

Background: HCV uses glycan shielding to mask epitopes recognized by neutralizing antibodies.
Results: The structure of a human antibody bound to an HCV E2 epitope revealed how it penetrates a shield created by glycosylation shifting.
Conclusion: Antibody binding induces an epitope conformation that accommodates multiple glycans.
Significance: The structure provides a template for engineering E2 to elicit protective antibodies able to overcome glycosylation shifting.
Keywords: Antibody, Crystal Structure, Glycosylation, Hepatitis C Virus (HCV), Vaccine, Neutralization
Abstract
Hepatitis C virus (HCV) is a major cause of liver cirrhosis and hepatocellular carcinoma. A challenge for HCV vaccine development is to identify conserved epitopes able to elicit protective antibodies against this highly diverse virus. Glycan shielding is a mechanism by which HCV masks such epitopes on its E2 envelope glycoprotein. Antibodies to the E2 region comprising residues 412–423 (E2412–423) have broadly neutralizing activities. However, an adaptive mutation in this linear epitope, N417S, is associated with a glycosylation shift from Asn-417 to Asn-415 that enables HCV to escape neutralization by mAbs such as HCV1 and AP33. By contrast, the human mAb HC33.1 can neutralize virus bearing the N417S mutation. To understand how HC33.1 penetrates the glycan shield created by the glycosylation shift to Asn-415, we determined the structure of this broadly neutralizing mAb in complex with its E2412–423 epitope to 2.0 Å resolution. The conformation of E2412–423 bound to HC33.1 is distinct from the β-hairpin conformation of this peptide bound to HCV1 or AP33, because of disruption of the β-hairpin through interactions with the unusually long complementarity-determining region 3 of the HC33.1 heavy chain. Whereas Asn-415 is buried by HCV1 and AP33, it is solvent-exposed in the HC33.1-E2412–423 complex, such that glycosylation of Asn-415 would not prevent antibody binding. Furthermore, our results highlight the structural flexibility of the E2412–423 epitope, which may serve as an immune evasion strategy to impede induction of antibodies targeting this site by reducing its antigenicity.
Introduction
Hepatitis C virus (HCV),2 a member of the Flaviviridae family of positive-stranded RNA viruses, infects at least 2% of the world population and is a major cause of liver cirrhosis, liver failure, and hepatocellular carcinoma (1, 2). The global burden is estimated at 170 million infected individuals with an annual increase of 3–4 million new infections (3). The recent introduction of successful HCV-specific direct-acting antiviral agents represents a major advance in HCV therapy (4). However, the high cost of direct-acting antiviral agents precludes their availability to the large majority of HCV-infected individuals living in developing countries. In addition, health care workers with occupational risk for blood-borne pathogens and injection drug users will remain at risk for repeated exposure to HCV, even after successful direct-acting antiviral agents treatment. Thus, there is a compelling need for a preventive HCV vaccine.
The key challenge in HCV vaccine development is to overcome the high diversity of this virus and its potential to escape from host immune responses. HCV is composed of a nucleocapsid core enveloped by a lipid bilayer in which two surface glycoproteins, E1 and E2, are anchored. Entry of HCV into hepatocytes is mediated by interactions between the E1E2 heterodimer and at least four cellular receptors: the tetraspanin CD81 (5), scavenger receptor class B type 1 (6), and the tight junction proteins occludin (7) and claudin 1 (8). An effective vaccine must include conserved epitopes of E1E2 that are able to elicit broadly neutralizing antibodies. Accordingly, much effort has been focused on the identification of conserved regions of E1E2 mediating virus neutralization through the isolation and characterization of mAbs from HCV-infected individuals and experimentally immunized mice (9). The large majority of these mAbs recognize E2, which is more immunogenic than E1. In addition, E2 contains binding sites for CD81 and scavenger receptor class B type 1 (5–8).
Immunogenic determinants on the E2 glycoprotein are roughly segregated into the hypervariable region 1 (10–13) and at least five clusters of overlapping linear and nonlinear epitopes, designated antigenic domains A–E (14–17). Hypervariable region 1 is a major decoy that elicits isolate-specific neutralizing antibodies from which the virus can readily escape (11–13). Antigenic domain A is associated with non-neutralizing antibodies and constitutes another major decoy (15, 16, 18). By contrast, some antigenic domain B epitopes and most D and E epitopes elicit antibodies that are broadly neutralizing among the major HCV genotypes and subtypes (15–17). Antibodies to domain C epitopes neutralize HCV with more restricted genotype and subtype profiles (19).
Antigenic domain E is composed of overlapping linear epitopes located within amino acids 412–423 (QLINTNGSWHIN) of E2 (E2412–423) (20, 21). This region is involved in HCV binding to the CD81 entry receptor (22, 23), which likely explains why it is so highly conserved among >5,500 E2 sequences in the GenBankTM database. Accordingly, antibodies to E2412–423 hold considerable promise for vaccine development. Broadly neutralizing mAbs targeting this region have been isolated from immunized mice (22, 24–27) and HCV-infected individuals (14, 28). These antibodies, which block the interaction of E2 with CD81, include the rodent mAbs AP33 (22) and 3/11 (26, 29) and human mAbs HCV1 (24) and HC33.1 (14, 28).
The extracellular domains of both E1 and E2 are heavily glycosylated, with up to 5 sites on E1 and up to 11 sites on E2 modified by N-linked glycosylation (30). Some of these glycans are critical for protein folding and/or HCV infectivity (31). In addition, a number of studies have highlighted the role of N-linked glycosylation in shielding E2 epitopes from recognition by broadly neutralizing antibodies (31–33), as observed for HIV and influenza virus (34). In a particularly revealing recent example, deep sequencing analysis of HCV resistance to neutralization by mAbs HCV1 and AP33 identified mutations at asparagine 417 of E2 (N417S and N417T), an N-glycosylation site within the E2412–423 epitope recognized by these mAbs, as responsible for virus escape (35). Comparison of the glycosylation status of E2 containing these resistance mutations revealed a glycosylation shift from Asn-417 to Asn-415 in the N417S and N417T E2 proteins. Crystal structures of the wild-type E2412–423 epitope bound to HCV1 and AP33 showed that, whereas the side chain of Asn-417 points away from the antibody, Asn-415 is buried within the peptide-antibody interface (35–38). Consequently, attachment of a glycan at Asn-415, but not at Asn-417, would create major steric clashes with HCV1 and AP33, resulting in abrogation of antibody binding and HCV escape, as observed in resistance selection studies (29, 35, 39). Notably, liver transplantation patients with HCV who were treated with HCV1 showed rebounds in viral load at various times following transplantation (39). These rebounds were consistently associated with mutations in the E2412–423 epitope known to confer resistance to HCV1, in particular N417S.
