

Abstract
The development of chronic and recurrent Staphylococcus aureus infections is associated with the emergence of slow-growing mutants known as small-colony variants (SCVs), which are highly tolerant of antibiotics and can survive inside host cells. However, the host and bacterial factors which underpin SCV emergence during infection are poorly understood. Here, we demonstrate that exposure of S. aureus to sublethal concentrations of H2O2 leads to a specific, dose-dependent increase in the population frequency of gentamicin-resistant SCVs. Time course analyses revealed that H2O2 exposure caused bacteriostasis in wild-type cells during which time SCVs appeared spontaneously within the S. aureus population. This occurred via a mutagenic DNA repair pathway that included DNA double-strand break repair proteins RexAB, recombinase A, and polymerase V. In addition to triggering SCV emergence by increasing the mutation rate, H2O2 also selected for the SCV phenotype, leading to increased phenotypic stability and further enhancing the size of the SCV subpopulation by reducing the rate of SCV reversion to the wild type. Subsequent analyses revealed that SCVs were significantly more resistant to the toxic effects of H2O2 than wild-type bacteria. With the exception of heme auxotrophs, gentamicin-resistant SCVs displayed greater catalase activity than wild-type bacteria, which contributed to their resistance to H2O2. Taken together, these data reveal a mechanism by which S. aureus adapts to oxidative stress via the production of a subpopulation of H2O2-resistant SCVs with enhanced catalase production.
INTRODUCTION
Staphylococcus aureus is a frequent cause of chronic and recurrent infections, which often involve the emergence of slow-growing mutants known as small-colony variants (SCVs) (1–14).
The majority of SCVs isolated from clinical samples are auxotrophic for hemin, menadione, or thymidine due to mutations in the hem or men operons or in thyA, respectively (2, 4, 15–19). However, SCVs with mutations conferring resistance to fusidic acid or which arise via mutation in succinate dehydrogenase have also been identified, and there also appear to be isolates with a transient SCV phenotype, which are likely not mutants (12, 20, 21). SCVs with mutations in heme or menaquinone biosynthetic pathways have defective electron-transport chains, which confers resistance to aminoglycoside antibiotics such as gentamicin (1, 6, 8, 9).
Previous work has shown that gentamicin-resistant SCVs emerge in replicating populations in the absence of environmental stress via stochastic mutations but frequently revert to the wild type (WT) via the acquisition of suppressor mutations (15, 17, 22). However, while a few factors have been identified that select for the SCV phenotype, there is also evidence that environmental stimuli can trigger the emergence of SCVs in S. aureus populations, although the mechanism(s) by which this occurs is unknown (12, 23, 24).
In addition to aminoglycoside resistance, SCVs that arise via the loss of the electron transport chain are more tolerant than wild-type bacteria of other classes of bactericidal antibiotics (8, 9, 25–29). Furthermore, SCVs exhibit other phenotypic characteristics which may promote survival in host tissues, including elevated rates of host cell invasion and intracellular survival, enhanced capsule production, and robust biofilm formation (5, 12, 13, 30–32). Several of these phenotypes are ascribed to a combination of decreased Agr activity and enhanced SigB activity, which results in strong expression of surface proteins and an absence of cytolysin production (16, 33–35).
However, there is one aspect of the biology of electron-transport chain defective SCVs that appears to be at odds with a role in chronic infection: an apparently reduced level of defense against oxidative stress. This is important because the generation of reactive oxygen species (ROS) such as O2− and H2O2 by neutrophils is a crucial host defense mechanism against S. aureus (36, 37). To combat ROS, S. aureus uses a number of defensive molecules, including catalase (KatA), superoxide dismutases (SodA/M), and the golden pigment staphyloxanthin (36–44). Despite the importance of these defenses for wild-type S. aureus survival in the host, SCVs have been reported to produce significantly reduced levels of staphyloxanthin and heme auxotrophs are deficient in catalase, which would be expected to make them more susceptible to ROS generated by neutrophils and thus clearance from host tissues (9, 19, 28, 36, 45). Therefore, the aim of this work was to determine the effect of ROS on the emergence and persistence of electron-transport chain defective SCVs within S. aureus populations and establish the degree to which SCVs are sensitive to oxidative stress.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains used in the present study are listed in Table 1. S. aureus was cultured in tryptic soy broth at 37°C with shaking as described previously (22). Broth cultures were inoculated with bacteria from stationary phase (105 CFU ml−1), followed immediately by oxidants or ciprofloxacin and incubated for 16 h at 37°C with shaking at 180 rpm. Bacteria were used in stationary phase since this is when pigmentation of the wild type is greatest (36). Transposon mutants were cultured in the presence of erythromycin (10 μg ml−1), but subsequent assays were performed in the absence of the antibiotic to reduce off-target effects. CFU counts were determined by serial dilution and plating of aliquots onto tryptic soy agar (TSA) or Columbia blood agar (CBA) with or without gentamicin (2 μg ml−1). SCVs were defined as gentamicin-resistant (MIC > 2 μg ml−1) bacteria that produced small, slow-growing, nonhemolytic or weakly hemolytic, and nonpigmented or weakly pigmented colonies on blood agar. We did not study other types of SCVs, such as those resistant to sulfonamides or fusidic acid. DNA from transposon mutants was transduced into wild-type SH1000 by transduction with ϕ11 as described previously, and transductants bearing the inserted transposon were selected for on TSA containing erythromycin (10 μg ml−1) (46).
TABLE 1.
Bacterial strains used in this study
| Bacterial strain | Relevant characteristicsa | Source or reference |
|---|---|---|
| E. coli | ||
| DC10B | DNA cytosine methyltransferase deficient | 50 |
| DC10B/pCN34 | DC10B transformed with pCN34 | 49; this study |
| DC10B/pCL55 | DC10B transformed with pCL55 | 47 |
| DC10B/phemB | DC10B transformed with pCL55 containing the promoter of the hem operon fused to the coding sequence of hemB | |
| DC10B/pmenD | DC10B transformed with pCL55 containing the promoter of the men operon fused to the coding sequence of menD | |
| DC10B/pumuC | DC10B transformed with pCN34 containing the promoter and coding region of umuC | |
| S. aureus | ||
| SH1000 | Functional rsbU+ derivative of NCTC 8325-4 | 66 |
| SCV2 | SH1000-derived Gmr SCV without auxotrophy for Men, Hem, Thy, CO2, or fatty acids; isolated in the absence of oxidants | This study |
| SCV4 | SH1000-derived Gmr SCV without auxotrophy for Men, Hem, Thy, CO2, or fatty acids; isolated in the absence of oxidants | |
| SCV9 | SH1000-derived Gmr SCV with auxotrophy for Hem; isolated in the absence of oxidants | |
| SCV13 | SH1000-derived Gmr SCV with auxotrophy for CO2; isolated in the absence of oxidants | |
| SCV14 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the absence of oxidants; single nucleotide deletion in menB, leading to premature stop codon after 28 amino acids | |
| SCV15 | SH1000-derived Gmr SCV with auxotrophy for fatty acids; isolated in the absence of oxidants | |
| SCV17 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the absence of oxidants | |
| SCV20 | SH1000-derived Gmr SCV with auxotrophy for CO2; isolated in the absence of oxidants | |
| SCV21 | SH1000-derived Gmr SCV without auxotrophy for Men, Hem, Thy, CO2, or fatty acids; isolated in the absence of oxidants | |
| SCV1036 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the absence of oxidants | |
| SCV1045 | Single nucleotide deletion in menA, leading to a premature stop codon after 184 amino acids | |
| SCV1047 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the absence of oxidants; single nucleotide polymorphism in menF, resulting in A367D substitution of a highly conserved alanine | |
| SCV1057 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of H2O2; single nucleotide deletion in menE, leading to a premature stop codon after 205 amino acids | |
| SCV1058 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of H2O2; single nucleotide deletion in menB, leading to a premature stop codon after 113 amino acids | |
| SCV1060 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of H2O2; single nucleotide polymorphism in menE, leading to a premature stop codon after 165 amino acids | |
| SCV1072 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of paraquat; single nucleotide polymorphism in menE, leading toa premature stop codon after 373 amino acids | |
| SCV1072 katA::Tn | SCV1072 transduced with DNA from NE1366, resulting in inactivation of catalase; Eryr | |
| SCV1077 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of paraquat; single nucleotide polymorphism in menF, resulting in T79K substitution | |
| SCV1080 | SH1000-derived Gmr SCV with auxotrophy for Men; isolated in the presence of paraquat; single nucleotide polymorphism in aroB, resulting in H241Q substitution of a highly conserved histidine likely involved in metal binding | |
| SH1000t | SH1000-derived Tcr strain geh::pTM304 | 22 |
| MJH502 | SH1000 sigB::Tc | 66 |
| SH331 | SH1000 rexA::Tn | This study |
| SH445 | SH1000 umuC::Tn | |
| SH805 | SH1000 recA::Tn | |
| SH1012 | SH1000 rexB::Tn | |
| SH1366 | SH1000 transduced with DNA from NE1366, resulting in inactivation of catalase; Eryr | This study |
| SH1866 | SH1000 dinB::Tn | |
| SH445/pCN34 | SH1000 umuC::Tn transformed with pCN34 | |
| SH445/pumuC | SH1000 umuC::Tn transformed with pumuC | |
| USA300 LAC | LAC strain of the USA300 CA-MRSA lineage | 85 |
| USA300 hemB | USA300 in which hemB has been deleted | 35 |
| USA300 hemB geh::pCL55 | USA300 hemB mutant with pCL55 integrated into the geh locus | This study |
| USA300 hemB geh::phemB | USA300 hemB mutant with phemB integrated into the geh locus, restoring wild-type phenotype | |
| USA300 menD | USA300 in which menD has been deleted | 35 |
| USA300 menD geh::pCL55 | USA300 menD mutant with pCL55 integrated into the geh locus | This study |
| USA300 menD geh::pmenD | USA300 menD mutant with pmenD integrated into the geh locus, restoring wild-type phenotype | |
| USA300 JE2 | USA300 cured of plasmids | 61 |
| NE331 | USA300 JE2 rexA::Tn | |
| NE445 | USA300 JE2 umuC::Tn | |
| NE805 | USA300 JE2 recA::Tn | |
| NE1012 | USA300 JE2 rexB::Tn | |
| NE1366 | USA300 JE2 katA::Tn | |
| NE1866 | USA300 JE2 dinB::Tn | |
| CX003SCV | Clinical Men-auxotroph SCV | This study |
| CX003WT | Revertant of CX003SCV with wild-type phenotype | |
| CX004SCV | Clinical Men auxotroph SCV | |
| CX004WT | Revertant of CX004SCV with wild-type phenotype | |
| CX005SCV | Clinical Men auxotroph SCV | |
| CX005WT | Revertant of CX005SCV with wild-type phenotype | |
| CX006SCVM | Clinical Men auxotroph SCV | |
| CX006SCVH | Clinical Hem auxotroph SCV | |
| CX006WT | Revertant of CX005SCVM with wild-type phenotype | |
| CX009SCV | Clinical Hem auxotroph SCV | |
| CX009WT | Revertant of CX009SCV with wild-type phenotype | |
| Wood | Wild-type | NCTC 7121 |
| MRSA252 | Wild-type | 86 |
Gmr, gentamicin resistance; Tcr, tetracycline resistance; Eryr, erythromycin resistance; Hem, hemin; Men, menadione; Thy, thymidine.
