

Background: We studied the interplay between base excision repair (BER) of 8-oxoguanine (GO) and mismatch repair (MMR).
Results: BER and MMR interact during the processing of GO/A but not GO/C mispairs.
Conclusion: BER of GO-containing lesions appears to be regulated.
Significance: BER intermediates were believed to be unavailable to other pathways of DNA metabolism. This hypothesis may be incorrect.
Keywords: Base Excision Repair (BER), DNA Damage, DNA Mismatch Repair, DNA Repair, Oxidative Stress
Abstract
Replicative DNA polymerases are high fidelity enzymes that misincorporate nucleotides into nascent DNA with a frequency lower than [1/105], and this precision is improved to about [1/107] by their proofreading activity. Because this fidelity is insufficient to replicate most genomes without error, nature evolved postreplicative mismatch repair (MMR), which improves the fidelity of DNA replication by up to 3 orders of magnitude through correcting biosynthetic errors that escaped proofreading. MMR must be able to recognize non-Watson-Crick base pairs and excise the misincorporated nucleotides from the nascent DNA strand, which carries by definition the erroneous genetic information. In eukaryotes, MMR is believed to be directed to the nascent strand by preexisting discontinuities such as gaps between Okazaki fragments in the lagging strand or breaks in the leading strand generated by the mismatch-activated endonuclease of the MutL homologs PMS1 in yeast and PMS2 in vertebrates. We recently demonstrated that the eukaryotic MMR machinery can make use also of strand breaks arising during excision of uracils or ribonucleotides from DNA. We now show that intermediates of MutY homolog-dependent excision of adenines mispaired with 8-oxoguanine (GO) also act as MMR initiation sites in extracts of human cells or Xenopus laevis eggs. Unexpectedly, GO/C pairs were not processed in these extracts and failed to affect MMR directionality, but extracts supplemented with exogenous 8-oxoguanine DNA glycosylase (OGG1) did so. Because OGG1-mediated excision of GO might misdirect MMR to the template strand, our findings suggest that OGG1 activity might be inhibited during MMR.
Introduction
To improve the fidelity of DNA replication, mismatch repair (MMR)2 must be able to detect base/base mismatches and small insertion/deletion loops that escaped the proofreading activity of the replicative polymerases and direct their excision and resynthesis activity to the nascent strand, which carries by definition the erroneous genetic information. In Gram-negative bacteria such as Escherichia coli, the newly synthesized strand remains transiently unmethylated on adenines within GATC sequences. Mismatch-activated MutS-MutL complex licenses MutH to incise the unmethylated GATC, which allows the loading of the UvrD helicase together with one of several exonucleases. This leads to the degradation of the error-containing strand from the MutH-catalyzed nick toward and some distance past the mismatch to generate a single-stranded gap that is subsequently filled in by polymerase III. The remaining nick is sealed by DNA ligase (1–3).
In eukaryotes, DNA methylation is not used in strand discrimination during MMR. Instead, the nascent strand is distinguished from the template by transient discontinuities such as gaps between Okazaki fragments in the lagging strand. The leading strand contains no such discontinuities other than the 3′ terminus. Because the only MMR-associated exonuclease identified to date, EXO1, has an obligate 5′ to 3′ polarity, it appeared unlikely that MMR would use the 3′ terminus of the primer strand for initiation. However, seminal work from Modrich and co-workers (4, 5) demonstrated that association of the mismatch-activated MutLα (a heterodimer of MLH1 and PMS2) with proliferating cell nuclear antigen bound at the 3′ terminus activates a cryptic endonuclease in the PMS2 subunit, which introduces nicks into the newly synthesized strand and thus provides EXO1 with entry sites where 5′ to 3′ degradation of the error-containing strand can initiate.
While studying the involvement of MMR in somatic hypermutation, which is triggered by activation-induced cytidine deaminase that converts cytosines to uracils at the immunoglobulin locus of activated B cells (6), we discovered that the MMR system can hijack strand breaks generated by the base excision repair (BER) system during uracil removal for the purpose of strand discrimination (7). This was unexpected because BER was thought to be a concerted process in which the strand break generated by apurinic endonuclease (APE1) after removal of the aberrant base is not available for other processes of DNA metabolism (8). In later experiments, we showed that breaks generated during the RNase H2-catalyzed removal of ribonucleotides misincorporated into DNA during replication can also be utilized by MMR as strand discrimination signals (9).
The above experiments suggested that strand breaks arising during different processes of DNA metabolism may be hijacked by MMR. Although this might lead to improved replication fidelity when breaks in the nascent DNA strand were involved, the opposite would be true if breaks in the template strand were used to direct the MMR process. To gain novel insight into this phenomenon, we set out to examine the interplay between MMR and oxidative DNA damage metabolism because BER-mediated processing of oxidized DNA could potentially take place on both nascent and template strands.