In sharp contrast to mAbs HCV1 and AP33, HC33.1, which was isolated from an HCV-infected blood donor, can neutralize virus bearing the E2 N417S and N417T adaptive mutations with the glycosylation shift to Asn-415 (14, 28). Moreover, infectious HCV virions containing the N417S mutation displayed increased sensitivity to neutralization by HC33.1 and related antibodies (28), suggesting that the glycosylation shift actually enhanced binding. To understand how HC33.1 is able to penetrate the glycan shield of HCV created by this shift, we determined the crystal structure of this broadly neutralizing human mAb in complex with its E2412–423 epitope.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The HC33.1 antibody was expressed as a single-chain Fv fragment (scFv) by in vitro folding from inclusion bodies produced in Escherichia coli. The scFv construct consisted of the heavy chain variable (VH) region (residues Glu-1–Ser-127) connected to the light chain variable (VL) region (residues Gln-1–Leu-110) by an 18-residue linker (GSTGGGGSGGGGSGGGGS). The HC33.1 scFv was cloned into the expression vector pET-26b (Novagen) and expressed as inclusion bodies in BL21(DE3) E. coli cells (Novagen). Bacteria were grown at 37 °C in LB medium to an absorbance of 0.6–0.8 at 600 nm and induced with 1 mm isopropyl-β-d-thiogalactoside. After incubation for 3 h, the bacteria were harvested by centrifugation and resuspended in 50 mm Tris-HCl (pH 8.0) containing 0.1 m NaCl and 2 mm EDTA; cells were disrupted by sonication. Inclusion bodies were washed extensively with 50 mm Tris-HCl (pH 8.0) and 2% (v/v) Triton X-100 and then dissolved in 8 m urea, 50 mm Tris-HCl (pH 8.0), and 10 mm DTT. For in vitro folding, inclusion bodies were diluted into ice-cold folding buffer containing 1 m l-arginine HCl, 50 mm Tris-HCl (pH 8.0), 1 mm EDTA, 3 mm reduced glutathione, and 0.9 mm oxidized glutathione, to a final protein concentration of 60 mg/liter. After 72 h at 4 °C, the folding mixture was concentrated 50-fold, dialyzed against 50 mm MES (pH 6.0), and centrifuged to remove aggregates. Correctly folded HC33.1 scFv was then purified using sequential Superdex 75 HR and MonoQ columns (GE Healthcare).
Crystallization and Data Collection
For crystallization of the HC33.1-E2412–423 complex, HC33.1 scFv (10 mg/ml) was mixed with E2412–423 peptide (GenScript) in a 1:5 molar ratio. The complex crystallized in sitting drops at room temperature in 20% (w/v) PEG 8000, 3% (v/v) 2-methyl-2,4-pentanediol, and 0.1 m imidazole (pH 6.5). For data collection, crystals were transferred to a cryoprotectant solution of mother liquor containing 20% (v/v) glycerol, prior to flash cooling in a nitrogen stream. X-ray diffraction data were collected in-house at 100 K using a Rigaku R-AXIS IV++ area detector. The HC33.1-E2412–423 crystal belongs to space group P212121 with one complex molecule per asymmetric unit. Diffraction data were indexed, integrated, and scaled with the program CrystalClear (Rigaku). The data set is 94.9% complete to 2.0 Å resolution with Rmerge = 7.9%. Data collection statistics are shown in Table 1.
TABLE 1.
Data collection and structure refinement statistics
| HC33.1–E2412–423 | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Unit cell (Å) | a = 36.1, b = 53.7, c = 107.2 |
| Resolution (Å)a | 50–1.99 (2.06–1.99) |
| Observations | 132,497 |
| Unique reflections | 14,203 |
| Completeness (%)a | 94.9 (88.9) |
| Mean I/σ(I)a | 13.3 (4.3) |
| Rsym (%)a,b | 7.9 (38.9) |
| Refinement | |
| Resolution range (Å) | 50–2.0 |
| Rwork (%)c | 22.3 |
| Rfree (%)c | 27.2 |
| Protein atoms | 1,962 |
| Water atoms | 80 |
| RMSDs from ideality | |
| Bond lengths (Å) | 0.010 |
| Bond angles (o) | 1.61 |
| Ramachandran statistics (%) | |
| Favorable | 86.5 |
| Additional | 13.0 |
| Generous | 0.0 |
| Forbidden | 0.5 |
a The values in parentheses are statistics for the highest resolution shell.
b Rsym = Σ|Ij − <I>|/ΣIj, where Ij is the intensity of an individual reflection, and <I> is the average intensity of that reflection.
c Rwork = Σ||Fo| − |Fc||/Σ|Fo|, where Fc is the calculated structure factor. Rfree is as for Rwork but calculated for a randomly selected 5.0% of reflections not included in the refinement.
Structure Determination and Refinement
The structure of the HC33.1-E2412–423 complex was solved by molecular replacement with Phaser (40) using an ensemble search model constructed by combining aligned structures of the anti-severe acute respiratory syndrome spike protein scFv 80R (Protein Data Bank accession code 2GHW) (41), the anti-gankyrin scFv F5 (Protein Data Bank accession code 4NIK) (42), and the anti-influenza hemagglutinin scFv F10 (Protein Data Bank accession code 3FKU) (43). After model building for HC33.1 scFv was almost complete, extra density corresponding to the E2412–423 peptide was very clear, and peptide residues were built manually without any ambiguity. All refinements were carried out using CNS1.1 (44), including iterative cycles of simulated annealing, positional refinement, and B factor refinement, interspersed with model rebuilding into σA-weighted Fo − Fc and 2Fo − Fc electron density maps using XtalView (45). Refinement statistics are summarized in Table 1. Stereochemical parameters were evaluated with PROCHECK (46).
Modeling of Epitope Mutants and Glycans
Rosetta version 2.3 was used to perform a computational alanine scan and escape mutant simulations of the E2412–423 epitope using the interface mutagenesis module (47), removing the N-terminal arginine residue from the HC33.1-E2412–423 complex structure prior to modeling to avoid any influence of nonepitope residues on results. Extra side chain rotamers were included to ensure sufficient sampling (-ex1 -ex2 -ex3), and a separate simulation for non-alanine mutants was performed incorporating backbone and off-rotamer side chain minimization of the complex before and after mutation (-min_interface -int_bb -int_chi). Glycans were modeled using the GlyProt web server (48), using oligomannose glycans because high mannose glycans were found to be predominant in native E2 (49). As with the Rosetta simulations, the engineered N-terminal arginine residue was removed from the peptide in the complex structure prior to input into the server.
Protein Data Bank Accession Code
Coordinates and structure factors for the HC33.1-E2412–423 complex have been deposited under accession code 4XVJ.
