche, Meylan, France) with Sybr Select Master Mix (Applied Biosystems, Foster City, CA, USA). Gene-specific primers for real-time PCR (Table1) were designed using Primer3 Software (Rozen & Skaletsky, 2000). Differences in transcript level were determined using the cycle threshold method, in accordance with the manufacturer's instructions. Specificity of amplification was checked by melting curve analysis and gene expression was normalized to expression of three housekeeping genes: Gapdh (glyceraldehyde-3-phosphate dehydrogenase), Gusb (β-glucuronidase) and Hprt (hypoxanthine phosphoribosyltransferase 1), according to the formula 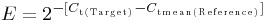 , where Ct is the threshold cycle. Amplicon sizes (70–106 bp) and AT content (47–55%) were chosen to allow comparison between relative expression values obtained for each gene (Colborn et al. 2008).
, where Ct is the threshold cycle. Amplicon sizes (70–106 bp) and AT content (47–55%) were chosen to allow comparison between relative expression values obtained for each gene (Colborn et al. 2008).
Table 1.
Sequence of primers used for quantitative RT-PCR
| Gene name | GenBank accession number | Protein name | forward primer (5′ to 3′) | reverse primer (5′ to 3′) |
|---|---|---|---|---|
| scn1A | NM_018733.2 | Nav1.1 | TTGCAAGGGGCTTCTGTTTA | AGGTCCACAAACTCCGTCAC |
| scn2A | NM_001099298.2 | Nav1.2 | GGGTTGCATATGGTTTCCAA | CCCAAGGCATTTGCAGTTA |
| scn3A | NM_018732.3 | Nav1.3 | TCCTCAGTAGTGGTGCTTTGG | GATGTAAGTGAAGACTTTGTCAGCA |
| scn4A | NM_133199.2 | Nav1.4 | GAAAACCATCACGGTCATCC | TCCGAGAGCTTTTTCACAGAC |
| scn5A | NM_021544.4 | Nav1.5 | GCCAGATCTCTATGGCAACC | TTGCCCTTATTCAGCACGAT |
| scn8A | NM_001077499.2 | Nav1.6 | CTGGTGCTGGTTGGACTTC | GCCCAGGGCATTAGCTATAA |
| scn9A | NM_018852.2 | Nav1.7 | AAAGCAGGTGGGACAAAGG | CTCTCCTTGGCACTCTTTGAG |
| scn10a | NM_009134.3 | Nav1.8 | GCTGAGCCTATCAATGCAGA | ACTTGGCAGCATGGAAATCT |
| scn11A | NM_011887.3 | Nav1.9 | TGGGTAGCTTATGGCTTCAAA | CTATGAGGCTTGTGAGGGAGA |
| scn1B | NM_011322.2 | β1 | TGCTACAAGAAGATTGCTGCTG | AATGGCCAGGTATTCTGAGG |
| scn2B | NM_001014761.2 | β2 | CAGGAGTGTAACAATTGCACAGA | GGGTTCCCTGAGAACTCCAC |
| scn3B | NM_153522.2 | β3 | CCCCTAGCTTCTCTAGTGCTCA | CTGTCTCCGAGGGTACTTCTACA |
| scn4B | NM_001013390.2 | β4 | TGAAGAAGACCCGAGAGAAGA | ACCCGTTCTCTGTGTTGTCA |
| Gusb | NM_010368.1 | Gus | CTCTGGTGGCCTTACCTGAT | CAGTTGTTGTCACCTTCACCTC |
| Hprt | NM_013556.2 | Hprt | AGGACCTCTCGAAGTGT | ATTCAAATCCCTGAAGTACTCAT |
| Gapdh | NM_008084.2 | Gapdh | CCGGGGCTGGCATTGCTCTC | GGGGTGGGTGGTCCAGGGTT |
For protein extraction, adrenal medullary and brain tissues were dissociated using lysis buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, 1 mm sodium orthovanadate and 10 mm NaF as phosphatase inhibitors, 10 mm β-glycerophosphate and 1% Triton X-100), supplemented with complete mini protease inhibitor tablets (Roche Applied Science, Laval, Quebec, Canada). Adrenal medullary homogenates were centrifuged for 5 min at 2000×g. The resulting supernatants were collected and centrifuged for 30 min at 15,000×g. Protein concentrations were determined using the Dc Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA). Equal amount of protein samples (10 μg for adrenal medulla and 30 μg for brain) were heated for 5 min at 95°C, separated on a 7% polyacrylamide gel and transferred onto a nitrocellulose membrane. To ensure equal loading of protein samples, blots were cut in two at ∼60 kDa. The lower parts of the blots were probed with a specific anti-β-actin antibody (Mono/Poly BD Biosciences Clontech, Palo Alto, CA, USA; 1:5000). The upper parts of the blots were first probed for the Nav1.3 N-terminal (Sigma-Aldrich, clone 3F3; 1:500) and the Nav1.7 C-terminal (University of California, Davis/National Institutes of Health NeuroMab Facility, clone N68/6; 1:500). Blots were incubated with a peroxydase-conjugated secondary antibodies and the signal was detected using the ECL-Plus Chemiluminescence kit (Amersham Biotech, Little Chalfont, UK). Labelled blots were then exposed to Fujifilm Medical X-Ray Film to visualize antibody binding with a luminescent image analyser (LAS-3000; Fujifilm, Tokyo, Japan). Intensities of Nav1.3 and Nav1.7 channel bands were normalized to those of β-actin and quantified using Image Gauge software (Fujifilm).
Statistical analysis
Data are given as the mean ± SEM for the number (n) of cells. Statistical significance was estimated with paired Student's t tests in case two groups of measurements had to be compared and with a one-way ANOVA followed by post hoc Bonferroni analysis in case more than two groups had to be compared with one another. Data were found statistically significant when P ≤ 0.05. Statistical analysis was performed with SPSS software, version 20.0 (IBM Corp., Armonk, NY, USA). Data analysis was performed with pClamp and Origin software (OriginLab Corporation, Northampton, MA, USA).
Results
Nav1.3 and Nav1.7 channel expression in MCCs
We first addressed, by real-time PCR, the transcriptional expression profile of all known Nav channel isoforms. The data showed a differential expression of the six mRNAs (scn1a, scn2a, scn3a, scn4s, scn8a and scn9a) encoding Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6 and Nav1.7 TTX-sensitive Nav channels respectively (Fig.1A, left). Among the detectable isoforms (26 < Ct < 30), the rank order of the relative mRNAs abundance was scn3A > scn9a > scn2a > scn1a. Scn3a mRNA level was ∼7-fold higher compared to scn9a (***P < 0.001). Scn4a cDNA was amplified with a Ct of 35 and thus was considered not significantly expressed. Scn8a was undetectable. Messenger RNAs encoding all TTX-resistant Nav channels were detected at very low levels (Ct of 34 for scn5a and 35 for scn10a and scn11a) and therefore were considered absent from the mouse adrenal medulla tissue. We also examined the expression profile of the Nav channel auxiliary β subunits (Fig.1A, right). Scn1b, 2b and 3b (β1–3 subunits were significantly expressed (26 < Ct < 28), whereas scn4b mRNA (β4 subunit) was considered absent (Ct of 35). Scn1b and scn2b subunits displayed comparable mRNA expression levels and scnb3 mRNA was found to be 1.7-fold more abundant than the scnb1 subunit (**P < 0.01). The expression of Nav1.3 and 1.7 was also investigated at the protein level by immunoblotting using specific antibodies. The western blots (macro-dissected adrenal medulla extracts) showed a strong immunoreactivity for a single band with an apparent molecular weight of ∼230 kDa (Nav1.3) and ∼220 kDa (Nav1.7) (Fig.1B).
Figure 1. Expression profile of Nav channel α and β-subunit genes and immunodetection of Nav1.3 and Nav1.7 proteins in the mouse adrenal medulla.
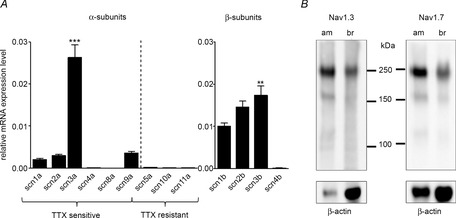
A, RT-qPCR data illustrating that, among all Nav channel genes, only TTX-sensitive isoforms are expressed in the adrenal medulla, with major expression for mRNAs encoding Nav1.3 and Nav1.7 (left: ***P < 0.001 for scn3a compared to scn9a). Except for scn4b (β4-subunit), all β-subunit mRNAs are present (right: **P < 0.01 for scn3b compared to scn1b). Significance tests between groups of data were performed using one-way ANOVA followed by a post hoc Bonferroni test. Data are the mean ± SEM of six independent samples. B, western blot analysis of proteins extracted from macrodissected mouse adrenal medulla (am, 10 μg) and from mouse brain (b, 30 μg). Note that a 3-fold higher amount of proteins was used for the brain as a result of the weak expression of Nav1.3 and Nav1.7 channels, as reported previously (Sangameswaran et al. 1997; Catterall et al. 2005). When probed for Nav1.3 and Nav1.7 proteins, a single band is immunodetected at 230 kDa (Nav1.3) and 220 kDa (Nav1.7) in both tissues, with stronger signal intensities in the adrenal medulla (upper membranes). Immunoblotting of β-actin was used as a control (lower membranes).
Altogether, quantitative RT-PCR experiments and western blots indicate that voltage-gated Na+ channels expressed in the mouse adrenal medullary tissue are probably formed by combination of β1–3 subunits with Nav α-subunits, with a predominance of Nav1.3 and Nav1.7. These data are in agreement with previously reported electrophysiological studies showing that MCC Nav channels are sensitive to TTX (Mahapatra et al. 2011; Vandael et al. 2012).
