

Background: The Saccharomyces cerevisiae Spt-Ada-Gcn5 acetyltransferase (SAGA) complex regulates transcription through chromatin modification and other mechanisms.
Results: The overall structure of SAGA and the arrangement of all subunits within this complex were determined.
Conclusion: SAGA is flexible and is composed of core modules that support peripheral catalytic modules.
Significance: Understanding the structural mechanisms of SAGA multifunctionality improves the understanding of other chromatin-modifying complexes.
Keywords: Acetyltransferase, Chromatin Modification, Deubiquitylation (Deubiquitination), Electron Microscopy (EM), Mass Spectrometry (MS), Protein Cross-linking, Structural Biology, Yeast, Macromolecular Assembly
Abstract
The Spt-Ada-Gcn5 acetyltransferase (SAGA) complex is a highly conserved, 19-subunit histone acetyltransferase complex that activates transcription through acetylation and deubiquitination of nucleosomal histones in Saccharomyces cerevisiae. Because SAGA has been shown to display conformational variability, we applied gradient fixation to stabilize purified SAGA and systematically analyzed this flexibility using single-particle EM. Our two- and three-dimensional studies show that SAGA adopts three major conformations, and mutations of specific subunits affect the distribution among these. We also located the four functional modules of SAGA using electron microscopy-based labeling and transcriptional activator binding analyses and show that the acetyltransferase module is localized in the most mobile region of the complex. We further comprehensively mapped the subunit interconnectivity of SAGA using cross-linking mass spectrometry, revealing that the Spt and Taf subunits form the structural core of the complex. These results provide the necessary restraints for us to generate a model of the spatial arrangement of all SAGA subunits. According to this model, the chromatin-binding domains of SAGA are all clustered in one face of the complex that is highly flexible. Our results relate information of overall SAGA structure with detailed subunit level interactions, improving our understanding of its architecture and flexibility.
Introduction
Transcription is a highly regulated process involving the stepwise recruitment of factors to the site of transcription to facilitate RNA polymerase II activity (1). In eukaryotic cells, DNA is packaged with nucleosomes, an octameric complex of histone proteins, to form chromatin. Chromatin serves as a steric barrier against transcription, and various post-translational modifications of histones play important roles in regulating both the chromatin landscape and the recruitment of transcription factors. Histone acetylation, a modification that correlates with an “open” chromatin conformation and increased transcription, is mediated by several multisubunit histone acetyltransferase (HAT)2 complexes (2). These HAT complexes are often recruited to specific genes by DNA-binding transcriptional activator proteins such as Gal4 and Gcn4 in yeast Saccharomyces cerevisiae (3).
The Spt-Ada-Gcn5 Acetyltransferase (SAGA) complex is a highly conserved HAT complex that activates the transcription of stress response genes in yeast (4). As the largest HAT complex, SAGA consists of 19 core subunits that associate into a stable assembly of ∼1.8 MDa in overall mass, with Gcn5 serving as the catalytic subunit for acetylating histone H3 (Table 1) (5). The other subunits of SAGA confer additional functionalities to the complex. These subunits are functionally organized into four distinct modules: the HAT module, the deubiquitination (DUB) module, the SPT module, and the TAF module (6). Within the DUB module, Ubp8 catalyzes the deubiquitination of histone H2B, an important step in the progression of transcription activation (7). SPT module subunits Spt3 and Spt8 enable SAGA to bind TATA-binding protein (TBP), an important general transcription factor in the formation of the transcriptional preinitiation complexes (8), and regulate transcription of different genes (9, 10). Tra1, another SPT module subunit, is targeted by different transcriptional activators to recruit SAGA to certain genes (3). The TAF module contains five histone fold-containing Taf proteins, shared with the TFIID general transcription factor complex, that serve important roles in maintaining the integrity of the complex (11).
TABLE 1.
Subunits of yeast SAGA
| Module | Subunit | Molecular mass |
|---|---|---|
| kDa | ||
| HAT module | Ada2a | 50.6 |
| Ada3 | 79.3 | |
| Gcn5b | 51.1 | |
| Sgf29 | 29.4 | |
| DUB module | Sgf11 | 11.3 |
| Sgf73a | 72.9 | |
| Sus1 | 11.1 | |
| Ubp8b | 53.6 | |
| Spt module | Ada1 | 54.5 |
| Spt3 | 38.8 | |
| Spt7 | 152.6 | |
| Spt8 | 66.2 | |
| Spt20 | 67.8 | |
| Tra1 | 433.2 | |
| Taf module | Taf5 | 89.0 |
| Taf6 | 57.9 | |
| Taf9 | 17.3 | |
| Taf10 | 23.0 | |
| Taf12 | 61.1 |
a Anchors module to complex.
b Catalytic subunit.
Several studies have sought to elucidate the structural features of SAGA. Single-particle EM analysis by Wu et al. (12) provided the first step toward understanding the overall structure of this complex by generating the first three-dimensional reconstruction of SAGA, and localizing nine core subunits using antibody labeling techniques. Two-dimensional analysis in this study also revealed that a subpopulation of SAGA particles possesses an additional region of density, which can adopt different conformations. A recent study mapping the DUB module to an EM structure of SAGA also observed this subpopulation (13). More recently, an EM study on human TFIID showed that this complex undergoes massive rearrangement that alters both the position and the connectivity of an entire lobe (14). Even though SAGA shares multiple core subunits with TFIID, whether the observed structural flexibility is a shared property of these complexes is not known. Furthermore, at the time of the first EM study, the H2B deubiquitination activity of SAGA and the subunits associated with this catalytic activity have not been identified (7). Finally, apart from the DUB module, there is a dearth of high resolution structural information for the other SAGA subunits, rendering our structural understanding of the complex incomplete.
By developing a modified purification strategy that enhances the stability of SAGA, we uncovered the remarkable conformational flexibility of this complex using single-particle EM. Systematic subunit deletions and mutations approaches enabled us to further dissect the role of the different modules in mediating structural rearrangement. By combining established EM-based labeling methods with chemical cross-linking of proteins coupled with mass spectrometry analysis (CXMS), we mapped and validated the spatial location of all core components of SAGA, including subunits of the DUB module. Collectively, our data enabled us to generate a model for the molecular organization of SAGA and to gain insights into the physiological relevance of its conformational flexibility.
EXPERIMENTAL PROCEDURES
S. cerevisiae Strain Construction
Haploid strains BY4741, BJ2168, and SF1 backgrounds were used to construct all the strains in this study (Table 2). Tandem affinity purification (TAP)- and GFP-tagged strains were constructed using PCR-amplified inserts from plasmids pBS1539 and pFA6a-GFP(S65T)-kanMX6, respectively, essentially as described (15, 16). Deletion strains were similarly constructed using PCR-amplified inserts from pFA6a-kanMX6 (16). FLAG-tagged strains were also constructed using PCR-amplified inserts from pMK144–3×FLAG-kanMX6. Proper integration of inserts was checked by colony PCR (17), and expression of tags was evaluated using Western blot with α-TAP antibody (Thermo Scientific), α-GFP antibody (Roche), or α-FLAG M2 antibody (Sigma). Standard methods for S. cerevisiae culturing and lithium acetate-mediated transformations were used.
TABLE 2.
S. cerevisiae strains used in this study
| Strain name | Genotype |
|---|---|
| SF1 | MATa ade2-1 can1-100 his3-11 leu2-3,112 trp1 ura3-1 |
| SF1 Spt7-TAP | MATa SPT7-TAP::URA3 ade2-1 can1-100 his3-11 leu2-3,112 trp1 ura3-1 |
| BJ2168 | MATa leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP | MATa SPT7-TAP::URA3 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Δada2 | MATa ada2::KanMX6 SPT7-TAP::URA3 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Δsgf73 | MATa sgf73::KanMX6 SPT7-TAP::URA3 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Gcn5-GFP | MATa SPT7-TAP::URA3 GCN5-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Ada2-GFP | MATa SPT7-TAP::URA3 ADA2-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Sgf29-GFP | MATa SPT7-TAP::URA3 SGF29-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Spt3-GFP | MATa SPT7-TAP::URA3 SPT3-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Spt8-GFP | MATa SPT7-TAP::URA3 SPT8-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Spt20-GFP | MATa SPT7-TAP::URA3 SPT20-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Taf5-GFP | MATa SPT7-TAP::URA3 TAF5-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Taf9-GFP | MATa SPT7-TAP::URA3 TAF9-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BJ2168 Spt7-TAP Taf10-GFP | MATa SPT&-TAP::URA3 TAF10-GFP::KanMX6 leu2 trp1 ura3-52 prb1-1122 pep4-3 prc1-407 gal2 |
| BY4741 | MATa his3Δ1 leu2Δ20 met15Δ0 ura3Δ0 |
| BY4741 Sgf73-TAP | MATa SGF73-TAP::URA3 his3Δ1 leu2Δ20 met15Δ0 ura3Δ0 |
| BY4741 Sgf73-TAP Spt7(1-1180) | MATa SGF73-TAP::URA3 spt7-1180::KanMX6 his3Δ1 leu2Δ20 met15Δ0 ura3Δ0 |
Purification of Native SAGA
Native SAGA was purified by a traditional TAP purification (12, 15) or a modified version substituting the calmodulin binding step with GraFix (18). In particular, TAP-tagged yeast cells were grown to an A600 of ∼2.5, harvested by centrifugation, and the cell pellets were frozen at −80 °C. 20–25 g of frozen cells were resuspended in ∼80 ml of lysis buffer (40 mm HEPES, pH 7.4, 350 mm NaCl, 10% glycerol, 0.1% Tween 20, 1 mm PMSF, 50 mm NaF, 0.1 mm Na3VO4, 2 mm benzamidine, and EDTA-free protease inhibitor (Roche)) and lysed by bead beating. The lysate was then centrifuged at 30,000 × g for 30 min. Clarified lysates were incubated with 500 μl of IgG-Sepharose (GE Healthcare) at 4 °C for 1.5 h. The IgG resin was washed with IPP150 buffer (40 mm HEPES, pH 7.4, 150 mm NaCl, 0.2% Nonidet P-40, and 10% glycerol) and resuspended in 750 μl of TEV-C buffer (40 mm HEPES, pH 7.4, 150 mm NaCl, 0.1% Nonidet P-40, 0.5 mm EDTA, 10% glycerol, and 1 mm DTT). Bound proteins were eluted by tobacco etch virus protease cleavage at 16 °C for 1.5 h. 2 μl of 10 mg/ml RNase A was added to 500 μl of the eluate and incubated on ice for 30 min. 2 × 200 μl of the eluate were overlaid on two linear 15–30% glycerol gradients, with one containing 0.00–0.05% glutaraldehyde cross-linker prepared using the Gradient Station (Biocomp). The gradients were spun at 58,800 × g for 16.5 h at 4 °C. The gradients were then fractionated using the Gradient Station. Fractions from gradients without cross-linker were TCA-precipitated for silver stain SDS-PAGE analysis, and corresponding fractions with cross-linker, concentrated using 100,000 molecular mass cutoff concentrators (Millipore) if necessary, were used for further EM analysis.