USA300-derived hemB and menD mutants (35) were complemented by cloning the appropriate gene, under the control of the native promoter of the relevant operon, into integrative plasmid pCL55 (47). For hemB, the promoter region (48) was amplified by using the primer pair Hem Prom For (CCTTTCGTCTTCAACGTATATTCATTGACCCG) and Hem Prom Rev (GTCTATCAAATTTCATGTTCAATTCCTCCTAGG). For menD, the 331 bases upstream of menF, the first gene in the men operon, were amplified by using the primer pair Men Prom For (CCCTTTCGTCTTCAATGAATACAAAACCTCTTTAAATC) and Men Prom Rev (CTTTATGATTTCCCATATAAAAGCGATCTCCTGCC). Amplicons containing promoter regions were fused with coding sequences by using the Gibson assembly protocol (NEB). DNA overhangs were built into primers (indicated in boldface) to facilitate recombination. The hemB gene was amplified by using the primer pair Hem For (GGAGGAATTGAACATGAAATTTGATAGACATAG) and Hem Rev (TACCGAGCTCGAATTCACCTTAATTATCTAAATAGC), while menD was amplified by using the primer pair Men For (GGAGATCGCTTTTATATGGGAAATCATAAAGCAG) and Men Rev (ACCGAGCTCGAATTCTTATAATGTGTCATGAATCATTTC). The vector, pCL55, was amplified by using primers with overhangs to facilitate Gibson assembly. For hemB constructs, pCL55 was amplified by using the primer pair pCL55 Hem For (AGATAATTAAGGTGAATTCGAGCTCGGTACC) and pCL55 Hem Rev (AATGAATATACGTTGAAGACGAAAGGGCCTC), and for menD constructs, the primer pair pCL55 Men For (CATGACACATTATAAGAATTCGAGCTCGGTAC) and pCL55 Men Rev (AGAGGTTTTGTATTCATTGAAGACGAAAGGG) was used.
The SH1000 umuC::Tn mutant was complemented with the umuC coding sequence (including the promoter region) using pCN34 (49). The umuC gene and the promoter region was amplified by using the primer pair umuC For (AAAGGATCCCGGCGTCAGTTACTTCGC) and umuC Rev (AAAGGATCCCGTATCGCGACGCACTAC), which included BamHI restriction sites (underlined) to enable ligation into BamHI-digested pCN34. Vector without the umuC coding sequence served as a control. The successful generation of constructs was confirmed by DNA sequencing. Vectors were constructed in Escherichia coli strain DC10B and transformed directly into S. aureus strains (50). In the case of pCL55, plasmid integration was confirmed by PCR. DC10B was cultured in LB broth containing ampicillin (100 μg ml−1) where necessary to select for plasmid maintenance (50). S. aureus strains containing plasmids were cultured in the presence of 10 μg of chloramphenicol ml−1 (pCL55) or 90 μg of kanamycin ml−1 (pCN34) and washed in phosphate-buffered saline (PBS) to remove antibiotics, and experiments were performed in the absence of antibiotics to avoid off-target effects.
Hydrogen peroxide quantification.
The concentration of H2O2 in culture medium was determined by using a Pierce quantitative peroxide assay kit according to the manufacturer's instructions.
SCV stability assays.
The stability of SCV isolates was determined as described previously (22). SCV colonies (n = 30 to 50) on TSA plates containing 2 μg of gentamicin ml−1 were subcultured by streaking them onto antibiotic-free TSA using a sterile pipette tip, followed by incubation at 37°C for 48 h. Subsequently, subcultured bacteria were scored for reversion. If all colonies in the subcultured streak retained the SCV phenotype, then that SCV was scored as stable. If all of the colonies had the WT phenotype, the streak was scored as unstable. SCVs that generated a mixture of SCV and WT phenotype were categorized as partially stable (22).
Phenotype-switching assay.
To understand the relative contributions of phenotype-switching and replication to determining the size of the SCV population, we used a previously described assay (22). Briefly, inocula of 105 CFU tetracycline-sensitive wild-type SH1000 S. aureus and 10 CFU SH1000t tetracycline-resistant SCVs were cultured in the absence or presence of oxidants. The total CFU were quantified by plating serial dilutions on TSA plates. SCVs were isolated on TSA plates containing gentamicin, as described above. Subsequently, 100 SCV colonies were picked and patched onto TSA plates containing tetracycline to determine the percentage of SCVs that were resistant to the antibiotic. This assay determines the percentage of the final SCV population that arose from wild-type or SCV bacteria in the inoculum. Previous work has shown that the tetracycline-resistant strain does not suffer a fitness cost under the conditions used (22).
SCV reversion assay.
Individual SCV colonies were picked from TSA plates containing gentamicin (2 μg ml−1) and resuspended in 150 μl of PBS. Aliquots (50 μl) of each bacterial suspension were then spread over TSA plates containing paraquat (0.1 mM), ciprofloxacin (0.05 μg ml−1), or neither before incubation for 24 h at 37°C. Subsequently, plates were examined for the presence of colonies of wild-type bacteria (large, pigmented colonies).
Mutation rate analyses.
S. aureus strains were cultured in 3 ml of TSB after inoculation from agar plates. Cultures were diluted to 105 CFU ml−1 in 30 parallel 1-ml cultures (this was the smallest inoculum that allowed bacterial growth in the presence of H2O2) and grown to stationary phase at 37°C with shaking. Total CFU counts were determined in 10 randomly selected cultures by plating of serial dilutions onto TSA without antibiotics. Each culture was then plated onto TSA containing rifampin (100 μg ml−1), followed by incubation for 24 h at 37°C. The number of resistant colonies was counted, and mutation rates with confidence intervals were calculated by using the maximum-likelihood setting of the FALCOR mutation rate calculator (51, 52). The statistical significances of differences between the mutation rate in the absence and presence of H2O2 were determined by using a Student t test as described in equation 5 of FALCOR (51, 52).
Hydrogen peroxide killing assays.
S. aureus cells in late exponential phase (when pigmentation is strongest) were washed by sequential rounds of centrifugation and resuspension in PBS before subsequent adjustment to a final concentration of ∼106 CFU ml−1 in PBS. Bacterial suspensions (10 μl) were added to the wells of a microtiter plate, and H2O2 was added to 30 mM for SH1000-derived isolates or 25 mM for USA300-derived strains (this concentration was chosen because preliminary assays indicated that they were the lowest required to achieve >1-log killing of the wild-type over 1 h [data not shown]). The microtiter plate was incubated at 37°C in the dark for 15 to 60 min. Surviving bacteria were enumerated by serial dilution in PBS and plating onto CBA (which naturally contains catalase to neutralize residual H2O2).
Catalase activity assay.
S. aureus was grown and washed as described above for hydrogen peroxide killing assays before 107 CFU were added to 1 ml of PBS containing 100 μM H2O2. The concentration of H2O2 was measured over time by using a Pierce quantitative peroxide assay kit in accordance with the manufacturer's instructions and the use of a standard plot.
Whole-genome sequencing.
DNA was extracted from wild-type SH1000 and derived SCVs using lysostaphin and phenol-chloroform extraction (46). Purified DNA was sheared into fragments of ∼150 bp and sequenced using an Illumina MiSeq DNA sequencer. The sequences obtained yielded >100-fold coverage.
RESULTS
Culture of S. aureus in the presence of hydrogen peroxide leads to a specific and dose-dependent increase in the size of the SCV subpopulation.
To determine the effect of oxidative stress on the size of the gentamicin-resistant SCV subpopulation, S. aureus was cultured in the presence of increasing concentrations of H2O2, paraquat, or diamide, which have previously been shown to trigger distinct changes in the staphylococcal proteome (53).
We used an inoculum size (<105 ml−1) that was predicted to not contain SCVs due to their low frequency in the population. Therefore, SCVs that appeared in the cultures were generated by the acquisition of mutations in wild-type cells (22).
Increasing concentrations of H2O2 or paraquat, but not diamide, led to dose-dependent increases in the size of the gentamicin-resistant SCV subpopulation, which was up to 50-fold greater than in the absence of oxidative stress (Fig. 1A, B, and C). Similar effects of H2O2 (1 mM) and paraquat (5 mM) on the gentamicin-resistant SCV subpopulation were also observed for genetically diverse S. aureus strains USA300 LAC, Wood, and MRSA252 (Fig. 1D).
FIG 1.