MCC Nav channels are TTX-sensitive and fully inactivating
Voltage-clamp recordings of Na+ currents at −10 mV were carried out to confirm TTX sensitivity, as well as the activation and inactivation properties of MCC Nav channels. TTX was perfused in gradually increasing concentrations and the degree of Na+ current block with respect to control was fitted with a dose–response curve (Fig.2A). Half-maximal block was obtained at 6.7 ± 0.8 nM (n = 11) and saturation set at ∼300 nm. This confirms the quantitative RT-PCR data and also confirms that all α-subunits expressed in MCCs (in particular the two dominant ones: Nav1.3 and Nav1.7) are TTX-sensitive. Accordingly, we used subtraction of current traces measured in the presence of 300 nm TTX from control traces to obtain pure Na+ currents. From this point on, we use Nav to refer to a mixture of Nav1.3 and Nav1.7 channels.
Figure 2. Nav channels in MCCs are transient and fully blocked by TTX.
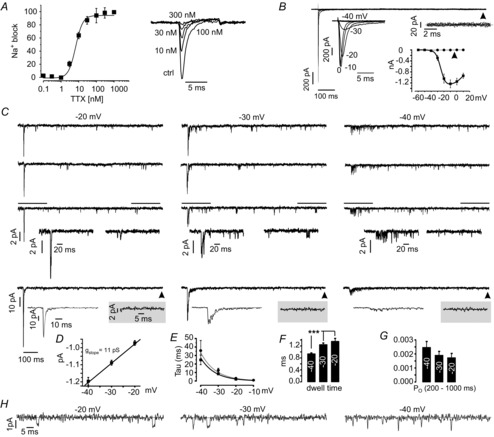
A, TTX dose–response curve representing the mean of 11 cells, fitted by a sigmoidal function with IC50 = 7 nm. Traces are normalized against control values. Currents were triggered by step depolarizations to −10 mV from Vh = −70 mV every 5 s. B, whole-cell Nav currents after subtraction of TTX-insensitive currents evoked by depolarizing pulses lasting 1 s in steps of 10 mV from Vh = −70 mV (n = 8). Left inset: transient segment of Nav currents at an expanded time scale. Right inset: an enlargement of the current at steady-state indicated by the arrow. Bottom inset: I/V relationship for the transient component (continuous line) and the steady-state component (dotted line). C, three representative single channel traces recorded from perforated microvesicles pulled off from six MCCs at −20, −30 and −40 mV. Bottom trace represents the ensemble currents obtained by summing-up 10 consecutive single channel traces. Decay of the ensemble currents were fitted by a single exponential function (grey line superimposing the current trace). Grey windows show ensemble currents at steady-state indicated by the arrow. D, single channel slope conductance obtained by linear fit through the mean unitary current amplitudes estimated within the last 800 ms of the recording at −40 mV (226 openings), −30 mV (243 openings) and −20 mV (111 openings). F, time constants of Nav current inactivation for whole-cell currents (referred to B, thick line) and ensemble currents (referred to C, dotted line) obtained from single exponential fits. F, single channel mean dwell times for the indicated potentials, starting at Vh = −70 mV. G, estimated mean open probability of Nav channels during the steady state normalized to the total amount of channels in the patch. H, Single channel traces obtained at the indicated test potentials at expanded time scale (100 ms).
So far, the activation properties of Na+ channels have been described for bovine and rat CCs (Fenwick et al. 1982; Lou et al. 2003; Wada et al. 2008). Nav channel activation properties have been studied also in MCCs (Mahapatra et al. 2011), although many aspects remain unexplored. Of particular interest in excitable cells is the presence of persistent Na+ currents, as well as resurgent Na+ currents (Raman & Bean, 1997; Bean, 2007). To test for the existence of persistent currents, we opted for depolarization steps of 1 s in duration, representing the average time that a MCC spends between two consecutive spontaneous APs. It is known that, besides the persistent nature of slow inactivating Na+ currents, the activation kinetics of these currents are typically shifted to a more negative Vm (French et al. 1990; Magistretti et al. 1999). These two features are thus ideally suited for a role in generating spontaneous membrane oscillations and can be exploited to help distinguish ‘persistent’ from ‘transient’ current components. All of the cells tested (n = 8) showed no direct signs of a persistent Na+ current component (Fig.2B). For potentials above −20 mV, time constants of channel inactivation (τinact) did not exceed 1 ms, whereas, for −40 and −30 mV, we obtained τinact = 35.5 ± 11.9 ms and 12.7 ± 2.2 ms, respectively (Fig.2E). Currents at steady-state did not show the typical bell-shaped current–voltage relationship (Fig.2B, arrow) as was observed for the transient current component, which rose steeply with voltage and had half-maximal activation (V½) at −21 mV. This suggests that we are mainly dealing with noise fluctuations near the baseline rather than steady-currents conducted by voltage-gated channels. Nevertheless, whole-cell Na+ currents were too noisy at steady state (1 s) to provide conclusive evidence in favour of the absence of persistent Na+ currents. Because MCCs typically exhibit high input resistances (2–5 GΩ), small non-inactivating currents could lead to sizeable membrane depolarizations, and thus should be carefully evaluated. Based on our recordings, we estimated that these currents could not be larger than 1–2 pA.
Single Nav channel recordings in perforated micro-vesicles
To better test for the presence of a persistent Na+ current in MCCs, we studied the properties of single Na+ channels in perforated micro-vesicles that were pulled out from a perforated-patch configuration (Fig.2C). This procedure leads to excellent voltage control and allows validation of the entity of the measured channels by selective blockers, maintaining the intracellular environment intact (Levitan & Kramer, 1990). Typically, micro-vesicles (n = 6 cells) were obtained containing six to 15 Nav channels. The total number of channels in a patch was estimated by the maximal number of superimposed channel openings at potentials of maximal activation (−10 mV) (Carbone & Lux, 1986). Levels were set based on unitary channel current amplitude that was revealed for each potential at steady state and threshold based statistics were performed (Carabelli et al. 2001; Carabelli et al. 2002). Most unitary events occurred soon after the onset of the depolarizing pulse and reopenings were observed for the whole duration of the recording. Unitary events became infrequent and dwell times were rather short lived (Fig.2C and H). Based on the unitary current amplitudes at −40, −30 and −20 mV (distributed around a Gaussian function), we estimated a single channel conductance of 11 pS (Fig.2D), which is in good agreement with single Nav channel recordings from bovine CCs (BCCs) (13 pS; Fenwick et al. 1982) and chick dorsal root ganglion neurons (11 pS; Carbone & Lux, 1986). Dwell time was measured at steady state (200–1000 ms) because individual channel openings could be accurately followed in this time interval. Altogether, the number of openings that were included in the analysis were 226, 243 and 111 for voltage steps to −40, −30 and −20 mV, respectively. Concerning the mean dwell time, single openings significantly increased in duration (***P < 0.001, ANOVA, post hoc Bonferroni) at more depolarized potentials (−30 mV and −20 mV) compared to −40 mV (Fig.2F). This is in good agreement with the reported values in BCCs (Fenwick et al. 1982) and dorsal root ganglion neurons (Carbone & Lux, 1986) if our MCCs data were shifted by −10 mV to −15 mV to account for the different recording conditions (perforated vesicle vs. outside-out patch). With respect to the Nav channels of BCCs, the mean dwell time in MCCs was always below the inactivation time constant (τinact) of the ensemble current (mean dwell time 1.35 ms and mean τinact 2.0 ms at −20 mV) (Fig.2E and F).
One disadvantage of using perforated micro-vesicles is the rare occurrence of obtaining single ion channel recordings. This issue limits the possibility to measure with accuracy the channel open probability (PO). Because we were interested in persistent Na+ currents (late openings), we measured PO at steady state and divided for the total number of Nav channels estimated at −10 mV where all available Nav channels are expected to be open and the whole-cell current is maximal. PO obtained in this way was extremely low (PO < 0.0025 with n = 6 at −40 mV; Fig.2G), suggesting that Na+ channels are more transient than persistent. Even if, in the worst case, the openings at steady-state would represent only one single ion channel, open probabilities would not exceed 0.01, which would still be too low to be considered persistent. In stellate cells of the enthorinal cortex, persistent openings have been reported to result in PO values in the range of 0.1–0.9 (Magistretti et al. 1999).
Finally, the ensemble currents were constructed by summing up 10 consecutive traces as shown in Fig.2C (bottom). Again, we found a well-defined transient component with no signs of persistent inward currents at 1 s depolarization (shadowed traces in Fig.2C, bottom). The decay phase of the ensemble current was well fitted by a single exponential with τinact similar to the whole-cell recordings (Fig.2E).
Nav channel inactivation in MCCs reduces the size of ‘window currents’
The availability of voltage-gated Nav channels depends critically on the holding potential (Vh) that the cell is clamped to. Given the transient nature of Nav channels and the low PO, a critical factor that will determine the final steady-state current is how many channels will enter a Vh-dependent state of slow (ultraslow) inactivation. Accordingly, we performed voltage-clamp experiments in which we simply stepwise depolarized the cell up to +60 mV (ΔV = 10 mV) from a Vh varying from −100 mV to −30 mV (Fig.3). As expected, the maximal current density significantly decreased by ∼3-fold (**P < 0.01) when the cell was maintained at Vh = −40 mV compared to −100 mV (Fig.3B). Maximal current densities plotted vs. Vh revealed a clear sigmoidal relationship (Fig.4B) with half-available Nav channels at −47.9 ± 1.9 mV. At Vh = −40 mV, the availability of Nav channels reduces further to only 30%. This phenomenon will clearly reduce the Nav current amplitudes at critically low pacemaker potentials between −50 mV and −40 mV.
Figure 3. Holding potential dependent availability of MCC Nav channels and window currents.