For antibody binding and cross-linking mass spectrometry analyses, SAGA was purified using anti-FLAG affinity chromatography. 3×FLAG-tagged Spt7 yeast cell pellets were obtained as before. Frozen pellets are pooled and ground using a freezer mill (SPEX SamplePrep 6870) under liquid nitrogen temperatures. 35–40 ml of the finely ground cell lysate was resuspended in ∼80 ml of lysis buffer. The lysate was centrifuged at 30,000 × g for 30 min. Clarified lysates were incubated with 500 μl of α-FLAG M2 resin (Sigma) for 1 h. The resin was washed three times with 5 ml of lysis buffer without inhibitors and once with 5 ml of reduced salt (150 mm) lysis buffer without inhibitors. The resin was resuspended in 1 ml of reduced salt lysis buffer containing 2.5 μg/ml RNase A and incubated at 4 °C for 30 min. The resin was washed twice with 5 ml of reduced salt lysis buffer. Bound proteins were eluted twice in 500 μl of reduced salt lysis buffer without inhibitors and 0.5 mg/ml 3×FLAG peptides.
Electron Microscopy
Negatively stained specimens for two-dimensional analysis were prepared from purified SAGA as described previously (19). Samples for three-dimensional analysis were prepared using the carbon sandwich negative staining technique to improve stain embedding and to minimize sample flattening (19). Samples were visualized using a Tecnai Spirit transmission electron microscope (FEI) operated at an accelerating voltage of 120 kV. Images were taken at a nominal magnification of 49,000× using an FEI Eagle 4K charge-coupled device camera at a defocus value of −1.2 μm under low dose conditions. For tilt pair data collection, the same parameters were used, and two images, one at 60° tilt and one untilted, were taken from the same specimen area. 2 × 2 image pixels were then averaged for a final pixel size of 4.7 Å.
Image Processing
For two-dimensional analysis, individual particle images were interactively selected using Boxer (20). The selected particles were then windowed into 128 × 128-pixel images, rotationally and translationally aligned, and subjected to K-means classification to generate class averages using the SPIDER image processing suite (21). For GFP tagging analysis, we used a preliminary round of unsupervised classification and averaging to visualize regions with possible additional density. These averages were used to create masks that focused on the vicinity of the additional density, and the areas within the masks were used to reclassify the input particle images. Averages obtained from this method were compared with untagged SAGA of a similar conformation via subtraction analysis. Images of untagged SAGA were subtracted from images of tagged SAGA, and the resulting difference image was threshold to find signals that were either 3 or 4 standard deviations from the mean pixel value.
For determining the de novo three-dimensional reconstructions of SAGA, 22,518 pairs of particle images were first selected using WEB (21). Particles in the untilted set were windowed into 128 × 128-pixel images, aligned, and classified into 50 classes using SPIDER. Two class averages that correspond to each of the three conformations were merged. Three-dimensional reconstructions were generated from the tilted particles of these combined averages using the backprojection and angular refinement algorithms in SPIDER. The final resolutions of the three-dimensional models were estimated by the Fourier shell correlation function using the 0.5 Fourier shell correlation criterion. The curved, arched, and donut reconstructions had resolutions of 41.7, 45.3, and 38.9 Å, respectively. Molecular docking analysis and model construction was performed using UCSF Chimera (22).
Conformation Population Analysis
Conformation population analysis similar to a previous study was conducted (23). Particle measurements were done using ImageJ (24). 100 class averages from the Spt7-TAP wild type and sgf73 strains were used. Only averages with unambiguous outlines corresponding to SAGA were analyzed. The length of the cleft formed in the arched conformation of SAGA and the shortest distance between the distal end of the tail and the shoulder were measured. Three independent measurements were made, and the averages were used to create combined scatter and bar plots.
Antibody Labeling of SAGA
Non-cross-linked SAGA purified from Sgf73-TAP-tagged strain was incubated with 10 μg/ml α-TAP antibody (Thermo Scientific) at room temperature for 10 min and used for single-particle EM analysis. Particles with bound antibodies were selected for two-dimensional analysis as detailed above.
Chemical Cross-linking and Mass Spectrometry Analysis
FLAG-purified SAGA was concentrated from 2 ml to ∼100 μl using 100,000 molecular mass cutoff concentrators (Millipore) and were cross-linked with disuccinimidyl suberate as described (25), precipitated with 4 volumes of cold acetone, washed once with cold acetone, air-dried, and then dissolved in 8 m urea, 100 mm Tris, pH 8.5. After 5 mm Tris-(2-carboxyethyl)phosphine reduction and 10 mm iodoacetic acid alkylation, the samples were digested with Lys-C at a 1:100 enzyme:substrate ratio at 37 °C for 4 h. The samples were 4-fold diluted with 100 mm Tris, pH 8.5, before they were digested with trypsin (1:50, enzyme:substrate) at 37 °C. After 12 h, formic acid was added to a final concentration of 5% to stop digestion. The samples were cleared by centrifugation at 14,000 rpm for 10 min and desalted with a homemade 250-μm × 1-cm C18 reverse phase column. Desalted peptides were loaded onto a 75-μm - 10-cm analytical column packed with 1.8 μm, 120 Å UHPLC-XB-C18 resin (Welch Materials Inc.) and separated over a 107-min linear gradient from 100% buffer A (0.1% formic acid) to 30% buffer B 100% acetonitrile, 0.1% formic acid), and then a 10-min gradient from 30 to 80% buffer B and maintaining at 80% buffer B for 6 min before returning to 100% buffer A in 5 min and ending with a 9-min 100% buffer A wash. The flow rate was 200 nl/min. The Easy-nLC 1000 UPLC was coupled to a Q Exactive mass spectrometer (Thermo-Fisher Scientific). The MS parameters were as follows: top 20 most intense ions in a survey full scan were selected for MS2 by HCD dissociation; r = 140,000 in full scan, r = 17,500 in HCD scan; AGC targets were 1e6 for FTMS full scan, 5e4 for MS2; minimal signal threshold for MS2 = 4e4; precursors of charge states +1, +2, >+8, or unassigned were excluded; normalized collision energy = 30 for HCD; peptide match is preferred.
Expression and Purification of Recombinant Transcriptional Activators
Coding regions of Gcn4, TBP, and Gal4 activation domain (residues 768–881) were PCR-amplified from yeast genomic DNA (26). The PCR products were cloned into the NdeI/EcoRI sites of pET28b-HMT vector. BL21* (Life Technologies) Escherichia coli expression strain transformed with the resulting constructs were grown to an A600 of 0.5 and induced with 1 mm isopropyl β-d-thiogalactopyranoside for either 3 h at 37 °C or overnight at 16 °C. The bacteria were then harvested by centrifugation and stored at −80 °C. For each purification, a frozen cell pellet was resuspended 10 ml/g in lysis buffer (40 mm HEPES, pH 8.0, 500 mm NaCl, and 2 mm PMSF). The cells were lysed by sonication, and the lysate was clarified by centrifugation at 30,000 × g. The clarified lysates were then incubated with 500 μl of nickel-nitrilotriacetic acid-Sepharose (Thermo Scientific) for 30 min at 4 °C. The resin was washed three times with 5 ml of lysis buffer and then twice with 5 ml of lysis buffer with 50 mm imidazole. Bound proteins were eluted in five rounds, using 1 ml of lysis buffer with 250 mm imidazole. Fractions containing the desired protein were concentrated using a 10,000 molecular mass cutoff concentrators (Millipore) and further purified by gel filtration chromatography (GE Healthcare). Peak fractions containing pure activator proteins were flash frozen in liquid nitrogen and stored at −80 °C.