RESULTS
Overview of the HC33.1-E2412–423 Complex
We expressed the HC33.1 antibody as an scFv by in vitro folding from bacterial inclusion bodies. The scFv construct consisted of the VH region connected to the VL region by an 18-residue linker. The HC33.1 scFv was crystallized with a 13-mer peptide (RQLINTNGSWHIN) representing the E2412–423 epitope to which an N-terminal arginine was added to increase solubility. We determined the structure of the HC33.1-E2412–423 complex to 2.0 Å resolution by molecular replacement using an ensemble search model built from three scFv structures (41–43) (Table 1 and Fig. 1, A and E). Clear electron density corresponding to the E2412–423 peptide was found in the binding site, as evident from the 2Fo − Fc electron density map (Fig. 2). All scFv residues were visible, except for six residues in the linker connecting VH and VL. The antibody-peptide interface was unambiguous.
FIGURE 1.
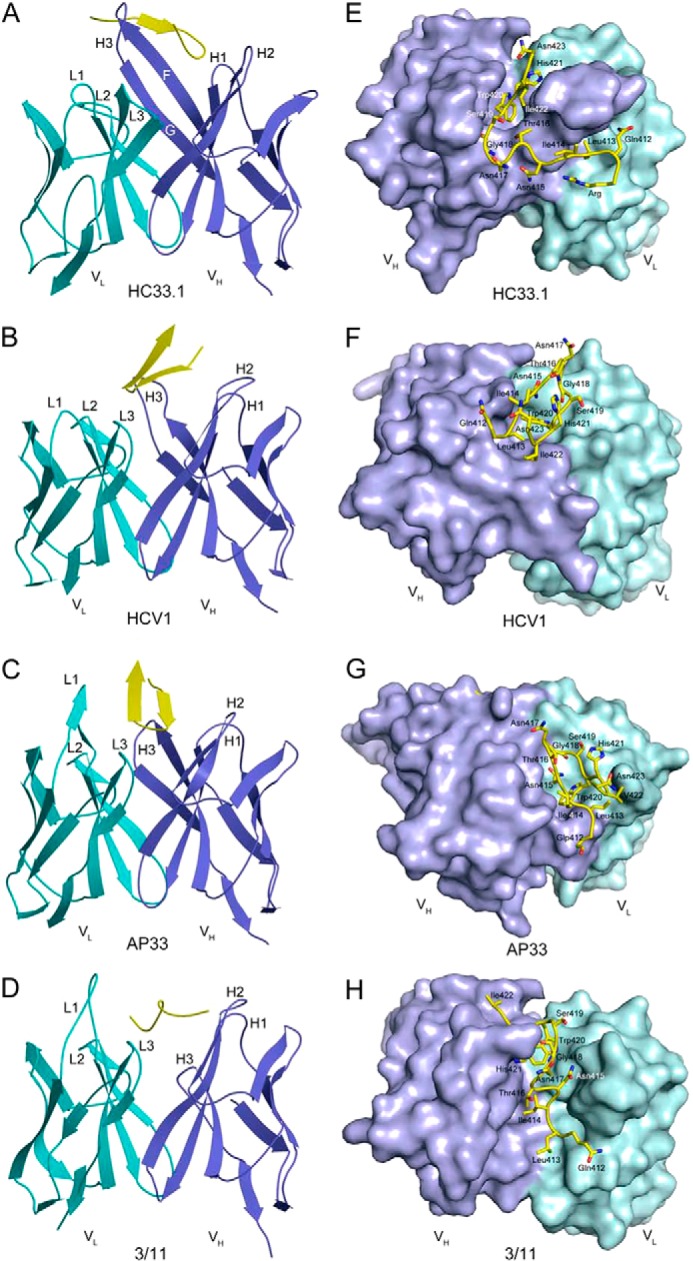
Structure of the HC33.1-E2412–423 complex and comparison with other antibody-E2412–423 complexes. A, ribbon diagram of the HC33.1-E2412–423 complex (side view). Cyan, VL; blue, VH; yellow, E2412–423. VLCDR loops are labeled L1–L3; VHCDR loops are labeled H1–H3. β-Strands F and G of the VH domain are labeled. B, structure of the HCV1-E2412–423 complex (Protein Data Bank accession code 4DGY). The bound peptide adopts a β-hairpin conformation. C, structure of the AP33-E2412–423 complex (Protein Data Bank accession code 4GAJ). D, structure of the 3/11-E2412–423 complex (Protein Data Bank accession code 4WHT). The peptide adopts an extended conformation. E–H, top view of the HC33.1-E2412–423 complex (E) compared with the HCV1-E2412–423 (F), AP33-E2412–423 (G), and 3/11-E2412–423 (H) complexes. For each antibody, the VL molecular surface is cyan, and the VH molecular surface is blue. The E2412–423 peptide is drawn in stick format with carbons atoms in yellow, nitrogen atoms in blue, and oxygen atoms in red.
FIGURE 2.
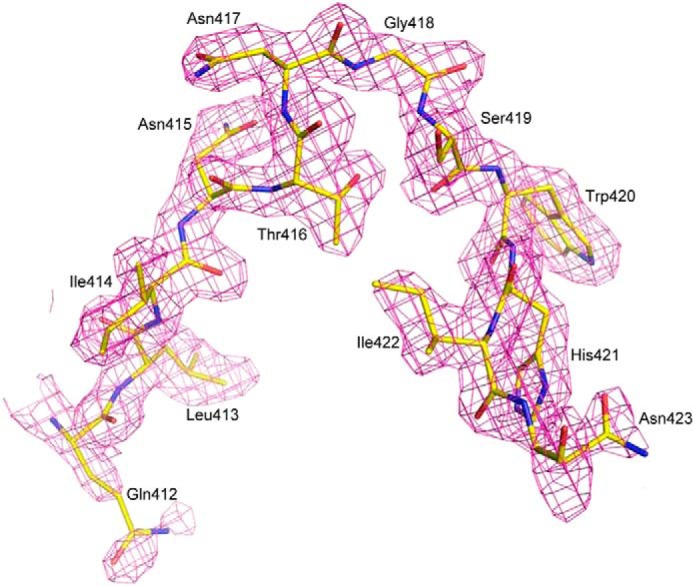
Electron density for the bound E2412–423 peptide in the HC33.1-E2412–423 complex. Density from the final 2Fo − Fc map at 2.0 Å resolution is contoured at 1 σ.