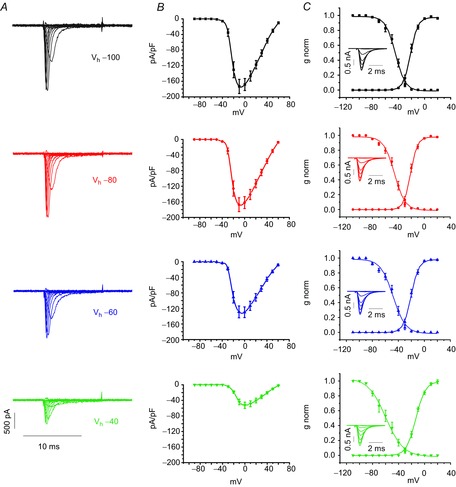
A, Representative Nav currents evoked from varying Vh as indicated: −100 mV (black), −80 mV (red), −60 mV (blue) and −40 mV (green). Cells were maintained for 10 s at Vh before recording; intersweep intervals of 5 s were applied. Traces shown were obtained after subtracting the TTX-insensitive current traces measured in the presence of 300 nm TTX. B, I–V relationship (n = 26) for Nav current densities measured at the indicated Vh. C, normalized conductance (n = 26) (derived from the data shown in B and normalized steady-state inactivation (n = 16) measured at −10 mV for the indicated Vh. Boltzmann equations were used for fitting both activation and inactivation curves. Insets depict representative traces that show steady-state inactivation of Nav currents at the indicated Vh. Half-maximal values (in mV) and slope factors of activation (in mV) obtained from the fit are −21.9 and 5.5 for Vh = −100 mV; −21.3 and 5.0 for −80 mV, 20.3 and 5.5 for −60 mV, and −13.4 and 6.0 for −40 mV, respectively. Half-maximal inactivation (in mV) and slope factors of inactivation (in mV) are −44.6 and 7.6 at Vh = −100 mV, −46.0 and 8.2 at −80 mV, −48.0 and 9.5 at −60 mV, and −59.7 and 13.4 at −40 mV, respectively.
Figure 4. Holding potential dependent reduction of window currents and channel availability in MCCs.
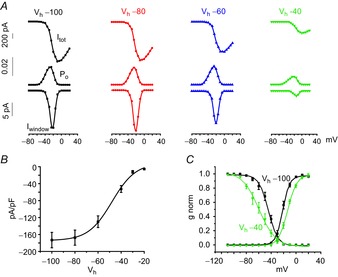
A, top: whole-cell currents; middle: open probabilities (Po) obtained by multiplying activation and inactivation curves shown in Fig.3c; bottom: window currents calculated by multiplying the whole-cell currents (top) by Po (middle). The Vh values of the experiment are indicated at the top: −100 mV (black), −80 mV (red), −60 mV (blue) and −40 mV (green). B, availability of Nav current densities with respect to the indicated holding potentials (n = 26). Data were fit by a Boltzmann equation. The half-maximal value and slope factor of inactivation are −47.9 mV and 10.5 mV. C, overlap of activation and inactivation curves at Vh = −100 mV (black) and Vh = −40 mV (green), derived from Fig.3.
To quantify the current flowing at steady-state potentials, we calculated the corresponding ‘window current’ (Fig.4A) by measuring activation and steady-state inactivation at diverse Vh (Fig. 3C). The latter was obtained by anticipating the test pulse at −10 mV (Imax) by a pre-pulse lasting 100 ms. Multiplying the activation curve by the inactivation curve gave the PO(V) (Fig.4A, middle) that, when multiplied by the whole-cell current (upper traces), led to the inward ‘window current’ vs. voltage (Fig.4A, bottom). As expected, we found rather low PO values at −40 mV (0.0035) compared to Vh = −100 mV (0.009). The closer the values for half-maximal activation and half-maximal inactivation, the bigger the overlap between the relative activation and inactivation curve and the bigger the window current. We observed that the gap between half-maximal activation and inactivation became bigger as we fixed Vh to gradually more positive values. The discrepancy between both relative activation and inactivation doubled when we compared Vh = −100 mV with Vh = −40 mV (−21.4 ± 0.3 mV and −44.6 ± 0.7 mV for Vh = −100 mV and −13.4 ± 0.3 mV and −59.7 ± 1.2 mV for Vh = −40 mV) (Fig.4C). Not unexpectedly, the window current that we measured decreased from −15.6 pA at Vh = −100 mV to −2.2 pA at Vh = −40 mV. The fact that the window current is bell shaped and almost zero aro-und −20 mV, further indicates that inactivation is complete towards more positive potentials. The inactivation curve of a ‘persistent current’ would not fall to zero but rather would give robust currents at positive potentials (given the high relative channel availability at these voltages) (French et al. 1990). Altogether, channel availability and activation/inactivation properties of Nav current in MCCs at physiologically relevant Vh (−50 mV, −40 mV) do not favour sizeable steady-state currents that could drive spontaneous membrane oscillations.
Recovery from fast inactivation and onset of slow inactivation of Nav channels
Nav channels contributing to rhythmic firing should be able to recover sufficient Na+ current between two consecutive APs to guarantee stable AP waveforms. We thus assayed the onset of recovery from fast inactivation by comparing the timing at Vh = −90 mV with two physiological Vh representing the resting state (Vh = −50 mV) and a depolarized state that would occur during acute stress response (Vh = −40 mV; Fig.5A). Recovery from fast inactivation in all three conditions occurred in a bi-exponential mode and striking differences were notable. Recovery was slow and remarkably incomplete (50%) after 1 s when Vh was set at −40 mV, whereas it was fast and fully complete (100%) at Vh = −90 mV (Fig.5A). The time constant of the dominant fast component (τfast) decreased from 23.7 ms (Vh = −40 mV) to 4.9 ms (Vh = −90 mV). A pulse of moderate duration (100 ms) starting from Vh = −40 mV thus leads to a loss of approximately half of the available Nav channels that failed to recover.
Figure 5. Inactivation properties of MCC Nav channels.
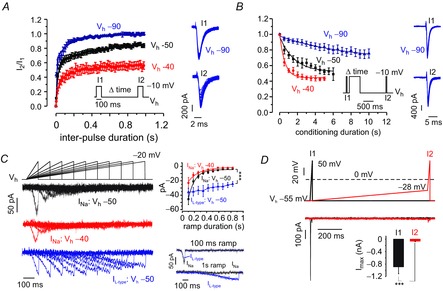
A, recovery from steady-state inactivation of Nav currents from Vh = −90 mV (blue, n = 13), −50 mV (black, n = 14) and −40 mV (red, n = 10). Current amplitudes at the test pulse (I2) were normalized against current amplitudes at the preceding pulse (I1). Averaged data were fit by double exponentials with fast and slow components of different amplitude (Afast, Aslow) and time constants (τfast, τslow). τfast, τslow and amplitudes (in parenthesis) were: 4.8 ms (0.6) and 156 ms (0.4) at −90 mV, 21.3 ms (0.6) and 501 ms (0.3) at −50 mV, 23.7 ms (0,34) and 229 ms (0.22) at −40 mV. B, onset of closed-state slow inactivation during Vh = −90 mV (blue, n = 10), −50 mV (black, n = 5) and −40 mV (red, n = 6). Current amplitudes during the test pulse (I2) were compared with currents at onset (I1). The conditioning pulse was followed by a recovery period of 1.2 s to allow recovery from fast inactivation. Data were fit by a single exponential function of different time constant (τ) and amplitude (A). τ and A (in parenthesis) were 7.8 s (0.32) at Vh = −90 mV, 1.6 s (0.45) at −50 mV and 0.58 s (0.54) at −40 mV. C, Nav and L-type currents elicited by ramp depolarizations starting from Vh = −50 mV (black) or Vh = −40 mV (red). Nav currents are the result of subtracting control traces from the TTX-resistant component. L-type currents (blue) are the results of subtracting control traces from nifedipine-insensitive currents. Inset to the right: mean ± SEM of peak Na+ and L-type currents at different ramp duration. D, comparison of Nav currents evoked by a 20 ms ramp in the presence (I2, red trace) or absence (I1, black trace) of a preceding slow pacemaker such as ramp of 1 s duration subsequent to the indicated protocol on top (***P < 0.001, paired Student's t test). The black and red voltage-clamp protocols indicated on top were separated by 5 s. The starting voltage of the ramp (−55 mV) is the mean AHP peak of the Aps, whereas −28 mV is the mean spike threshold value (Vandael et al. 2012).
In addition to fast inactivation, Nav channels can transit into a phase of slow inactivation from which the recovery can take several seconds (Cantrell & Catterall, 2001). Thus, the question arises as to whether the incomplete recovery from inactivation at Vh = −40 mV is the result of a switch of channels to such a state of slow inactivation. To answer this question, we constructed a protocol where slow inactivation was induced by a strong depolarization to −10 mV, lasting up to 10 s. The Nav current before this pulse (I1) was then compared with the current available after the conditioning step (I2). Between the conditioning pulse and the second test pulse (I2), the cell was maintained at Vh for 1.2 s to allow for the recovery from fast inactivation (Fig.5B). The data shown in Fig.5B were fit with a 1st order exponential function with time constants that declined with increasing Vh. Nav current decay measured in this way was 14-fold faster for Vh = −40 mV compared to Vh = −90 mV and 3-fold faster compared to Vh = −50 mV (τ = 7.8 ± 1.6 s for Vh = −90; 1.64 ± 0.3 s for Vh = −50 and 0.58 ± 0.63 s for Vh = −40 mV). Thus, at physiologically relevant holding potentials, a larger fraction of channels enter states from which recovery from inactivation is slower. This phenomenon is of relevance when CCs are depolarized during stress responses. The incomplete recovery from fast inactivation and the fast onset of slow inactivation can thus produce a drastic reduction of available Nav channels at −40 mV and −50 mV.