Activator Pulldown Experiments and Western Blotting
Approximately 50 ng/μl of FLAG-purified SAGA was mixed with 300 μg/ml HMP and HMP-tagged Gcn4, Gal4AD, and TBP in binding buffer (40 mm HEPES, pH 7.4, 150 mm NaCl, 10% glycerol, 0.1% Tween 20, 0.5 mm DTT, and 1 mm PMSF). The mixture was incubated with 50 μl of amylose resin (New England Biolabs) for 30 min at 4 °C. The resin was collected using centrifugal spin columns and washed twice with 200 μl of binding buffer. Bound proteins were eluted with 200 μl of binding buffer with 100 mm maltose. Eluates were then analyzed by SDS-PAGE and Western blot using the following antibodies: mouse α-FLAG antibody (Sigma), mouse α-His antibody (ABM), and α-mouse HRP antibody (Sigma).
Activator Binding Localization Experiments
FLAG and glycerol gradient-purified SAGA was mixed with 4–8 μg/ml of purified activators and incubated at room temperature for 30 min before negative-stained EM specimens were prepared.
RESULTS
Improved Procedure for Isolating Native S. cerevisiae SAGA
The TAP procedure is an established method for isolating native SAGA from yeast containing genomically tagged SPT7-TAP (12, 15). We combined the first part of TAP with the GraFix technique, which involves subjecting the complex to limited glutaraldehyde cross-linking during glyverol gradient ultracentrifugation (18). GraFix has been shown to increase EM image quality and particle homogeneity. To ensure that SAGA is amenable to glycerol gradient ultracentrifugation, we analyzed fractions from a corresponding glycerol gradient lacking glutaraldehyde by SDS-PAGE. SAGA migrates to a single fraction with minimal contaminants (Fig. 1A). Subsequent MS analysis confirmed that this fraction contains all 19 core SAGA subunits (Table 3).
FIGURE 1.
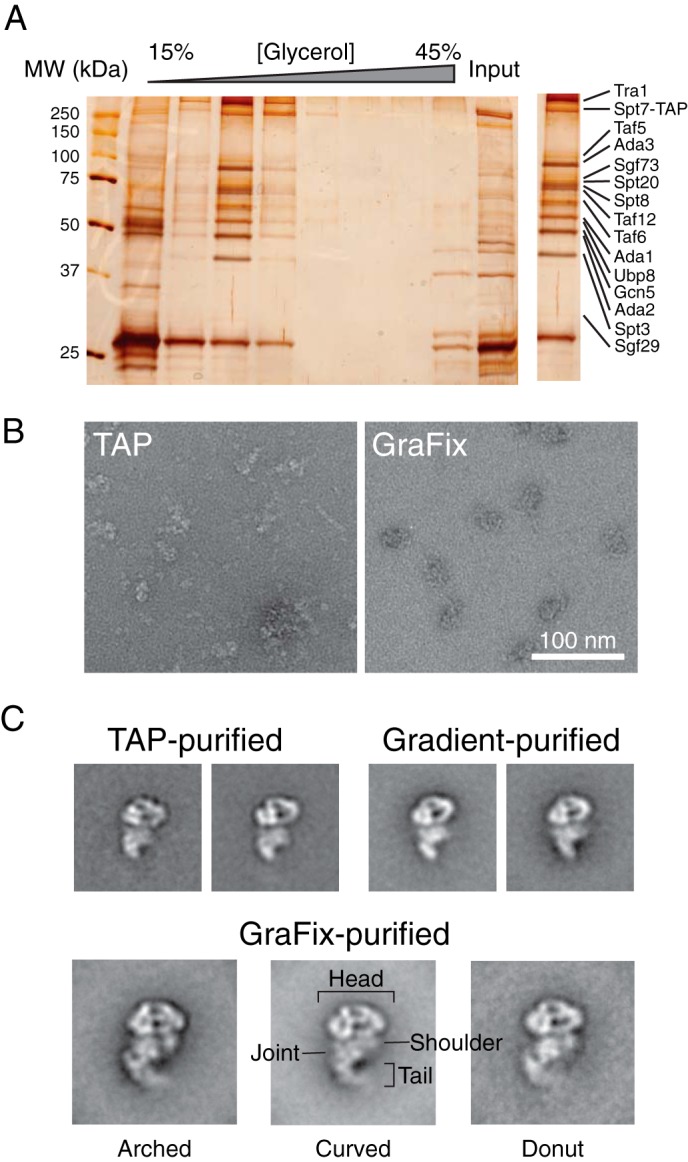
Purification of S. cerevisiae SAGA and negative stain EM analysis. A, SDS-PAGE analysis of yeast SAGA purified from IgG-Sepharose and 15–30% glycerol gradient ultracentrifugation. Protein bands were visualized by silver staining, and the inset on the right represents the fraction that was used for EM and mass spectrometric analysis (Table 3). B, a representative raw image of negatively stained TAP-purified or GraFix-purified SAGA. C, representative two-dimensional class averages of SAGA purified by TAP, glycerol gradient ultracentrifugation without cross-linker, and GraFix. The three GraFix class averages correspond to the three observed conformations. The side length of every class average panel is 60 nm. MW, molecular mass.
TABLE 3.
Mass spectrometry analysis of purified SAGA subunit composition
| Subunit | Molecular mass |
WT |
ada2Δ |
sgf73Δ |
|||
|---|---|---|---|---|---|---|---|
| Peptide | Coverage | Peptide | Coverage | Peptide | Coverage | ||
| kDa | n | % | n | % | n | % | |
| Tra1 | 432.9 | 59 | 21.1 | 23 | 7.7 | 18 | 6.5 |
| Spt7 | 152.5 | 37 | 35.4 | 17 | 17.3 | 11 | 12.1 |
| Taf5 | 88.9 | 38 | 59 | 30 | 49.9 | 33 | 49.8 |
| Ada3 | 79.2 | 30 | 52.6 | 0 | NAa | 19 | 42.9 |
| Sgf73 | 72.8 | 20 | 44.4 | 16 | 39 | 0 | NA |
| Spt20 | 67.8 | 26 | 58 | 26 | 59.9 | 30 | 66.9 |
| Spt8 | 66.1 | 21 | 46.7 | 20 | 47.5 | 18 | 39.9 |
| Taf12 | 61 | 23 | 53.6 | 20 | 42.9 | 23 | 49.5 |
| Taf6 | 57.9 | 19 | 51 | 18 | 46.5 | 22 | 53.3 |
| Ada1 | 54.4 | 19 | 48.4 | 17 | 46.1 | 21 | 55.9 |
| Ubp8 | 53.6 | 23 | 55 | 15 | 35.5 | 1 | 2.9 |
| Gcn5 | 51 | 21 | 60.8 | 2 | 5.7 | 19 | 51.7 |
| Ada2 | 50.5 | 19 | 57.6 | 0 | NA | 17 | 57.1 |
| Spt3 | 38.8 | 9 | 30.9 | 7 | 24.6 | 11 | 35.6 |
| Sgf29 | 29.4 | 10 | 47.1 | 2 | 10.4 | 10 | 46.7 |
| Taf10 | 23 | 8 | 63.1 | 5 | 47.1 | 7 | 49 |
| Taf9 | 17.3 | 4 | 36.9 | 4 | 39.5 | 4 | 42.7 |
| Sgf11 | 11.3 | 1 | 17.2 | 1 | 17.2 | 0 | NA |
| Sus1 | 11.1 | 2 | 32.3 | 4 | 64.6 | 0 | NA |
a NA, not available.
We next examined the purified SAGA using negative stain electron microscopy. We observed particles of similar size and shape as those observed by Wu et al. (12) from samples purified by the conventional two-step TAP method and by non-cross-linked glycerol gradient ultracentrifugation. However, the samples from the TAP purification and glycerol gradient purification showed a high degree of subunit dissociation (Fig. 1B). Meanwhile, samples that underwent GraFix treatment display not only significantly reduced the level of sample heterogeneity but also preserved fine structural features of individual SAGA particles (Fig. 1B).
SAGA Adopts Three Distinct Conformations
To gain further insights into the structural properties of SAGA, we applied a two-dimensional single-particle EM approach that involves classifying manually selected particles according to similarities in overall morphology and aligning and calculating an average image of the particles constituting each class. The class averages of TAP-purified and glycerol gradient purified SAGA without cross-linker were practically identical to each other and to previously published images (Fig. 1C) (12). However, class averages of SAGA purified using GraFix showed improved image quality and better resolved features (Fig. 1C). These class averages, calculated from 7,753 GraFix-purified particles, showed that SAGA consists of a prominent globular “head” and a long and slender “tail” separated by a “torso” region. The torso region can be further subdivided into two halves: a “joint” that is connected to the tail, and the “shoulder” that does not make a direct connection with the tail. Interestingly, the prominent extended tail that we observed is only found in a small population of particles in the previous EM analysis of SAGA, whereas we observed the tail in 91% of our particles. We attributed this discrepancy to the fact that the tail portion of SAGA is more labile and has a tendency to dissociate from the complex upon purification and/or during the negative staining specimen preparation procedure.