The conformation of the E2412–423 peptide bound to HC33.1 (Fig. 3A) is very different from the β-hairpin conformation of this same peptide bound to HCV1 (36) or AP33 (37, 38) (Fig. 3B) or to the humanized and affinity-matured mAbs MRC10.v362 (derived from AP33) and hu5B3.v3 (35). It is also distinct from the extended conformation of E2412–423 observed in the complex with mAb 3/11 (Fig. 3C) (50). This rat antibody, like HCV1 or AP33 but unlike HC33.1, cannot recognize E2 with a glycosylation shift from Asn-417 to Asn-415 (29). In the HC33.1-E2412–423 complex (Fig. 1, A and E), residues 414–415 of the peptide form an anti-parallel β-sheet with β-strand F of the VH domain, whereas the rest of the peptide adopts a coil conformation. In the HCV1-E2412–423 (Fig. 1, B and F) and AP33-E2412–423 (Fig. 1, C and G) complexes, by contrast, these residues form part of the first strand of the β-hairpin (36–38). In the HC33.1-E2412–423 structure, the N-terminal coil preceding the β-strand of the bound peptide (residues 412–413) extends behind the FG β-sheet of the HC33.1 VH domain (Fig. 1A). The peptide makes a turn at residues 416–419, surrounding the F strand of VH, whereas its C terminus sits loosely in front of the FG β-sheet. The peptide interacts predominantly with the HC33.1 H chain, which mediates 90% of the 186 total contacts. Interactions with the L chain are restricted to the side chains of Leu-413, Trp-420, and His-421 at the N and C termini of E2412–423. HC33.1 has an unusually long VH complementarity-determining region 3 (CDR3) (22 residues compared with 14 and 18 residues for AP33 and HCV1, respectively). In particular, the C terminus of strand F and N terminus of strand G, which constitute part of this VHCDR3, are each 3–4 residues longer in HC33.1 than in HCV1 or AP33. The extended F strand permits E2412–423 to “clip” onto the antibody through formation of the anti-parallel β-sheet described above (Fig. 1A). VHCDR1 and VHCDR2 mainly contact the β-turn of the bound peptide.
FIGURE 3.

Conformation of the E2412–423 epitope bound to HC33.1 and other broadly neutralizing antibodies. A, structure of E2412–423 in complex with HC33.1. Only backbone atoms of the peptide are shown: carbon atoms in yellow, nitrogen atoms in blue, and oxygen atoms in red. Hydrogen bond stabilizing the backbone conformation is drawn as dotted lines. The N and C termini are labeled (N-ter and C-ter, respectively). B, structure of E2412–423 in complex with HCV1. C, structure of E2412–423 in complex with 3/11. D, superposition of E2412–423 epitopes from complexes with HC33.1 (yellow), HCV1 (green), and AP33 (magenta) through residues 416–419 of the peptide β-turn.
Conformation of the E2412–423 Epitope
Because the conformation of E2412–423 bound to HC33.1 (Fig. 3A) is distinct from the β-hairpin or extended conformations of this peptide bound to other mAbs (Fig. 3, B and C), it would be of interest to know the conformation of unbound E2412–423 in the context of the native E2 protein. However, residues 412–420 were disordered in one of the two recently reported E2 core structures (51) and were not included in the crystallization construct used for the other E2 structure (52). Accordingly, we used several prediction methods to assess the preferred unbound conformation(s) of the E2412–423 epitope. Peptide structure prediction using the PEP-FOLD server, an ab initio peptide folding method, resulted in models with a β-hairpin conformation very similar to that in the HCV1 and AP33 complexes (36–38) with a type I′ β-turn at residues 416–419 and a backbone root mean square distance (RMSD) of 2.3 Å between the HCV1-bound epitope and the top model. The I-TASSER server, with homologous templates excluded to prevent usage of E2412–423-containing structures as templates, produced a model that was nearly identical to the HCV1-bound β-hairpin (1.0 Å RMSD). Given these modeling results, in addition to the existence of the β-hairpin in the majority of mAb-bound structures, E2412–423 likely adopts a β-hairpin conformation in the E2 protein and maintains a similar structure after binding to AP33 or HCV1. However, this β-hairpin structure can be disrupted through interactions with certain antibodies, such as 3/11 (50) or HC33.1.
We next asked whether any epitope substructures are shared between the β-hairpin structure bound by HCV1 or AP33 and the HC33.1-bound structure. Indeed, secondary structural analysis identified a conserved β-turn conformation at residues 416–419 (TNGS) of E2412–423 in complex with each of these mAbs. Superposing the HC33.1-bound peptide onto the HCV1-bound peptide through residues 416–419 gave an RMSD of 0.35 Å in the position of their Cα atoms (RMSD = 0.34 for a comparison of the HC33.1- and AP33-bound peptides), indicating close similarity (Fig. 3D). An analogous comparison of the HC33.1-bound and 3/11-bound E2412–423 epitopes failed to identify regions of comparable substructural similarity. This result, together with the β-strand preceding the 416–419 turn in the HC33.1, HCV1, and AP33, but not 3/11, complexes (Fig. 1, A–D), implies that the HC33.1-bound conformation of E2412–423 is structurally intermediate between the HCV1/AP33- and 3/11-bound conformations.
In the β-hairpin structure of E2412–423 bound to HCV1 (36) or AP33 (37, 38), the anti-parallel β-strands are stabilized by main chain-main chain hydrogen bonds between Ile-414 and His-421 and between Thr-416 and Ser-419 (Fig. 3B). By contrast, in the HC33.1-E2412–423 complex, Ile-414 forms main chain-main chain hydrogen bonds with VHSer-103 in β-strand F of HC33.1, resulting in an anti-parallel β-sheet that not only stabilizes the antibody-peptide interaction but also disrupts the β-hairpin conformation of E2412–423 that likely predominates in the E2 protein (Fig. 1, A and E).
The HC33.1-E2412–423 Interface
The HC33.1-E2412–423 complex buries a total solvent-accessible surface area of 1744 Å2, with 11 peptide residues contacting 23 antibody residues. The interface is characterized by high shape complementarity, based on a calculated shape correlation statistic (Sc) (53) of 0.85 (Sc = 1.0 for interfaces with geometrically perfect fits). For clarity, the HC33.1-E2412–423 interface may be subdivided into three regions, according to the distribution of contacting residues on the peptide. In region 1, N-terminal residues 412–416 (QLINT) interact exclusively with VHCDR3, except for Leu-413, which also contacts VLCDR1 Tyr-33 and VLCDR2 Asp-51 (Fig. 4A). Region 2, comprising residues 417–419 (NGS) at the peptide β-turn, interacts with VHCDR1 and VHCDR2 (Fig. 4B). In region 3, C-terminal residues 420–423 (WHIN) contact HC33.1 over a broader area that includes 11 residues from VLCDR3, VH framework region 2, VHCDR2, VH framework region 3, and VHCDR3 (Fig. 4C).
FIGURE 4.