Slow inactivation of Nav channels during slow ramp commands
So far, we considered only square pulse protocols that do not represent the spontaneous membrane oscillations at rest. We thus tested the Nav current behaviour during slow gradual depolarizing pulses (ramp commands of 0.1 to 1 s) that mimic MCCs interspike intervals (ISI). The ramps reached −20 mV, starting from two different Vh values (−40 mV and −50 mV). Currents evoked in this manner in both conditions were then compared (Fig.5C). In line with the above findings, current amplitude was bigger (1.8-fold) when ramps of 100 ms duration were triggered from Vh = −50 mV compared to −40 mV. With increasing ramp duration, currents decayed in an exponential manner both for Vh = −50 mV (τ = 134 ± 15 s) and Vh = −40 mV (τ = 160 ± 23 s). For ramps lasting 1 s, the remaining currents had mean amplitudes of −3.3 ± 1.2 pA for Vh = −50 mV and −2.5 ± 0.8 pA for Vh = −40 mV (Fig.5C). Note that, when the same protocol (Vh = −50 mV) was performed, the LTCCs were 8-fold bigger after ramps lasting 1 s (−25 ± 3.6 pA, Fig.5C and inset). Together with the more negative activation kinetics, it is clear that LTCCs possess more suitable features to contribute to pacemaker depolarization than Nav channels do. Nav currents activate at ∼20 mV more depolarized potentials with respect to LTCCs (Mahapatra et al. 2011) and inactivate fully during long ramp commands, as shown in the bottom-right inset of Fig.5C.
It thus appears that the slow depolarization preceding the AP will carry a rather small (although not negligible) current and will probably inactivate the Na+ current sustaining the spike upstroke. To test this hypothesis, we constructed a protocol where a fast ramp (20 ms) starting from spike threshold (−28 mV) and reaching +50 mV (I2) was anticipated by a 1 s ramp (ISI) that covers the pacemaker potential (i.e. from its most negative and most positive average values) (−55 mV and −28 mV). These values represent the previously reported mean after-hyperpolarization peak (AHP = −53 mV) and mean spike threshold (−28 mV) (Vandael et al. 2012). The resulting current was then compared with a ramp of 20 ms duration (I1) elicited from −55 mV to +50 mV (Fig.5D). Both ramp depolarizations were separated by intersweep intervals of 5 s to ensure complete recovery of Nav channels from fast inactivation. Nav currents were 14-fold smaller during I2 as compared to I1 (−65 ± 17 pA vs. −889 ± 223 pA, n = 8; ***P < 0.001, t test). This suggests that, during the pacemaker potential, massive Nav channel inactivation occurs (Fig.5D).
Loss of Nav channels during depolarization profoundly modifies the AP shape
Given the profound effects of Vh on Nav channel availability, we assayed how this phenomenon affects MCCs firing and AP-waveform properties (Fig.6). Experiments were performed in current-clamp mode without current injection. Cells under control conditions fired at 0.98 ± 0.08 Hz and Vrest was −46 ± 1.3 mV (n = 12). By injecting inward current to bring Vrest near −40 mV (−38.4 ± 1.0 mV), the firing frequency increased almost 3-fold (***P < 0.001, t test) accompanied by a significant widening of the AP half-width (2.5-fold; ***P < 0.001) and robust decrease of the peak overshoot (by ∼25 mV; ***P < 0.001) (Fig.6A and B, right). Averaged APs under control conditions and after current injection were used to calculate dV/dt, which was then visualized graphically vs. Vm in a phase plane plot (Fig.6B, left). A massive reduction of dV/dtmax (∼5 fold; ***P < 0.001, t test) is evident. This parameter represents the point where the AP rising phase is fastest during the up-stroke and is associated with Nav channel density (Bean, 2007). Altogether, these changes point towards a strong decrease of Nav channel contribution to the AP at depolarized Vh. To confirm this, we performed AP-clamp experiments, using trains of APs measured under control conditions and after depolarization (Fig.6C). Clean Nav currents were obtained by final subtraction of control recordings from traces measured in the presence of 300 nm TTX (Fig.6C). Comparison of Nav currents under control conditions (Vh = −50 mV) and depolarized Vrest (Vh = −40 mV) showed marked reductions of Nav current amplitudes after each AP: ∼60% at −40 mV and ∼40% at −50 mV, respectively (*P < 0.05) (Fig.6C, right)
Figure 6. Chromaffin cell stimulation leads to Nav channel inactivation and AP widening.
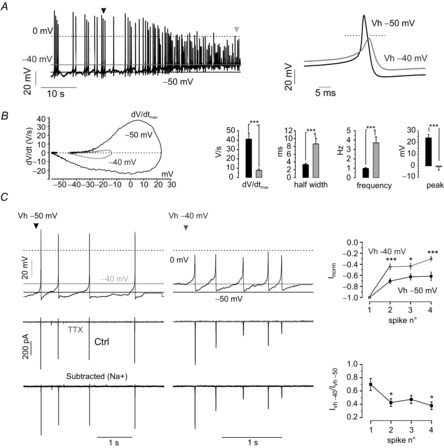
A, representative response of a current clamped MCC to a 10 mV steady depolarization. Right: overlap of a control AP (black) with an AP obtained after the imposed depolarization (grey). B, Phase plane plots (dV/dt vs. V) from the same cell constructed by averaging n = 4 APs at Vh = −50 and n = 5 APs at −40 mV, respectively. Right: effects of a 10 mV depolarization on spike parameters (***P < 0.001, paired Student's t test). Half-width was measured as the duration of the AP at half of the total amplitude (from the after-hyperpolarization peak to overshoot peak). C, Nav currents measured by AP-clamp using APs at rest (left) and APs that originate from a depolarized resting potential (right). Middle: control trace in black and TTX (300 nm) resistant component in grey. Bottom: pure Nav currents after subtraction for TTX-insensitive currents. Top-right inset: normalized current amplitudes against the first current peak amplitude of the spike train. The grey line and dots refer to Nav currents triggered at depolarized resting potentials (−40 mV, n = 12), whereas the black line and squares represent Nav currents that flow during spikes at rest (n = 12) (*P < 0.05, ***P < 0.001, paired Student's t test). Bottom-right inset: the ratio between Nav currents triggered by control APs and APs at depolarized resting potentials for each consecutive spike (*P < 0.05, ANOVA followed by a post hoc Bonferroni analysis).
Nav channel blockade by TTX leads to a switch from tonic to burst-like firings
As cell depolarization leads to a reduction in the contribution of Nav channels to the AP, we planned a series of experiments in which increasing fractions of Nav channels were pharmacologically blocked to test how Nav current availability may impact on firing patterns and spike parameters (Fig.7). TTX was tested on spontaneously firing MCCs and applied at the same concentration used in voltage-clamp experiments (Fig.2B). As previously reported (Vandael et al. 2010; Mahapatra et al. 2011), TTX did not impede the cell from generating spontaneous APs, not even at saturating concentrations (300 nm; Fig.7A). As expected, we observed a dose-dependent reduction of the AP peak (IC50 = 9.0 ± 1.9 nm) and a dose-dependent increase of the half-width (IC50 = 18.7 ± 4.3 nm) (Fig.7C and D). We also measured the coefficient of variance (CV = SD mean–1) for the ISI duration as an indication of the regularity of the firing pattern. The CV during control firing was 0.8 ± 0.01 and increased up to 1.1 ± 0.1 in the presence of 300 nm TTX, indicating that, in the absence of Nav channels, the firing pattern is clearly more irregular.
Figure 7. TTX leads to a dose-dependent switch to burst firing of MCCs.
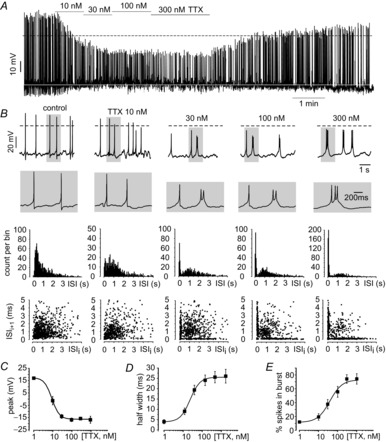
A, representative current clamp trace showing the action of increasing TTX concentration. B, top: a better view of the effects of diverse TTX concentrations on the AP waveforms and firing patterns at slower and faster (grey windows) time scales. Periods of activity were taken from (a). Middle: ISI distribution for the indicated TTX concentrations. Lower: joint ISI plots obtained by plotting each ISI on the x-axis (ISIi) against its succeeding ISI duration (ISIi+1) on the y-axis. Random firing in the absence or presence of 10 nm TTX leads to a cloudy pattern, whereas burst firing patterns at an elevated TTX concentration (30–300 nm) leads to an L-shaped distribution. C, dose–response curve of AP peak vs. TTX concentration. Data were fit with a sigmoid function with saturating value at 17.5 mV (IC50 = 9 nm). D, dose–response curve of AP half-width vs. TTX concentration. Data were fit with a sigmoid function with saturating value at 26.2 ms (IC50 = 18.7 nm). E, dose–response curve of the fraction of spikes fired in bursts vs. TTX concentration. Data were fit with a sigmoid function with saturating value at 76% (IC50 = 39.9 nm).
A closer look at the firing patterns after Nav channel blockade shows the tendency of MCCs to fire its APs preferentially in bursts, especially at saturating doses of TTX (Fig.7B). The switch from tonic to burst-like firing is particularly evident in the distributions of the ISI duration (Fig.7B, centre). Under control conditions, the distribution is nearly random (low CV), whereas, at increasing TTX concentrations, two separate Gaussian distributions with distinct peaks become evident. The first peak (brief durations) represents the ISI between two consecutive spikes in a burst (intra-burst interval), whereas the second peak at longer times represents the ISI between bursts (inter-burst interval). The centres of the Gaussian for the intraburst interval varied from 77.1 ms to 57.8 ms by increasing TTX from 30 nm to 300 nm, whereas the Gaussian of the interburst intervals centred at 1.1 s to 1.3 s.
When we subsequently plotted each ISI against its preceding ISI duration, we could distinguish a clear difference between control and Nav block-induced firing patterns (Fig.7B, bottom). In these so called joint-interspike-interval plots, irregular ‘tonic’ firing patterns give rise to a cloudy distribution of events, whereas ‘burst’ firing gives rise to an ‘L-shaped’ distribution. The L-shaped joint ISI can be ascribed to the prevalence of altering a long with a peculiarly brief ISI during a burst. Together with a change in the ISI distribution, there was also a dose-dependent percentage increase (IC50 of 38.9 ± 9.8 nm) of spikes fired in bursts with increasing doses of TTX (Fig.7E). The parameter that was most sensitive to TTX was the AP amplitude (IC50 = 9.0 nm; Fig.7C), followed by the half-width (IC50 = 18.7 nm; Fig.7D) and the percentage of spikes fired in bursts (Fig.7E). Out of 16 cells, the overall firing frequency was increased after Nav channel blockade compared to control, whereas, in three cells, we observed a decrease.