Most strikingly, our analysis revealed that the head, torso, and tail regions of SAGA are all conformationally flexible. In particular, the tail region can curve and sample a broad range of space. The coordinated movement of multiple densities within SAGA is remarkable because of large distances covered; the tip of the SAGA tail traverses over 50 Å between different conformations. From the gallery of class averages, three major types of conformations could be distinguished based on the arrangement of the mobile tail region with respect to the rest of the complex (Fig. 1C). In the “donut” conformation, the tail curls up with its tip pointing toward the shoulder of the torso to generate an almost complete circular structure at the bottom half of the complex. In the “arched” conformation, the tail retracts from the shoulder to generate a kink and a pronounced deep cleft at the back of the torso. In the “curved” conformation, the tail adopts a gentle curvature with its tip projected away from the shoulder. Interestingly, the different tail arrangements are accompanied by changes in the morphology of the SAGA head and shoulder regions. We observed multiple class averages that would fall into intermediate states among the three major conformations. Collectively, our two-dimensional analysis suggests that SAGA is structurally dynamic, and its conformational changes involve coordinated movements and rearrangements of different subunits and modules within the complex.
Removal of Key Subunits Affects the Conformational Flexibility of SAGA
We next examined the effects of subunit and module deletions on the conformational plasticity of SAGA. Previous studies have shown that deleting the ADA2 gene dislodges the HAT module from SAGA while leaving the rest of the complex intact (6). Our mass spectrometry analysis confirmed this finding by showing that subunits of the HAT module are absent in SAGA isolated from the ada2Δ yeast strain (Table 3). Subsequent EM analysis of SAGA devoid of Ada2 revealed that the tail region is severely shortened in all class averages, suggesting that the HAT module likely constitutes a distal segment of the tail (Fig. 2B). Despite the reduced size of the tail region, the absence of the HAT module does not diminish the conformational flexibility of SAGA. In fact, the shoulder region of this mutant SAGA shows even greater mobility, translocating away from the head and toward the tail. Thus, although the HAT module does not influence the ability of SAGA to adopt different conformations, its absence induces additional structural variability in other regions of SAGA.
FIGURE 2.

Effects of mutations on SAGA flexibility. SAGA containing subunit deletions were analyzed using single-particle EM for loss of density and changes in flexibility. A–C, class averages from wild type, ada2Δ, and truncated Spt7(1–1080) SAGA arranged to display their range of flexibility. Yellow circles show electron densities that are absent from the mutants compared with wild type. Schematics of SAGA demonstrates the movement of regions and missing densities within the complex. D and E, conformational population analysis of wild type and Δsgf73 SAGA. Description of the measurements used are shown in D. The bar graphs represent the total number of particles measured for each value on the corresponding axis.
SALSA/SLIK is an alternate form of yeast SAGA that lacks the Spt8 subunit because of a C-terminal truncation of the Spt7 subunit (27). To emulate SALSA/SLIK, we generated a yeast strain containing a truncation of the SPT7 gene from residue 1180 to the C terminus. Negative stain two-dimensional EM revealed that this SALSA/SLIK complex lacks the shoulder region, but the mutation does not affect the conformational flexibility of the tail (Fig. 2C). Spt8 likely comprises a significant portion of the shoulder region and is a peripheral subunit because its absence does not dramatically alter the rest of this complex.
Deletion of the SGF73 gene has been previously shown to dissociate the DUB module from SAGA (6). Although we confirmed that SAGA isolated from the sgf73Δ strain lacks the DUB module, the two-dimensional class averages of this mutant complex show no apparent loss of density (Fig. 2D and Table 3). We did observe an increased number of particles with the HAT module absent, suggesting that SGF73 deletion modestly destabilizes the complex.
A centrally located DUB module would render the loss of density more difficult to observe, explaining our observation. Intriguingly, the sgf73Δ SAGA mutant appears to have a lower propensity than wild type SAGA to adopt the donut conformation. To more accurately assess the shift in occupancy of the different conformation states between wild type SAGA and the sgf73Δ mutant complex, we defined the three major conformations based on two measured parameters: the shoulder to tail distance and the cleft length (Fig. 2D). More specifically, we defined the donut conformation to have a shoulder to tail distance under 20 Å and the arched conformation to have a cleft length greater than 15 Å, with precedence given to the former. Based on this analysis, we found that 28% of wild type SAGA adopted the donut conformation, compared with only 4% by the sgf73t mutant complex. The DUB module is therefore necessary for SAGA to efficiently adopt the donut conformation, either by stabilizing the rearrangements involved in the movement of the tail or physically mediating the connection between the shoulder and the tail. Our results show that different SAGA modules contribute to the flexibility of the complex in varying degrees.
Three-dimensional Reconstructions of SAGA in Three Conformations
The intrinsic heterogeneity of SAGA and the low yields from our endogenous purification procedure precluded high resolution cryo-EM analysis. Instead, we generated de novo three-dimensional reconstructions of SAGA using the random conical tilt approach (28). This approach was chosen because SAGA adopts a preferred orientation on the carbon support layer of the EM grids, precluding the use of common line techniques that require comprehensive angular representation. We selected tilted particles corresponding to the curved, arched, and donut conformations and used them to calculate three-dimensional reconstructions. Despite the intrinsic flexibility of the complex, we were able to visualize the structural rearrangements associated with the conformational shifts (Fig. 3, A–C). Notably, we observed that the tail undergoes a large degree of rearrangement between the three conformations. The arched conformation showed disconnected density in the middle of the tail, an observation that is indicative of a particularly heterogeneous region. Furthermore, the shoulder and its adjacent head region also displayed substantial rearrangements, with multiple shifting densities. In the donut conformation, the shoulder region splits into two separate densities and shifted away from each other. Despite its limiting resolution, our three-dimensional analysis demonstrated that transition between the three conformations requires large scale structural rearrangement of the subunits within the complex. A recent study by Durand et al. (13) also generated a three-dimensional reconstruction of SAGA purified without the use of GraFix. The overall configuration EM maps of Durand et al. correspond most closely with our SAGA donut reconstruction (Fig. 3D), although the precise distribution of densities varies slightly between the two. The different cross-linking method employed by their study may cause different conformations to be stabilized, resulting in the dissimilarities between the reconstructions.
FIGURE 3.
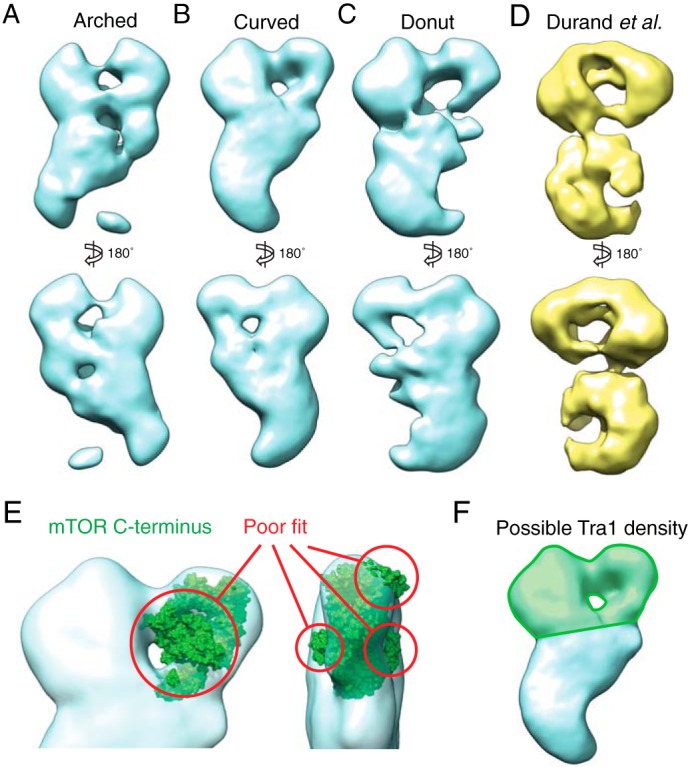
Three-dimensional reconstructions of the three SAGA conformations. A–C, models were generated de novo from both tilted and untilted particles that correspond to each SAGA conformation using the random conical tilt method. D, three-dimensional reconstruction of SAGA purified without gradient fixation by Durand et al. (Electron Microscopy Data Bank code 2693). E, the mTOR C-terminal crystal structure (Protein Data Bank code 4JSV), which shares a high degree of secondary structure similarity with the Tra1 C terminus, does not fit within the region of the head of SAGA previously proposed to contain Tra1. F, the region that likely contains the entire Tra1 structure is highlighted in green.