Hydrogen peroxide exposure leads to a specific, dose-dependent increase in the size of the SCV subpopulation. (A to C) S. aureus SH1000 was cultured in increasing concentrations of H2O2 (A), paraquat (B), or diamide (C) for 16 h, and the sizes of the total and SCV populations were determined. (D) The effect of H2O2 (1 mM) or paraquat (5 mM) on the size of the SCV subpopulations of strains USA300 LAC (LAC), Wood, and MRSA252 (252) were also determined. None of the oxidants used affected the size of the total population at the concentrations used (data not shown). (E) Wild-type S. aureus SH1000 was incubated in the absence (−) or presence (+) of paraquat and/or catalase, and the sizes of the total (open bars) and SCV populations (closed bars) were determined. (F) Colony morphology of wild-type S. aureus SH1000 (top left panel) and a representative menadione-auxotrophic SH1000-derived SCV (SCV1072) isolated from a culture containing paraquat (top right panel). The lack of pigment in the SCV seen on agar plates (top right panel) was also seen after liquid culture (bottom left panel). Culture of this SCV isolate in the presence of menadione restored pigmentation, indicating deficiencies in menaquinone production (bottom right panel) (2). Values which are significantly different (P < 0.05 [Student t test]) from oxidant-free conditions are indicated (*). These data represent the mean averages of 12 independent cultures. Error bars represent the standard deviations of the mean.
Paraquat generates superoxide radicals, which S. aureus can convert to H2O2 via superoxide dismutases (43). To determine whether the effect of paraquat on SCV numbers was due to H2O2 or superoxide production, S. aureus was cultured with paraquat in the presence or absence of purified bovine catalase (10 μg ml−1). The presence of exogenous catalase abrogated the effect of paraquat on SCV subpopulation expansion, indicating that H2O2 production, rather than superoxide, was responsible for the increased SCV frequency (Fig. 1E). However, it is possible that superoxide enhances H2O2-mediated damage by increasing free iron levels in the cell (54).
To investigate the nature of the recovered SCVs, representative colonies from independent cultures containing H2O2 or paraquat (n = 6) were examined, and each was found to have a typical SCV phenotype, with reduced pigmentation, and were classified as menadione auxotrophs (Fig. 1F). Whole-genome sequencing of a selection of each of the independently isolated menadione-auxotrophs (two from TSB only, three from TSB plus H2O2, and three from TSB plus paraquat) revealed mutations in genes in the menaquinone biosynthetic pathway (aroB and menABDEF) (Table 1) (55). These mutations were similar to those reported previously in clinical isolates and confirm that the SCV phenotype was due to genetic changes rather than to epigenetic effects or the physiological response of the bacterium to oxidative stress (15, 17).
SCVs emerge during H2O2-induced bacteriostasis.
To understand how H2O2 modulates the size of the SCV subpopulation, we monitored the population dynamics of S. aureus during growth in the absence or presence of H2O2. As reported previously, wild-type S. aureus grew rapidly in the absence of H2O2 and produced a small SCV subpopulation during the early exponential phase (Fig. 2A) (22). In contrast, there was no change in total CFU counts in the presence of 1 mM H2O2, resulting in an extended lag phase that lasted until the H2O2 concentration was reduced to <400 μM (presumably due to the action of catalase and/or alkyl hydroperoxidase [40]). Once the H2O2 concentration was reduced, S. aureus replication began at a similar rate to that seen in the absence of H2O2 (Fig. 2A).
FIG 2.

SCV emergence in S. aureus populations exposed to a bacteriostatic concentration of H2O2. (A) The numbers of wild-type (circles) and SCV (squares) CFU ml−1 were measured over time in the absence (open symbols) or presence (closed symbols) of 1 mM H2O2 (left axis). The concentration of H2O2 was also measured over time (dashed line, right axis). (B) The growth of a mixed population of phenotypically stable SCVs in the absence (open symbols) or presence (closed symbols) of H2O2 was also measured over time, together with the concentration of H2O2 (dashed line, axes as for panel A). (C) Growth of a single menadione-auxotrophic SCV (SCV1072) over time in the absence (open symbols) or presence (closed symbols) of H2O2. The concentration of H2O2 is indicated by the dashed line and the right-hand axis. The data points represent the mean average of 12 independent cultures. Error bars represent the standard deviations of the mean.
We hypothesized that the elevated rate of SCV emergence in the presence of H2O2 was either due to switching of wild-type bacteria into SCVs or, despite the small inoculum size, the replication of a very few SCVs present in the inoculum. To test whether SCVs could replicate in the presence of H2O2, we inoculated broth containing 1 mM H2O2 with a mixed population of phenotypically stable SCVs (including those auxotrophic for menadione, hemin, fatty acids, and those without identified auxotrophy) that represent the composition of SCVs found in cultures not exposed to oxidants and then monitored growth. Similar to wild-type bacteria, the growth of the SCV population was inhibited by 1 mM H2O2, leading to an extended lag phase relative to SCV growth in the absence of H2O2 (Fig. 2B). However, as seen for the wild-type population, once the concentration of H2O2 fell to ∼400 μM, SCV replication began (Fig. 2B). Because menadione-auxotrophic SCVs were the predominant SCV type isolated from cultures exposed to H2O2, we undertook a similar experiment to that described in Fig. 2B using a stable menadione auxotroph isolated from a culture exposed to paraquat (SCV1072). As for the wild-type and the mixed SCV inoculum, SCV1072 did not initiate replication until the H2O2 concentration had fallen to ∼400 μM (Fig. 2C). Therefore, SCV replication is not a viable explanation for the appearance of SCVs at early time points in cultures exposed to H2O2, when the oxidant is at concentrations inhibitory to staphylococcal growth.
SCV emergence in the presence of H2O2 is dependent upon mutagenic DNA repair.
Because mutations have been shown to occur in stressed, nonreplicating E. coli cells via DNA double-strand break repair and the SOS response, which is strongly induced in S. aureus upon exposure to H2O2, we hypothesized that this may provide a mechanism for the emergence of SCVs under growth-inhibitory conditions (56–60). To test this, we utilized the NARSA transposon library to identify genes that were important for mutagenic DNA repair in the USA300 background (61). Wild-type and transposon mutants deficient in genes associated with DNA repair and the SOS response, including recombinase A (recA::Tn), error-prone polymerases IV or V (dinB::Tn, umuC::Tn), and rexAB (functionally equivalent to recBCD in E. coli), were grown in the absence or presence of H2O2. Several mutants, including recA and rexAB mutants, displayed increased sensitivity to H2O2, confirming a role in repair of damage caused by oxidative stress (data not shown). However, this increased sensitivity required a lower concentration of H2O2 (0.05 mM) to be used in these experiments compared to that of Fig. 1 and 2. Nonetheless, even at these reduced concentrations, H2O2 resulted in an increase in SCV frequency of ∼10-fold in wild-type S. aureus populations (Fig. 3A). In contrast, H2O2 exposure had no effect on the size of the SCV subpopulations of the umuC::Tn, recA::Tn, rexA::Tn, or rexB::Tn mutants relative to cultures without oxidant (Fig. 3A), indicating that double-strand break repair and the SOS response is required for SCV emergence during H2O2 exposure but not in its absence. In contrast, the mutant lacking functional dinB (which is not part of the S. aureus SOS regulon [62]) had only a slight defect in H2O2-induced SCV formation (Fig. 3A).
FIG 3.

Expansion of the SCV subpopulation in response to H2O2 requires error-prone polymerase V under the control of the SOS regulon. (A) Frequency of SCVs in populations of WT S. aureus USA300 or transposon mutants lacking functional polymerase V (umuC::Tn), RecA (recA::Tn), polymerase IV (dinB::Tn), or RexAB (rexA::Tn and rexB::Tn) in the absence (open bars) or presence of H2O2 (filled bars). (B) Mutation rate of strains detailed in panel A grown in the absence (open bars) or presence (closed bars) of H2O2. (C and D) As for panels A and B but with strains constructed in the SH1000 background. In addition, panels C and D show data from the umuC::Tn mutant transformed with pCN34 only or pCN34 containing the umuC gene and promoter region (pumuC). (E) Total (WT) and SCV CFU counts from a SH1000-derived sigB mutant (MJH502) grown in the absence (CTL) or presence of paraquat (para). (F) Mutation rate of wild-type SH1000 (WT) and an SH1000-derived sigB mutant (sigB) grown in the absence (open bars) or presence (closed bars) of H2O2. The data in panels A, C, and E represent the mean averages of 12 independent cultures, and error bars represent the standard deviation of the mean. Values in panels B, D, and F represent the mutation rate as determined by fluctuation analysis, and error bars represent the 95% confidence intervals. Values which are significantly different (P < 0.05 [Student t test corrected for multiple comparisons via the Bonferroni method]) in the presence of H2O2 by comparison to those obtained in the absence of oxidants are indicated (*).
These findings were concordant with measurements of the mutation rate in S. aureus grown with or without H2O2, which showed that H2O2 exposure increased the mutation rate >5-fold in wild-type S. aureus but had no effect on the mutation rate of strains defective for rexAB or polymerase V (Fig. 3B). There was a modest (<3-fold) increase in the mutation rate of the recA::Tn mutant, but this was still significantly reduced compared to the wild-type (Fig. 3B). In contrast, there was no decrease in H2O2-induced mutation in S. aureus lacking polymerase IV (dinB) (Fig. 3B).
To ensure that these findings also applied to the SH1000 genetic background, DNA from recA::Tn, rexA::Tn rexB::Tn, dinB::Tn, and umuC::Tn was transduced into SH1000. Each of the DNA repair mutants behaved in a very similar manner to that described above for the USA300 mutants. Specifically, the mutants were defective for H2O2-induced SCV formation or mutation, with the exception of dinB::Tn (Fig. 3C and D). Complementation of the umuC coding sequence, under the control of the native promoter, to the umuC::Tn mutant restored H2O2-induced mutation and SCV formation, while the umuC::Tn mutant transformed with vector alone was defective for H2O2-induced mutation and SCV formation (Fig. 3C and D).