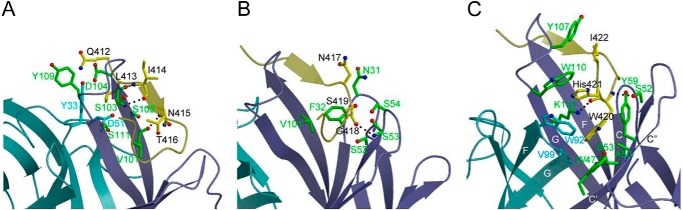
The HC33.1-E2412–423 binding interface. A, close-up view of the interactions between region 1 of E2412–423 (yellow) and the VL (cyan) and VH (blue) domains of HC33.1. The side chains of contacting residues are shown in ball and stick representation with carbon atoms in yellow (E2412–423), cyan (VL), or green (VH); nitrogen atoms in blue; and oxygen atoms in red. Hydrogen bonds are drawn as dotted black lines. B, interactions between region 2 of E2412–423 and HC33.1, showing contacts made by the peptide β-turn. C, interactions between region 3 of E2412–423 and HC33.1. The side chain of Trp-420 binds in a β-barrel formed by VL strands F and G and VH strands C, C′, C″ F, and G (labeled).
In a previous study (14), alanine-scanning mutagenesis was used to identify three E2412–423 residues as critical for binding of the E2 glycoprotein to HC33.1: Leu-413, Gly-418, and Trp-420. The HC33.1-E2412–423 complex readily explains this result, because these epitope residues constitute key anchor points in the structure. Thus, the side chains of Leu-413 and Trp-420 point directly into the HC33.1 binding site, with Leu-413 92% buried and Trp-420 completely buried upon complex formation. Indeed, computational alanine scanning (47) of the HC33.1-E2412–423 complex structure identified Leu-413 and Trp-420 as critical binding residues (Table 2). The isobutyl group of Leu-413 is oriented parallel to the aromatic ring of VLCDR1 Tyr-33, with which it makes extensive hydrophobic interactions (Fig. 4A). In addition, the Cδ1 atom of Leu-413 contacts the side chain of VLCDR2 Asp-51, whereas its Cδ2 atom contacts the main chains of VHCDR3 Ser-102 and Ser-103 in the unusually long β-strand F of HC33.1. Together, these four antibody residues form a tight, mainly hydrophobic cage encasing Leu-413, which probably accounts for the extreme sensitivity of HC33.1 to even conservative substitutions (e.g. Ile) at this position (28). Computational simulations of this mutant (Table 2) showed substantial predicted affinity loss, whether or not nearby residues were permitted to minimize to accommodate the Ile side chain; only the Cγ2 methyl group fit into the pocket noted above, yielding a loss of favorable HC33.1 contacts.
TABLE 2.
Rosetta ΔΔG scores of E2412–423 alanine and HC33.1 escape mutants
| Mutant | ΔΔG scorea |
|---|---|
| Q412A | 0.4 |
| L413A | 2.4 |
| I414A | 0.5 |
| N415A | 0.1 |
| T416A | 1.3 |
| N417A | −0.1 |
| G418A | −0.6 |
| S419A | 0.9 |
| W420A | 5.4 |
| H421A | 1.9 |
| I422A | 1.0 |
| N423A | 0.0 |
| L413I | 1.7 (1.9) |
| N417T | 0 (0.1) |
| S419N | 0.6 (0.4) |
a The Rosetta ΔΔG score was calculated with rotamer-based packing of the mutant residue, and for non-alanine mutants, backbone, and off-rotamer side chain, minimization of mutant and interface residues was also performed (in parentheses). Predicted substantially destabilizing mutants (ΔΔG scores >1.5) are shown in bold type.
The side chain of Trp-420 binds deep in a β-barrel composed of VH strands C, C′, C″, F, and G and VL strands F and G (Fig. 4C). It makes numerous hydrophobic interactions with side chains of residues lining the β-barrel, in particular Trp-47, Tyr-59, and Lys-112 from VH strands C′, C″,and G, respectively, and Trp-92 and Val-99 from the VL FG loop. These interactions would be mostly lost upon alanine substitution of Trp-420, in agreement with the finding that this mutation abolished binding of E2 to HC33.1 (14). Additionally, Trp-420 makes two main chain-side chain hydrogen bonds with HC33.1: Trp-420 N-Oγ VHCDR2 Ser-52 and Trp-420 O-Nζ VHCDR3 Lys-112 (Fig. 4C).
Like Trp-420, Gly-418 is completely buried in the HC33.1-E2412–423 interface, where it makes 21 close contacts with the backbones of VHCDR1 Asn-31 and Phe-32 and of VHCDR2 Ser-52, Ser-53, and Ser-54 (Fig. 4B). Modeling the structure of the G418A mutant indicated that the alanine methyl group could in principle be accommodated at this position without substantially clashing with HC33.1, as reflected by its predicted ΔΔG (Table 2). However, Gly-418 is located at the tip of the type I′ β-turn of the E2412–423 peptide (Fig. 3D), and glycine is a highly preferred amino acid at this position for this type of β-turn (54). Accordingly, substitution of Gly-418 by other residues could adversely affect folding of the β-turn, which is wedged between the VHCDR1 and VHCDR2 loops (Fig. 4B).
Accommodation of the Glycosylation Shift to Asn-415 of E2412–423
Escape of HCV from neutralization by the E2412–423-specific mAbs HCV1, AP33, and 3/11 is the result of adaptive mutations at Asn-417 (N417S and N417T) that cause a glycosylation shift from Asn-417 to Asn-415 in the E2 protein (29, 35, 39). In marked contrast to these mAbs, HC33.1 can penetrate the glycan shield at Asn-415 originating from the glycosylation shift (14, 28). We do not expect that the N417S or N417T mutation would affect the overall conformation of the E2412–423 epitope or the orientation of Asn-415 in the complex with HC33.1. First, the side chains of Asn-417 and Asn-415 are each ∼60% solvent-exposed in the HC33.1-E2412–423 structure. Second, serine and threonine, like asparagine, are polar residues whose side chains prefer to project into solvent.
Structures of the wild-type E2412–423 epitope bound to HCV1, AP33, and 3/11 showed that, whereas the side chain of Asn-417 points away from the antibody (as does that of Asn-423, an additional glycosylation site within this epitope), Asn-415 is buried in the antibody-peptide interface (Fig. 1, F–H) (35–38, 50). Therefore, attachment of a glycan at Asn-415 (but not at Asn-417 or Asn-423) would create untenable steric clashes with HCV1, AP33, and 3/11 (Fig. 5, A and B), abolishing antibody recognition.
FIGURE 5.
