

Background: Ca2+-independent phospholipase A-β (iPLAβ) has been repeatedly implicated as a homeostatic enzyme.
Results: Activity of iPLAβ correlates inversely with the hydrophobicity of the phospholipid substrate.
Conclusion: Efflux of the phospholipid substrate from a membrane mainly determines the activity of iPLAβ.
Significance: Results support the role of iPLAβ as a homeostatic enzyme that responds to increased phospholipid efflux proposed to occur at particular “critical” compositions.
Keywords: Homeostasis, Mass Spectrometry (MS), Membrane, Membrane Bilayer, Phospholipase A
Abstract
The A-type phospholipases (PLAs) are key players in glycerophospholipid (GPL) homeostasis and in mammalian cells; Ca2+-independent PLA-β (iPLAβ) in particular has been implicated in this essential process. However, the regulation of this enzyme, which is necessary to avoid futile competition between synthesis and degradation, is not understood. Recently, we provided evidence that the efflux of the substrate molecules from the bilayer is the rate-limiting step in the hydrolysis of GPLs by some secretory (nonhomeostatic) PLAs. To study whether this is the case with iPLAβ as well, a mass spectrometric assay was employed to determine the rate of hydrolysis of multiple saturated and unsaturated GPL species in parallel using micelles or vesicle bilayers as the macrosubstrate. With micelles, the hydrolysis decreased with increasing acyl chain length independent of unsaturation, and modest discrimination between acyl positional isomers was observed, presumably due to the differences in the structure of the sn-1 and sn-2 acyl-binding sites of the protein. In striking contrast, no significant discrimination between positional isomers was observed with bilayers, and the rate of hydrolysis decreased with the acyl chain length logarithmically and far more than with micelles. These data provide compelling evidence that efflux of the substrate molecule from the bilayer, which also decreases monotonously with acyl chain length, is the rate-determining step in iPLAβ-mediated hydrolysis of GPLs in membranes. This finding is intriguing as it may help to understand how homeostatic PLAs are regulated and how degradation and biosynthesis are coordinated.
Introduction
Glycerophospholipid (GPL)2 homeostasis is crucial for mammalian cells as indicated by the fact that they maintain the GPL compositions of their membranes within close limits (1), and major compositional deviations are lethal in mice (2). The key processes maintaining GPL homeostasis are biosynthesis, acyl chain remodeling and degradation (3). Regarding degradation, several studies have provided evidence that A-type phospholipases (PLAs) are key players in this process (3–9). Particularly compelling evidence for this comes from studies in which the synthesis of a GPL class was boosted by overexpression of the rate-limiting synthetic enzyme. For instance, overexpression of CTP:phosphocholine cytidylyltransferase in HeLa cells increased PC synthesis by 4–5-fold, but the PC content of the cells did not increase significantly. Instead, a marked increase of glycerophosphocholine, a deacylation product of PC, was detected in the cells and also the medium (4, 10). Forcing the synthesis of PE or PS also did not significantly increase the cellular content of these GPLs, but rather it increased their degradation mediated by PLAs (11, 12). Conversely, when the synthesis of a GPL is inhibited, its degradation decreases in proportion, thus maintaining GPL homeostasis. For example, when the synthesis of PE was inhibited by mutating the rate-limiting enzyme ethanolamine phosphotransferase, the PE content of the cells did not decrease due to reduced degradation (13). In CHO mutants with a partially inactive choline kinase-α, the rate of PC synthesis was reduced 4-fold, but the PC content was normal (14). Collectively, those data demonstrate that in mammalian cells GPL synthesis and degradation are strictly coordinated, and they provide strong evidence that the cells contain homeostatic PLA enzyme(s) that selectively degrade the GPL species that are present in excess.
Mammalian cells express numerous PLA proteins (15, 16), but those involved in GPL homeostasis have not been firmly established. However, select members of the Ca2+-independent PLA (iPLA) subfamily, particularly iPLAβ and iPLAδ, have been implicated in this process (17–20). For instance, it was shown that the activity of iPLAβ increased 2-fold in CHO cells when PC synthesis was boosted by overexpressing CTP:phosphocholine cytidylyltransferase (5) and that treatment of cells with bromoenol lactone, an inhibitor of iPLAβ, suppressed PC degradation (4). In addition, we have recently found that knockdown of iPLAβ in HeLa cells markedly inhibits the turnover of the major GPL classes as studied by heavy isotope-labeled precursors.3 Notably, for efficient homeostasis, the catalytic activity of iPLAβ should be regulated, because an alteration in its expression is obviously too slow to avoid energy-wasting compositional fluctuations (21). In essence this means that iPLAβ must be able to sense the GPL composition of a membrane to “know” whether a particular GPL is present in excess or not. Although the activity of iPLAβ might be regulated indirectly, i.e. by other homeostatic proteins (22), there is no evidence that such a regulation plays a role in GPL homeostasis. Thus, the simplest and the most attractive model is that the membrane GPL composition directly modulates the activity of this enzyme, like it regulates the activity of biosynthetic enzymes such as CT (23, 24) and PS synthase (25, 26). Consistent with this model, we have recently provided evidence that the activity of some secretory, nonhomeostatic PLAs correlates with the upward movement of the GPL substrate from the membrane bilayer (efflux propensity) (27). This finding is intriguing, because efflux propensity has been predicted to change abruptly at particular membrane GPL compositions (21).
To study whether substrate efflux propensity regulates the activity of the homeostatic iPLAβ as well, we employed a high throughput mass spectrometry-based assay that allowed us to study the hydrolysis of numerous GPL species simultaneously. The key result is that, when using vesicle bilayers as the macrosubstrate, the rate of iPLAβ-mediated hydrolysis decreased strongly and monotonously with increasing substrate hydrophobicity, thus strongly suggesting that the activity of iPLAβ is by and large determined by the ease of efflux of its substrate from the bilayer. Because efflux of a GPL is predicted to increase strongly when it is present in excess (i.e. over its optimal concentration), these data strongly support the role of iPLAβ in GPL homeostasis of mammalian cells.
EXPERIMENTAL PROCEDURES
Lipids and Other Chemicals
l-α-1-Palmitoyl-2-arachidonyl-[1-14C]PC (58 μCi/μmol) was purchased from PerkinElmer Life Sciences, and unlabeled GPLs, lyso-PCs, and sphingomyelins (SM) were obtained from Avanti Polar Lipids (Alabaster, AL), and d3-methyl iodide was from Cambridge Isotope Laboratories (Andover, MA). Ethanolamine, phospholipase D (Streptomyces species), Nonidet P-40 (IGEPAL), and EDTA-free protease inhibitor mixture were obtained from Sigma. Silica gel 60 thin layer plates and HPLC-grade solvents were from Merck; nickel-nitrilotriacetic acid-agarose was from Qiagen (Germantown, MD); Strep-Tactin Superflow resin and desthiobiotin were from IBA-Life Sciences (Goettingen, Germany); bromoenol lactone and methoxyarachidonyl fluorophosphonate inhibitors were from Cayman Chemicals (Ann Arbor, MI); bovine serum albumin standard and Coomassie Brilliant Blue R-250 solution were from Bio-Rad (Sundyberg, Sweden). Penicillin, streptomycin, fetal bovine serum (FBS), and fungizone were from Gibco Life Technologies (Bleiswijk, Netherlands). The sn-1–16:0/sn-2-Cn-PC and sn-1-Cn/sn-2–16:0-PC species were synthesized by acylating sn-1–16:0-LPC with the anhydride of a Cn-fatty acid or sn-1-Cn-LPC with the anhydride of 16:0-fatty acid, respectively. d9-Labeled PC species were synthesized from their corresponding phosphatidylethanolamines (PE) by methylation with d3-methyl iodide as described earlier (28), except that benzene was replaced with toluene. The purity of GPLs was confirmed by TLC and mass spectrometry, and their concentrations were determined by phosphate assay (29).
Cloning, Expression, and Purification of Human iPLAβ
Full-length iPLAβ cDNA from the clone DKFZp434A102Q (ImaGenes, GenBankTM AL080187.1) was amplified by PCR using the forward primer 5′-TTTGGTACCATGCAGTTCTTTGGACGC-3′ and the reverse primer 5′-TTTACCTGGGGGTGAGAGCAGCAGCT-3′. The product was digested with KpnI and AgeI and ligated into the pMT expression vector (Invitrogen) that contains the Drosophila metallothionein-inducible promoter and a C-terminal His6 tag. Next, the sequence coding for a C-terminal His10 tag was appended to the iPLAβ sequence by PCR using pMT-iPLAβ-His6 as template. The forward primer was 5′-GTCGGTCCGCCACCATGCAGTTCTTTGGCCGC-3′ containing the RsrII cleavage site, and the reverse was 5′-CTGGTACCTTAGTGGTGGTGGTGATGGTGATGATGATGATGTTCGAAACGACCTTCGATGGGTGAGAGCAGCAGCT-3′ containing the BstBI and KpnI cleavage sites. The RsrII- and KpnI-digested PCR product was ligated into pFastBac1-vector (pFB1, Gibco Life Technologies) resulting in pFB1-iPLAβ-His10. Replacing the BstBI-KpnI fragment of pFB1-iPLAβ-His10 by the annealed oligonucleotides 5′-CGAATGGAGCCACCCGCAGTTCGAGAAAGGAGGAGGAAGCGGAGGAGGAAGCGGAGGAGGAAGCTGGAGCCACCCGCAGTTTGAAAAATAGGTAC-3′ and 5′-CTATTTTTAAACTGCGGGTGGCTCCAGCTTCCTCCTCCGCTTCCTCCTCCGCTTCCTCTCCTTTCTCGAACTGCGGGTGGCTCCATT-3′ resulted in the pFB1-iPLAβ-Strep vector. The inserted sequences and the reading frames of the constructs were verified by sequencing. The Bac-to-Bac baculoviral expression system (Gibco Life Technologies) was then utilized for bacmid preparation and CellFectin-mediated transfection of Sf9 cells to produce the recombinant baculovirus.