Altogether, we did not observe a significant difference (*P > 0.05) for the firing frequency when control conditions (0.9 ± 0.1 Hz) were compared with firing that persisted after Nav channel blockade (1.2 ± 0.2 Hz). During burst firing, however, brief periods of high frequency firing (14.8 ± 1.2 Hz; n = 16) were altered by relatively long gaps of no activity. In addition, spikes fired in bursts emerge from depolarization plateaus (−38.7 ± 1.3 mV up to −33.1 ± 1.2 mV) that last for the whole burst duration. It is thus possible that the Ca2+ influx is particularly high during such firing patterns and an increased degree of catecholamine secretion takes place.
Reduced Nav availability inverts the net current during the AHP from outward to inward
Figure 7 shows clearly a reduction of Na+ current amplitude upon cell depolarization, leading to broad and low amplitude spikes that oscillate in a voltage range where the driving force on Ca2+ ions is high. Less Nav channel availability leads to less KV and BK channel activation because cells do not reach positive membrane potentials during the AP peak and this in turn will result in a slower return to baseline potentials. Of particular interest is the effect of this phenomenon on the currents flowing during the AHP. A possible switch from a net outward to a net inward current could explain the development of a sufficiently low AHP that drives the burst of APs when Nav channel availability is drastically reduced.
To better understand this issue, we performed AP-clamp experiments in which we aimed to unravel the balance between outward K+ and inward Ca2+ currents during APs measured under three conditions: (i) control firing patterns; (ii) depolarized Vh potentials; and (iii) burst firing in 300 nm TTX (Fig.8). Ca2+-activated K+ currents (IKCa) were isolated by subtraction of the whole-cell Ca2+ current (measured in 135 mm TEA, 2 mm Ca2+ and 300 nm TTX) and the KV current (IKV) (Tyrode standard with 200 μm of Cd2+ and TTX) from the total current measured in Tyrode standard with 300 nm TTX. Compared to control spikes, we observed a 4-fold reduction of IKV flowing during spikes that started from a depolarized membrane potential (**P < 0.01, n = 9) and a 25-fold reduction during the burst doublets in the presence of 300 nm TTX (***P < 0.001, n = 9). Note that, during the APs burst, IKV is almost absent (Fig.8C, red trace) and IKCa is drastically reduced (Fig.8C, blue trace; *P < 0.05, n = 9). The net current during the spikes remained outward, justifying the effective repolarizing phase under all three conditions. However, marked changes were observed during the AHP. Although the net current during the AHP of control spikes was outward, the current during the intraburst interval was net inward and carried by Ca2+ (Fig.8B, inset). It thus results that incoming Ca2+, together with a lack of sufficient outward K+ current, is the trigger of burst firing when Nav channels are blocked. Broadening of APs during cell depolarization and bursts of APs in the presence of TTX caused a 2- to 10-fold prolongation of Ca2+ currents (Fig.8A–C, bottom insets) that, despite the lower amplitudes, could carry 2- and 3.5-fold more Ca2+ charges inside the cell, respectively (Fig.8C, inset).
Figure 8. Reducing Nav availability inverts net current covering the AHP from outward to inward.
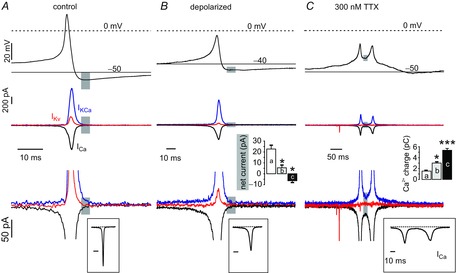
A, top: AP-clamp experiment showing representative ctrl spike. (middle) Kv currents (red traces) were measured in a Tyrode standard solution with TTX (300 nm) and Cd2+ (200 μm). Ca2+ currents (black) were measured in the presence of TTX (300 nm) and high extracellular TEA (135 mm). Ca2+-activated K+ currents (blue) were obtained by subtracting from a control recording in Tyrode standard with TTX (300 nm) the KV and the Ca2+ current. Bottom: close up. The grey rectangle indicates the AHP phase and the respective currents that sustain it. B and C, as for A, using a single spike stimulus protocol triggered from −40 mV and a spike doublet fired after complete block of Nav currents with 300 nm TTX. B, inset: net current amplitudes measured during the AHP phase indicated by the grey windows. C, inset: Ca2+ charge entering the cell during the AHP phase calculated by the integrating the corresponding Ca2+ inward current (*P < 0.05; ***P < 0.001; n = 9; ANOVA followed by a post hoc Bonferroni analysis).
A minority of MCCs exhibits spontaneous burst firing
Spontaneous burst firing (zero current injected) was also observed in a minority of cells (14%; 39 out of 278) that were measured in current clamp (Fig.9A). A train of high frequency spikes emerged on a ‘slow-wave’ depolarization plateau with somewhat different shape and duration but comparable frequency of those induced by TTX. Burst durations were 2.6-fold longer (0.37 ± 0.08 s vs. 0.14 ± 0.03 s with TTX; ***P < 0.001, unpaired t test) but occurred at almost the same frequency (0.56 ± 0.09 Hz vs. 0.48 ± 0.15 Hz). As a result of Nav channel availability, the first spike overshoot was always larger compared to the subsequent APs. Spikes showed a gradual reduction in peak amplitude (from +16.3 mV to −10.11 mV; ***P < 0.001, paired t test), a sequential increase in half-width duration (from 2.9 ms to 6.8 ms; ***P < 0.001) and reduction in AHP amplitude (from −41 mV to −34.4 mV; ***P < 0.001) (Fig.9B). The intraburst ISI duration and thus also the intraburst instantaneous firing frequency increased strongly towards the end of the burst (from 170.3 ms to 65.3 ms and from 8.9 Hz to 21.2 Hz, respectively; ***P < 0.001). Again we hypothesized that Nav channel inactivation leads to a reduction in the AP peak, which leads to less KV activation and thus spike broadening. The low amplitude spikes could furthermore explain the gradual reduction in the AHP that helps shape the burst envelope. To quantify this, we performed AP-clamp recordings using bursts as voltage clamp commands (Fig.9C–E). When considering Na+-currents, we found that the amplitudes significantly (***P < 0.001, n = 6) decreased by 15-fold when comparing the currents elicited during the first and last spike of the burst (Fig.9F). This is not unexpected given that a narrow ISI does not allow sufficient time for recovery from inactivation. In addition, gradually decreasing AHP and spike broadening lead to an increased depolarization and thus more complete Nav inactivation. A decrease of outward currents also occurred. KV currents reduced by 4.4-fold (***P < 0.001, n = 18), whereas BK current amplitudes decreased by 5.6-fold (***P < 0.001, n = 7) comparing currents during the first and last spike (Fig.9F). Ca2+ current amplitudes reduced by 3-fold (***P < 0.001, n = 17) but, when we measured the area under the curve, we found a 1.7-fold increase (***P < 0.001) in Ca2+ charge (Fig.9F). In line with the idea of Ca2+ building up with each and every spike, we found a sequential and significant increase in SK current amplitudes (***P < 0.001, n = 7) (Fig.9F). It is worth noting that the decrease of Nav, Kv, BK and Cav current amplitude during consecutive APs is a result not only of inactivation, but also the decreasing amplitude of the APs in the command waveform.
Figure 9. Spontaneous burst firing in a minority of MCCs.
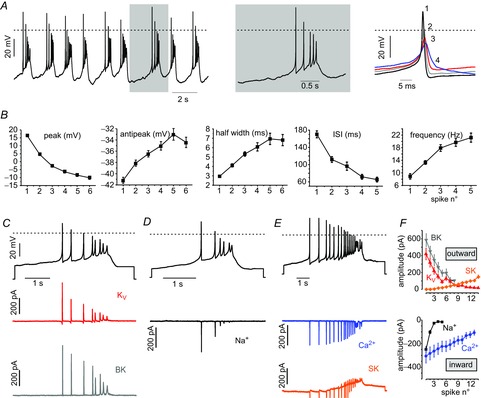
A, left: representative trace of spontaneous trains of APs fired in burst-like mode. Centre: a single burst at an expanded time scale. Right: overlap of consecutive APs of a burst. Numbers indicate the position/sequence in the burst. B, evolution of the AP peak, AHP amplitude, half-width, interspike interval and instantaneous firing frequency of consecutive spikes in a burst. Mean values were obtained for each bursting cell (n = 39) and then averaged. C, AP-clamp experiment measuring KV (n = 18) and BK (n = 7) currents. Top: burst recorded and used as voltage command (black trace), KV currents are shown in red and BK currents in grey. D and E, as for C but the currents isolated were Ca2+ (blue, n = 17), SK (orange, n = 7) and Na+ (black, n = 6). F, top, BK, SK and Kv outward current amplitudes vs. the spike number of the burst. Bottom, as for top but for the Na+ and Ca2+ inward currents (***P < 0.001, paired Student's t test).