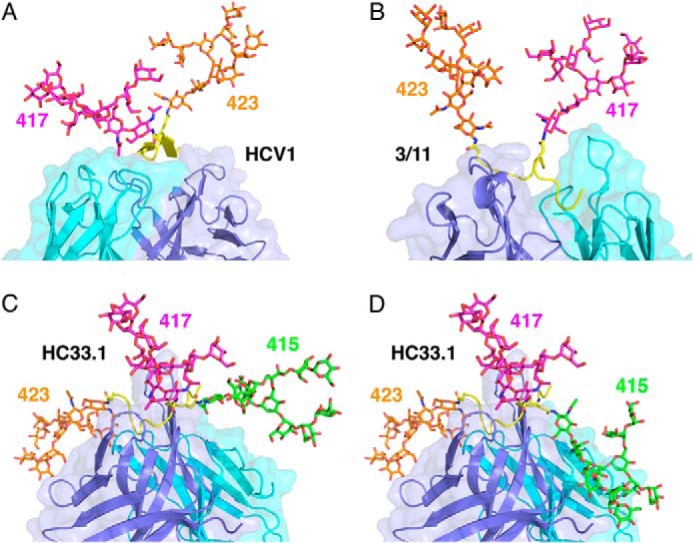
Accommodation of the glycosylation shift in E2412–423. A, N-linked glycans attached to Asn-417 (magenta) and Asn-423 (orange) in the HCV1-E2412–423 complex. A glycan at Asn-415 would be buried in the interface. VL is cyan, and VH is blue. B, N-linked glycans attached to Asn-417 and Asn-423 in the 3/11-E2412–423 complex. As in the HCV1-E2412–423 complex, a glycan at Asn-415 would be buried in the interface. C, compatibility of the HC33.1-E2412–423 complex with N-linked glycans attached to Asn-415 (green), Asn-417 (magenta), and Asn-423 (orange). D, N-linked glycans attached to Asn-415, Asn-417, and Asn-423 in the HC33.1-E2412–423 complex, showing an alternative conformation of the Asn-415 glycan compared with that in Fig. 5C.
The ability of HC33.1 to accommodate the glycosylation shift to Asn-415 is explained by the unique conformation of E2412–423 induced (or captured) by this mAb, rather than by HC33.1 binding in a different way to the β-hairpin or extended conformation of E2412–423 recognized by HCV1, AP33, or 3/11. In the HCV1-E2412–423 and AP33-E2412–423 complexes, the buried Asn-415 residue is part of the first strand of the E2412–423 β-hairpin (Fig. 1, F and G) (36–38), whereas in the HC33.1-E2412–423 complex, Asn-415 is part of a strand that forms an anti-parallel β-sheet with strand F of VH, such that the Asn-415 side chain flanks the antibody and is 58% solvent-accessible (Fig. 1E). A glycan chain can be attached to the side chain amide of Asn-415, based on modeling using the GlyProt server (48), indicating that glycosylation of E2 at Asn-415 would not prevent binding of HC33.1 (Fig. 5C). Indeed, a glycan at this position could mediate additional productive interactions with the mAb, resulting in higher affinity, as suggested by the finding that the neutralization potency of HC33.1 against glycan-shifted HCV virions is >10-fold greater than against wild-type virions (28). Although the simulated oligomannose glycan at Asn-415 was largely directed away from the antibody because of Asn-415 side chain movement by the GlyProt server (48), we generated an alternate conformation of the Asn-415 glycan by using the E2412–423 structure alone as input to GlyProt (removing mAb HC33.1) (Fig. 5D). This resulted in a modeled Asn-415 side chain conformation closer to the antibody-flanking conformation observed in the crystal structure and attached glycan with substantial antibody contacts, including ones with VHCDR1, VHCDR3, and the L chain framework.
Importantly, the side chains of Asn-417 and Asn-423 are also solvent-exposed (60 and 95%, respectively) in the HC33.1-E2412–423 structure, which would permit attachment of glycan chains to these residues as well, without interfering with mAb binding. Modeling of the Asn-417 and Asn-423 glycans using GlyProt (48) supports this (Fig. 5C). Thus, HC33.1 binding to the flexible E2412–423 epitope induces a conformation that allows accommodation of multiple glycans in the interface, whether at Asn-417 and Asn-423 in wild-type E2 or at Asn-415 and Asn-423 in glycosylation-shifted E2 mutants.
DISCUSSION
We have identified the structural basis for human antibody recognition of HCV E2412–423 that is capable of neutralizing clinically observed glycan shift mutants in this linear epitope. This provides new insights into the capability of antibodies such as HC33.1 to effectively target HCV, thereby overcoming previously identified escape mechanisms through mutation or glycan attachment at E2 residue 415 (35). Based on initial structures of mAbs bound to E2412–423, it appeared that this epitope was restricted to a β-hairpin conformation (36–38). However, our study has revealed a third distinct conformation of E2412–423, intermediate between a β-hairpin and coil (50), that highlights the remarkable structural heterogeneity of this epitope.
Along with sequence variability and N-glycosylation, conformational flexibility has been proposed as a mechanism used by viruses such as HIV and influenza to evade humoral immunity (34). Conformational flexibility is thought to pose a barrier to effective elicitation of broadly neutralizing antibodies to a viral site because antibodies are unable to simultaneously target many possible epitope conformations (55, 56). For example, murine norovirus was found to escape neutralization by antibodies against the capsid protein by undergoing mutations in two flexible loops of the capsid that switch their conformation to one with poor stereochemical complementarity to neutralizing antibodies (57).
Our results and those of others (50, 51, 58) suggest that HCV may also employ structural flexibility as an immune evasion strategy. The crystal structure of the HCV E2 core revealed that ∼60% of all residues are either disordered or in loops, implying considerable overall flexibility (51). Neutralizing and non-neutralizing mouse mAbs specific for an E2 epitope comprising residues 427–446 were found to bind distinct conformations of this epitope that determined recognition specificity (58). Here we have shown that the E2412–423 epitope can adopt at least three different conformations, which may contribute to reducing its immunogenicity in HCV-infected individuals (14).
Although structural flexibility may render E2412–423 less antigenic, such flexibility could also be disadvantageous to HCV, insofar as conformational variability provides the immune system with multiple potential targets for antibody neutralization. Thus, if the E2412–423 epitope were restricted to the β-hairpin conformation recognized by HCV1-like antibodies, it could not be targeted by HC33.1-like antibodies. As a consequence, the immune system would be unable to counter HCV variants that have undergone the Asn-415 glycosylation shift.
The HC33.1-E2412–423 structure explains the ability of HC33.1 to accommodate glycans at Asn-417 and Asn-423 in wild-type E2 or at Asn-415 and Asn-423 in glycosylation-shifted E2 mutants. A possible additional glycan has been proposed for the mutation S419N, which was observed in the context of other mutations, notably N417T, in a cell-based study of HC33.1 escape mutants (28). In combination, the S419N and N417T mutations may generate a new glycan at position 419 because of creation of a Thr-Xaa-Asn glycosylation sequon (59). Although far less common than the Asn-Xaa-Ser/Thr sequon, the Thr-Xaa-Asn sequon is one of several reported alternative motifs for N-glycosylation. Although the GlyProt server (48) does not recognize this sequon, in silico mutagenesis of S419N and N417T permitted modeling of a glycan at Asn-419. As expected from the high solvent exposure (60%) of the wild-type Ser-419 side chain in the HCV1-E2412–423 structure (Fig. 1E), this glycan, if present, is not predicted to abolish antibody binding completely. Indeed, N417S/S419N escape variants could still be neutralized by HC33.1, albeit at higher antibody concentrations, and HC33.1 retained significant binding to recombinant E1E2 bearing these mutations (28). Apart from affecting interactions with HC33.1 directly, a glycan at Asn-419 may influence the structure or conformational dynamics of the E2412–423 epitope, as noted in studies of peptide dynamics (60), or interact with other regions of E2. Another possibility is that the side chain change of Ser-419 to Asn has a direct effect on HC33.1 recognition; this would be supported by the hydrogen bond of Ser-419 with Asn-54 on the HC33.1 H chain, as well as some predicted loss of affinity upon in silico mutation (Table 2).