To express the iPLAβ-StrepIII protein, Sf9 cell suspension (500 ml, ∼106 cells/ml) was infected with the recombinant baculovirus at the multiplicity of 1. After incubation for 72 h at 26 °C, the cells were washed with ice-cold PBS and resuspended in 50 ml of a lysis buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm ATP, 0.5 mm EDTA, 0.5% Nonidet P-40, 5% glycerol) containing an EDTA-free protease inhibitor mixture. The cells were then lysed by sonication (six times for 1 min with 30-s intervals, 40% duty cycle) and centrifuged at 50,000 × g for 1 h to pellet the membranes. The supernatant was collected and poured into a Poly-Prep column (Bio-Rad) containing buffer W (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 5% glycerol) equilibrated Strep-Tactin Superflow resin. The column was then washed with 5 column volumes of buffer W, and iPLAβ-StrepIII was eluted five times with 1 ml of buffer E (= buffer W + 2.5 mm desthiobiotin).
To prepare tag-free iPLAβ, the Strep-Tactin column fractions with high PLA activity were pooled and incubated with the Factor Xa protease (Thermo Scientific) at a Factor Xa to protein ratio of 1:100 for 2 h at 20 °C. After inhibiting any further proteolysis with 1 mm PMSF, the sample was applied to a DG-10 desalting column (Bio-Rad) to exchange the incubation buffer for buffer A (20 mm Tris-HCl, pH 8.0). The sample was then loaded onto a buffer A-equilibrated HiTrap Q HP (GE Healthcare) ion-exchange column (1 ml bed volume) coupled to an AKTA-FPLC system (GE Healthcare). The elution was performed with a gradient of 0–1 m NaCl in buffer A at a rate of 1 ml/min with detection at 280 nm. Fractions were pooled based on the elution profile, concentrated on an ultrafiltration device (Amicon Ultra-30k, Millipore), and assayed for PLA activity. The fractions with the highest activity were pooled and stored at −80 °C in the presence of 60% (v/v) glycerol.
Immunoblotting
To assess their iPLAβ content, the transfected cells were pelleted, washed with ice-cold PBS, resuspended in a lysis buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 0.1% SDS, 1.0% Triton X-100, 1.0% deoxycholate, 5 mm EDTA), and left on ice for 30 min. The lysate was centrifuged for 3 min at 800 × g at 4 °C, and 20 μg of supernatant protein was run on a 10% SDS-polyacrylamide gel. The proteins were then transferred onto a PVDF membrane (Millipore) and treated with 5% defatted dry milk in 50 mm Tris-buffered NaCl (150 mm) containing 0.1% Tween 20. The recombinant iPLAβ was detected with an anti-iPLAβ (Santa Cruz Biotechnology, 1:100 dilution) or a Streptactin HRP-conjugated antibody (Bio-Rad, 1:10,000 dilution). After treatment with ECL Plus detection reagent kit (GE Healthcare), the blot was scanned with Starion FLA-9000 image scanner (Fujifilm) and visualized using Image Reader FLA-9000 software.
Extraction of HeLa Microsomal GPLs
HeLa cells were grown on eight 14-cm dishes until ∼80% confluent (30). After rinsing with ice-cold PBS, cells were scraped off and centrifuged at 1500 × g for 3 min. After resuspending in 1 ml of a lysis buffer (25 mm Hepes-NaOH, pH 7.4, 500 mm sucrose, 10 mm KCl, 1.5 mm MgCl2) followed by incubation on ice for 30 min, the cells were broken with a Dounce homogenizer followed by 10 passes through a 27-gauge needle. The intact cells and nuclei were removed by centrifugation at 1200 × g for 5 min, and the supernatant was centrifuged at 50,000 × g for 1.5 h at 4 °C to pellet the microsomes. After suspending the microsomes in 1 ml of a buffer constituting 100 mm Hepes-NaOH, pH 7.4, 0.5 m sucrose, any aggregates present were removed by centrifugation at 800 × g for 10 min. The supernatant containing the microsomes was transferred to a Kimax tube, and lipids were extracted (31), and after determination of the GPL content (29), the extract was stored at −20 °C.
Assay of PLA Activity
PLA activity of the recombinant iPLAβ was determined using a radioactivity assay. The macrosubstrate was prepared by mixing 10,000 cpm of 1-palmitoyl-2-arachidonyl-[1-14C]PC, unlabeled 16:0/18:1-PC, and 16:0/18:1-PA (250/25 nmol) in chloroform followed by removal of the solvent under a N2 stream and then under high vacuum for 1 h followed by addition of 0.5 ml of assay buffer (50 mm sodium phosphate, pH 7.4, 10 mm ATP, 0.5% Triton X-100) and vortexing for 2 min at room temperature. To start the reaction, an aliquot of purified iPLAβ was added, and the mixture was incubated for 30 min at 37 °C. Aliquots of 50 μl were removed at intervals and mixed with 2 ml of ice-cold methanol to stop the reaction, and the lipids were extracted according to Ref. 31, except that 0.1 m HCl was used to maximize lipid recovery. The extract was analyzed on TLC using hexane/ethyl ether/acetic acid (70:30:1, v/v) as the eluent, and radiolabeled PC, LPC, and fatty acid were detected with Starion FLA-9000 image scanner (FujiFilm), visualized using ImageReader FLA-9000 software, and quantified with the MultiGauge analysis software.
Substrate Specificity of iPLAβ
A mixture of synthetic GPLs or total microsomal lipids, 16:0/18:1-PA and 21:00-SM as an internal standard, were mixed in chloroform and dried under a N2 stream and then under high vacuum for 1 h. Butylated hydroxytoluene (1 mol %) was included as an antioxidant when the mixture contained unsaturated GPLs. To prepare small unilamellar vesicles (SUVs), the dried lipids were dissolved in 20 μl of ethanol and injected into 0.5 ml of the assay buffer (50 mm sodium phosphate, pH 7.4, 10 mm ATP) using a constant-speed Hamilton syringe. To prepare large unilamellar vesicles (LUVs), lipids dried in a glass tube were overlaid with 1 ml of the assay buffer and placed in a water bath at 4 °C for 1 min and then in a water bath at 50 °C for 1 min. This cooling-heating cycle was repeated four times. The lipid suspension was vortexed for 1 min at room temperature, then extruded 10 times through two stacked 1.2-μm polycarbonate membranes, and then 10 times through two 0.4-μm membranes using the Lipex extruder (Vancouver, Canada) pressurized with argon. The SUV or LUV bilayers were incubated in the presence of purified iPLAβ at 37 °C; 50-μl aliquots were removed at intervals and mixed with 2 ml of ice-cold methanol to stop the reaction. After addition of 0.8 ml of water and 4 ml of chloroform, vortexing, and removal of the upper phase, the lower phase was washed three times with the theoretical upper phase (31) and taken to dryness under a N2 stream. The residue was reconstituted in 200 μl of chloroform/methanol (1:2, v/v) and stored at −20 °C. To study the hydrolysis in micelles, a lipid mixture was dried as above and dispersed in the assay buffer with 1% Nonidet P-40 detergent, and the assay was conducted as above.
Effect of the Headgroup of GPL on Its Spontaneous Intervesicle Translocation
Spontaneous intermembrane translocation of 18:1/18:1 GPLs with varying headgroups was measured as described previously (32). Briefly, negatively charged donor vesicles consisting of an equimolar mixture of 18:1/18:1-PC, -PE, -PS, -PG, -PA, -PI (1335 nmol GPL), d9-labeled 22:1/22:1-PC donor marker (15 nmol) + tetra-18:1 cardiolipin (150 nmol), and uncharged acceptor vesicles consisting of 16:0/18:1-PC were both prepared in a buffer constituting 10 mm Hepes-NaOH, pH 7.0, 25 mm KCl, 0.5 mm EDTA by probe sonication in an ice-water bath. The donor and acceptor vesicles (1.5 and 7.5 μmol of total GPL, respectively) were coincubated at 37 °C; aliquots were withdrawn at intervals and applied to a DEAE-Sephacel (Amersham Biosciences) minicolumn to trap the negatively charged donor vesicles. To quantify the 18:1/18:1 GPLs transferred to the acceptor vesicles (in the flow-through), a standard for each GPL class was added before lipid extraction and mass spectrometric analysis.