In E. coli, stress-induced mutation requires both the SOS response and a second signal via the RpoS sigma factor, which is part of the general stress response (63). Therefore, we considered the possibility that this may also be the case in S. aureus, especially since previous work has indicated that the alternative sigma factor SigB is required for SCV emergence in the presence of antibiotics (64, 65). However, the absence of SigB did not prevent an SCV population increase in the presence of 0.1 mM paraquat (Fig. 3E), and the sigB mutant was not defective for a paraquat-induced increase in the mutation rate (Fig. 3F). Therefore, SigB does not appear to be required for oxidative-stress-induced mutation in S. aureus. It should be noted, however, that we were unable to test higher concentrations of paraquat or H2O2 at any concentration used in Fig. 1 due to the increased sensitivity of the sigB mutant to oxidative stress (66). Taken together, these data demonstrate that components of the SOS response trigger switching from the wild-type to SCV phenotype via mutagenic DNA repair, which explains the emergence of SCVs in the presence of H2O2.
H2O2 selects for phenotypically stable SCVs, enhancing population expansion via replication.
The data presented in Fig. 2A demonstrate that the SCV subpopulation emerged in the presence of H2O2 and continued to expand after the concentration of the oxidant fell below the growth-inhibitory concentration. However, it was not clear whether SCV population expansion at growth-permissive concentrations of H2O2 was predominantly due to the replication of a few SCVs generated by mutagenic DNA repair or was due to a very high rate of phenotype-switching from the wild-type to the SCV phenotype.
To investigate this, culture medium with or without oxidants was inoculated with ∼105 CFU tetracycline-sensitive (Tets) wild-type S. aureus ml−1 and ∼10 CFU SCVs from tetracycline-resistant SH1000 (Tetr) ml−1 and grown for 24 h. It should be noted that the mixed SCV subpopulation arose in cultures that had not been exposed to oxidants.
As expected, the size of the SCV subpopulation in cultures containing oxidants was greater than those without oxidants (Fig. 4A). In the absence of oxidative stress, the percentage of SCVs that were tetracycline resistant fell from 100% in the inoculum to ca. 20% in the mature culture, indicating that 80% of the final SCV subpopulation had arisen via phenotype switching from the tetracycline-sensitive wild-type population (Fig. 4B), i.e., SCVs revert at high frequency in the absence of oxidative stress. In contrast, in the presence of H2O2 or paraquat, the percentage of tetracycline-resistant SCVs at 24 h were ca. 60% each, indicating that these oxidative stresses select for maintenance of the SCV phenotype and that SCV reversion to the wild type does not occur at a high frequency in the presence of oxidants (Fig. 4B).
FIG 4.

Hydrogen peroxide selects for the SCV phenotype. (A) Tryptic soy broth aliquots were inoculated with 105 CFU tetracycline-sensitive wild-type S. aureus SH1000 and 10 CFU tetracycline-resistant SCVs in the absence (TSB) or presence of either H2O2 or paraquat (para), and the sizes of the total (open bars) and SCV (filled bars) populations were determined after 16 h of culture. (B) After 16 h of culture, the percentage of SCVs that were resistant to tetracycline was determined. Values significantly (P < 0.05 [Student t test]) different from those obtained with bacteria cultured in the absence of oxidants (TSB) are indicated (*). Bars represent the mean averages of 12 independent cultures. Error bars represent the standard deviations of the mean. (C, D, and E) The relative stability of SCVs isolated from cultures without (TSB) (C) or with H2O2 (D) or paraquat (para) (E) were determined by using a previously described assay (22). Individual SCVs were classified as stable (stab), partially stable (part), or unstable (unst) as described in Materials and Methods. The data points represent a single independent culture. The percentag of SCVs classified as unstable was significantly lower in cultures containing oxidants than those without (an asterisk [*] indicates a significant difference relative to TSB without oxidants). (F and G) SCVs that were incubated either in the absence (F) or presence (G) of a subinhibitory concentration of H2O2 were assessed for phenotypic stability. Each data point represents a single independent culture. SCVs that were exposed to H2O2 or paraquat were significantly more stable than those incubated in TSB alone. Each data point represents a single culture (10 from each condition). Significant differences between each stability category (P < 0.05 [Student t test corrected for multiple comparisons via the Bonferroni method]) are indicated (*).
It has been shown previously that SCV replication is associated with an increase in phenotypic stability, since unstable SCVs revert to the wild-type (22). To test whether oxidants select for SCV stability, we used a previously described stability assay (22) and found that SCVs that arose in cultures exposed to H2O2 or paraquat were significantly more stable than those that arose in broth only (Fig. 4C, D, and E). We then generated a pool of SCVs that had arisen spontaneously in cultures not exposed to oxidants and then grew them in the absence or presence of H2O2. Oxidative stress resulted in significantly increased SCV stability, demonstrating that H2O2 selects for SCV stability regardless of whether SCVs arose via the SOS response or spontaneously (Fig. 4F and G). Therefore, the exposure of SCVs to oxidants results in enhanced stability, which reduces reversion to the wild-type and thus enables SCV population expansion via replication.
Nonoxidative SOS induction promotes SCV reversion to the wild type.
The data presented in Fig. 4 strongly suggest that H2O2 selects for phenotypically stable SCVs, which was surprising because activation of the SOS response would be expected to increase the frequency of suppressor mutations which promote SCV reversion to the wild-type phenotype. This suggests that the selective pressure exerted by oxidants on SCVs is great enough to overcome the increased mutation rate caused by induction of the SOS response.
However, we considered two alternative explanations for the enhanced stability of SCVs exposed to oxidative stress: that SCVs generated by the SOS response are inherently more stable than those that arise spontaneously or that the SOS response cannot trigger reversion of SCVs to wild-type bacteria.
To test these possibilities, we used the antibiotic ciprofloxacin, which induces a very similar DNA damage repair to that described upon H2O2 exposure (56, 62). Exposure of wild-type but not umuC::Tn mutant bacteria to a subinhibitory concentration of ciprofloxacin led to an increase in SCV frequency, confirming that induction of the SOS response promotes SCV emergence via mutagenic DNA repair (Fig. 5A). However, the SCVs triggered by ciprofloxacin were no more stable than those which emerged in the absence of the antibiotic (Fig. 4C and 5B). This demonstrates that SCVs generated via the SOS response are not inherently more stable than those that arise spontaneously during bacterial replication. We then tested whether the SOS response can promote SCV reversion to the wild type by exposing a panel of SCVs with various levels of stability to ciprofloxacin or paraquat. In five of the seven SCVs examined, ciprofloxacin exposure promoted the frequency of reversions, indicating that SOS induction can indeed promote SCV reversion to the wild type (Fig. 5C). In contrast, exposure of each of the SCVs to paraquat using the same assay either had no effect or reduced SCV reversion frequency (Fig. 5C). Therefore, while both ciprofloxacin and paraquat stress trigger the SOS mutagenic repair pathway, only the antibiotic promotes bidirectional switching between the wild type and SCVs. In contrast, oxidative stress triggers wild-type-to-SCV switching but selects against SCV reversion to the wild type.
FIG 5.
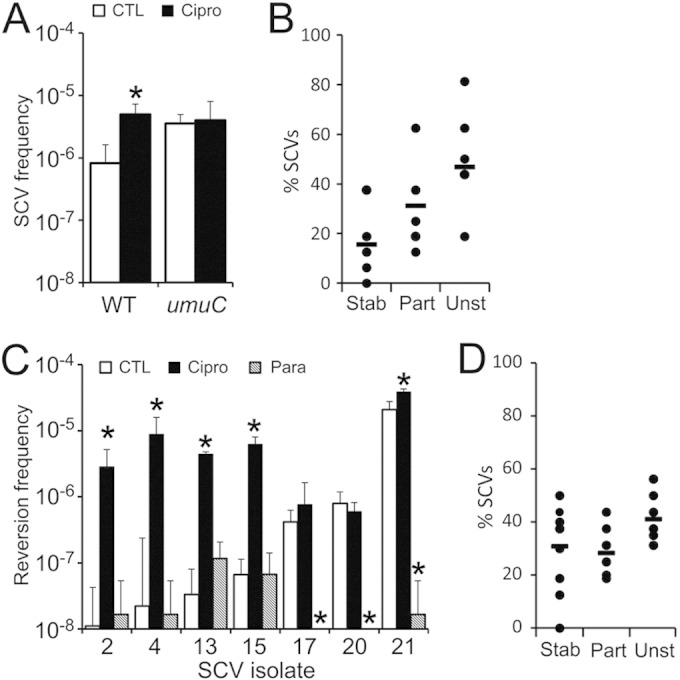
Ciprofloxacin promotes bidirectional phenotype-switching. (A) Wild-type SH1000 and a umuC mutant were grown in the absence (□) or presence (■) of a subinhibitory concentration of ciprofloxacin for 16 h, and the frequency of gentamicin-resistant SCVs was determined. The data represent the mean average of 10 independent cultures, and error bars represent the standard deviations. Values which differ from those seen in media lacking ciprofloxacin are highlighted (*). (B) Stability of SCV isolates that arose in wild-type SH1000 populations in the presence of ciprofloxacin (n = 6). The data are presented and analyzed as described in the legend to Fig. 4. (C) The frequency of SCV reversion to the wild type was determined in the absence (CTL) or presence of ciprofloxacin (Cipro) or paraquat (Para). Reversion rates that differ from the those found on media without supplements are highlighted (*). (D) Stability of SCVs that arose in the umuC background in the absence of oxidants or ciprofloxacin (n = 8).
Finally, we examined whether loss of mutagenic DNA repair affected SCV stability in the absence of SOS-inducing stresses. This revealed that SCVs generated by the umuC::Tn mutant in the SH1000 background were as stable as those that arise in the wild type, demonstrating that the SOS response does not play a role in SCV emergence or reversion in the absence of genotoxic stresses (Fig. 4C and 5D).
SCVs are less susceptible to H2O2 than parental strains.