Tra1 Occupies a Substantial Portion of the Head Region of SAGA
Upon examination of the SAGA three-dimensional reconstructions, we sought to evaluate the proposal by Wu et al. (12) that the Tra1 subunit resides within one half of the head region. Further evidence of this localization comes from the NuA4 HAT complex EM structure, which bears a striking resemblance to the head region of SAGA, while sharing only the Tra1 subunit (29). At 400 kDa, Tra1 is the largest subunit of SAGA and is thought to be responsible for recruitment of this complex to its target genes (3). Tra1 is a pseudokinase that belongs to the phosphatidylinositol 3-kinase-related kinase family of extraordinarily large protein kinases, whose members share a common domain organization: extensive tandem HEAT repeats at its N-terminal region, followed by the FAT, kinase, and FATC domains at the C-terminal region (30). Although there is no high resolution structural data available for Tra1, the crystal structure of the 1,174 C-terminal residues of mTOR, a phosphatidylinositol 3-kinase-related kinase protein that shares a high degree of secondary structure similarity to the predicted C-terminal domain of Tra1, has been reported (31). We used this crystal structure to evaluate the proposed location of Tra1. We found that even at less than one-third the size of full-length Tra1, the mTOR C-terminal domain was too large to fit within the region proposed by Wu et al. (Ref. 12 and Fig. 3D). Furthermore, a low resolution crystal structure of another phosphatidylinositol 3-kinase-related kinase protein, DNA-PKcs, shows a globular region with slender projections that form a ring (32), reminiscent of the arrangement of electron density within the SAGA head. Although our three-dimensional reconstructions are of insufficient resolution for further computational docking analysis, we believe that based on size alone, these comparisons demonstrate that Tra1 likely occupies a large proportion of both lobes of the prominent head region (Fig. 3E).
Comprehensive EM-based Mapping of Subunit Localization
Using an antibody-based approach, Wu et al. (12) deduced the positions of several subunits within SAGA. To validate these subunit locations in light of our ability to visualize SAGA with a fully extended tail and to further expand this analysis, we applied a proven labeling approach that involves introducing C-terminal GFP tags to different SAGA subunits (33). We purified the corresponding GFP-tagged SAGA complexes and located the additional electron density introduced by GFP by negative stain two-dimensional EM method.
Our earlier ADA2 deletion experiment suggested that the HAT module makes up the tail region of SAGA. In agreement with this result, SAGA containing GFP-tagged Ada2, Gcn5, or Sgf29 all display additional density centered about the tail region (Fig. 4A). Our analysis showed that Gcn5 localizes to the tip of the tail region, whereas Wu et al. (12) localized this subunit within the shoulder region. This discrepancy may be explained by the instability and potential dissociation of the tail region of non-cross-linked SAGA. The localization of the HAT activity of SAGA to the most mobile region of the complex provides a tantalizing explanation for its ability to act on a wide range of chromatin templates.
FIGURE 4.
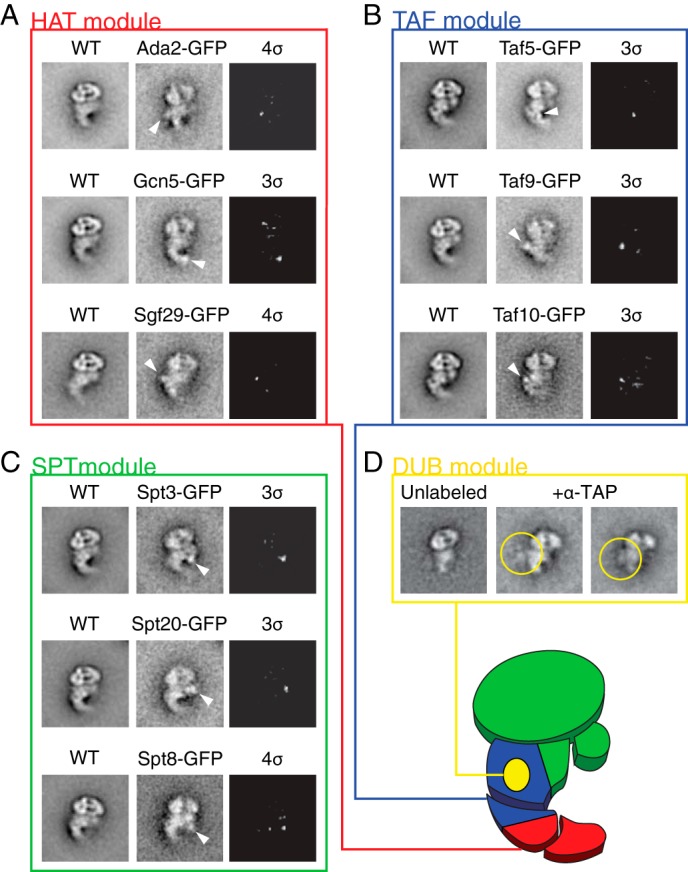
Localization of SAGA domains and subunits. A–C, class averages of untagged WT SAGA and SAGA containing GFP-tagged subunits were compared to determine the approximate locations of the tagged subunits. White arrows denote the additional density on the GFP-tagged image. The third images of each set are difference maps between tagged and untagged SAGA, thresholded to show only signals 3 or 4 standard deviations (σ) above the average pixel value. D, to localize the DUB module, SAGA purified using Sgf73-TAP subunit was incubated with α-TAP antibody and visualized using single-particle EM. Yellow circles denote density attributed to the antibody. The proposed locations of the modules are shown on the schematic of SAGA in the lower right corner of the figure.
We next investigated the localization of the TAF module subunits Taf5, Taf9, and Taf10. SAGA containing GFP tag fused to these three subunits resulted in additional electron densities observed near the torso joint region (Fig. 4B). Although SAGA is thought to contain two copies of Taf5, Taf6, Taf9, and Taf12, we did not observe two unambiguous densities for GFP-tagged Taf5 and Taf9 (12). This is likely due to a central location of the second copy of each protein, where other protein density can obstruct the visualization of the second GFP density. The TAF module therefore resides within the torso region of SAGA. Because SAGA containing a truncated tail still displays extensive flexibility, the central location of the TAF module supports our proposal that the shared TFIID subunits mediate a large degree of the conformational changes of the complex.
We next targeted the Spt3, Spt8, and Spt20 subunits of the SPT module. Consistent with our truncated Spt7 findings, the corresponding GFP-tagged SAGA complexes all displayed an additional density near the shoulder region and central torso of SAGA (Fig. 4C). The flexibility of the shoulder region, where both Spt3 and Spt8 reside, may be necessary to accommodate TBP binding and release from these subunits. Based on the size and number of the SPT module subunits, some of these proteins likely occupy the torso region near the TAF module subunits. Previous EM studies placed Spt20 on the opposite end of SAGA from Tra1 (12), a location that disagrees with our central localization. However, our observation is consistent with the subunit depletion study of Lee et al. (6) that proposed Tra1 and Spt20 to be in close proximity to each other.
When we applied the same experimental approach to map the location of subunits of the DUB module, we were unable to find any additional density clearly attributable to GFP. Furthermore, we were also unable to unambiguously fit the published crystal structure of the DUB module into the three SAGA three-dimensional reconstructions because of their low resolutions (34, 35). As an alternative approach, we applied an antibody labeling method that involves incubating SAGA purified from an SGF73-TAP strain with the anti-TAP antibody. The large size and characteristic shape of antibodies are more clearly distinguishable compared with GFP. We identified a large additional density corresponding to the antibody adjacent to the torso, near the proposed TAF module location (Fig. 4D), suggesting that the DUB module shares this region with both the TAF module and parts of the SPT module. The central location of the DUB module adjacent to the conformationally flexible TAF core is consistent with its role in facilitating the donut conformation. We summarized the results of our localization studies in Fig. 4E and divided SAGA into regions where each module is likely located.
Cross-linking Mass Spectrometry Analysis of apo-SAGA
Our EM-based localization studies enabled us to determine the spatial relationship among the different modules of SAGA. However, the relatively low resolution of these experiments precluded further understanding of the molecular organization of SAGA. Chemical CXMS is a powerful technique that can deduce the subunit connectivity of multiprotein complexes with precision to the amino acid residue level (25). Notably, two peptides of different subunits can be cross-linked only when they are located on or adjacent to the interface between the two proteins. We applied CXMS to comprehensively map the various different subunit interfaces of the SAGA complex. We incubated SAGA purified from FLAG-tagged Spt7 strain with disuccinimidyl suberate, a bifunctional cross-linker that reacts with primary amines, trypsin-digested the complex, and analyzed the resulting peptides using liquid chromatography coupled with LC-MS/MS. We searched the MS/MS data using the program pLink (25) and identified 78 unique intersubunit and 185 unique intrasubunit cross-links (supplemental Table S1). Our cross-linking results are represented in Fig. 5, emphasizing the interconnectivity between modules. Very recently, Han et al. (36) applied the CXMS approach to analyze the molecular architecture of SAGA in complex with TBP. In addition to confirming many cross-links that they identified, we were able to find interlinks involving Sgf11 and Sus1. This is likely due to better preservation of zinc finger domains in the DUB module by the FLAG affinity purification procedure in the absence of the chelating agent EGTA. Furthermore, we found unique cross-links connecting Tra1 to Spt7 and Taf6. We validated our CXMS results using available high resolution structures of SAGA subunits, combined with the ∼30 Å theoretical Cα-Cα cross-linking distance that disuccinimidyl suberate is capable of bridging (34, 37–40). We found that 20 of 21 cross-linked residue pairs were under 30 Å of each other, providing a high degree of confidence for the cross-links detected (supplemental Table S1).
FIGURE 5.