Mass Spectrometric Analysis of GPLs
After addition of NH4OH (4% final concentration), the lipid sample was infused at 6 μl/min to a Micromass Quattro Micro triple-quadrupole mass spectrometer operated as described previously (33). GPL species were detected using headgroup-specific precursor or neutral-loss scanning (34). The spectra were exported to Microsoft Excel, and the individual GPL species were quantified using LIMSA software (35). Concentrations of the individual lipid species were plotted against time, and the relative rate constants of hydrolysis were obtained by fitting a first order exponential decay model to the data. The maximum hydrolyzable fraction at t∞ was constrained to 1.0 with micelles, to 0.67 with SUVs, and to 0.5 with LUVs (27).
LC-MS with selective reaction monitoring (SRM) was used when the hydrolysis of microsomal GPLs was investigated. Waters ACQUITY Ultra Performance LC system equipped with a Waters ACQUITY BEH C18 column (1.0 × 100 mm) was used to separate the molecular species using gradient elution. Solvent A was acetonitrile/H2O (60:40) with 10 mm ammonium formate and 1% NH4OH, whereas solvent B was isopropyl alcohol/acetonitrile (90:10) containing 10 mm ammonium formate and 1% NH4OH. The flow rate was 0.13 ml/min, and the column temperature was 60 °C. Solvent B was set to 40% at injection and increased linearly to 100% in 14 min, remained at this value for 3 min, decreased back to 40% in 1 min, and then remained there until the end of the gradient at 20 min. The eluent was directed to the ESI source of Waters Quattro Premier triple-quadrupole mass spectrometer operated in the positive ion mode. For SRM transitions, proton adducts of the PC, PE, PS, and PI species were selected as the precursors, whereas the product ion was either the headgroup (PC and PI) or the diacylglycerol fragment (PE and PS). For quantification of the microsomal lipids 20:1/20:1-PC, 22:1/22:1-PE, 20:1/20:1-PS, and 16:0/16:0-PI, standards were added during the single-phase stage of extraction. The SRM chromatograms were integrated, and the relative concentrations of the individual GPL species were calculated using the QuanLynx software (Waters).
Other Procedures
Protein concentration was determined using Lowry (36) or a fluorometric method (37) with BSA as the standard. SDS-PAGE was carried out according to Laemmli (38), and the proteins were stained with Coomassie Blue (39).
RESULTS
Expression and Purification of Human iPLAβ
We initially expressed iPLAβ with a C-terminal His6 tag in Drosophila S2 cells and then subjected it to purification on a Ni2+-affinity column. Although the preparation was not fully homogeneous, iPLAβ-His6 was the main band when visualized on an SDS-polyacrylamide gel and most likely responsible for all the PLA activity present in the preparation. However, to exclude that any of the contaminating protein impurities might bias the specificity data, we constructed iPLAβ with a C-terminal His10 or StrepIII tag (see “Experimental Procedures”). Although the His10 tag did not help to purify the enzyme beyond that achieved with the His6 version, iPLAβ-StrepIII was almost pure after affinity chromatography on a Strep-Tactin column. To exclude the possibility that even the minor impurities remaining could bias the hydrolysis data, the StrepIII tag was cleaved off with the Factor Xa protease, and the tag-free iPLAβ was purified on a cation-exchange column. As shown in Fig. 1, an essentially homogeneous protein was obtained. The specificity data shown in this study was obtained with this homogeneous enzyme, but identical results were obtained with iPLAβ-His6 as well (data not shown). Accordingly, neither the tag nor the degree of purity of iPLAβ had a detectable effect on the results.
FIGURE 1.

SDS-PAGE analysis of tag-free iPLAβ after cation-exchange chromatography. The tag was removed from iPLAβ-StrepIII by selective proteolysis with Factor Xa, and the reaction mixture was submitted to cation-exchange chromatography as described under “Experimental Procedures.” A, right column fraction containing all PLA activity was analyzed by SDS-PAGE, and the gel was stained with Coomassie Blue. B, left column shows the PLA-containing fraction stained with anti-iPLAβ antibody (1:100 dilution). The left column in A and the right column in B show the marker protein and molecular weights.
High Throughput Assay of iPLAβ Substrate Specificity
To obtain information on how the structure of the acyl chain and the headgroup structure influences GPL hydrolysis by iPLAβ, we employed a high throughput mass spectrometry-based assay devised previously (30). This assay allows one to determine the rates of hydrolysis of a multitude of GPL species simultaneously. Four different mixtures of synthetic GPL species as well as the GPLs extracted from HeLa cell microsomes were studied here. The first mixture (“PC-mix”) consisted of a total of 25 species, i.e. (i) 10 disaturated species with the length of both acyl chains varying from 13 to 22 carbons; (ii) two monounsaturated species (14:0/18:1 and 16:0/18:1); (iii) six species with two double bonds, including 16:0/18:2 and five dimonounsaturated species with acyl chain lengths varying between 14 to 22 carbons (14:1/14:1, 16:1/16:1, 18:1/18:1, 20:1/20:1, and 22:1/22:1); (iv) two species with four double bonds (16:0/20:4 and 18:0/20:4); (v) three species with six double bonds (18:3/18:3, 16:0/22:6, and 18:0/22:6); (vi) one with eight double bonds (20:4/20:4); and (vii) one with 12 double bonds (22:6/22:6). The second mixture (16:0/Cn-PCs) consisted of 17 saturated PC species with a 16:0 sn-1 chain and an sn-2 chain of 6–24 carbons, and the third mixture (d9-Cn/16:0-PCs) consisted of the positional isomers of the second one. The fourth mixture consisted of six 18:1/18:1 GPLs each with a different polar headgroup (i.e. 18:1/18:1-PC, -PE, -PS, -PG, -PI, and -PA). 21:0-SM (10 mol %) was included as a nonhydrolyzable internal standard in all the mixtures. The lipid mixtures were presented to the enzyme either in the form of SUV or LUV bilayers or in detergent micelles.
Effects of the Acyl Chain Length and Unsaturation on PC Hydrolysis in Micelles
We first studied the hydrolysis of the 25 PC species (PC-mix) incorporated in detergent micelles. In the case of fully saturated species, the rate of hydrolysis decreased with increasing chain length with some minor deviations from a monotonic behavior (Fig. 2). An analogous trend was observed for the dimonounsaturated species. An inhibitory effect of increasing chain length was observed for the other PC sets as well. Also, the number of double bonds had a strong effect on the rate of hydrolysis. This is particularly notable for the species with a total of 36, 38, or 40 acyl chain carbons. For example, 36:6 (18:3/18:3) species was hydrolyzed ∼12.3-fold faster than the 36:0 (18:0/18:0) species, and the 40:8 (20:4/20:4) species ∼14.7-fold faster than the 40:0 (20:0/20:0) species. We propose that the major positive effect of unsaturation in these cases is, at least in part, due to increased affinity of the substrate for the active site of the enzyme (cf. 30).
FIGURE 2.
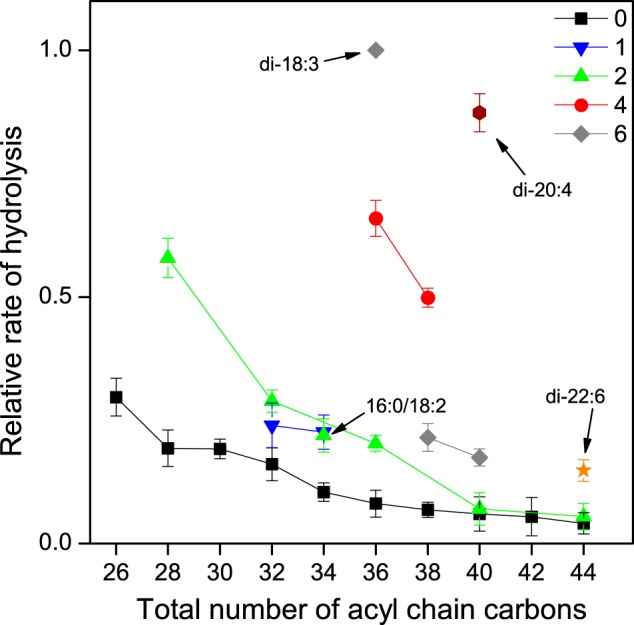
Hydrolysis of saturated and unsaturated PC species of varying chain length in Nonidet P-40 micelles. PC-mix, 16:0/18:1-PA, and 21:0-SM (250:25:25 nmol) were dispersed in 0.5 ml of the assay buffer containing 1% Nonidet P-40 and incubated with 8.0 μg of iPLAβ for 30 min at 37 °C. Samples of 50 μl were drawn at intervals, and the relative rates of hydrolysis were determined as indicated under “Experimental Procedures.” The relative rate of hydrolysis is plotted against the total number of acyl chain carbons for several series containing a fixed number of acyl chain double bonds. The sets with only a single member are indicated by an arrow. The specific activity for total PC hydrolysis was 18.2 nmol/μg protein. Data are mean ± S.D. of four independent experiments.