From the standpoint of vaccine design, the HC33.1-E2412–423 structure provides critical information regarding a well conserved site and a new point of viral vulnerability. Having demonstrated how in principle antibodies can effectively target this epitope without including residue 415 in the interface core, vaccine design efforts can attempt to present this epitope structure to the immune system, rather than the likely preferred β-hairpin. Given that additive neutralization was observed between antibodies targeting this site (including HC33.1) and antibodies targeting another well conserved site that also contains residues critical for binding CD81 (14), simultaneously inducing robust immune responses to these epitope sites while minimizing the chances of escape (including glycan shifting to Asn-415) is key to an effective HCV vaccine.
Acknowledgment
We thank Sneha Rangarajan for critical reading of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant AI036900 (to R. A. M.). This work was also supported by MPower Maryland.
The atomic coordinates and structure factors (code 4XVJ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HCV
- hepatitis C virus
- VH
- heavy chain variable region
- VL
- light chain variable region
- CDR
- complementarity-determining region
- RMSD
- root mean square distance.
REFERENCES
- 1. Shepard C. W., Finelli L., Alter M. J. (2005) Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5, 558–567 [DOI] [PubMed] [Google Scholar]
- 2. Lavanchy D. (2009) The global burden of hepatitis C. Liver Int. 29, 74–81 [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization. Hepatitis C. Fact sheet number 164. http://www.who.int/mediacentre/factsheets/fs164/en/#content (2013) WHO website (online)
- 4. Schinazi R., Halfon P., Marcellin P., Asselah T. (2014) HCV direct-acting antiviral agents: the best interferon-free combinations. Liver Int. 34, 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A. J., Houghton M., Rosa D., Grandi G., Abrignani S. (1998) Binding of hepatitis C virus to CD81. Science 282, 938–941 [DOI] [PubMed] [Google Scholar]
- 6. Scarselli E., Ansuini H., Cerino R., Roccasecca R. M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. (2002) The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ploss A., Evans M. J., Gaysinskaya V. A., Panis M., You H., de Jong Y. P., Rice C. M. (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evans M. J., von Hahn T., Tscherne D. M., Syder A. J., Panis M., Wölk B., Hatziioannou T., McKeating J. A., Bieniasz P. D., Rice C. M. (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805 [DOI] [PubMed] [Google Scholar]
- 9. Angus A. G., Patel A. H. (2011) Immunotherapeutic potential of neutralizing antibodies targeting conserved regions of the HCV envelope glycoprotein E2. Future Microbiol. 6, 279–294 [DOI] [PubMed] [Google Scholar]
- 10. Kato N., Sekiya H., Ootsuyama Y., Nakazawa T., Hijikata M., Ohkoshi S., Shimotohno K. (1993) Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol. 67, 3923–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. von Hahn T., Yoon J. C., Alter H., Rice C. M., Rehermann B., Balfe P., McKeating J. A. (2007) Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132, 667–678 [DOI] [PubMed] [Google Scholar]
- 12. Dowd K. A., Netski D. M., Wang X. H., Cox A. L., Ray S. C. (2009) Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136, 2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bankwitz D., Steinmann E., Bitzegeio J., Ciesek S., Friesland M., Herrmann E., Zeisel M. B., Baumert T. F., Keck Z. Y., Foung S. K., Pécheur E. I., Pietschmann T. (2010) Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 84, 5751–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keck Z., Wang W., Wang Y., Lau P., Carlsen T. H., Prentoe J., Xia J., Patel A. H., Bukh J., Foung S. K. (2013) Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J. Virol. 87, 37–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keck Z. Y., Li T. K., Xia J., Bartosch B., Cosset F. L., Dubuisson J., Foung S. K. (2005) Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. J. Virol. 79, 13199–13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keck Z. Y., Op De Beeck A., Hadlock K. G., Xia J., Li T. K., Dubuisson J., Foung S. K. (2004) Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. J. Virol. 78, 9224–9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keck Z. Y., Xia J., Wang Y., Wang W., Krey T., Prentoe J., Carlsen T., Li A. Y., Patel A. H., Lemon S. M., Bukh J., Rey F. A., Foung S. K. (2012) Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 8, e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hadlock K. G., Lanford R. E., Perkins S., Rowe J., Yang Q., Levy S., Pileri P., Abrignani S., Foung S. K. (2000) Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 74, 10407–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Owsianka A. M., Tarr A. W., Keck Z. Y., Li T. K., Witteveldt J., Adair R., Foung S. K., Ball J. K., Patel A. H. (2008) Broadly neutralizing human monoclonal antibodies to hepatitis C virus E2 glycoprotein. J. Gen. Virol. 89, 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tarr A. W., Urbanowicz R. A., Jayaraj D., Brown R. J., McKeating J. A., Irving W. L., Ball J. K. (2012) Naturally occurring antibodies that recognize linear epitopes in the amino terminus of the hepatitis C virus E2 protein confer noninterfering, additive neutralization. J. Virol. 86, 2739–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarr A. W., Owsianka A. M., Jayaraj D., Brown R. J., Hickling T. P., Irving W. L., Patel A. H., Ball J. K. (2007) Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 88, 2991–3001 [DOI] [PubMed] [Google Scholar]
- 22. Owsianka A., Tarr A. W., Juttla V. S., Lavillette D., Bartosch B., Cosset F. L., Ball J. K., Patel A. H. (2005) Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 79, 11095–11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Owsianka A. M., Timms J. M., Tarr A. W., Brown R. J., Hickling T. P., Szwejk A., Bienkowska-Szewczyk K., Thomson B. J., Patel A. H., Ball J. K. (2006) Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80, 8695–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broering T. J., Garrity K. A., Boatright N. K., Sloan S. E., Sandor F., Thomas W. D., Jr., Szabo G., Finberg R. W., Ambrosino D. M., Babcock G. J. (2009) Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J. Virol. 83, 12473–12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flint M., Maidens C., Loomis-Price L. D., Shotton C., Dubuisson J., Monk P., Higginbottom A., Levy S., McKeating J. A. (1999) Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73, 6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsu M., Zhang J., Flint M., Logvinoff C., Cheng-Mayer C., Rice C. M., McKeating J. A. (2003) Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U.S.A. 100, 7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sabo M. C., Luca V. C., Prentoe J., Hopcraft S. E., Blight K. J., Yi M., Lemon S. M., Ball J. K., Bukh J., Evans M. J., Fremont D. H., Diamond M. S. (2011) Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J. Virol. 85, 7005–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keck Z. Y., Angus A. G., Wang W., Lau P., Wang Y., Gatherer D., Patel A. H., Foung S. K. (2014) Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids. PLoS Pathog. 10, e1004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dhillon S., Witteveldt J., Gatherer D., Owsianka A. M., Zeisel M. B., Zahid M. N., Rychłowska M., Foung S. K., Baumert T. F., Angus A. G., Patel A. H. (2010) Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J. Virol. 84, 5494–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goffard A., Dubuisson J. (2003) Glycosylation of hepatitis C virus envelope proteins. Biochimie 85, 295–301 [DOI] [PubMed] [Google Scholar]