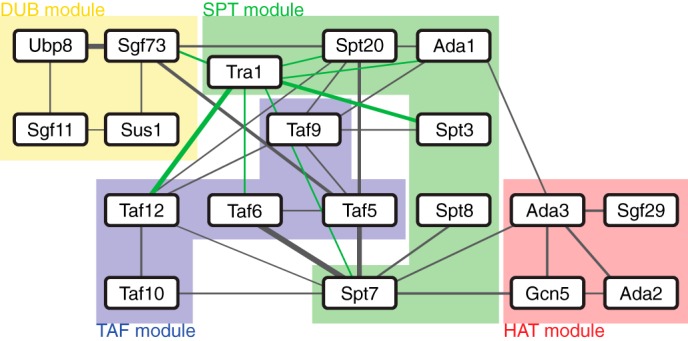
Network of SAGA subunit interconnectivity determined using cross-linking mass spectrometry. Lines denote cross-links, and the line thickness is proportional to the number of cross-link spectra detected between two subunits. Cross-links involving Tra1 are shown in green for clarity. Subunits are grouped into their respective domains, as shown by the labeled highlights.
Results from our CXMS analysis suggest that the TAF module in combination with the SPT subunits Spt7, Spt20, and Tra1 form a central core containing highly interconnected subunits, with the remaining subunits peripherally attached to this core. These findings are consistent with our EM-based analysis of deletion mutants and GFP localization, which suggests that the TAF and SPT modules are centrally located within the complex. Interestingly, although almost all of the SPT module subunits cross-link to members of the TAF module and Tra1, these subunits appear to constitute two distinct groups because Spt7 and Spt8 do not cross-link to Spt3 or Spt20. This suggests that Spt7-Spt8 and Spt3-Spt20 are present on separate faces of SAGA, sandwiching the TAF module. On the upper edge of this “sandwich,” Tra1 makes contact with the two separate groups of SPT subunits, whereas the HAT module similarly bridges the two groups on the opposite edge. Meanwhile, aside from Sgf73, no cross-links were detected between the DUB module subunits and the rest of SAGA, suggesting that Ubp8, Sgf11, and Sus1 face outwards from the complex. Sgf73, which anchors the DUB module to SAGA, cross-links to Spt20 and Taf5, suggesting that it is situated on the Spt3-Spt20 face of the SPT-TAF-SPT sandwich.
Interaction Interface with Transcriptional Activators
Using CXMS, we generated a detailed linkage map of all 19 SAGA subunits. Armed with this information, we sought to probe the functional interfaces of SAGA through investigating the binding locations of transcriptional activators. We purified recombinant His-maltose-binding protein (HMP)-tagged Gcn4, TBP, and the Gal4 activation domain (Gal4AD), and showed, by pulldown experiments, that these recombinant proteins bound FLAG-purified SAGA (Fig. 6A). We analyzed purified SAGA bound to the HMP-tagged activators by negative stain EM and observed that SAGA bound to Gcn4 or Gal4-AD contains additional electron densities near the globular half of the head region of SAGA near the head-torso junction (Fig. 6B). Because Gcn4 and Gal4 are known to bind Tra1 (3), this observation is consistent with our proposal that Tra1 spans both halves of the SAGA head region. We believe that the slight difference in position of the extra density between Gcn4 and Gal4 can be attributed to the flexibility of the linker between the activator and the HMP tag, as opposed to binding two different sites on Tra1. Meanwhile, HMP-tagged TBP bound SAGA contains additional electron density near the shoulder region where we proposed that its binding partners, Spt3 and Spt8, are located. In contrast to the globular head region, this region undergoes a large degree of conformational rearrangement. Because the transcriptional effects of Spt3 and Spt8 binding to TBP are chromatin context-specific, the flexibility of the region may facilitate this modulation of activity. Taken together, the results from our activator binding experiments further reinforce our proposed organization of the SAGA subunits as well as revealing new possibilities in the physiological role of the flexibility of the complex.
FIGURE 6.
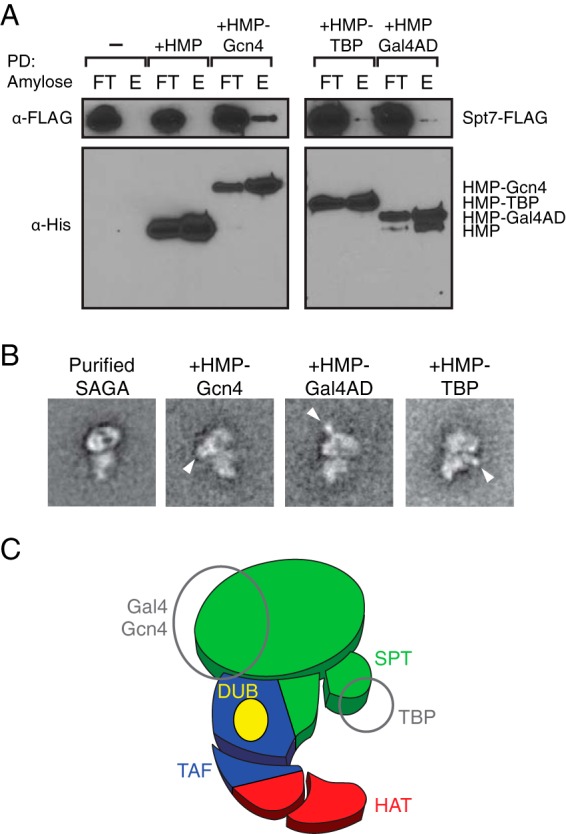
Localization of transcriptional activator binding sites. A, SAGA binds the recombinantly expressed HMP-tagged transcriptional activators. Amylose resin pulldowns (PD) of FLAG-purified SAGA mixed with HMP, HMP-Gcn4, HMP-TBP, and HMP-Gal4 activation domains (AD) were probed using anti-FLAG antibody. FT, flow-through; E, elution. B, class averages of negative-stained SAGA bound to transcriptional activators, without glutaraldehyde cross-linking. White arrows denote additional density attributed to the HMP-tagged activators. C, summary of activator binding locations shown on a modular schematic of SAGA.
Model of SAGA Subunit Arrangement in the Context of the EM map
By combining results from our EM-based GFP labeling, CXMS, and transcriptional activator binding experiments, we generated a model for the spatial arrangement of the 19 SAGA subunits within our EM reconstruction (Fig. 7). We approximated each subunit as spheres based on their molecular weights and the average density of proteins. The CXMS results provided distance restraints between pairs of subunits, whereas the localization analyses served to map specific modules to regions of density.
FIGURE 7.

Spatial arrangement of SAGA subunits. Results from EM-based subunit localization experiments and cross-linking mass spectrometry were combined with the curved SAGA three-dimensional reconstruction to propose an overall arrangement of all 19 complex subunits. The volumes of spheres represent the molecular mass of each subunit (DUB module crystal structure, Protein Data Bank code 3M99).
We included two copies of Taf5, Taf6, Taf9, and Taf12 and arranged them in the same fashion as the subunits within the recently studied human TFIID core (41). We were unable, however, to confirm that both copies are present in SAGA. Our TAF subunit GFP tagging analyses did not show two additional densities within the same class average, which may be due to the alignment algorithms only focusing on one label at the expense of the other or one label being obscured by other protein density. Despite this, we believe that the composition of TFIID and the fact that the molecular weight of SAGA far exceeds the sum of all its subunits support the dimerization argument. The large size of the entire TAF module can be encompassed within SAGA if it is oriented along the long axis of the complex, with a small portion contained within the globular head region. This placement provides a broad interaction surface to every other region of SAGA, consistent with the role of the TAF module as the backbone of the rest of the complex.
Our analysis of Tra1 being too large to fit within one lobe of the head led us to place spheres corresponding to its N- and C-terminal domains within the two regions of the head. The N-terminal domain of Tra1 consists of a long stretch of HEAT repeat motifs that have the propensity to form superhelical structures thought to generate a flexible scaffold for protein binding (42). Because we observed a larger degree of rearrangement in the region of the head adjacent to the shoulder, we believe that the Tra1 N terminus localizes there. We placed the smaller, likely more globular C-terminal domain of Tra1 in the globular head region, adjacent to one arm of the TAF module. Interestingly, this region corresponds to the Gcn4 and Gal4 binding region. In an earlier UV cross-linking study, Gcn4 was shown to be cross-linked to both Tra1 and Taf12, supporting the proposed arrangement of these two subunits within the SAGA head region (43).
The CXMS results suggest that within the SPT module, Spt7 resides on the opposite side of the TAF module as Spt20 and the DUB module. We positioned the DUB module, which includes the Ubp8-Sgf11-Sus1-Sgf73 N-terminal crystal structure and a sphere corresponding to the C-terminal region of Sgf73, on the Spt20 face oriented outwards, consistent with the peripheral connectivity of the module to SAGA. A recent EM structure of SAGA suggested that the DUB module localizes to a density within the torso of SAGA (13). Our reconstructions do not contain a density that correspond to the one encompassing the DUB module in this other SAGA EM structure. This may be due to the gradual cross-linking of the GraFix treatment capturing a different set of conformations than the direct incubation of TAP-purified SAGA with glutaraldehyde used in this other study. Despite this difference, the two studies agree on the relative location of the DUB module in the context of the fully assembled SAGA complex.