Effect of sn-1 Versus sn-2 Chain Length of PC on Hydrolysis in Micelles
To assess the contribution of the individual acyl chains on the rate of hydrolysis of iPLAβ, we employed two sets of saturated PC species in which the length of either the sn-1 (Cn/16:0-PCs) or the sn-2 (16:0/Cn-PCs) acyl chain varied from 6 to 24 carbons, while the length of the acyl chain at the other sn position was constant at 16 carbons. Because the Cn/16:0-PC species contained a d9-labeled choline headgroup, whereas the 16:0/Cn-PC species were unlabeled, both sets could be present together thus eliminating the possibility of any matrix bias. As shown in Fig. 3, variation of the sn-1 versus sn-2 chain length had distinct effects on the rate of hydrolysis. When the length of the sn-1 chain increased, the rate of hydrolysis was first essentially independent of the length until C12 and then decreased in an approximately logarithmic manner. The ratio of the fastest to the slowest rate was ∼14.5. In contrast, when the length of the sn-2 chain increased, the rate first increased from C7 to C8 and then declined in a rather monotonous manner. The fastest species was hydrolyzed ∼23.7 times more rapidly than the slowest one, a difference that is significantly higher than in the case of Cn/16:0-PC set. Most probably, the observed differences between the two sets of positional isomers are due to the different properties of the sn-1 versus sn-2 acyl-binding sites of iPLAβ.
FIGURE 3.
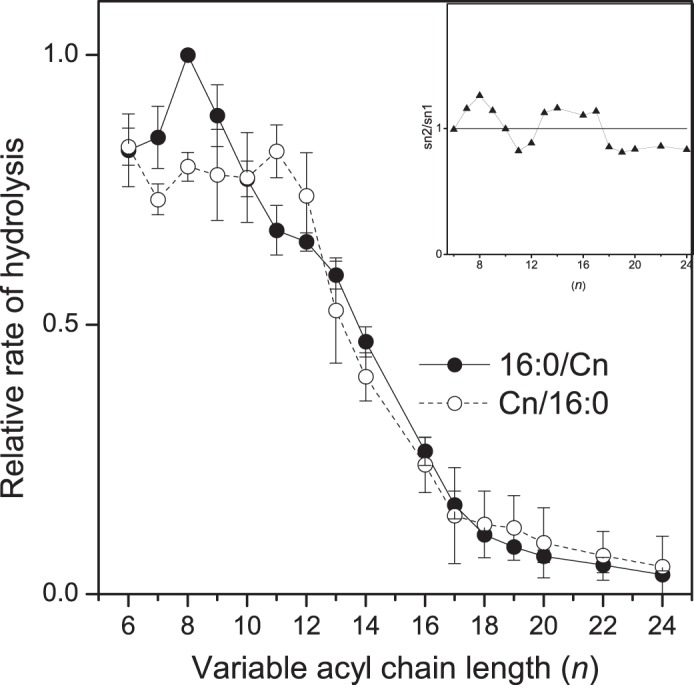
Hydrolysis of 16:0/Cn-PC and Cn/16:0-PC species versus the length of the variable acyl chain in Nonidet P-40 micelles. d9-16:0/Cn-PCs, Cn/16:0-PCs, 16:0/18:1-PA, and 21:0-SM (125/125/25/25 nmol) were dispersed in 0.5 ml of assay buffer containing 1% Nonidet P-40 and incubated with 9.5 μg of iPLAβ for 30 min at 37 °C. See legend of Fig. 2 for other details. Inset shows an isomer preference plot obtained by pairwise division of the rate of 16:0/Cn-PC hydrolysis by that of Cn/16:0-PC. Dotted line at y = 1.0 indicates the absence of any isomer preference. Pairwise Student's t test showed that the differences between isomer pairs was significant only when n was 8 (t value = 10.9496, two-tailed p value = 0.0016, n = 4, 95% confidence interval (CI)) or 11 carbons (t value = −3.0906, two-tailed p value = 0.0214, n = 4, 95% CI). The specific activity for total PC hydrolysis was 20.78 nmol/μg protein. Data are mean ± S.D. of four independent experiments.
Effects of the Acyl Chain Length and Unsaturation on PC Hydrolysis in SUV Bilayers
We next studied hydrolysis of the PC-mix using vesicle bilayers as the macrosubstrate. Independent of acyl chain unsaturation, the rate of hydrolysis decreased monotonously with increasing chain length (Fig. 4). Notably, however, the effect of chain length was far more pronounced than with micelles. For instance, the most rapidly hydrolyzed saturated species (13:0/13:0) was hydrolyzed ∼50-fold faster than the slowest one (22:0/22:0). In the case of the diunsaturated species, the fastest species (14:1/14:1) was hydrolyzed ∼20-fold faster than the slowest one (22:1/22:1). The corresponding differences in micelles were ∼7.2 and ∼10.6, respectively. Furthermore, the rate increased with an increasing number of double bonds for the species with an equal number of total acyl chain carbons. These data are consistent with the proposition that substrate efflux (hydrophobicity) is the key determinant in the hydrolysis of membrane-bound PC species by iPLAβ.
FIGURE 4.
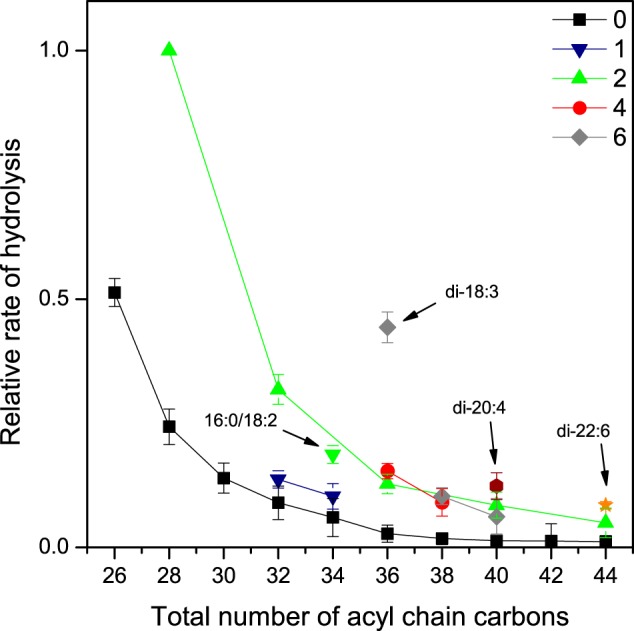
Hydrolysis of saturated and unsaturated PC species of varying chain length in SUV bilayers. Vesicles consisting of PC-mix, 16:0/18:1-PA, and 21:0-SM (250:25:25 nmol) were incubated with 10.5 μg of iPLAβ in 0.5 ml of assay buffer for 30 min at 37 °C. See legend of Fig. 2 for other details. Specific activity for total PC hydrolysis was 11.2 nmol/μg protein. Data are mean ± S.D. of four independent experiments.
Effect of sn-1 Versus sn-2 Chain Length of PC on Hydrolysis in SUV Bilayers
To study how the variation of the length of the sn-1 or sn-2 chains independently affect the rate of hydrolysis in bilayers, d9-16:0/Cn-PCs and Cn/16:0-PCs were incorporated together in SUVs that were then incubated with iPLAβ. As shown in Fig. 5, increase in the length of either the sn-1 or the sn-2 chain reduced the rate of hydrolysis dramatically, i.e. the difference between the fastest and the slowest species was ∼125-fold in both isomer sets. In contrast with micelles, no detectable differences in the rate of hydrolysis were observed for the positional isomers. Such a monotonous and acyl sn-position independent decrease of hydrolysis further supports the notion that efflux of the GPL substrate from the bilayer is the rate-limiting step in hydrolysis of membrane-bound substrates by iPLAβ.
FIGURE 5.
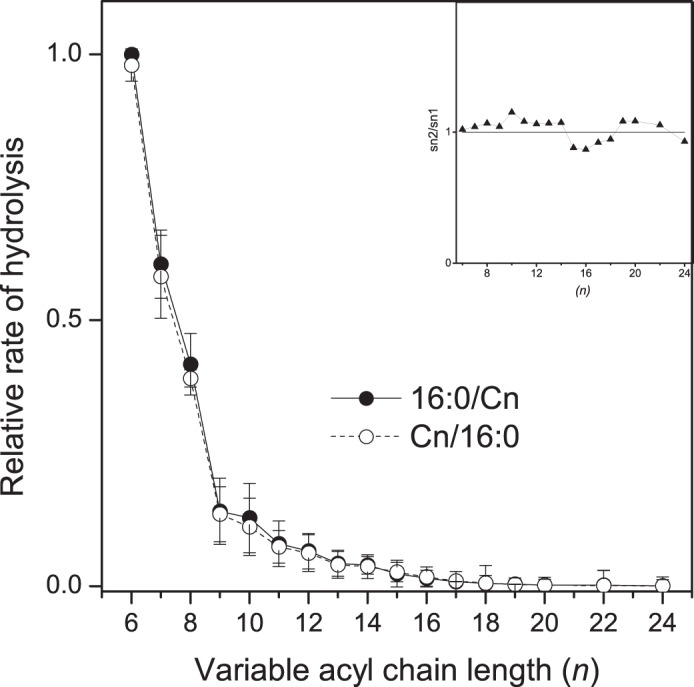
Hydrolysis of 16:0/Cn-PC and Cn/16:0-PC species versus the length of the variable acyl chain in SUV bilayers. Vesicles consisting of d9-16:0/Cn-PCs, Cn/16:0-PCs, 16:0/18:1-PA, and 21:0-SM (125:125:25:25 nmol) were incubated with 11.0 μg of iPLAβ in 0.5 ml assay buffer for 30 min at 37 °C. See legend of Fig. 2 for other details. Inset shows an isomer preference plot obtained as indicated in the legend of Fig. 3. Pairwise Student's t test showed that the apparent differences observed were statistically insignificant. The specific activity for total PC hydrolysis was 15.3 nmol/μg protein. Data are mean ± S.D. of four independent experiments.