Block of Nav channels boosts MCCs exocytosis
Bursts of APs originating from plateau potentials of −35 mV, and lasting for hundreds of milliseconds, thus go hand in hand with an increase of inflowing Ca2+ charge (Figs8C and 9E and F). One can hypothesize that the block of TTX-sensitive Nav channels paradoxically leads to an increased Ca2+-dependent secretion of catecholamines in MCCs as a result of both the sustained depolarization (slow-wave) and the increased AP frequency during bursts. To test this, we combined current-clamp recordings with carbon fibre amperometry to reveal fast quantal release of catecholamines during MCCs firing patterns (for 40 s) at rest and after Nav block (Fig.10) (Carabelli et al. 2007b). Cells were hyperpolarized to −70 mV to cease spontaneous activity and to prevent Ca2+ accumulation at the onset of the recording. Subsequently, inhibition was relieved and spontaneous activity was measured for 40 s. As expected, in the presence of TTX, all cells switched from a tonic (Fig.10A) into a burst-firing mode (Fig.10B). Amperometric signals associated with vesicle fusion and catecholamine release were detected in both cases but at a significantly different frequency. Block of Nav channels by TTX caused an almost 2.5-fold increase of the rate of vesicle release with respect to control (*P < 0.05, n = 14) (Fig.10D, bottom) with no significant changes to the waveform of amperometric spikes (Fig.10D, top). The parameters associated with the peak amplitude (Imax), time to peak (tp), half-width (t1/2), rise time (m), total quantity of charge released (Q) and cubic root of Q (Q1/3) (as an estimate of vesicle size) remained unchanged (*P > 0.05, t test), regardless of Nav channel blockade and the switch of cell firing mode (Fig.10D). We also determined the time course of cumulative secretion (Fig.10C) and calculated the mean quantity of cumulative charges recorded during a 40 s recording and found a 3.7-fold increase of the cumulative charge (Fig.10D). As shown previously (Marcantoni et al. 2009), this can be explained by the increase of the amperometric spike frequency (2.5-fold), which may sum to a small net charge increase of the single secretory events not sufficiently large to be detected (Fig.10D). Given that the quantity of Ca2+ charges is linearly related to the amount of released vesicles in CCs (Carabelli et al. 2003; Carabelli et al. 2007a), the 2.5-fold increased frequency of amperometric spikes and the 3.7-fold increase of cumulative released charges in the presence of TTX are in very good agreement with the 3.5-fold increase of Ca2+-entry during bursts of APs (Fig.8C).
Figure 10. Burst firing associated with Nav channel blockade boosts MCC exocytosis.
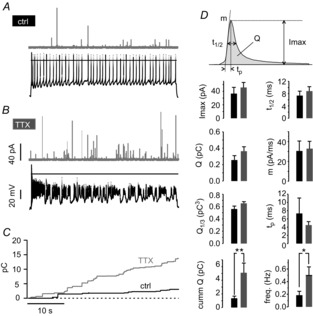
A, example of combined recording of APs (black traces) and amperometric events (grey traces) by carbon fibre amperometry in the control. B, representation of combined recording of AP bursts after TTX application (same cell as shown in A) and amperometric events by amperometry. C, overlap of cumulative secretion plots obtained from amperometric measurements shown in A and B for control (black curve) and during TTX application (grey curve). D, comparison of amperometric spike parameters (see top), frequency and cumulative charge between control (black bars) and TTX-treated cells (grey bars) (*P < 0.05 and **P < 0.01, n = 14; paired Student's t test).
On average, five APs are required to elicit an exocytotic event in RCCs under control conditions (1.1 Hz APs vs. 0.2 Hz amperometric spikes) (Zhou & Misler, 1995). We found that the same occurs in MCCs. Specifically, we found no strict correlation between APs and amperometric events under both control conditions or during burst firing induced by TTX. During burst firing, periods of elevated vesicle release sometimes occurred during the gaps that separated the bursts rather than during the bursts themselves. It is thus evident that Ca2+ has to diffuse in the cell to trigger most of the exocytotic events. It is important to emphasize that our data do not clarify whether increased secretion in the presence of TTX is mainly a result of Ca2+-entry during the slow-wave depolarization or a result of the increased frequency of APs riding on top of the slow wave. Both phenomena occur simultaneously and cause increased Ca2+-influx, whose separation requires specific protocols.
Discussion
We have provided evidence indicating that Nav channels are not mandatory for generating spontaneous APs in MCCs. APs persist in the presence of saturating doses of TTX and the contribution of persistent or resurgent Nav channels to pacemaking is null. We have shown that, similar to TTX, small depolarizations from rest drastically lower Nav channel availability, mostly as a result of the incomplete recovery from fast inactivation and fast onset of closed-state inactivation. Similar to TTX, this causes drastic changes in AP-shape and firing patterns that increase Ca2+-entry and catecholamine release. Because Nav channel down-regulation is implicated in the control of input–output relationships in several neurons, a key implication of our findings is that gradual loss of Nav channels may drastically change electrical excitability of neuroendocrine cells (Cantrell & Catterall, 2001).
Expression profile of the fast-inactivating TTX-sensitive Nav channels of MCCs
Previously, scn9a (Nav1.7) was considered to be the principal Nav α-subunit gene expressed in the adrenal medulla (Klugbauer et al. 1995; Morinville et al. 2007; Wada et al. 2008; Yanagita et al. 2011). Our expression data confirm the presence of scn9A in MCCs but, unexpectedly, uncover a 7.3-fold higher expression level of scn3A transcripts (Nav1.3). Both isoforms are confirmed at the protein level and it cannot be excluded that Nav1.1 and Nav1.2 are expressed despite their low degree of transcripts. The absence of the mRNA of TTX-resistant Nav isoforms such as scn5A (Nav1.5), scn10A (Nav1.8) and scn11A (Nav1.9) supports our data suggesting that saturating doses of TTX block all Nav currents (IC50 ∼7 nm). Half-maximal Nav blockade by TTX is comparable with that reported for Nav1.7 and Nav1.3 (IC50 ∼4 nm) (Sangameswaran et al. 1997; Catterall et al. 2005), which are the main Nav channels expressed by MCCs. Our Nav activation data (V1/2 = −21 mV) on MCCs fit with those reported for BCCs (Fenwick et al. 1982) and RCCs (Lou et al. 2003; Wada et al. 2008) and are also in good agreement with the reported values for Nav1.3 and Nav1.7 α-subunits expressed in HEK cells (V1/2 = −26 mV; Catterall et al. 2005). Based on their V1/2 of activation and inactivation and their kinetics of recovery from fast inactivation and onset of slow closed-state inactivation, Nav1.3 and Nav1.7 are almost indistinguishable, except for the slower recovery of inactivation of Nav1.7 with respect to Nav1.3 (26 ms vs. 11 ms at Vh = −100 mV and 113 ms vs. 60 ms at Vh = −80 mV) (Cummins et al. 2001). These latter values, however, are too small to be associated to the slow component of the recovery of inactivation of Nav channels in MCCs (156 ms to 501 ms; Fig.5A), whose cause remains to be clarified. Compared to Nav1.2, both channels display a marked slow entry rate in the closed-state inactivation, which makes both of them suitable for sustaining robust responses to slowly depolarizing inputs. Together with an absence of reported values on the single channel conductance of Nav1.3 (Catterall et al. 2005), this makes it almost impossible to associate the functional Nav channel properties of MCCs to either Nav1.3 or Nav1.7. This issue would certainly benefit from analysis of the Nav currents of MCCs in Nav1.3 (Nassar et al. 2006) and Nav1.7 null mice (Nassar et al. 2004).
Our whole-cell data and single-channel recordings confirm that MCC Nav currents are not persistent. Sustained Nav currents are typically associated with the Nav1.6 isoform (scn8a) (Raman & Bean, 1997), which activates at potentials as negative as −70 mV (Taddese & Bean, 2002) and sustains tonic-firing at elevated frequencies (20–100 Hz) in several neurons (Smith et al. 1998; Rush et al. 2005; Enomoto et al. 2007; Mercer et al. 2007). MCCs do not fire at elevated frequencies and lack scn8a. In addition, the auxiliary subunit scn4b (Navβ4) is linked to resurgent Na+ currents (Bant & Raman, 2010). Neither scn4b, nor resurgent Na+ currents were revealed in MCCs.
Nav1.3/Nav1.7 channel contribution to MCC autorythmicity
Early reports showed that Nav channels are critically involved in driving AP firing in gerbil and human CCs (Biales et al. 1976). Recording conditions in these studies were nevertheless strikingly different from ours (perforated-patch in 2 mm Ca2+ vs. sharp-electrode impalement in 10 mm Ca2+) and cells were more depolarized (−40 mV to −10 mV) than MCCs (−50 mV to −45 mV). A similar approach on RCCs resulted in a Vrest –49 mV and TTX strongly interfered with AP generation (Brandt et al. 1976). The present assumption is that Nav1.7 represents the main Nav channel isoform in bovine and rat adrenal glands (Klugbauer et al. 1995; Morinville et al. 2007; Wada et al. 2008). Nav1.7 possesses slow closed-state inactivation during very negative ramp depolarizations, a feature that favours its contribution to autorythmicity (Lou et al. 2003). In the present study, we report that Nav1.3 is highly expressed in MCCs and, similar to Nav1.7, undergoes strong inactivation during prolonged depolarizations (Cummins et al. 2001). MCC Nav channels can inactivate considerably during ramp-depolarizations, mimicking the velocity of the interspike depolarization from physiological Vh values (Fig.5C and D).
An important factor that influences Nav channel availability in MCCs is the relatively depolarized Vh. Between −50 mV and −40 mV, Nav availability ranges from 50% to 30%. We report, moreover, the drastic changes in recovery from inactivation at physiologically relevant Vh. At −40 mV, recovery of inactivation remains fast (τ = 24 ms) but is strikingly incomplete as a result of the transition into a slow-inactivation state. The AP amplitude and the velocity of the spike upstroke strongly depend on the amount of available Nav channels. The ‘vulnerability’ of MCC Nav channels to inactivation is indeed reflected in profound changes of AP shape when cells are depolarized to −40 mV. At such depolarized potentials, the Nav window-current is peculiarly small because of a shift of the inactivation curve to more negative potentials. Opposing shifts of both activation and inactivation curves (as occurs in MCCs when changing Vh from −100 mV to −40 mV) are expected to slow down the frequency of tonic firing, indicating their effective contribution to pacemaking. This is true for dopaminergic neurons of the substantia nigra, where Nav channels contribute to pacemaking (Tucker et al. 2012) but opposite to that found in MCCs in which firing is accelerated. The fact that saturating doses of TTX do not interfere with spontaneous AP generation is conclusive evidence that Nav channels are not required for MCC pacemaking. Activation threshold for Nav channels occurs at more positive voltages than the MCC ISI (−46 mV), suggesting that a channel such as the L-type Cav1.3 is required to bridge the gap (Marcantoni et al. 2010; Vandael et al. 2012).