Previous work has suggested that SCVs should be more susceptible to H2O2 than wild-type bacteria due to the lack of staphyloxanthin pigment and reduced catalase activity in heme auxotrophs (19, 36, 37, 45). However, since H2O2 selected for the SCV phenotype we considered the possibility that SCVs are in fact less sensitive to oxidative stress than wild-type bacteria. To test this, the survival of wild-type SH1000 in the presence of 30 mM H2O2 was compared to a phenotypically stable SCV isolate that arose in the presence of paraquat (SCV1072). This revealed that survival of the SCV was significantly greater than that of the wild type (Fig. 6A). Further analyses of three stable menadione-auxotrophic SCVs from independent cultures containing either H2O2 or paraquat revealed that each SCV isolate was significantly more resistant to H2O2 killing than the WT strain (Fig. 6B). Because culture in the presence of H2O2 or paraquat may have selected for mutations that confer elevated resistance to oxidative stress, three additional, independently isolated menadione-auxotrophic SCVs, which arose in broth without oxidants, were assessed. These showed similarly high levels of resistance to H2O2 killing (Fig. 6B), suggesting that H2O2 resistance is an intrinsic property of menadione-auxotrophic SCVs.
FIG 6.

SCVs are more resistant to H2O2 than wild-type bacteria. (A) The survival of wild-type SH1000 S. aureus (○) or a derived SCV isolate (□) during exposure to 30 mM H2O2 was determined by quantifying CFU. Survival of the SCV was significantly greater (P < 0.05) than that of the wild type at the 45- and 60-min time points. (B) Survival of three independently isolated, menadione-auxotrophic SCVs from cultures without (TSB) or with H2O2 or paraquat (para) was determined after 60 min of exposure to 30 mM H2O2. The survival of the wild type after 60 min is shown for comparison. The survival of all SCV isolates was significantly greater than that of the wild type. (C) Survival of independently isolated SCVs with auxotrophy for hemin (H), CO2 (C), menadione (M), fatty acids (F), or where auxotrophy has not been established (?) after 60 min in 30 mM H2O2. (D) Survival of wild-type or various, independently isolated menadione-auxotrophic SCVs from cultures without oxidants, grown in the absence (−) or presence (+) of menadione. Supplementation of SCV but not wild-type cultures with menadione significantly reduced survival in the presence of H2O2. (E) Growth of S. aureus SH1000 in the presence of the electron-transport chain inhibitor HQNO (Gro) promotes resistance to H2O2 relative to growth in TSB only (Ctl) or growth in TSB, followed by addition of HQNO to bacteria 5 min prior to H2O2 exposure (Post). (F) Survival of the USA300 wild-type strain and derived deletion mutants lacking hemB or menD after incubation in 25 mM H2O2. Survival of the SCVs was significantly greater than WT. (G) Survival after incubation in 25 mM H2O2 of USA300 hemB and menD mutant strains transformed either with pCL55 or PCL55 containing the hemB (phemB) or menD (pmenD) coding sequences. Survival of complemented strains was significantly lower than that of mutants transformed with vector alone (pCL55). (H) Survival of a clinical menadione-auxotrophic SCV (CX003SCV) and derived revertant (CX003WT) with the wild-type phenotype after incubation in 30 mM H2O2. Survival of the SCV was significantly greater than wild-type after 60 min. Significance was determined by using a Student t test corrected for multiple comparisons via the Bonferroni method and declared significant when P < 0.05.
To determine whether resistance to H2O2 killing was related to the auxotrophic phenotype a panel of SCVs, isolated from gentamicin-containing media, with various or unknown auxotrophies was assessed for resistance to H2O2 killing. All of these isolates were significantly more resistant to H2O2 killing than the wild-type strain (Fig. 6C).
We also considered the possibility that gentamicin-resistant SCVs may consistently accumulate mutations which decrease susceptibility to H2O2. To test this, menadione-auxotrophic SCV isolates were cultured in the absence or presence of menadione, and their susceptibility to H2O2 killing was determined. Culture of menadione-auxotrophic SCVs in the presence of menadione produced bacteria that were as sensitive as the wild-type parental strain to H2O2, indicating that secondary mutations are not responsible for the elevated H2O2 resistance (Fig. 6D). It should be noted that menadione has been used as a redox cycling agent to generate superoxide within bacteria. However, the concentrations typically used in such studies are ∼1,000-fold greater than those used here, and there was no significant effect on the viability of the wild-type bacteria in the presence of H2O2 (2, 67). To further test whether reduced susceptibility to H2O2 was solely due to defects in the electron transport chain, S. aureus wild type was cultured in the presence of the Pseudomonas exoproduct HQNO, which blocks the electron transport chain of Gram-positive bacteria and confers an SCV phenotype upon S. aureus (68). Culture of S. aureus in the presence of HQNO produced bacteria that were resistant to H2O2 killing (Fig. 6E). However, the presence of HQNO alone did not alter H2O2 resistance of S. aureus which had been cultured in the absence of the exoproduct (Fig. 6E). Therefore, simply blocking the electron transport chain is not protective against H2O2. Rather, resistance is most likely due to the physiological adaptation of S. aureus to loss of the electron transport chain.
To ensure that these findings were of clinical relevance, we assessed the survival of hemB and menD deletion mutants constructed in the USA300 community-associated MRSA strain. In keeping with the data for SH1000, the survival of wild-type USA300 in the presence of H2O2 was significantly lower than that of isogenic hemB or menD mutants (Fig. 6F). Complementation of either mutant with the relevant coding sequence restored the wild-type growth phenotype (data not shown) and resulted in decreased survival in H2O2, whereas vector alone did not affect growth or survival (Fig. 6G). Finally, increased H2O2 resistance was demonstrated in a clinical menadione-auxotrophic SCV isolate (CX003SCV), relative to a wild-type revertant (CX003WT) (Fig. 6H). Therefore, resistance to H2O2 is an inherent property of electron-transport chain-deficient SCVs that very likely contributes to their ability to persist within host tissues during chronic infections.
Elevated catalase activity in SCVs partially explains enhanced H2O2 resistance.
In addition to staphyloxanthin, catalase is a major staphylococcal defense against H2O2 and has been reported to be expressed at higher levels in clinical SCVs than wild-type S. aureus (33, 40–42). However, heme-auxotrophic SCVs cannot generate functional catalase and SCVs isolated from the lungs of patients with cystic fibrosis have been reported to have reduced catalase activity (5, 45).
To resolve the question of catalase activity in electron-transport chain-deficient SCVs, we measured the ability of wild-type S. aureus SH1000, menadione-auxotrophic SCV isolate SCV1072 and heme-auxotrophic SCV9 to degrade H2O2. This revealed significantly elevated catalase activity in the menadione-auxotrophic SCV relative to the wild type, while the catalase activity of the heme-auxotrophic SCV was significantly impaired relative to wild type (Fig. 7A). Similarly, a clinical menadione-auxotrophic SCV isolate had significantly higher catalase activity than a revertant isolate with the wild-type phenotype (Fig. 7B). In keeping with these data, analyses of an additional four clinical SCV isolates and matching revertants revealed that menadione-auxotrophic SCVs degraded significantly more H2O2 than revertants, whereas heme auxotrophs exhibited defective catalase activity (Fig. 7C).
FIG 7.

Most SCVs display elevated catalase production, which partially explains their decreased susceptibility to H2O2. (A) The H2O2 concentration was measured over time during incubation with SH1000 wild type, menadione-auxotrophic SCV1072 (1072) or heme-auxotrophic SCV9 (9). (B) H2O2 degradation by a clinical menadione-auxotrophic SCV isolate (SCV) and a revertant with wild-type (WT) phenotype. (C) H2O2 degradation after 30 min of incubation by clinical SCV isolates auxotrophic for menadione (M) or hemin (H) and paired revertant isolates. (D) H2O2 degradation after 30 min of incubation by SH1000-derived SCV isolates with diverse or unidentified auxotrophies, including CO2 (C), menadione (M), fatty acids (F), or where auxotrophy has not been established (?) or does not exist. Wild-type (WT) S. aureus SH1000 is included as a control. (E) H2O2 degradation by wild-type USA300 and menD (menD) and hemB (hemB) mutants. (F) H2O2 degradation by menD (menD) and hemB (hemB) mutants transformed with plasmids containing the deleted genes (pmenD or phemB) or vector only (pCL55). (G) H2O2 levels after incubation with SH1000 katA::Tn (katA::Tn) or SCV1072 katA::Tn (1072 katA::Tn) or in the absence of bacteria (No bact.). (H) Survival of wild-type (WT) SH1000, menadione-auxotrophic SCV1072 (1072), SH1000 katA::Tn (katA::Tn), or SCV1072 katA::Tn (1072 katA::Tn) after incubation in 30 mM H2O2 for 1 h. All data points represent the means of four independent experiments performed in duplicate. Error bars represent the standard deviations of the mean. Values that are significantly different from the wild type are denoted (*), with the exception of panel F, in which a significant difference from strains expressing catalase is denoted. Significance was determined by using a Student t test corrected for multiple comparisons via the Bonferroni method and declared significant when P < 0.05.
Next, we examined catalase activity in a panel of gentamicin-resistant SCVs with diverse auxotrophies or no identified auxotrophy isolated from broth cultures in the absence of oxidants. In every case, the catalase activity of the SCV was greater than that of the wild type, although significant variation was observed between isolates (Fig. 7D). Finally, to demonstrate that selection for SCVs does not itself select for elevated catalase activity, we measured catalase in isogenic menD and hemB mutants and the wild-type USA300 parent strain. Consistent with the previous data, this revealed that the menD deletion mutant had significantly higher levels of catalase activity than wild-type USA300, while the hemB deletion mutant was unable to degrade H2O2 (Fig. 7E) (45). Complementation of the hemB and menD mutants with the relevant wild-type coding sequence restored catalase activity to wild-type levels (Fig. 7F).
To confirm that the degradation of H2O2 was due to catalase, rather than alkyl-hydroperoxidase or other peroxidases, we transduced SH1000 WT and SCV1072 with DNA from a USA300 katA::Tn mutant (40). Both strains were completely devoid of catalase activity, confirming the role of catalase (KatA) in the H2O2 breakdown (Fig. 7G).