Effect of GPL Polar Headgroup Structure on Hydrolysis
We next investigated the effect of the GPL polar head on the rate of hydrolysis by iPLAβ in micelles as well as SUV and LUV bilayers. Hydrolysis of six different 18:1/18:1 GPLs presented together on the enzyme was compared. With micelles, the best substrate was PE that was hydrolyzed ∼3-fold faster than PG and PA. The other GPLs were hydrolyzed at intermediate rates (Fig. 6A). In striking contrast, the GPL species were hydrolyzed similarly in SUV bilayers (Fig. 6B), although PG was hydrolyzed slightly faster and PI slightly slower than the rest. Very similar results were obtained with LUVs (Fig. 6C). These results are consistent with the notion that with bilayers substrate efflux is rate-limiting, but with micelles differences in steric and/or other interactions of the headgroup with the enzyme catalytic site play a more important role.
FIGURE 6.
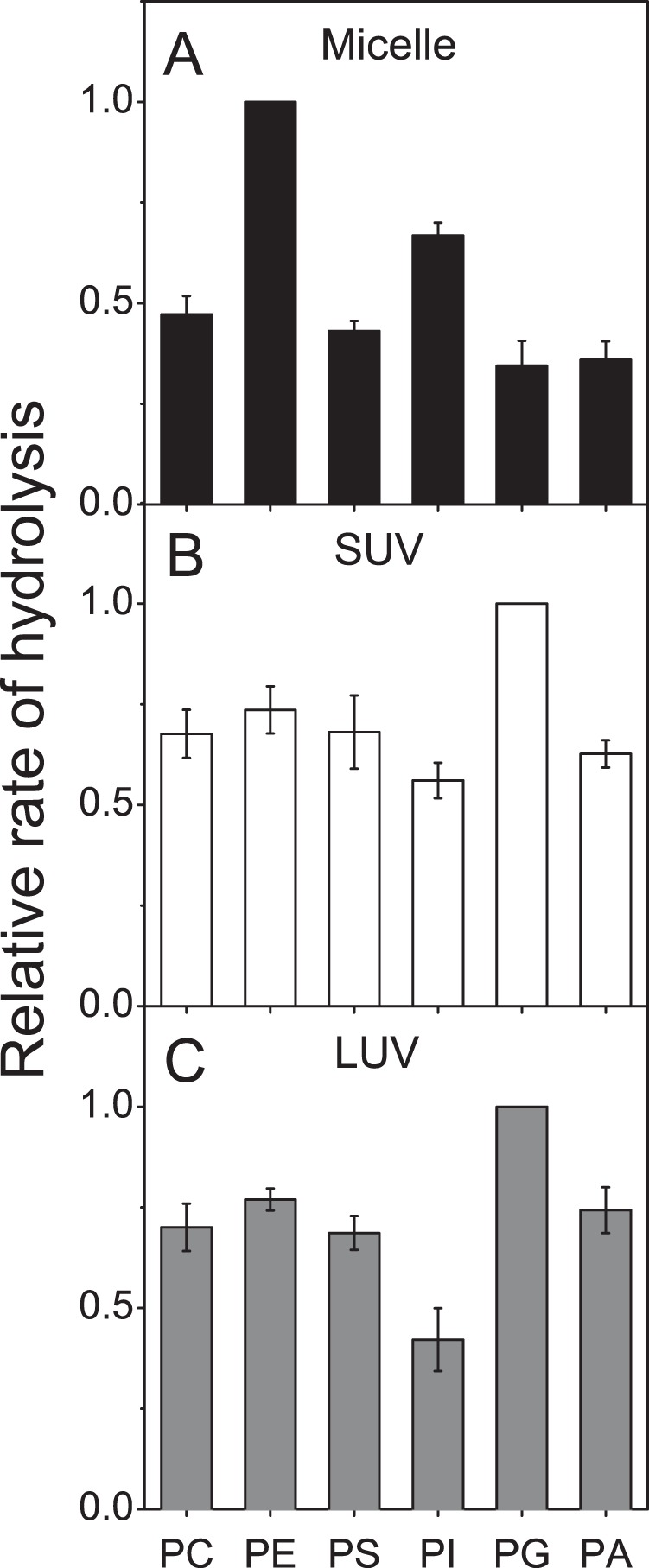
Effect of the headgroup on the hydrolysis of GPL in micelles, SUV, and LUV bilayers. GPLs, 18:1/18:1-PC, -PE, -PS, -PI, -PG, and -PA (250 nmol of total GPL) incorporated to micelles, SUVs, or LUVs were incubated with iPLAβ (9.0, 12.0, or 28.0 μg, respectively) in 0.5 ml of assay buffer for 30 min at 37 °C. Samples of 50 μl were drawn at intervals, and the relative rate of hydrolysis was determined as indicated under “Experimental Procedures.” The specific activity was 24.99, 18.74, and 8.04 nmol 18:1/18:1-PE hydrolyzed per μg protein in micelles, SUVs, and LUVs, respectively. The values have been normalized to that of the most rapidly hydrolyzed GPL in each system. Data are mean ± S.D. of four independent experiments.
Effect of the Headgroup on the Intervesicle Transfer Rate (Efflux)
Although it seemed likely that the efflux of GPLs from a bilayer is rather independent of the headgroup structure, we determined this experimentally by measuring the relative rates of spontaneous intervesicle transfer of different headgroup variants. As shown in Fig. 7, all six GPLs translocated at rather similar rates from the donor to acceptor vesicles. Because the intervesicle transfer rate can be equated with the rate of efflux (32), these data indicate the headgroup has only a modest effect on the rate of efflux from a bilayer. This result is again consistent with the idea that the rate of hydrolysis of a bilayer-bound GPL by iPLAβ (Fig. 6) mainly depends on its efflux propensity.
FIGURE 7.
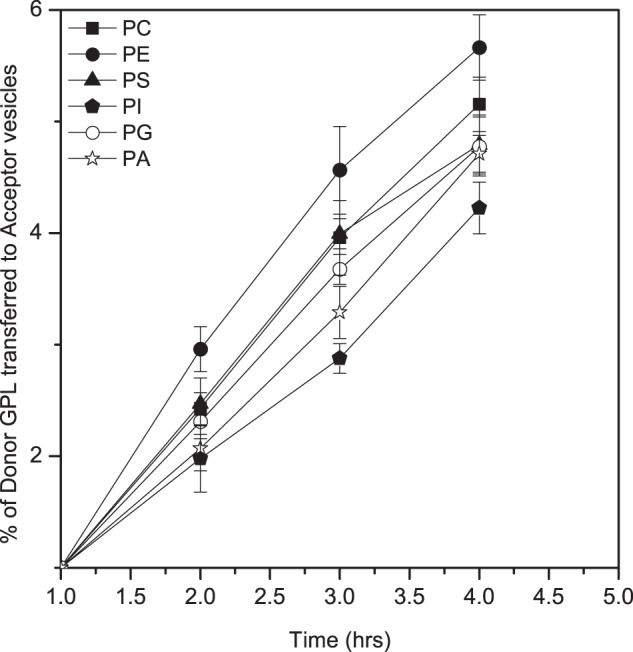
Spontaneous intervesicle transfer of GPLs with different polar headgroup. Donor vesicles (1335 nmol) consisting of 18:1/18:1-PC, -PE, -PS, -PG, -PA, and -PI in equal molar ratio, d9-labeled 22:1/22:1-PC (15 nmol), and tetra-18:1 cardiolipin (150 nmol) were mixed with a 5-fold excess of acceptor vesicles consisting of 16:0/18:1-PC, and the mixture was incubated at 37 °C. Samples were taken at intervals, and the amount of each 18:1/18:1 GPL transferred to the acceptor vesicles was determined as detailed under “Experimental Procedures.” The data are mean ± S.D. from four independent experiments.
Hydrolysis of Microsomal GPL Species in SUV Bilayers
To study the specificity of iPLAβ with a more natural macrosubstrate, we extracted lipids from HeLa cell microsomes and reconstituted them in SUV bilayers. Hydrolysis of 14 PC, 14 PE, 11 PS, and 9 PI molecular species could be reliably analyzed using LC-MS with SRM detection (see “Experimental Procedures”). As shown in Fig. 8A, an increase in total acyl chain length of PC systematically inhibited the hydrolysis independent of the number of double bonds. However, increasing the number of double bonds increased the rate of hydrolysis when the total acyl chain length was constant. Parallel results were observed for the PE, PS, and PI species (Fig. 8, B–D), i.e. increasing acyl chain length systematically inhibited hydrolysis, while increasing unsaturation increased it when the chain length was constant. In addition, the headgroup structure (among PC, PE, PS, and PI) had an insignificant effect on the rate of hydrolysis when the total acyl chain length and unsaturation were constant (data not shown). In conclusion, the data for multiple natural GPL species strongly support the proposition that iPLAβ-mediated hydrolysis of GPLs closely correlates with their efflux propensities.
FIGURE 8.