- 31. Goffard A., Callens N., Bartosch B., Wychowski C., Cosset F. L., Montpellier C., Dubuisson J. (2005) Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79, 8400–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Falkowska E., Kajumo F., Garcia E., Reinus J., Dragic T. (2007) Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81, 8072–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Helle F., Goffard A., Morel V., Duverlie G., McKeating J., Keck Z. Y., Foung S., Penin F., Dubuisson J., Voisset C. (2007) The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 81, 8101–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Julien J.-P., Lee P. S., Wilson I. A. (2012) Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol. Rev. 250, 180–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pantua H., Diao J., Ultsch M., Hazen M., Mathieu M., McCutcheon K., Takeda K., Date S., Cheung T. K., Phung Q., Hass P., Arnott D., Hongo J. A., Matthews D. J., Brown A., Patel A. H., Kelley R. F., Eigenbrot C., Kapadia S. B. (2013) Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 425, 1899–1914 [DOI] [PubMed] [Google Scholar]
- 36. Kong L., Giang E., Robbins J. B., Stanfield R. L., Burton D. R., Wilson I. A., Law M. (2012) Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. U.S.A. 109, 9499–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kong L., Giang E., Nieusma T., Robbins J. B., Deller M. C., Stanfield R. L., Wilson I. A., Law M. (2012) Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J. Virol. 86, 13085–13088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Potter J. A., Owsianka A. M., Jeffery N., Matthews D. J., Keck Z. Y., Lau P., Foung S. K., Taylor G. L., Patel A. H. (2012) Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J. Virol. 86, 12923–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung R. T., Gordon F. D., Curry M. P., Schiano T. D., Emre S., Corey K., Markmann J. F., Hertl M., Pomposelli J. J., Pomfret E. A., Florman S., Schilsky M., Broering T. J., Finberg R. W., Szabo G., Zamore P. D., Khettry U., Babcock G. J., Ambrosino D. M., Leav B., Leney M., Smith H. L., Molrine D. C. (2013) Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am. J. Transplant. 13, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Storoni L. C., McCoy A. J., Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 [DOI] [PubMed] [Google Scholar]
- 41. Hwang W. C., Lin Y., Santelli E., Sui J., Jaroszewski L., Stec B., Farzan M., Marasco W. A., Liddington R. C. (2006) Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J. Biol. Chem. 281, 34610–34616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robin G., Sato Y., Desplancq D., Rochel N., Weiss E., Martineau P. (2014) Restricted diversity of antigen binding residues of antibodies revealed by computational alanine scanning of 227 antibody-antigen complexes. J. Mol. Biol. 426, 3729–3743 [DOI] [PubMed] [Google Scholar]
- 43. Sui J., Hwang W. C., Perez S., Wei G., Aird D., Chen L. M., Santelli E., Stec B., Cadwell G., Ali M., Wan H., Murakami A., Yammanuru A., Han T., Cox N. J., Bankston L. A., Donis R. O., Liddington R. C., Marasco W. A. (2009) Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 45. McRee D. E. (1999) XtalView/Xfit: A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 46. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: A program to check the stereo chemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 47. Kortemme T., Baker D. (2002) A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. U.S.A. 99, 14116–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bohne-Lang A., von der Lieth C. W. (2005) GlyProt: in silico glycosylation of proteins. Nucleic Acids Res. 33, W214–W219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iacob R. E., Perdivara I., Przybylski M., Tomer K. B. (2008) Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J. Am. Soc. Mass Spectrom. 19, 428–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meola A., Tarr A. W., England P., Meredith L. W., McClure C. P., Foung S. K., McKeating J. A., Ball J. K., Rey F. A., Krey T. (2015) Structural flexibility of a conserved broadly neutralizing epitope in hepatitis C virus glycoprotein E2. J. Virol. 89, 2170–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kong L., Giang E., Nieusma T., Kadam R. U., Cogburn K. E., Hua Y., Dai X., Stanfield R. L., Burton D. R., Ward A. B., Wilson I. A., Law M. (2013) Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khan A. G., Whidby J., Miller M. T., Scarborough H., Zatorski A. V., Cygan A., Price A. A., Yost S. A., Bohannon C. D., Jacob J., Grakoui A., Marcotrigiano J. (2014) Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lawrence M. C., Colman P. M. (1993) Shape complementarity at protein-protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 54. Hutchinson E. G., Thornton J. M. (1994) A revised set of potentials for β-turn formation in proteins. Protein Sci. 3, 2207–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Myszka D. G., Sweet R. W., Hensley P., Brigham-Burke M., Kwong P. D., Hendrickson W. A., Wyatt R., Sodroski J., Doyle M. L. (2000) Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U.S.A. 97, 9026–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pantophlet R., Burton D. R. (2006) GP120: target for neutralizing HIV-1 antibodies. Annu. Rev. Immunol. 24, 739–769 [DOI] [PubMed] [Google Scholar]
- 57. Kolawole A. O., Li M., Xia C., Fischer A. E., Giacobbi N. S., Rippinger C. M., Proescher J. B., Wu S. K., Bessling S. L., Gamez M., Yu C., Zhang R., Mehoke T. S., Pipas J. M., Wolfe J. T., Lin J. S., Feldman A. B., Smith T. J., Wobus C. E. (2014) Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J. Virol. 88, 4543–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deng L., Ma L., Virata-Theimer M. L., Zhong L., Yan H., Zhao Z., Struble E., Feinstone S., Alter H., Zhang P. (2014) Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc. Natl. Acad. Sci. U.S.A. 111, 10690–10695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schulz B. L. (2012) Beyond the Sequon: Sites of N-Glycosylation. In Glycosylation (Petrescu S., ed) pp. 21–40, InTech, Rijeka, Croatia [Google Scholar]
- 60. Imperiali B., Rickert K. W. (1995) Conformational implications of asparagine-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 92, 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]