Next, we sought to determine whether catalase activity explained the enhanced resistance of menadione-auxotrophic SCVs to H2O2 (Fig. 6A and B). Wild-type SH1000, SCV1072, SH1000 katA::Tn, and SCV1072 katA::Tn were each exposed to 30 mM H2O2 for 1 h, and the survival was determined. Strains deficient in catalase showed increased sensitivity to H2O2 (Fig. 7H). However, SCV1072 katA::Tn was not as sensitive to H2O2 as SH1000 katA::Tn, indicating that elevated catalase activity only partially explains the resistance of menadione-auxotrophic SCVs to H2O2 (Fig. 7H).
Taken together, these data indicate that enhanced catalase activity is common to most electron transport chain-deficient SCVs, with the exception of those that cannot synthesize heme. However, additional factors beyond catalase contribute to the resistance of SCVs to H2O2, particularly in heme auxotrophs.
DISCUSSION
S. aureus is responsible for a raft of chronic and recurrent infections despite triggering a potent immune response and antibiotic therapy (69, 70). During the course of infection, S. aureus frequently acquires mutations which promote survival in host tissues, including those that confer a small-colony variant phenotype. The data presented in this report reveal that these mutations increase in frequency in response to one of the major ROS produced by neutrophils, H2O2, via the SOS response. These data support previous work showing that increases in the mutation rate following DNA damage are due to the action of specific repair machinery, rather than the DNA damage itself (57–60).
The ability of bacteria to transiently increase mutation rates in response to environmental stress increases the probability of beneficial (adaptive) mutations that enhance survival (58, 71). Certainly, the emergence of electron transport chain-deficient SCVs in response to oxidative stress appears to be beneficial to S. aureus due to their resistance to oxidative stress and enhanced catalase production, which may enhance survival and/or replication of wild-type bacteria via detoxification of H2O2 (Fig. 6 and 7). In addition, SCVs have a number of other phenotypic properties which might promote persistence in host tissues, including intracellular survival, strong biofilm formation, and a high degree of antibiotic tolerance (5, 12, 13, 30–32). Therefore, a single inactivating mutation in the menaquinone biosynthetic pathway has a profound effect on the phenotype of S. aureus, changing it from a fast-growing, toxin-producing pathogen to a much less pathogenic and slow-growing variant that is able to persist within host tissues for extended periods. However, the close correlation between mutation rate (as determined by mutations at the rpoB locus) and SCV emergence indicates that the men operon is probably not a mutation hot spot, at least with respect to H2O2-associated mutations.
In E. coli, stress-induced mutation involves the low-fidelity polymerases IV and V. Although the ability of a bacterium to increase the mutation rate is a beneficial tool, the principal function of these polymerases is the replication of damaged DNA in a process known as “trans-lesion synthesis,” the low-fidelity nature of the polymerase enabling it to bypass DNA lesions at the cost of a high-frequency of base pair mismatches (72). However, it is not clear whether the increased mutation rate associated with polymerases IV and V is simply a consequence of DNA repair or part of a coevolved mechanism to promote the mutation rate during times of DNA-damaging stress and thus increase the likelihood of beneficial mutations arising.
In S. aureus, trans-lesion synthesis appears to make a small contribution to S. aureus resistance to oxidative stress since the umuC::Tn mutant lacking polymerase V (but not dinB/polymerase IV) was slightly more sensitive to H2O2 than the wild type (data not shown). These data fit with previous work which shows that the expression of umuC, but not dinB, is increased in response to H2O2 (56). Therefore, in S. aureus, the expression of polymerase V appears to facilitate efficient repair of H2O2-mediated DNA damage. However, this does not rule out the possibility that polymerase V is part of a coevolved mechanism to increase the mutation rate in response to environmental stress. For example, umuC is one of the most strongly expressed genes in response to various genotoxic stresses, and this may result in greater polymerase V production than is strictly necessary to repair the damaged DNA (56, 62, 73).
Although H2O2 exposure led to large increases in SCV frequency, this was not solely due to an elevated mutation rate but also to the subsequent replication of emergent SCVs. H2O2 selected for the SCV phenotype, which may reflect the enhanced resistance of SCVs to H2O2, coupled with enhanced catalase production. Therefore, with the possible exception of heme auxotrophs, which lack catalase activity, gentamicin-resistant SCVs appear to be well equipped to persist in environments with a high burden of ROS. This correlates with the clinical evidence that SCVs are able to persist in host tissues, resisting clearance by immune cells that expose the pathogen to the oxidative burst (1–14, 36, 37).
The discovery of enhanced catalase activity in non-heme-auxotrophic SCVs is in keeping with a transcriptomic study of clinical SCV isolates, which reported enhanced katA expression (33). Also in keeping with previous work, proteomics analysis of clinical and in vitro selected heme-auxotrophic SCVS revealed reduced catalase than in corresponding wild-type bacteria (74). Therefore, it appears that loss of the electron transport chain results in enhanced expression of katA, leading to elevated catalase activity, except where heme biosynthesis is defective (33, 74). The reason why catalase activity is elevated in menadione auxotrophic SCVs is under investigation but may reflect the significantly altered metabolic profile of these mutants, which results in altered production of virulence factors and defense molecules such as staphyloxanthin (discussed below) (8, 9, 14, 19, 28, 30, 36, 45). It could, therefore, be hypothesized that enhanced catalase activity is a compensatory mechanism for the loss of staphyloxanthin, but this remains to be tested.
Although there appear to be a number of different pathways by which electron transport chain-deficient SCVs can arise (resulting in diverse auxotrophies), cultures exposed to H2O2 consistently generated menadione auxotrophs. Since menadione auxotrophs were no more resistant to H2O2 and produced similar levels of catalase to other SCVs (with the exception of heme auxotrophs), this is most likely explained by the increased likelihood of this variant arising relative to others. Specifically, menadione-auxotrophic SCVs can arise via inactivating mutations anywhere in the menaquinone biosynthetic pathway, whereas other types of SCV might only arise via mutations in much smaller loci. In support of this hypothesis, in cultures not exposed to H2O2, menadione-auxotrophic SCVs were the most abundant (40%), followed by heme auxotrophs (35%). Therefore, it appears that H2O2 selects for catalase producing SCVs, of which menadione auxotrophs are the most abundant, over the catalase-deficient heme auxotroph.
While SCVs are resistant to oxidative stress and have many phenotypic properties which promote survival in host tissues, these come at the cost of slow growth and loss of exotoxin production (2, 4, 6, 9, 13, 35). Therefore, S. aureus populations must provide a balance between fast-growing, toxin-producing wild-type bacteria which are essential for the establishment of infection and slow-growing non-toxin-producing SCVs, which are able to resist threats such as oxidative stress or antibiotics. Indeed, such a strategy parallels the formation of antibiotic tolerant persister cells (75, 76). Balaban et al. showed that persister cells arise stochastically during growth (type I) and that the frequency increases in response to specific environmental stresses such as subinhibitory concentrations of antibiotics (type II) (75, 77). The production of persister cells prior to antibiotic exposure is hypothesized to be a bet-hedging strategy to ensure the population against exposure to lethal concentrations of antimicrobials that would otherwise eradicate the entire population (75–79).
Although SCVs arise via mutation and persister cells via changes in the physiological state of cells, both events are stochastic in nature and the frequency of these events is influenced by genetic factors and there are, therefore, clear parallels in their emergence within populations (77). We have previously shown that SCVs emerge constitutively in replicating S. aureus cultures (type I) and in this report demonstrate that a specific environmental stress enhances SCV emergence and population size via the action of specific gene products (type II) (22). Therefore, we hypothesize that SCVs comprise a bet-hedging strategy against lethal oxidative and antibiotic stress in a similar way to persisters ensuring populations against bactericidal antibiotics. A key part of such an ensurance policy is the ability to restore the population of wild-type bacteria, which SCVs can do via the repair of mutations or acquisition of suppressor mutations that restore the function of mutated gene products (15, 17). In addition, activation of the SOS mutagenic repair pathway via subinhibitory ciprofloxacin (but not oxidative stress) can promote SCV reversion to the wild type.
The very high resistance of SCVs to concentrations of H2O2 that are lethal to the wild type was a surprising finding given the reduced pigmentation (and catalase levels in the hemin auxotroph). The SCVs that arose under oxidative stress were gentamicin resistant and consistently auxotrophic for menadione, indicating loss of menaquinone biosynthesis and thus interruption of the electron transport chain (2, 9).
The ability of electron transport chain-deficient bacteria to resist H2O2 is in apparent contrast to previous work which showed that blockage of the electron transport chain of E. coli using KCN, or disruption of the menA gene, resulted in increased susceptibility to H2O2 (80). Loss of the electron transport chain in E. coli led to a significant increase in reducing power inside the cell, which propagates the highly damaging Fenton reaction by reducing iron (80). Our experiments with HQNO demonstrate that SCV resistance to H2O2 is not simply a function of a defective electron transport chain. Rather, it is only when S. aureus has been cultured in the absence of a functional electron transport chain that it is able to survive subsequent H2O2 challenge. Although this is partially due to catalase activity, additional factors promote the resistance of electron transport chain-deficient S. aureus to H2O2. For example, S. aureus can avoid redox stress during loss of the electron transport chain by switching to fermentative metabolism via the redox-regulatory element Rex (81–83). Metabolic and transcriptomic analyses of SCVs reveal a huge increase in lactate and alcohol dehydrogenase activity, and this maintains redox balance in the cell, preventing an accumulation of reducing power (14, 30, 83). Furthermore, it is possible that fermentative metabolism renders SCVs more resistant to H2O2 killing by reducing the need for iron-containing metabolic enzymes in the cytoplasm, as well as cytochromes. In support of this hypothesis, wild-type S. aureus exposed to H2O2 increase expression of genes associated with fermentation and a Staphylococcus epidermidis mutant lacking a functional TCA cycle displayed elevated resistance to H2O2 killing (54, 55, 84).