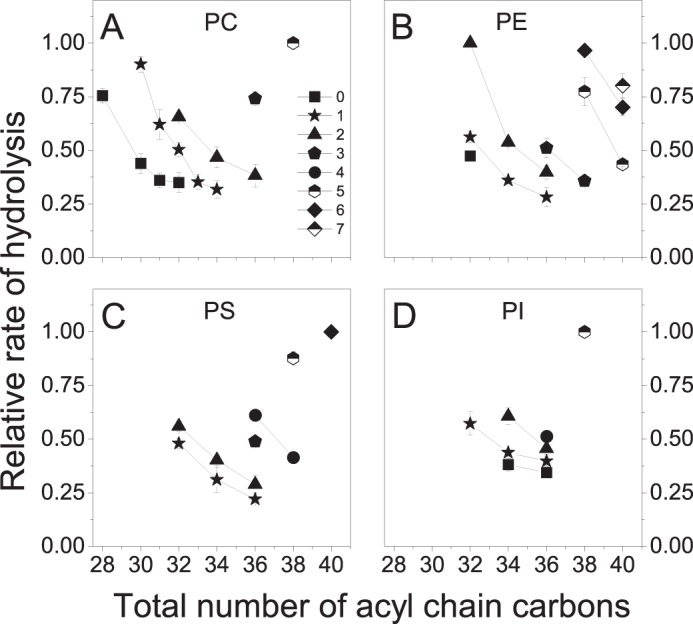
Hydrolysis of microsomal GPL species in SUV bilayers. Microsomal lipids (250 nmol of GPL) extracted from HeLa cells were reconstituted into SUV bilayers and incubated with 8.0 μg of iPLAβ in 0.5 ml of assay buffer for 30 min at 37 °C. The relative rates of hydrolysis were determined as described under “Experimental Procedures.” Data for the most significant PC, PE, PS, and PI species are shown in A–D, respectively. Legend of A indicates the total number of acyl chain double bonds in the GPL sets. The data are mean ± S.D. from four independent experiments.
DISCUSSION
In the case of soluble PLAs like iPLAβ, several steps have been proposed to be necessary for the hydrolysis of a membrane-bound GPL molecule: 1) binding of the enzyme to the membrane; 2) efflux (at least partially) of the substrate molecule from the bilayer; 3) accommodation of the substrate in the catalytic site of the enzyme; and 4) cleavage of the acyl-glycerol ester bond (40–42). However, when studying the substrate specificity of a PLA using the present approach, only steps 2 and 3 need to be considered because all GPL species are present simultaneously and are structural homologues, i.e. have an identical chemical structure at the site of cleavage. The requirement for substrate efflux derives from the (unproven) assumption that soluble PLAs may not penetrate the membrane significantly, and thus the catalytic site remains above the membrane surface during their action (40, 43). As indicated by recent hydrogen/deuterium exchange and homology modeling studies (44, 45), this may apply for iPLAβ as well. It was proposed that the substrate has to be extracted (i.e. efflux) from the membrane to be hydrolyzed by iPLAβ, but this could not be proven due to limitations of the methodology used (46).
Our previous study on three nonhomeostatic secretory PLAs, i.e. those from bee or snake venom and pig pancreas, strongly suggested that efflux of the substrate is the rate-determining step of hydrolysis of membrane-bound GPLs (27). This conclusion was based on the following: (i) the hydrolysis of homologous PC species in vesicles decreased strongly and monotonously with increasing length of the acyl chains; (ii) identical effect was observed when the length of the sn-1 versus sn-2 chain was varied, i.e. there was no discrimination between the sn positional isomers; and (iii) the rate of hydrolysis correlated closely with the rate of spontaneous intervesicle diffusion that depends on the rate of efflux of the PC species. Importantly, very different results were obtained with detergent micelles, i.e. the rate of hydrolysis decreased monotonously with the length of the sn-1 chain but not with that of the sn-2 chain. For the latter, an optimum length was observed, and the effects of the chain length and the number of double bonds varied markedly between the three enzymes, most probably reflecting differences in the structure of the catalytic site. Collectively, those data indicated that with micelles, specific interactions of the substrate with the substrate-binding site on the enzyme significantly contribute to the rate of hydrolysis, although with bilayers, efflux is the main rate-limiting factor. This difference between bilayers and micelles is most probably derived from different packing and intermolecular interactions in these aggregates. In micelles, the intermolecular packing is far more loose than in bilayers as shown for example by NMR studies (47) and MD simulations (48). Such loose intermolecular packing makes the rate of GPL efflux from micelles several orders of magnitude faster as compared with vesicle bilayers (49) and thus probably not rate-limiting in PLA-mediated hydrolysis. In contrast, tight intermolecular packing of the GPL molecules in bilayers strongly impedes their efflux, thus making it rate-limiting.
Here, we show that iPLAβ behaves similarly (but not identically) to the previously studied secretory PLAs that are structurally unrelated and nonhomeostatic (15, 16). Most importantly, the rate of hydrolysis of bilayer-bound PC species by iPLAβ decreased monotonously with the length of the acyl chains independent of the number of double bonds, and practically no hydrolysis of saturated species took place when the total number of acyl chain carbons exceeded 38 (Fig. 4). In contrast, increasing the number of acyl chain double bonds increased the rate of hydrolysis markedly. Because the substrate efflux rate decreases progressively with increasing acyl chain length and increases with the number of double bonds (see above and Ref. 50), these data strongly support the notion that efflux of the substrate from the bilayer is the rate-determining step in the hydrolysis of GPLs by iPLAβ. Consistently, increasing the length of the sn-1 or sn-2 chain of PC in bilayers inhibited the hydrolysis identically (Fig. 5), unlike in micelles where statistically significant differences were observed (Fig. 3). Also, the finding that the headgroup had a significant effect in micelles, but little if any in vesicles (Fig. 6), is consistent with the efflux being the rate-limiting step with bilayers, because efflux propensity is largely determined by the acyl chain length and unsaturation (51, 52).
Our previous study on three secretory PLAs showed significant differences in the effect of the sn-1 versus the sn-2 chain length on the rate of hydrolysis with micelles as the macrosubstrate, i.e. the rate of hydrolysis decreased monotonously with the length of the sn-1 chain, although for sn-2 chain an optimum at C8 to C10 was observed for each of the three enzymes. Such data suggest that there is a binding site for the sn-2 chain but not for the sn-1 chain in those enzymes and that this site interacts only with the first 8–10 carbons of the sn-2 chain (27). This conclusion is consistent with the fact that these PLAs hydrolyze the sn-2 ester bond but not the sn-1 (53, 54). For iPLAβ, the differences between the sn-1 and sn-2 chain were much smaller, albeit statistically significant (Fig. 3). This result is consistent with the fact that, in contrast to venom PLAs, iPLAβ is not specific for the sn-2 or sn-1 ester bond but hydrolyzes both (55, 56). For both the sn-1 and sn-2 chain, the rate started to drop strongly when the chain length exceeded 11–13 carbons, which indicates that only the corresponding C-terminal carbons in the acyl chains interact with the substrate-binding site of iPLAβ.
Previously, few studies have investigated the substrate specificity of iPLAβ using a limited set of GPL species (41, 57, 58). Although some differences between GPL species were observed, the data are difficult to interpret because of the following: (i) mixed GPL/detergent micelles were employed in those studies, and (ii) the apparent specificity profile of iPLAβ can vary dramatically depending on the detergent/GPL ratio used (59). In addition, the charge of the micelle surface can vary depending on the GPL present, which could bias the data because binding of PLAs to a macrosubstrate is sensitive to the surface charge (60, 61). The present results and previous studies (27, 62–65) clearly show that the properties of the macrosubstrate can have a remarkable effect on the apparent specificity of PLAs.
Could efflux propensity regulate GPL homeostasis? Several studies have indicated that lipids in multicomponent bilayers tend to adopt regular rather than random lateral distributions. This putative behavior, which is formulated in the so-called superlattice/regular distribution model (21, 66), is relevant to GPL homeostasis for two reasons. First, it predicts the existence of a number of “critical” GPL bilayer compositions that are energetically more favorable than the intervening compositions, and therefore, there is an intrinsic tendency for the composition to settle in one of the critical ones. Second, if the composition deviates from a critical one (e.g. due to synthesis of a certain GPL “in excess”), packing defects will appear as the molecules present in excess cannot be equally accommodated in the bilayer lattice (i.e. the optimal stoichiometry is perturbed). Those molecules in excess are predicted to have an increased chemical activity and therefore increased efflux propensity (fugacity), which would make them susceptible for hydrolysis by homeostatic PLAs, as shown here for iPLAβ. When molecules present in excess have been degraded, hydrolysis is predicted to slow down markedly. This hypothetical model is reminiscent of that proposed to regulate the concentration of cholesterol in the plasma membrane of eukaryotic cells via the formation of putative stoichiometric cholesterol-sphingomyelin complexes (67). However, in the present case “critical compositions” with particular stoichiometries, rather than complexes, are assumed.
Such a critical composition-based regulation would be highly accurate and would thus minimize compositional fluctuations (hysteresis) that are energy-wasting due to futile competition between synthesis and degradation (3). Supporting this scenario, it is well established that many PLAs are strongly activated by bilayer packing defects (68–70), and a (venom) PLA seems to respond to predicted critical compositions (71). Importantly, such critical compositions could also regulate GPL biosynthesis (and thus coordinate it with degradation), because the GPL molecules present in excess should have increased chemical activity and could thus potently inhibit the synthetic enzymes. Notably, there is good evidence for such product inhibition in case of e.g. PS synthases 1 and 2 by PS (25, 26) and PI synthase (72).