Earlier EM studies on SAGA placed Spt20 to be on the end of the complex away from Tra1 (12), which our localization studies and other MS-based experiments argue against (6, 36). In our model, Spt20 is centrally located, adjacent to the SPT/TAF core, the DUB module, and Tra1. We placed Ada1 in two copies in close proximity to Taf12, because the two subunits heterodimerize through their histone fold domains (44). Spt7 is thought to bind both Spt8 and the Taf10 HFD through its C terminus (45). Our placement of the Spt8 subunit within the shoulder region adjacent to Taf10 accommodates Spt7 binding to both subunits and agrees with a recent EM study of SAGA (13). Furthermore, Spt3, Spt7, Spt8, and Spt20 are all known to be in close proximity to the TBP binding location (36). Our own TBP binding analysis showed the binding site to be near the shoulder region along one edge of the SPT-TAF-SPT sandwich, where all of the SPT subunits have access to the activator.
Both sides of the SPT module interact with Ada3, which serves as the major interface between the HAT module and SAGA. Both faces are accessible in the lower tail placement for the module, underneath the SPT-TAF-SPT sandwich. These results are in direct contention with the earlier EM-based model of SAGA, which placed the HAT module to be within the torso (12). We believe that our ability to preserve the tail region in the overwhelming majority of particles contributes to a more accurate investigation of the region. Conversely, the top of the sandwich provides access between Tra1 and its many cross-linking partners. The model we generated suggests a layered arrangement of SAGA modules, where the top layer contains Tra1; the middle layer contains the TAF, SPT, and DUB modules; and the bottom layer contains the HAT module.
DISCUSSION
The large size and sophisticated composition of SAGA pose immense technical challenges to characterizing its detailed molecular structure and subunit organization in relation to its multiple roles in transcriptional regulation. Using single-particle EM, Wu et al. (12) provided the first glimpse of the overall morphology of SAGA and delineated the location of several core components of this complex. Lee et al. (6) subsequently applied an approach combining systematic gene deletion and mass spectrometry to identify SAGA subunits that anchor the functional modules to the core complex. More recently, Han et al. (36) conducted CXMS analysis of SAGA in complex with TBP to determine the subunit interconnectivity. The work presented in this paper built on and further expanded these initial efforts. In particular, through developing an improved purification method that enhanced the stability of SAGA, we were able to more precisely visualize and analyze the different conformational states of SAGA and determined the contributions of the catalytic modules in mediating structural rearrangements. Our comprehensive localization strategy, which combines EM-based labeling and CXMS of SAGA, enabled us to construct a unifying model of the subunit organization of this complex, improving our understanding of the relationships between different activities within this complex.
Conformational Flexibility of SAGA
Our improved purification procedure significantly enhanced the stability of SAGA and enabled us to systematically analyze its conformational flexibility for the first time. In comparison to previously published EM studies of SAGA (12, 13), the gradual cross-linking of the GraFix method greatly preserved the presence of the extended tail of SAGA. The first SAGA EM study by Wu et al. (12) showed that the standard TAP procedure resulted in 35% of the particles not displaying the tail density. Meanwhile, a recent study by Durand et al. (13) applied glutaraldehyde cross-linking to TAP-purified SAGA, decreasing the percentage of tail-less SAGA to 25%. Our GraFix-purified SAGA reduced the number of dissociated tails to 9%, demonstrating the effectiveness of this treatment.
We proceeded to analyze the distribution of the different conformational states using single-particle EM methods. To investigate the degree of conformational rearrangement that SAGA undergoes, we generated three-dimensional reconstructions of each conformation. Although our reconstructions have slightly lower resolution than previously published structures (12, 13)—likely because of the GraFix treatment making fine conformational rearrangements resolvable and therefore rendering alignment more difficult—they effectively demonstrate the extensive rearrangements between the three conformations. We suggest that the structural plasticity of SAGA reflects the need to adapt quickly to interact with different substrates and cofactors to mediate different physiological functions in the cell.
SAGA is not the only chromatin-related complex that displays a large degree of conformational flexibility. The chromatin structure remodeling complex adopts an open and closed conformation, where a sizable domain rearranges about a cavity in a manner reminiscent of the SAGA tail (46). TFIID undergoes an even greater degree of conformational rearrangement, where an entire lobe undergoes over 100 Å of movement and alters its connectivity to the rest of the complex (14). Given that the structural core of SAGA is comprised largely of subunits shared with TFIID, it is tempting to speculate that the molecular mechanisms behind the conformational rearrangement might be conserved between these two related complexes. Although we were able to distinguish three major conformations adopted by SAGA, delineating the physiological roles of these conformations would require the ability to isolate SAGA locked in a distinct conformation, a technical challenge that will need to be overcome in future studies.
Several factors appear to affect the conformational flexibility of SAGA, with disruption of the DUB module preventing the donut conformation from forming and the removal of the HAT module causing the shoulder region to be much more mobile. Disruption of the DUB module decreases HAT activity while only marginally affecting its structural integrity. Interestingly, it has been shown that the acetyltransferase and deubiquitination activities of SAGA show significant cross-talk, interacting both genetically and catalytically (7, 47). Our finding that the absence of the DUB module affects the flexibility and presence of the tail region of SAGA, where the HAT module is located, may reveal an indirect cooperativity between the two catalytic activities.
Spatial Arrangement of SAGA Chromatin-binding Domains
We combined our three-dimensional SAGA reconstructions with subunit localization and interconnectivity data to generate a model detailing the spatial organization of all 19 SAGA subunits. We show that SAGA is likely arranged into three major layers, with the topmost layer housing Tra1, the middle layer containing the SPT-TAF-SPT sandwich and the DUB module, and the lower layer encompassing the HAT module. This layered arrangement of SAGA is supported by CXMS data from Han et al. (36) and our analyses, with very few subunits bridging the top and bottom layers.
Based on the model we constructed, subunits with chromatin-binding domains are clustered along one side of the complex in close proximity to each other (Fig. 8). These domains include the Gcn5 and Spt7 bromodomains, Sgf29 Tudor domain, Ada2 SANT and SWIRM domains, and the Spt8 WD40 domain, all of which have been shown to bind different chromatin templates (48). Their proximity suggests a major interaction surface with the chromatin template within a region of SAGA that shows a large degree of flexibility. Chromatin surrounding a transcribed gene is by definition dynamic, decorated with various post-translational modifications depending upon the state of the cell. SAGA activity transitions between the different phases of transcription, from acetylation during transcription initiation to deubiquitination during elongation. The highly diverse chromatin-binding domains of SAGA are capable of binding methylated and acetylated histones, suggesting considerable versatility in template recognition. Given that transcription involves extensive remodeling of nucleosome occupancy, SAGA must be able to compensate for variations in nucleosome positions.
FIGURE 8.
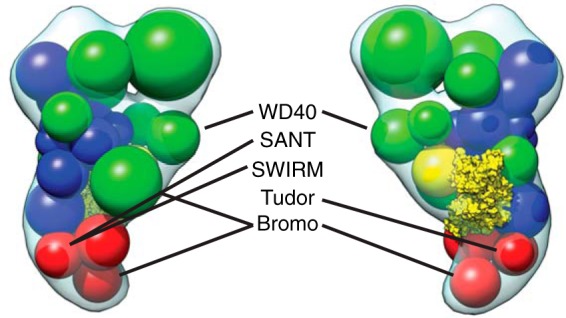
Chromatin-binding domains of SAGA cluster around one edge of the complex. SAGA subunits with domains known to bind histones are shown in the spatial arrangement model.
The extreme diversity of context-specific activities that SAGA fulfills, compounded with the recent proposition that the complex is active in all RNA polymerase II-mediated transcription (49), provides a compelling hypothesis for a highly flexible and adaptable chromatin interaction surface. In contrast, our finding that the activator binding surface is relatively static is likely due to the conserved acidic patches within the activation domains of transcriptional activators serve as universal adapters, delegating the task of chromatin binding to the DNA-binding domains of the activators (3). These hypotheses on the nature of SAGA flexibility predict that other chromatin-binding complexes will likely be flexible, a feature that is likely de-emphasized in single-particle EM studies where this property negatively affects achievable resolution. We believe that conformational variability of chromatin-binding complexes should be studied more closely, because such studies could provide intriguing new insights into the mechanism of action of these important regulators.
SAGA is a fascinating multifunctional complex that provides a paradigm to delineate the molecular mechanisms of fundamental transcriptional processes and to gain insights into how important chromatin-modifying complexes that exert exquisite epigenetic control over all eukaryotic gene expression. Further understanding of SAGA mechanisms of action would require higher resolution analysis of the full complex and individual subunits, using a joint approach of various structural techniques.
Supplementary Material
Acknowledgments
We thank H. Rho and S. Schaffner for technical assistance and J. Moon and L. J. Foster for assistance with mass spectrometry analysis.
This work was supported by Natural Sciences and Engineering Research Council of Canada Discovery Grant 418157, a Michael Smith Foundation for Health Research Career Investigator Award, a Canadian Institutes of Health Research New Investigator Award, an infrastructure grant from the Canadian Foundation for Innovation, and startup funds from the University of British Columbia.

This article contains supplemental Table S1.