Taken together, the data presented here reveal an additional strategy by which S. aureus can promote its survival under conditions of oxidative stress via the production of small-colony variants in response to H2O2 exposure. In addition to ensuring the population against potentially lethal oxidative stress, elevated SCV production is likely to promote persistent infection via reduced susceptibility to antibiotic therapy, increased biofilm formation, and enhanced intracellular persistence.
ACKNOWLEDGMENTS
A.M.E. gratefully acknowledges funding from the Royal Society and the Department of Medicine, Imperial College. K.L.P. is supported by a Ph.D. Scholarship from the Department of Medicine, Imperial College. D.A.-J. is supported by a Clinician Scientist Fellowship from the Medical Research Council UK.
James Imlay (University of Illinois) and Angela Nobbs (University of Bristol) are acknowledged for helpful discussions. Simon Foster (University of Sheffield), Tim Foster (Trinity College Dublin), Angelika Grundling (Imperial College), Malcolm Horsburgh (University of Liverpool), Ruth Massey (University of Bath), and Terry Roemer (Merck) are acknowledged for kindly providing strains or phage. Transposon mutants were supplied through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program under NIAID/NIH contract HHSN272200700055C. Genome sequencing was supported at the Sanger Institute by WT grant 098051.
REFERENCES
- 1.Abele-Horn M, Schupfner B, Emmerling P, Waldner H, Göring H. 2000. Persistent wound infection after herniotomy associated with small-colony variants of Staphylococcus aureus. Infection 2:53–54. doi: 10.1007/s150100050014. [DOI] [PubMed] [Google Scholar]
- 2.Acar JF, Goldstein FW, Lagrange P. 1978. Human infections caused by thiamine- or menadione-requiring Staphylococcus aureus. J Clin Microbiol 8:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal H, Verrall R, Singh SP, Tang YW, Wilson G. 2007. Small colony variant Staphylococcus aureus multiorgan infection. Pediatr Infect Dis J 26:269–271. doi: 10.1097/01.inf.0000256749.29244.67. [DOI] [PubMed] [Google Scholar]
- 4.Besier S, Ludwig A, Ohlsen K, Brade V, Wichelhaus TA. 2007. Molecular analysis of the thymidine-auxotrophic small colony variant phenotype of Staphylococcus aureus. Int J Med Microbiol 297:217–225. doi: 10.1016/j.ijmm.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Kahl B, Herrmann M, Everding AS, Koch HG, Becker K, Harms E, Proctor RA, Peters G. 1998. Persistent infection with small colony variant strains of Staphylococcus aureus in patients with cystic fibrosis. J Infect Dis 177:1023–1029. doi: 10.1086/515238. [DOI] [PubMed] [Google Scholar]
- 6.Kahl BC. 2014. Small colony variants (SCVs) of Staphylococcus aureus: a bacterial survival strategy. Infect Genet Evol 21:515–522. doi: 10.1016/j.meegid.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Kipp F, Ziebuhr W, Becker K, Krimmer V, Höbeta N, Peters G, Von Eiff C. 2003. Detection of Staphylococcus aureus by 16S rRNA directed in situ hybridization in a patient with a brain abscess caused by small colony variants. J Neurol Neurosurg Psychiatry 74:1000–1002. doi: 10.1136/jnnp.74.7.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proctor RA, van Langevelde P, Kristjansson M, Maslow JN, Arbeit RD. 1995. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin Infect Dis 20:95–102. doi: 10.1093/clinids/20.1.95. [DOI] [PubMed] [Google Scholar]
- 9.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 10.Sendi P, Rohrbach M, Graber P, Frei R, Ochsner PE, Zimmerli W. 2006. Staphylococcus aureus small colony variants in prosthetic joint infection. Clin Infect Dis 43:961–967. doi: 10.1086/507633. [DOI] [PubMed] [Google Scholar]
- 11.Seifert H, von Eiff C, Fätkenheuer G. 1999. Fatal case due to methicillin-resistant Staphylococcus aureus small colony variants in an AIDS patient. Emerg Infect Dis 5:450–453. doi: 10.3201/eid0503.990319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Löffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Eiff C, Becker K, Metze D, Lubritz G, Hockmann J, Schwarz T, Peters G. 2001. Intracellular persistence of Staphylococcus aureus small-colony variants within keratinocytes: a cause for antibiotic treatment failure in a patient with Darier's disease. Clin Infect Dis 32:1643–1647. doi: 10.1086/320519. [DOI] [PubMed] [Google Scholar]
- 14.von Eiff C, Peters G, Becker K. 2006. The small colony variant (SCV) concept: the role of staphylococcal SCVs in persistent infections. Injury 37:S26–S33. doi: 10.1016/j.injury.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Dean MA, Olsen RJ, Long SW, Rosato AE, Musser JM. 2014. Identification of point mutations in clinical Staphylococcus aureus strains that produce small colony variants auxotrophic for menadione. Infect Immun 82:1600–1605. doi: 10.1128/IAI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kriegeskorte A, Block D, Drescher M, Windmüller N, Mellmann A, Baum C, Neumann C, Lorè NI, Bragonzi A, Liebau E, Hertel P, Seggewiss J, Becker K, Proctor RA, Peters G, Kahl BC. 2014. Inactivation of thyA in Staphylococcus aureus attenuates virulence and has a strong impact on metabolism and virulence gene expression. mBio 5:e01447-14. doi: 10.1128/mBio.01447-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lannergård J, von Eiff C, Sander G, Cordes T, Seggewiss J, Peters G, Proctor RA, Becker K, Hughes D. 2008. Identification of the genetic basis for clinical menadione-auxotrophic small-colony variant isolates of Staphylococcus aureus. Antimicrob Agents Chemother 52:4017–4022. doi: 10.1128/AAC.00668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaaff F, Bierbaum G, Baumert N, Bartmann P, Sahl HG. 2003. Mutations are involved in emergence of aminoglycoside-induced small colony variants of Staphylococcus aureus. Int J Med Microbiol 293:427–435. doi: 10.1078/1438-4221-00282. [DOI] [PubMed] [Google Scholar]
- 19.von Eiff C, Heilmann C, Proctor RA, Woltz C, Peters G, Götz F. 1997. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J Bacteriol 179:4706–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaupp R, Schlag S, Liebeke M, Lalk M, Götz F. 2010. Advantage of upregulation of succinate dehydrogenase in Staphylococcus aureus biofilms. J Bacteriol 192:2385–2394. doi: 10.1128/JB.01472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norstrom T, Lannergard J, Hughes D. 2007. Genetic and phenotypic identification of fusidic acid-resistant mutants with the small-colony-variant phenotype in Staphylococcus aureus. Antimicrob Agents Chemother 51:4438–4446. doi: 10.1128/AAC.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards AM. 2012. Phenotype switching is a natural consequence of Staphylococcus aureus replication. J Bacteriol 194:5404–5412. doi: 10.1128/JB.00948-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massey RC, Buckling A, Peacock SJ. 2001. Phenotypic switching of antibiotic resistance circumvents permanent costs in Staphylococcus aureus. Curr Biol 11:1810–1814. doi: 10.1016/S0960-9822(01)00507-3. [DOI] [PubMed] [Google Scholar]
- 24.Vesga O, Groeschel MC, Otten MF, Brar DW, Vann JM, Proctor RA. 1996. Staphylococcus aureus small colony variants are induced by the endothelial cell intracellular milieu. J Infect Dis 173:739–742. doi: 10.1093/infdis/173.3.739. [DOI] [PubMed] [Google Scholar]
- 25.Baumert N, von Eiff C, Schaaff F, Peters G, Proctor RA, Sahl HG. 2002. Physiology and antibiotic susceptibility of Staphylococcus aureus small colony variants. Microb Drug Resist 8:253–260. doi: 10.1089/10766290260469507. [DOI] [PubMed] [Google Scholar]
- 26.Bates DM, von Eiff C, McNamara PJ, Peters G, Yeaman MR, Bayer AS, Proctor RA. 2003. Staphylococcus aureus menD and hemB mutants are as infective as the parent strains, but the menadione biosynthetic mutant persists within the kidney. J Infect Dis 187:1654–1661. doi: 10.1086/374642. [DOI] [PubMed] [Google Scholar]
- 27.Brouillette E, Grondin G, Lefebvre C, Talbot BG, Malouin F. 2004. Mouse mastitis model of infection for antimicrobial compound efficacy studies against intracellular and extracellular forms of Staphylococcus aureus. Vet Microbiol 101:253–262. doi: 10.1016/j.vetmic.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Garcia LG, Lemaire S, Kahl BC, Becker K, Proctor RA, Denis O, Tulkens PM, Van Bambeke F. 2013. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J Antimicrob Chemother 68:1455–1464. doi: 10.1093/jac/dkt072. [DOI] [PubMed] [Google Scholar]
- 29.Tsuji BT, von Eiff C, Kelchlin PA, Forrest A, Smith PF. 2008. Attenuated vancomycin bactericidal activity against Staphylococcus aureus hemB mutants expressing the small-colony-variant phenotype. Antimicrob Agents Chemother 52:1533–1537. doi: 10.1128/AAC.01254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seggewiss J, Becker K, Kotte O, Eisenacher M, Yazdi MR, Fischer A, McNamara P, Al Laham N, Proctor R, Peters G, Heinemann M, von Eiff C. 2006. Reporter metabolite analysis of transcriptional profiles of a Staphylococcus aureus strain with normal phenotype and its isogenic hemB mutant displaying the small-colony-variant phenotype. J Bacteriol 188:7765–7777. doi: 10.1128/JB.00774-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh R, Ray P, Das A, Sharma M. 2010. Enhanced production of exopolysaccharide matrix and biofilm by a menadione-auxotrophic Staphylococcus aureus small-colony variant. J Med Microbiol 59:521–527. doi: 10.1099/jmm.0.017046-0. [DOI] [PubMed] [Google Scholar]
- 32.Tuchscherr L, Heitmann V, Hussain M, Viemann D, Roth J, von Eiff C, Peters G, Becker K, Löffler B. 2010. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J Infect Dis 202:1031–1040. doi: 10.1086/656047. [DOI] [PubMed] [Google Scholar]