In conclusion, this study provides the first experimental evidence that the efflux of substrate from a bilayer is the rate-limiting factor in the hydrolysis of GPL molecules by iPLAβ, a putative homeostatic PLA. This finding is intriguing because it suggests, particularly when related to predictions of the superlattice model of lipid lateral distribution, an intriguing model as how iPLAβ or other homeostatic PLAs could recognize and hydrolyze the GPLs present in excess thus maintaining GPL homeostasis.
Acknowledgments
We thank Tarja Grundström for expert technical assistance, Satu Hänninen and Saawan Jha for their help with various issues, and other members of the group for useful comments on the manuscript.
This work was supported by research grants from Finnish Academy, Sigrid Juselius Foundation, and Magnus Ehnrooth Foundation (to P. S.).
M. Hermansson, S. M. Hänninen, K. Hokynar, and P. Somerharju, unpublished data.
- GPL
- glycerophospholipid
- PLA
- phospholipase
- SUV
- small unilamellar vesicle
- LUV
- large unilamellar vesicle
- LPC
- lysophosphatidylcholine
- iPLA
- Ca2+-independent phospholipase A
- SM
- sphingomyelin
- BEL
- bromoenol lactone
- 16:0/Cn-PC
- sn-1–16:0/ sn-2-Cn-phosphatidylcholine (n = 6–24)
- Cn/16:0-PC
- sn-1-Cn/sn-2–16:0-phosphatidylcholine (n = 6–24)
- PS
- phosphatidylserine
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- PI
- phosphatidylinositol
- PA
- phosphatidic acid
- PG
- phosphatidylglycerol
- SRM
- selective reaction monitoring.
REFERENCES
- 1. van Meer G., Voelker D. R., Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vance D. E., Vance J. E. (2009) Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J. Lipid Res. 50, S132–S137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hermansson M., Hokynar K., Somerharju P. (2011) Mechanisms of glycerophospholipid homeostasis in mammalian cells. Prog. Lipid Res. 50, 240–257 [DOI] [PubMed] [Google Scholar]
- 4. Baburina I., Jackowski S. (1999) Cellular responses to excess phospholipid. J. Biol. Chem. 274, 9400–9408 [DOI] [PubMed] [Google Scholar]
- 5. Barbour S. E., Kapur A., Deal C. L. (1999) Regulation of phosphatidylcholine homeostasis by calcium-independent phospholipase A2. Biochim. Biophys. Acta 1439, 77–88 [DOI] [PubMed] [Google Scholar]
- 6. Ghomashchi F., Naika G. S., Bollinger J. G., Aloulou A., Lehr M., Leslie C. C., Gelb M. H. (2010) Interfacial kinetic and binding properties of mammalian group IVB phospholipase A2 (cPLA2β) and comparison with the other cPLA2 isoforms. J. Biol. Chem. 285, 36100–36111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Tienhoven M., Atkins J., Li Y., Glynn P. (2002) Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J. Biol. Chem. 277, 20942–20948 [DOI] [PubMed] [Google Scholar]
- 8. Murakami M., Masuda S., Ueda-Semmyo K., Yoda E., Kuwata H., Takanezawa Y., Aoki J., Arai H., Sumimoto H., Ishikawa Y., Ishii T., Nakatani Y., Kudo I. (2005) Group VIB Ca2+-independent phospholipase A2γ promotes cellular membrane hydrolysis and prostaglandin production in a manner distinct from other intracellular phospholipases A2. J. Biol. Chem. 280, 14028–14041 [DOI] [PubMed] [Google Scholar]
- 9. Winkler H., Smith A. D., Dubois F., van den Bosch H. (1967) The positional specificity of lysosomal phospholipase A activities. Biochem. J. 105, 38C–40C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walkey C. J., Kalmar G. B., Cornell R. B. (1994) Overexpression of rat liver CTP:phosphocholine cytidylyltransferase accelerates phosphatidylcholine synthesis and degradation. J. Biol. Chem. 269, 5742–5749 [PubMed] [Google Scholar]
- 11. Lykidis A., Wang J., Karim M. A., Jackowski S. (2001) Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway. J. Biol. Chem. 276, 2174–2179 [DOI] [PubMed] [Google Scholar]
- 12. Stone S. J., Cui Z., Vance J. E. (1998) Cloning and expression of mouse liver phosphatidylserine synthase-1 cDNA. Overexpression in rat hepatoma cells inhibits the CDP-ethanolamine pathway for phosphatidylethanolamine biosynthesis. J. Biol. Chem. 273, 7293–7302 [DOI] [PubMed] [Google Scholar]
- 13. Polokoff M. A., Wing D. C., Raetz C. R. (1981) Isolation of somatic cell mutants defective in the biosynthesis of phosphatidylethanolamine. J. Biol. Chem. 256, 7687–7690 [PubMed] [Google Scholar]
- 14. Nishijima M., Kuge O., Maeda M., Nakano A., Akamatsu Y. (1984) Regulation of phosphatidylcholine metabolism in mammalian cells. Isolation and characterization of a Chinese hamster ovary cell pleiotropic mutant defective in both choline kinase and choline-exchange reaction activities. J. Biol. Chem. 259, 7101–7108 [PubMed] [Google Scholar]
- 15. Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. (2011) Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog. Lipid Res. 50, 152–192 [DOI] [PubMed] [Google Scholar]
- 16. Burke J. E., Dennis E. A. (2009) Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 23, 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Balsinde J., Balboa M. A. (2005) Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell. Signal. 17, 1052–1062 [DOI] [PubMed] [Google Scholar]
- 18. Chiu C. H., Jackowski S. (2001) Role of calcium-independent phospholipases (iPLA(2)) in phosphatidylcholine metabolism. Biochem. Biophys. Res. Commun. 287, 600–606 [DOI] [PubMed] [Google Scholar]
- 19. Manguikian A. D., Barbour S. E. (2004) Cell cycle dependence of group VIA calcium-independent phospholipase A2 activity. J. Biol. Chem. 279, 52881–52892 [DOI] [PubMed] [Google Scholar]
- 20. Glynn P. (2005) Neuropathy target esterase and phospholipid deacylation. Biochim. Biophys. Acta 1736, 87–93 [DOI] [PubMed] [Google Scholar]
- 21. Somerharju P., Virtanen J. A., Cheng K. H., Hermansson M. (2009) The superlattice model of lateral organization of membranes and its implications on membrane lipid homeostasis. Biochim. Biophys. Acta 1788, 12–23 [DOI] [PubMed] [Google Scholar]
- 22. Ben-Tekaya H., Kahn R. A., Hauri H. P. (2010) ADP ribosylation factors 1 and 4 and group VIA phospholipase A(2) regulate morphology and intraorganellar traffic in the endoplasmic reticulum-Golgi intermediate compartment. Mol. Biol. Cell 21, 4130–4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davies S. M., Epand R. M., Kraayenhof R., Cornell R. B. (2001) Regulation of CTP:phosphocholine cytidylyltransferase activity by the physical properties of lipid membranes: an important role for stored curvature strain energy. Biochemistry 40, 10522–10531 [DOI] [PubMed] [Google Scholar]
- 24. Fagone P., Jackowski S. (2013) Phosphatidylcholine and the CDP-choline cycle. Biochim. Biophys. Acta 1831, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuge O., Hasegawa K., Saito K., Nishijima M. (1998) Control of phosphatidylserine biosynthesis through phosphatidylserine-mediated inhibition of phosphatidylserine synthase I in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U.S.A. 95, 4199–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuge O., Saito K., Nishijima M. (1999) Control of phosphatidylserine synthase II activity in Chinese hamster ovary cells. J. Biol. Chem. 274, 23844–23849 [DOI] [PubMed] [Google Scholar]
- 27. Haimi P., Hermansson M., Batchu K. C., Virtanen J. A., Somerharju P. (2010) Substrate efflux propensity plays a key role in the specificity of secretory A-type phospholipases. J. Biol. Chem. 285, 751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel K. M., Morrisett J. D., Sparrow J. T. (1979) A convenient synthesis of phosphatidylcholines: acylation of sn-glycero-3-phosphocholine with fatty acid anhydride and 4-pyrrolidinopyridine. J. Lipid Res. 20, 674–677 [PubMed] [Google Scholar]
- 29. Bartlett E. M., Lewis D. H. (1970) Spectrophotometric determination of phosphate esters in the presence and absence of orthophosphate. Anal. Biochem. 36, 159–167 [DOI] [PubMed] [Google Scholar]
- 30. Kainu V., Hermansson M., Somerharju P. (2008) Electrospray ionization mass spectrometry and exogenous heavy isotope-labeled lipid species provide detailed information on aminophospholipid acyl chain remodeling. J. Biol. Chem. 283, 3676–3687 [DOI] [PubMed] [Google Scholar]
- 31. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 32. Jones J. D., Thompson T. E. (1990) Mechanism of spontaneous, concentration-dependent phospholipid transfer between bilayers. Biochemistry 29, 1593–1600 [DOI] [PubMed] [Google Scholar]