- HAT
- histone acetyltransferase
- SAGA
- Spt-Ada-Gcn5 acetyltransferase
- GraFix
- gradient fixation
- DUB
- deubiquitination
- TBP
- TATA-binding protein
- CXMS
- cross-linking of proteins coupled with mass spectrometry
- TAP
- tandem affinity purification
- HMP
- His-maltose-binding protein.
REFERENCES
- 1. Hahn S., Young E. T. (2011) Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rando O. J., Winston F. (2012) Chromatin and transcription in yeast. Genetics 190, 351–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown C. E. (2001) Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science 292, 2333–2337 [DOI] [PubMed] [Google Scholar]
- 4. Koutelou E., Hirsch C. L., Dent S. Y. (2010) Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 22, 374–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant P. A., Duggan L., Côté J., Roberts S. M., Brownell J. E., Candau R., Ohba R., Owen-Hughes T., Allis C. D., Winston F., Berger S. L., Workman J. L. (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 11, 1640–1650 [DOI] [PubMed] [Google Scholar]
- 6. Lee K. K., Sardiu M. E., Swanson S. K., Gilmore J. M., Torok M., Grant P. A., Florens L., Workman J. L., Washburn M. P. (2011) Combinatorial depletion analysis to assemble the network architecture of the SAGA and ADA chromatin remodeling complexes. Mol. Syst. Biol. 7, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henry K. W., Wyce A., Lo W.-S., Duggan L. J., Emre N. C., Kao C.-F., Pillus L., Shilatifard A., Osley M. A., Berger S. L. (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 17, 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buratowski S., Hahn S., Guarente L., Sharp P. A. (1989) Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 56, 549–561 [DOI] [PubMed] [Google Scholar]
- 9. Barbaric S., Reinke H., Hörz W. (2003) Multiple mechanistically distinct functions of SAGA at the PHO5 promoter. Mol. Cell. Biol. 23, 3468–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belotserkovskaya R., Sterner D. E., Deng M., Sayre M. H., Lieberman P. M., Berger S. L. (2000) Inhibition of TATA-binding protein function by SAGA subunits Spt3 and Spt8 at Gcn4-activated promoters. Mol. Cell. Biol. 20, 634–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grant P. A., Schieltz D., Pray-Grant M. G., Steger D. J., Reese J. C., Yates J. R., 3rd, Workman J. L. (1998) A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell 94, 45–53 [DOI] [PubMed] [Google Scholar]
- 12. Wu P.-Y., Ruhlmann C., Winston F., Schultz P. (2004) Molecular architecture of the S. cerevisiae SAGA complex. Mol. Cell. 15, 199–208 [DOI] [PubMed] [Google Scholar]
- 13. Durand A., Bonnet J., Fournier M., Chavant V., Schultz P. (2014) Mapping the deubiquitination module within the SAGA complex. Structure 22, 1553–1559 [DOI] [PubMed] [Google Scholar]
- 14. Cianfrocco M. A., Kassavetis G. A., Grob P., Fang J., Juven-Gershon T., Kadonaga J. T., Nogales E. (2013) Human TFIID binds to core promoter DNA in a reorganized structural state. Cell 152, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 16. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 17. Lõoke M., Kristjuhan K., Kristjuhan A. (2011) Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques. 50, 325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stark H. (2010) GraFix: stabilization of fragile macromolecular complexes for single particle cryo-EM. Methods Enzymol. 481, 109–126 [DOI] [PubMed] [Google Scholar]
- 19. Ohi M., Li Y., Cheng Y., Walz T. (2004) Negative staining and image classification: powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 21. Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 [DOI] [PubMed] [Google Scholar]
- 22. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 23. Taylor D. W., Ma E., Shigematsu H., Cianfrocco M. A., Noland C. L., Nagayama K., Nogales E., Doudna J. A., Wang H.-W. (2013) Substrate-specific structural rearrangements of human Dicer. Nat. Struct. Mol. Biol. 20, 662–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 26. Chien C. T., Bartel P. L., Sternglanz R., Fields S. (1991) The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. U.S.A. 88, 9578–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu P.-Y., Winston F. (2002) Analysis of Spt7 function in the Saccharomyces cerevisiae SAGA coactivator complex. Mol. Cell. Biol. 22, 5367–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radermacher M., Wagenknecht T., Verschoor A., Frank J. (1987) Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J. Microsc. 146, 113–136 [DOI] [PubMed] [Google Scholar]
- 29. Chittuluru J. R., Chaban Y., Monnet-Saksouk J., Carrozza M. J., Sapountzi V., Selleck W., Huang J., Utley R. T., Cramet M., Allard S., Cai G., Workman J. L., Fried M. G., Tan S., Côté J., Asturias F. J. (2011) Structure and nucleosome interaction of the yeast NuA4 and Piccolo-NuA4 histone acetyltransferase complexes. Nat. Struct. Mol. Biol. 18, 1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knutson B. A., Hahn S. (2011) Domains of Tra1 important for activator recruitment and transcription coactivator functions of SAGA and NuA4 complexes. Mol. Cell. Biol. 31, 818–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang H., Rudge D. G., Koos J. D., Vaidialingam B., Yang H. J., Pavletich N. P. (2013) mTOR kinase structure, mechanism and regulation. Nature 497, 217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sibanda B. L., Chirgadze D. Y., Blundell T. L. (2010) Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 463, 118–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yip C. K., Berscheminski J., Walz T. (2010) Molecular architecture of the TRAPPII complex and implications for vesicle tethering. Nat. Struct. Mol. Biol. 17, 1298–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Köhler A., Zimmerman E., Schneider M., Hurt E., Zheng N. (2010) Structural basis for assembly and activation of the heterotetrameric SAGA histone H2B deubiquitinase module. Cell 141, 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Samara N. L., Datta A. B., Berndsen C. E., Zhang X., Yao T., Cohen R. E., Wolberger C. (2010) Structural insights into the assembly and function of the SAGA deubiquitinating module. Science 328, 1025–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Han Y., Luo J., Ranish J., Hahn S. (2014) Architecture of the Saccharomyces cerevisiae SAGA transcription coactivator complex. EMBO J. 33, 2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leitner A., Joachimiak L. A., Bracher A., Mönkemeyer L., Walzthoeni T., Chen B., Pechmann S., Holmes S., Cong Y., Ma B., Ludtke S., Chiu W., Hartl F. U., Aebersold R., Frydman J. (2012) The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 20, 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bian C., Xu C., Ruan J., Lee K. K., Burke T. L., Tempel W., Barsyte D., Li J., Wu M., Zhou B. O., Fleharty B. E., Paulson A., Allali-Hassani A., Zhou J.-Q., Mer G., Grant P. A., Workman J. L., Zang J., Min J. (2011) Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 30, 2829–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trievel R. C., Rojas J. R., Sterner D. E., Venkataramani R. N., Wang L., Zhou J., Allis C. D., Berger S. L., Marmorstein R. (1999) Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc. Natl. Acad. Sci. U.S.A. 96, 8931–8936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Owen D. J., Ornaghi P., Yang J. C., Lowe N., Evans P. R., Ballario P., Neuhaus D., Filetici P., Travers A. A. (2000) The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 19, 6141–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bieniossek C., Papai G., Schaffitzel C., Garzoni F., Chaillet M., Scheer E., Papadopoulos P., Tora L., Schultz P., Berger I. (2013) The architecture of human general transcription factor TFIID core complex. Nature 493, 699–702 [DOI] [PubMed] [Google Scholar]
- 42. Neuwald A. F. (2000) HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res. 10, 1445–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herbig E., Warfield L., Fish L., Fishburn J., Knutson B. A., Moorefield B., Pacheco D., Hahn S. (2010) Mechanism of mediator recruitment by tandem Gcn4 activation domains and three Gal11 activator-binding domains. Mol. Cell. Biol. 30, 2376–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gangloff Y. G., Werten S., Romier C., Carré L., Poch O., Moras D., Davidson I. (2000) The human TFIID components TAF(II)135 and TAF(II)20 and the yeast SAGA components ADA1 and TAF(II)68 heterodimerize to form histone-like pairs. Mol. Cell. Biol. 20, 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gangloff Y.-G., Sanders S. L., Romier C., Kirschner D., Weil P. A., Tora L., Davidson I. (2001) Histone folds mediate selective heterodimerization of yeast TAFII25 with TFIID components yTAFII47 and yTAFII65 and with SAGA component ySPT7. Mol. Cell. Biol. 21, 1841–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leschziner A. E., Saha A., Wittmeyer J., Zhang Y., Bustamante C., Cairns B. R., Nogales E. (2007) Conformational flexibility in the chromatin remodeler RSC observed by electron microscopy and the orthogonal tilt reconstruction method. Proc. Natl. Acad. Sci. U.S.A. 104, 4913–4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee K. K., Florens L., Swanson S. K., Washburn M. P., Workman J. L. (2005) The deubiquitylation activity of Ubp8 is dependent upon Sgf11 and its association with the SAGA complex. Mol. Cell. Biol. 25, 1173–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee K. K., Workman J. L. (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295 [DOI] [PubMed] [Google Scholar]
- 49. Bonnet J., Wang C.-Y., Baptista T., Vincent S. D., Hsiao W.-C., Stierle M., Kao C.-F., Tora L., Devys D. (2014) The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 28, 1999–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.