- 33.Moisan H, Brouillette E, Jacob CL, Langlois-Bégin P, Michaud S, Malouin F. 2006. Transcription of virulence factors in Staphylococcus aureus small-colony variants isolated from cystic fibrosis patients is influenced by SigB. J Bacteriol 188:64–76. doi: 10.1128/JB.188.1.64-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitchell G, Fugère A, Pépin Gaudreau K, Brouillette E, Frost EH, Cantin AM, Malouin F. 2013. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS One 8:e65018. doi: 10.1371/journal.pone.0065018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pader V, James EH, Painter KL, Wigneshweraraj S, Edwards AM. 2014. The agr quorum-sensing system regulates fibronectin binding but not hemolysis in the absence of a functional electron transport chain. Infect Immun 82:4337–4347. doi: 10.1128/IAI.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. 2008. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clauditz A, Resch A, Wieland KP, Peschel A, Götz F. 2006. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect Immun 74:4950–4953. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clements MO, Watson SP, Foster SJ. 1999. Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J Bacteriol 181:3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cosgrove K, Coutts G, Jonsson IM, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol 189:1025–1035. doi: 10.1128/JB.01524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaupp R, Ledala N, Somerville GA. 2012. Staphylococcal response to oxidative stress. Front Cell Infect Microbiol 2:33. doi: 10.3389/fcimb.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horsburgh MJ, Ingham E, Foster SJ. 2001. In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and Is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J Bacteriol 183:468–475. doi: 10.1128/JB.183.2.468-475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. 2003. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 149:2749–2758. doi: 10.1099/mic.0.26353-0. [DOI] [PubMed] [Google Scholar]
- 44.Valderas MW, Hart ME. 2001. Identification and characterization of a second superoxide dismutase gene (sodM) from Staphylococcus aureus. J Bacteriol 183:3399–3407. doi: 10.1128/JB.183.11.3399-3407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayfield JA, Hammer ND, Kurker RC, Chen TK, Ojha S, Skaar EP, DuBois JL. 2013. The chlorite dismutase (HemQ) from Staphylococcus aureus has a redox-sensitive heme and is associated with the small colony variant phenotype. J Biol Chem 288:23488–23504. doi: 10.1074/jbc.M112.442335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. doi: 10.1016/0076-6879(91)04029-N. [DOI] [PubMed] [Google Scholar]
- 47.Lee CY, Buranen SL, Ye ZH. 1991. Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103:101–105. doi: 10.1016/0378-1119(91)90399-V. [DOI] [PubMed] [Google Scholar]
- 48.Kafala B, Sasarman A. 1997. Isolation of the Staphylococcus aureus hemCDBL gene cluster coding for early steps in heme biosynthesis. Gene 199:231–239. doi: 10.1016/S0378-1119(97)00372-7. [DOI] [PubMed] [Google Scholar]
- 49.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarkar S, Ma WT, Sandri GH. 1992. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85:173–179. doi: 10.1007/BF00120324. [DOI] [PubMed] [Google Scholar]
- 52.Hall BM, Ma C, Liang KK, Singh P. 2009. Fluctuation AnaLysis CalculatOR (FALCOR): a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf C, Hochgräfe F, Kusch H, Albrecht D, Hecker M, Engelmann S. 2008. Proteomic analysis of antioxidant strategies of Staphylococcus aureus: diverse responses to different oxidants. Proteomics 8:3139–3153. doi: 10.1002/pmic.200701062. [DOI] [PubMed] [Google Scholar]
- 54.Keyer K, Imlay JA. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci U S A 93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakeman CA, Hammer ND, Stauff DL, Attia AS, Anzaldi LL, Dikalov SI, Calcutt MW, Skaar EP. 2012. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol Microbiol 86:1376–1392. doi: 10.1111/mmi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang W, Small DA, Toghrol F, Bentley WE. 2006. Global transcriptome analysis of Staphylococcus aureus response to hydrogen peroxide. J Bacteriol 188:1648–1659. doi: 10.1128/JB.188.4.1648-1659.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al Mamun AA, Lombardo MJ, Shee C, Lisewski AM, Gonzalez C, Lin D, Nehring RB, Saint-Ruf C, Gibson JL, Frisch RL, Lichtarge O, Hastings PJ, Rosenberg SM. 2012. Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338:1344–1348. doi: 10.1126/science.1226683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galhardo RS, Hastings PJ, Rosenberg SM. 2007. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol 42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. 2000. The SOS response regulates adaptive mutation. Proc Natl Acad Sci U S A 97:6646–6651. doi: 10.1073/pnas.120161797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shee C, Gibson JL, Rosenberg SM. 2012. Two mechanisms produce mutation hot spots at DNA breaks in Escherichia coli. Cell Rep 2:714–721. doi: 10.1016/j.celrep.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cirz RT, Jones MB, Gingles NA, Minogue TD, Jarrahi B, Peterson SN, Romesberg FE. 2007. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J Bacteriol 189:531–539. doi: 10.1128/JB.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lombardo MJ, Aponyi I, Rosenberg SM. 2004. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics 166:669–680. doi: 10.1534/genetics.166.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mitchell G, Séguin DL, Asselin AE, Déziel E, Cantin AM, Frost EH, Michaud S, Malouin F. 2010. Staphylococcus aureus sigma B-dependent emergence of small-colony variants and biofilm production following exposure to Pseudomonas aeruginosa 4-hydroxy-2-heptylquinoline-N-oxide. BMC Microbiol 10:33. doi: 10.1186/1471-2180-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitchell G, Brouillette E, Séguin DL, Asselin AE, Jacob CL, Malouin F. 2010. A role for sigma factor B in the emergence of Staphylococcus aureus small-colony variants and elevated biofilm production resulting from an exposure to aminoglycosides. Microb Pathog 48:18–27. doi: 10.1016/j.micpath.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 66.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J Bacteriol 184:5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uziel O, Borovok I, Schreiber R, Cohen G, Aharonowitz Y. 2004. Transcriptional regulation of the Staphylococcus aureus thioredoxin and thioredoxin reductase genes in response to oxygen and disulfide stress. J Bacteriol 186:326–334. doi: 10.1128/JB.186.2.326-334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoffman LR, Déziel E, D'Argenio DA, Lépine F, Emerson J, McNamara S, Gibson RL, Ramsey BW, Miller SI. 2006. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 103:19890–19895. doi: 10.1073/pnas.0606756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen CJ, Su LH, Lin TY, Huang YC. 2010. Molecular analysis of repeated methicillin-resistant Staphylococcus aureus infections in children. PLoS One 5:e14431. doi: 10.1371/journal.pone.0014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peyrani P, Allen M, Seligson D, Roberts C, Chen A, Haque N, Zervos M, Wiemken T, Harting J, Christensen D, Ramirez R. 2012. Clinical outcomes of osteomyelitis patients infected with methicillin-resistant Staphylococcus aureus U S A-300 strains. Am J Orthop 41:117–122. [PubMed] [Google Scholar]
- 71.Rosenberg SM, Shee C, Frisch RL, Hastings PJ. 2012. Stress-induced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. Bioessays 34:885–892. doi: 10.1002/bies.201200050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sale JE, Lehmann AR, Woodgate R. 2012. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kriegeskorte A, König S, Sander G, Pirkl A, Mahabir E, Proctor RA, von Eiff C, Peters G, Becker K. 2011. Small colony variants of Staphylococcus aureus reveal distinct protein profiles. Proteomics 11:2476–2490. doi: 10.1002/pmic.201000796. [DOI] [PubMed] [Google Scholar]
- 75.Balaban NQ. 2011. Persistence: mechanisms for triggering and enhancing phenotypic variability. Curr Opin Genet Dev 21:768–775. doi: 10.1016/j.gde.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Lewis K. 2012. Persister cells: molecular mechanisms related to antibiotic tolerance. Handb Exp Pharmacol 211:121–133. doi: 10.1007/978-3-642-28951-4_8. [DOI] [PubMed] [Google Scholar]
- 77.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 78.Johnson PJ, Levin BR. 2013. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS Genet 9:e1003123. doi: 10.1371/journal.pgen.1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kussell E, Kishony R, Balaban NQ, Leibler S. 2005. Bacterial persistence: a model of survival in changing environments. Genetics 169:1807–1814. doi: 10.1534/genetics.104.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Woodmansee AN, Imlay JA. 2002. Reduced flavins promote oxidative DNA damage in non-respiring Escherichia coli by delivering electrons to intracellular free iron. J Biol Chem 277:34055–34066. doi: 10.1074/jbc.M203977200. [DOI] [PubMed] [Google Scholar]
- 81.Brekasis D, Paget MS. 2003. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J 22:4856–4865. doi: 10.1093/emboj/cdg453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hecker M, Reder A, Fuchs S, Pagels M, Engelmann S. 2009. Physiological proteomics and stress/starvation responses in Bacillus subtilis and Staphylococcus aureus. Res Microbiol 160:245–258. doi: 10.1016/j.resmic.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 83.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 84.Chittezham TV, Kinkead LC, Janssen A, Schaeffer CR, Woods KM, Lindgren JK, Peaster JM, Chaudhari SS, Sadykov M, Jones J, AbdelGhani SM, Zimmerman MC, Bayles KW, Somerville GA, Fey PD. 2013. A dysfunctional tricarboxylic acid cycle enhances fitness of Staphylococcus epidermidis during β-lactam stress. mBio 4:e00437-13. doi: 10.1128/mBio.00437-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 86.Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, Barron A, Bason N, Bentley SD, Chillingworth C, Chillingworth T, Churcher C, Clark L, Corton C, Cronin A, Doggett J, Dowd L, Feltwell T, Hance Z, Harris B, Hauser H, Holroyd S, Jagels K, James KD, Lennard N, Line A, Mayes R, Moule S, Mungall K, Ormond D, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Sharp S, Simmonds M, Stevens K, Whitehead S, Barrell BG, Spratt BG, Parkhill J. 2004. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A 101:9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]