- 33. Hermansson M., Uphoff A., Käkelä R., Somerharju P. (2005) Automated quantitative analysis of complex lipidomes by liquid chromatography/mass spectrometry. Anal. Chem. 77, 2166–2175 [DOI] [PubMed] [Google Scholar]
- 34. Heikinheimo L., Somerharju P. (2002) Translocation of phosphatidylthreonine and -serine to mitochondria diminishes exponentially with increasing molecular hydrophobicity. Traffic 3, 367–377 [DOI] [PubMed] [Google Scholar]
- 35. Haimi P., Uphoff A., Hermansson M., Somerharju P. (2006) Software tools for analysis of mass spectrometric lipidome data. Anal. Chem. 78, 8324–8331 [DOI] [PubMed] [Google Scholar]
- 36. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 37. Storrie B., Madden E. A. (1990) Isolation of subcellular organelles. Methods Enzymol. 182, 203–225 [DOI] [PubMed] [Google Scholar]
- 38. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 39. Fazekas de St Groth S., Webster R. G., Datyner A. (1963) Two new staining procedures for quantitative estimation of proteins on electrophoretic strips. Biochim. Biophys. Acta 71, 377–391 [DOI] [PubMed] [Google Scholar]
- 40. Gelb M. H., Jain M. K., Hanel A. M., Berg O. G. (1995) Interfacial enzymology of glycerolipid hydrolases: lessons from secreted phospholipases A2. Annu. Rev. Biochem. 64, 653–688 [DOI] [PubMed] [Google Scholar]
- 41. Gelb M. H., Min J. H., Jain M. K. (2000) Do membrane-bound enzymes access their substrates from the membrane or aqueous phase: interfacial versus non-interfacial enzymes. Biochim. Biophys. Acta 1488, 20–27 [DOI] [PubMed] [Google Scholar]
- 42. Cao J., Burke J. E., Dennis E. A. (2013) Using hydrogen/deuterium exchange mass spectrometry to define the specific interactions of the phospholipase A2 superfamily with lipid substrates, inhibitors, and membranes. J. Biol. Chem. 288, 1806–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lio Y. C., Dennis E. A. (1998) Interfacial activation, lysophospholipase and transacylase activity of group VI Ca2+-independent phospholipase A2. Biochim. Biophys. Acta 1392, 320–332 [DOI] [PubMed] [Google Scholar]
- 44. Hsu Y. H., Burke J. E., Li S., Woods V. L., Jr., Dennis E. A. (2009) Localizing the membrane binding region of Group VIA Ca2+-independent phospholipase A2 using peptide amide hydrogen/deuterium exchange mass spectrometry. J. Biol. Chem. 284, 23652–23661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hsu Y. H., Bucher D., Cao J., Li S., Yang S. W., Kokotos G., Woods V. L., Jr., McCammon J. A., Dennis E. A. (2013) Fluoroketone inhibition of Ca(2+)-independent phospholipase A2 through binding pocket association defined by hydrogen/deuterium exchange and molecular dynamics. J. Am. Chem. Soc. 135, 1330–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bucher D., Hsu Y. H., Mouchlis V. D., Dennis E. A., McCammon J. A. (2013) Insertion of the Ca(2+)-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput. Biol. 9, e1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wenk M. R., Alt T., Seelig A., Seelig J. (1997) Octyl-β-d-glucopyranoside partitioning into lipid bilayers: thermodynamics of binding and structural changes of the bilayer. Biophys. J. 72, 1719–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacCallum J. L., Tieleman D. P. (2006) Computer simulation of the distribution of hexane in a lipid bilayer: spatially resolved free energy, entropy, and enthalpy profiles. J. Am. Chem. Soc. 128, 125–130 [DOI] [PubMed] [Google Scholar]
- 49. Nichols J. W. (1988) Phospholipid transfer between phosphatidylcholine-taurocholate mixed micelles. Biochemistry 27, 3925–3931 [DOI] [PubMed] [Google Scholar]
- 50. Silvius J. R., Leventis R. (1993) Spontaneous interbilayer transfer of phospholipids: dependence on acyl chain composition. Biochemistry 32, 13318–13326 [DOI] [PubMed] [Google Scholar]
- 51. Gardam M. A., Itovitch J. J., Silvius J. R. (1989) Partitioning of exchangeable fluorescent phospholipids and sphingolipids between different lipid bilayer environments. Biochemistry 28, 884–893 [DOI] [PubMed] [Google Scholar]
- 52. Massey J. B., Hickson-Bick D., Via D. P., Gotto A. M., Jr., Pownall H. J. (1985) Fluorescence assay of the specificity of human plasma and bovine liver phospholipid transfer proteins. Biochim. Biophys. Acta 835, 124–131 [DOI] [PubMed] [Google Scholar]
- 53. Scott D. L., Otwinowski Z., Gelb M. H., Sigler P. B. (1990) Crystal structure of bee-venom phospholipase A2 in a complex with a transition-state analogue. Science 250, 1563–1566 [DOI] [PubMed] [Google Scholar]
- 54. Plesniak L. A., Yu L., Dennis E. A. (1995) Conformation of micellar phospholipid bound to the active site of phospholipase A2. Biochemistry 34, 4943–4951 [DOI] [PubMed] [Google Scholar]
- 55. Ackermann E. J., Kempner E. S., Dennis E. A. (1994) Ca2+-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J. Biol. Chem. 269, 9227–9233 [PubMed] [Google Scholar]
- 56. Wolf M. J., Gross R. W. (1996) Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J. Biol. Chem. 271, 30879–30885 [DOI] [PubMed] [Google Scholar]
- 57. Tang J., Kriz R. W., Wolfman N., Shaffer M., Seehra J., Jones S. S. (1997) A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 272, 8567–8575 [DOI] [PubMed] [Google Scholar]
- 58. Yang H. C., Mosior M., Ni B., Dennis E. A. (1999) Regional distribution, ontogeny, purification, and characterization of the Ca2+-independent phospholipase A2 from rat brain. J. Neurochem. 73, 1278–1287 [DOI] [PubMed] [Google Scholar]
- 59. Bayburt T., Yu B. Z., Lin H. K., Browning J., Jain M. K., Gelb M. H. (1993) Human nonpancreatic secreted phospholipase A2: interfacial parameters, substrate specificities, and competitive inhibitors. Biochemistry 32, 573–582 [DOI] [PubMed] [Google Scholar]
- 60. Gabriel N. E., Agman N. V., Roberts M. F. (1987) Enzymatic hydrolysis of short-chain lecithin/long-chain phospholipid unilamellar vesicles: sensitivity of phospholipases to matrix phase state. Biochemistry 26, 7409–7418 [DOI] [PubMed] [Google Scholar]
- 61. Robinson M., Waite M. (1983) Physical-chemical requirements for the catalysis of substrates by lysosomal phospholipase A1. J. Biol. Chem. 258, 14371–14378 [PubMed] [Google Scholar]
- 62. Pete M. J., Exton J. H. (1995) Phospholipid interactions affect substrate hydrolysis by bovine brain phospholipase A1. Biochim. Biophys. Acta 1256, 367–373 [DOI] [PubMed] [Google Scholar]
- 63. Lin Q., Higgs H. N., Glomset J. A. (2000) Membrane lipids have multiple effects on interfacial catalysis by a phosphatidic acid-preferring phospholipase A1 from bovine testis. Biochemistry 39, 9335–9344 [DOI] [PubMed] [Google Scholar]
- 64. Thuren T., Sisson P., Waite M. (1990) Hydrolysis of lipid mixtures by rat hepatic lipase. Biochim. Biophys. Acta 1046, 178–184 [DOI] [PubMed] [Google Scholar]
- 65. Chong P. L., Zhu W., Venegas B. (2009) On the lateral structure of model membranes containing cholesterol. Biochim. Biophys. Acta 1788, 2–11 [DOI] [PubMed] [Google Scholar]
- 66. Lange Y., Steck T. L. (2008) Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 47, 319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Op den Kamp J. A., Kauerz M. T., van Deenen L. L. (1975) Action of pancreatic phospholipase A2 on phosphatidylcholine bilayers in different physical states. Biochim. Biophys. Acta 406, 169–177 [DOI] [PubMed] [Google Scholar]
- 68. Burack W. R., Biltonen R. L. (1994) Lipid bilayer heterogeneities and modulation of phospholipase A2 activity. Chem. Phys. Lipids 73, 209–222 [DOI] [PubMed] [Google Scholar]
- 69. Huang H. W., Goldberg E. M., Zidovetzki R. (1996) Ceramide induces structural defects into phosphatidylcholine bilayers and activates phospholipase A2. Biochem. Biophys. Res. Commun. 220, 834–838 [DOI] [PubMed] [Google Scholar]
- 70. Liu F., Chong P. L. (1999) Evidence for a regulatory role of cholesterol superlattices in the hydrolytic activity of secretory phospholipase A2 in lipid membranes. Biochemistry 38, 3867–3873 [DOI] [PubMed] [Google Scholar]
- 71. Imai A., Gershengorn M. C. (1987) Regulation by phosphatidylinositol of rat pituitary plasma membrane and endoplasmic reticulum phosphatidylinositol synthase activities. A mechanism for activation of phosphoinositide resynthesis during cell stimulation. J. Biol. Chem. 262, 6457–6459 [PubMed] [Google Scholar]
- 72. Jenkins C. M., Han X., Mancuso D. J., Gross R. W. (2002) Identification of calcium-independent phospholipase A2 (iPLA2) β, and not iPLA2γ, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J. Biol. Chem. 277, 32807–32814 [DOI] [PubMed] [Google Scholar]