Physiological relevance of Nav currents in MCCs: switch from tonic to burst firing
Fast-firing cells typically exhibit very narrow APs with small overshoots and fast AP-repolarization (Bean, 2007). In this way, Nav inactivation is kept incomplete and contributes to fast-firing phenotypes (Carter & Bean, 2010). In the present study, we clearly show that, upon sustained depolarization, the AP peak of MCCs gradually drops and broadens, reducing the velocity of spike depolarization and repolarization. Such broad and low amplitude spikes are correlated with strongly reduced Na+ current amplitudes (by 50% compared to Vrest). Nav1.3/Nav1.7 blockade by TTX leads to similar dose-dependent changes of AP waveform. Indeed, TTX blocks both AP peak and Nav currents with almost the same IC50 (9 mm vs.7 nm). This is different from several neurons that, at Vrest –70 mV, possess high-densities of Nav channels and the AP-shape is less sensitive to TTX (Madeja, 2000). MCCs have a lower surplus of Nav1.3/Nav1.7 channels whose availability is thus extremely sensitive to slight changes in membrane potential or channel down-regulation.
An interesting finding of the present study is that saturating doses of TTX do not lead to a cessation of firing but rather trigger a switch from single irregular APs into burst firing. Crucial to burst firing is the inward/outward current balance in between spikes (Swensen & Bean, 2003) and we have shown that blocking Nav channels inverts the net outward K+ current during the AHP to a net inward Ca2+ current. Similar changes on AP shape have been observed during TTX application or K+ channel block by TEA on spontaneously active MCCs in the intact adrenal gland (Nassar-Gentina et al. 1988). Spontaneous burst firing not only occurs in a minority of MCCs (Fig.9), but also in a small fraction of MCCs of adrenal slices (Martinez-Espinosa et al. 2014). This latter study also shows that burst firing occurs in the majority of MCCs in which the β2-subunit of fast-inactivating BK channels is deleted. Thus, burst firing appears as an intrinsic firing mode in a subpopulation of MCCs and may be easily uncovered during altered cell conditions. The final output is an increased Ca2+-entry that boosts cell activity and secretion.
Firing modes regulate catecholamine release in CCs
We found an excellent correlation between the 3.5-fold enhanced Ca2+-entry during the TTX-induced burst firing and the 3.7-fold increased total charges of released catecholamines. This is expected given the linear-dependence of Ca2+-entry and catecholamine release in MCCs (Marcantoni et al. 2009) and RCCs (Carabelli et al. 2003), which derives from the weak coupling of Ca2+-channels to the release sites (Marcantoni et al. 2008). Whether the major source of Ca2+-influx required for secretion in MCCs is a result of the slow-wave depolarization or the increased AP frequency during bursts remains to be resolved. Nevertheless, it is interesting to recall that secretion in RCCs is more sensitive to stimulations in form of repeated bursts rather than single AP-trains of increasing frequency (Duan et al. 2003). In midbrain dopaminergic neurons, bursting stimulations are twice as potent in triggering secretion compared to regular spaced ones, although with the same average frequency (Gonon, 1988).
The action of TTX reported in the present study is unique. The toxin does not block the Ca2+-dependent release of catecholamines but rather increases it. In PC12 cells, catecholamine release is fully Ca2+-dependent and TTX preserves this rather than blocking it (Ritchie, 1979). In intact perfused rat adrenal glands, 300 nm TTX leads to a reduction of ∼43% of catecholamine secretion after transmural stimulation (Wakade, 1981). This apparent conflicting result may correlate with our data by assuming that the TTX concentration used was not sufficient to guarantee an efficient block of Nav channels in the whole-gland or that TTX could have partially blocked the Nav channels of presynaptic splanchnic nerve endings. In our hands, RCC pacemaking is more sensitive to Nav channel blockade (Martin et al. 2001). A species-dependent origin of these divergent results is probable.
The present study indicates that the loss or down-regulation of Nav channels is a critical process leading to alteration of the AP waveform. In extreme situations, this phenomenon might result in a switch to slow waves of burst firing that boost Ca2+-entry inside the cell. Under physiological conditions, this may occur as a result of plasma hyperkalaemia, acidosis or increased histamine levels to which CCs respond with sustained depolarizations, burst firing (Wallace et al. 2002; Inoue et al. 2008; Mahapatra et al. 2011) and enhanced circulating catecholamines (Clausen, 1983). Of particular interest in CC biology is the potential role of muscarinic receptors, whose activation causes prolonged membrane depolarizations, increased cell firing and sustained catecholamines release (Nassar-Gentina et al. 1988; Olivos & Artalejo, 2007). The action is mediated by the M1 muscarinic receptor and proceeds through the inhibition of TASK1-like channels (Inoue et al. 2008). Because muscarine activates the protein kinase C pathway in RCCs (Akaike et al. 1993), Nav channels will probably undergo the well known protein kinase C-dependent down-regulation mediated by muscarinic receptors (Carr et al. 2003). This, together with the 30% reduced availability of Nav channels at depolarized Vh, would critically increase the probability of MCCs switching to burst-firing patterns, boosting catecholamine release during ACh-driven acute stress responses.
Acknowledgments
We thank C. Franchino, L. Grimaud and B. Toutain for providing technical assistance.
Glossary
Abbreviations
- AHP
after-hyperpolarization
- AP
action potential
- BCC
bovine chromaffin cell
- CC
chromaffin cells
- DMEM
Dulbecco's modified Eagle's medium
- ISI
interspike interval
- LTCC
L-type calcium channel
- MCC
mouse chromaffin cell
- Nav
voltage-gated sodium channel
- RCC
rat chromaffin cell
- TTX
tetrodotoxin
Additional information
Competing interests
The authors have no competing interests.
Author contributions
D.H.F.V. contributed to the data collection of the whole-cell, single channel and amperometric experiments. M.M.O. and A.A. contributed to the data collection of part of the whole-cell recordings. V.C. contributed to the design, analysis and interpretation of amperometric measurements. Ch.L., Cl.L. and N.C.G. contributed to the design, collection, analysis and interpretation of the quantitative RT-PCR and immunoblotting. D.H.F.V. and E.C. contributed to the conception and design of the experiments, data analysis, and drafting the article, and also revised it critically for important intellectual content with the input of all co-authors. All authors have approved the final version of the manuscript submitted for publication.
Fundings
This work was supported by the Italian MIUR (PRIN 2010/2011 project 2010JFYFY2) and the University of Torino.
References
- Akaike A, Sasa M, Tamura Y, Ujihara H. Takaori S. Effects of protein kinase C on the muscarinic excitation of rat adrenal chromaffin cells. Jpn J Pharmacol. 1993;61:145–148. doi: 10.1254/jjp.61.145. [DOI] [PubMed] [Google Scholar]
- Bant JS. Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 2010;107:12357–12362. doi: 10.1073/pnas.1005633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP( The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Berkefeld H. Fakler B. Ligand-gating by Ca2+ is rate limiting for physiological operation of BK(Ca) channels. J Neurosci. 2013;33:7358–7367. doi: 10.1523/JNEUROSCI.5443-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biales B, Dichter M. Tischler A. Electrical excitability of cultured adrenal chromaffin cells. J Physiol. 1976;262:743–753. doi: 10.1113/jphysiol.1976.sp011618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt BL, Hagiwara S, Kidokoro Y. Miyazaki S. Action potentials in the rat chromaffin cell and effects of acetylcholine. J Physiol. 1976;263:417–439. doi: 10.1113/jphysiol.1976.sp011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR. Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Carabelli V, D'Ascenzo M, Carbone E. Grassi C. Nitric oxide inhibits neuroendocrine Ca(v)1 L-channel gating via cGMP-dependent protein kinase in cell-attached patches of bovine chromaffin cells. J Physiol. 2002;541:351–366. doi: 10.1113/jphysiol.2002.017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Giancippoli A, Baldelli P, Carbone E. Artalejo AR. Distinct potentiation of L-type currents and secretion by cAMP in rat chromaffin cells. Biophys J. 2003;85:1326–1337. doi: 10.1016/S0006-3495(03)74567-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Hernandez-Guijo JM, Baldelli P. Carbone E. Direct autocrine inhibition and cAMP-dependent potentiation of single L-type Ca2+ channels in bovine chromaffin cells. J Physiol. 2001;532:73–90. doi: 10.1111/j.1469-7793.2001.0073g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Marcantoni A, Comunanza V. Carbone E. Fast exocytosis mediated by T- and L-type channels in chromaffin cells: distinct voltage-dependence but similar Ca2+-dependence. Eur Biophys J. 2007a;36:753–762. doi: 10.1007/s00249-007-0138-2. [DOI] [PubMed] [Google Scholar]
- Carabelli V, Marcantoni A, Comunanza V, De Luca A, Diaz J, Borges R. Carbone E. Chronic hypoxia up-regulates alpha(1H) T-type channels and low-threshold catecholamine secretion in rat chromaffin cells. J Physiol. 2007b;584:149–165. doi: 10.1113/jphysiol.2007.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E. Lux HD. Sodium-channels in cultured chick dorsal-root ganglion neurons. Eur Biophys J. 1986;13:259–271. [Google Scholar]
- Carbone E. Lux HD. Single low-voltage-activated calcium channels in chick and rat sensory neurons. J Physiol. 1987;386:571–601. doi: 10.1113/jphysiol.1987.sp016552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA. Surmeier DJ. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- Carter BC. Bean BP. Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron. 2010;64:898–909. doi: 10.1016/j.neuron.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL. Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Cesetti T, Hernandez-Guijo JM, Baldelli P, Carabelli V. Carbone E. Opposite action of beta 1- and beta 2-adrenergic receptors on Ca(V)1 L-channel current in rat adrenal chromaffin cells. J Neurosci. 2003;23:73–83. doi: 10.1523/JNEUROSCI.23-01-00073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T( Adrenergic control of Na+-K+-homoeostasis. Acta Med Scand Suppl. 1983;672:111–115. doi: 10.1111/j.0954-6820.1983.tb01622.x. [DOI] [PubMed] [Google Scholar]
- Colborn JM, Byrd BD, Koita OA. Krogstad DJ. Estimation of copy number using SYBR Green: confounding by AT-rich DNA and by variation in amplicon length. Am J Trop Med Hyg. 2008;79:887–892. [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD. Waxman SG. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21:5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Diego AM, Gandia L. Garcia AG. A physiological view of the central and peripheral mechanisms that regulate the release of catecholamines at the adrenal medulla. Acta Physiol (Oxf) 2008;192:287–301. doi: 10.1111/j.1748-1716.2007.01807.x. [DOI] [PubMed] [Google Scholar]
- Desarmenien MG, Jourdan C, Toutain B, Vessieres E, Hormuzdi SG. Guerineau NC. Gap junction signalling is a stress-regulated component of adrenal neuroendocrine stimulus-secretion coupling in vivo. Nat Commun. 2013;4:2938. doi: 10.1038/ncomms3938. [DOI] [PubMed] [Google Scholar]
- Duan K, Yu X, Zhang C. Zhou Z. Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J Neurosci. 2003;23:11235–11243. doi: 10.1523/JNEUROSCI.23-35-11235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel D. Jonas P. Presynaptic action potential amplification by voltage-gated Na+ channels in hippocampal mossy fiber boutons. Neuron. 2005;45:405–417. doi: 10.1016/j.neuron.2004.12.048. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF. Chandler SH. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J Neurophysiol. 2007;98:710–719. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A. Neher E. Sodium and calcium channels in bovine chromaffin cells. J Physiol. 1982;331:599–635. doi: 10.1113/jphysiol.1982.sp014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ. Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AG, Garcia-De-Diego AM, Gandia L, Borges R. Garcia-Sancho J. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev. 2006;86:1093–1131. doi: 10.1152/physrev.00039.2005. [DOI] [PubMed] [Google Scholar]
- Gonon FG( Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Guérineau NC, Desarmenien MG, Carabelli V. Carbone E. Functional chromaffin cell plasticity in response to stress: focus on nicotinic, gap junction, and voltage-gated Ca2+ channels. J Mol Neurosci. 2012;48:368–386. doi: 10.1007/s12031-012-9707-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Harada K, Matsuoka H, Sata T. Warashina A. Inhibition of TASK1-like channels by muscarinic receptor stimulation in rat adrenal medullary cells. J Neurochem. 2008;106:1804–1814. doi: 10.1111/j.1471-4159.2008.05521.x. [DOI] [PubMed] [Google Scholar]
- Kidokoro Y. Ritchie AK. Chromaffin cell action potentials and their possible role in adrenaline secretion from rat adrenal medulla. J Physiol. 1980;307:199–216. doi: 10.1113/jphysiol.1980.sp013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V. Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan ES. Kramer RH. Neuropeptide modulation of single calcium and potassium channels detected with a new patch clamp configuration. Nature. 1990;348:545–547. doi: 10.1038/348545a0. [DOI] [PubMed] [Google Scholar]
- Li L, Bischofberger J. Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci. 2007;27:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou XL, Yu X, Chen XK, Duan KL, He LM, Qu AL, Xu T. Zhou Z. Na+ channel inactivation: a comparative study between pancreatic islet beta-cells and adrenal chromaffin cells in rat. J Physiol. 2003;548:191–202. doi: 10.1113/jphysiol.2002.034405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeja M. Do neurons have a reserve of sodium channels for the generation of action potentials? A study on acutely isolated CA1 neurons from the guinea-pig hippocampus. Eur J Neurosci. 2000;12:1–7. doi: 10.1046/j.1460-9568.2000.00871.x. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Ragsdale DS. Alonso A. High conductance sustained single-channel activity responsible for the low-threshold persistent Na(+) current in entorhinal cortex neurons. J Neurosci. 1999;19:7334–7341. doi: 10.1523/JNEUROSCI.19-17-07334.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra S, Marcantoni A, Vandael DHF, Striessnig J. Carbone E. Are Ca(v)1.3 pacemaker channels in chromaffin cells? Possible bias from resting cell conditions and DHP blockers usage. Channels. 2011;5:219–224. doi: 10.4161/chan.5.3.15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni A, Baldelli P, Hernandez-Guijo JM, Comunanza V, Carabelli V. Carbone E. L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium. 2007;42:397–408. doi: 10.1016/j.ceca.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Carabelli V, Comunanza V, Hoddah H. Carbone E. Calcium channels in chromaffin cells: focus on L and T types. Acta Physiol (Oxf) 2008;192:233–246. doi: 10.1111/j.1748-1716.2007.01815.x. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Carabelli V, Vandael DH, Comunanza V. Carbone E. PDE type-4 inhibition increases L-type Ca2+ currents, action potential firing, and quantal size of exocytosis in mouse chromaffin cells. Pflugers Archiv Eur J Physiol. 2009;457:1093–1110. doi: 10.1007/s00424-008-0584-4. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DHF, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J. Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2010;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AO, Mathieu MN, Chevillard C. Guerineau NC. Gap junctions mediate electrical signaling and ensuing cytosolic Ca2+ increases between chromaffin cells in adrenal slices: a role in catecholamine release. J Neurosci. 2001;21:5397–5405. doi: 10.1523/JNEUROSCI.21-15-05397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Yang C, Gonzalez-Perez V, Xia XM. Lingle CJ. Knockout of the BK beta2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow-wave burst activity. J Gen Physiol. 2014;144:275–295. doi: 10.1085/jgp.201411253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JN, Chan CS, Tkatch T, Held J. Surmeier DJ. Nav1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J Neurosci. 2007;27:13552–13566. doi: 10.1523/JNEUROSCI.3430-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinville A, Fundin B, Meury L, Jureus A, Sandberg K, Krupp J, Ahmad S. O'Donnell D. Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: expression in the autonomic and endocrine systems. J Comp Neurol. 2007;504:680–689. doi: 10.1002/cne.21484. [DOI] [PubMed] [Google Scholar]
- Nassar-Gentina V, Pollard HB. Rojas E. Electrical activity in chromaffin cells of intact mouse adrenal gland. Am J Physiol Cell Physiol. 1988;254:C675–C683. doi: 10.1152/ajpcell.1988.254.5.C675. [DOI] [PubMed] [Google Scholar]
- Nassar MA, Baker MD, Levato A, Ingram R, Mallucci G, McMahon SB. Wood JN. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol Pain. 2006;2:33. doi: 10.1186/1744-8069-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH. Wood JN. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101:12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivos L. Artalejo AR. Muscarinic excitation-secretion coupling in chromaffin cells. Acta Physiol (Oxf) 2007;192:213–220. doi: 10.1111/j.1748-1716.2007.01816.x. [DOI] [PubMed] [Google Scholar]
- Raman IM. Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie AK. Catecholamine secretion in a rat pheochromocytoma cell line: two pathways for calcium entry. J Physiol. 1979;286:541–561. doi: 10.1113/jphysiol.1979.sp012636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S. Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD. Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol. 2005;564:803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM. Hunter JC. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272:14805–14809. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- Segura F, Brioso MA, Gomez JF, Machado JD. Borges R. Automatic analysis for amperometrical recordings of exocytosis. J Neurosci Meth. 2000;103:151–156. doi: 10.1016/s0165-0270(00)00309-5. [DOI] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH. Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM. Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci: Off J Soc Neurosc. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese A. Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S. Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KR, Huertas MA, Horn JP, Canavier CC. Levitan ES. Pacemaker rate and depolarization block in nigral dopamine neurons: a somatic sodium channel balancing act. J Neurosci. 2012;32:14519–14531. doi: 10.1523/JNEUROSCI.1251-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A, Mahapatra S, Caro A, Ruth P, Zuccotti A, Knipper M. Carbone E. Ca(v)1.3 and BK channels for timing and regulating cell firing. Mol Neurobiol. 2010;42:185–198. doi: 10.1007/s12035-010-8151-3. [DOI] [PubMed] [Google Scholar]
- Vandael DHF, Zuccotti A, Striessnig J. Carbone E. Ca(V)1.3-driven SK channel activation regulates pacemaking and spike frequency adaptation in mouse chromaffin cells. J Neurosci. 2012;32:16345–16359. doi: 10.1523/JNEUROSCI.3715-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada A, Wanke E, Gullo F. Schiavon E. Voltage-dependent Na(v)1.7 sodium channels: multiple roles in adrenal chromaffin cells and peripheral nervous system. Acta Physiol (Oxf) 2008;192:221–231. doi: 10.1111/j.1748-1716.2007.01810.x. [DOI] [PubMed] [Google Scholar]
- Wakade AR( Studies on secretion of catecholamines evoked by acetylcholine or transmural stimulation of the rat adrenal gland. J Physiol. 1981;313:463–480. doi: 10.1113/jphysiol.1981.sp013676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DJ, Chen C. Marley PD. Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. J Physiol. 2002;540:921–939. doi: 10.1113/jphysiol.2001.013370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagita T, Satoh S, Uezono Y, Matsuo K, Nemoto T, Maruta T, Yoshikawa N, Iwakiri T, Minami K. Murakami M. Transcriptional up-regulation of cell surface Na V 1.7 sodium channels by insulin-like growth factor-1 via inhibition of glycogen synthase kinase-3beta in adrenal chromaffin cells: enhancement of 22Na+ influx, 45Ca2+ influx and catecholamine secretion. Neuropharmacology. 2011;61:1265–1274. doi: 10.1016/j.neuropharm.2011.07.029. [DOI] [PubMed] [Google Scholar]
- Zhou Z. Misler S. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem. 1995;270:3498–3505. [PubMed] [Google Scholar]