Depending on cell type, human genomes have been estimated to harbor steady-state levels of the major product of DNA oxidation, 2′-deoxy-8-oxoguanosine, ranging between 1000 and 100,000 residues (10–12). This nucleoside can be present in DNA in two distinct contexts: either paired with deoxycytidine or mispaired with deoxyadenosine because the base, 8-oxoguanine (GO), can adopt either an anti or a syn conformation about the glycosidic bond (13, 14). Thus, although oxidation of double-stranded DNA gives rise solely to GO/C pairs in which GO is in the anti conformation, the replicative polymerases α, δ, and ϵ could insert either a C opposite anti-GO to form a Watson-Crick-like GO/C base pair or an A opposite syn-GO to form a Hoogsteen GO/A mispair (15–17).
Oxidation also affects the nucleotide pool where it generates dGOTP. However, because this nucleotide is hydrolyzed by MutT homolog 1 (MTH1) protein to dGOMP (18–20), GO should not be incorporated into the nascent DNA strand. Thus, in newly replicated DNA, all GO residues should be in the template strand, and all As mispaired with GO should be in the nascent strand. This point is key to our understanding of the potential interplay between oxidative damage processing and MMR.
GO/C base pairs are addressed primarily by 8-oxoguanine DNA glycosylase (OGG1), a glycosylase/lyase that removes the oxidized base and cleaves the sugar-phosphate backbone to initiate a BER process that ultimately replaces the oxidized nucleotide with a dGMP (13). GO/As are not addressed by OGG1; they are recognized by the MutY homolog (MYH) glycosylase, which removes the mispaired adenines to initiate a BER process that inserts Cs opposite the oxidized guanines. The GO/Cs arising in this way can then be repaired later to G/C by OGG1-dependent BER (21, 22). Thus, if MMR were to use breaks generated during MYH-initiated BER of GO/As, these discontinuities would be in the nascent strand, and their hijacking by MMR might improve the efficiency of the latter process. In contrast, incisions made during OGG1-initiated BER of GO/Cs would be in the template strand where they would not only misdirect the mismatch repair process to the wrong strand but where they could also give rise to double strand breaks that could cause replication fork collapse.
In an attempt to learn whether MMR utilized strand breaks arising during MYH- and/or OGG1-initiated BER processes, we generated substrates containing a single nick (a bona fide strand discrimination signal), a GO/A mispair, or a GO/C pair and studied the efficiency and directionality of MMR-catalyzed repair of a G/T mismatch situated in the vicinity. We show that, in extracts of human cells or Xenopus laevis eggs, the MMR excision machinery can use strand breaks arising upon MYH-catalyzed removal of adenines from GO/A mispairs as initiation sites for exonucleolytic degradation of error-containing strands on circular heteroduplex substrates. A similar phenomenon was not observed either on GO/C-containing heteroduplexes or on substrates containing 2-hydroxyadenine (AO), another product of purine oxidation (12), even though AO/C and AO/G mispairs were reported previously to be addressed by MYH (23, 24).
EXPERIMENTAL PROCEDURES
Restriction Enzymes
All restriction enzymes were purchased from New England Biolabs.
Recombinant Proteins
GST-tagged MYH was expressed from the pET41a-MYH expression vector (a gift from Dr. Barbara van Loon) and partially purified using glutathione-Sepharose beads (GE Healthcare) as described previously (25). In a second purification step, eluates were loaded onto a HiTrapTM Heparin HP column (GE Healthcare), which was then washed with 10 column volumes of wash buffer (30 mm Tris-HCl, pH 8, 0.1 mm EDTA, 10% glycerol, 1 mm DTT, 50 mm NaCl) at a flow rate of 0.5 ml/min. Elution was performed using a salt gradient (50–600 mm NaCl). MYH-GST was eluted with 400–500 mm NaCl. The active fractions were identified using a MYH nicking assay, pooled, aliquoted, and stored at −80 °C. Recombinant APE1 was purchased from New England Biolabs. Recombinant OGG1 was a kind gift from Barbara van Loon. Recombinant OGG1-GST was purchased from Trevigen (4130-100-EB). Recombinant MutLα and MutSα were expressed and purified in our laboratory as described previously (26).
The expression constructs for human geminin (geminin-pET28) and p27 (p27-pET21) were a kind gift from Yoshi Hashimoto and Vincenzo Costanzo. Briefly, protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside in BL21 cells (Invitrogen) grown at 37 °C. The cell pellets were resuspended in 20 mm Tris-HCl, pH 7.5, 500 mm KCl, 10% glycerol, 1 mm β-mercaptoethanol, 0.1% Nonidet P-40, and PMSF for lysis. After cell disruption and centrifugation of cell debris and membranes, the soluble fraction containing 10 mm imidazole was loaded onto a nickel-chelating column, which was then washed with 5–25 mm imidazole. The protein was eluted with a gradient of 50–300 mm imidazole. Fractions containing the desired polypeptides were pooled and dialyzed against EB buffer (100 mm KCl, 2.5 mm MgCl2, 50 mm HEPES-KOH, pH 7.5, 10% glycerol).
Cell Culture
HCT116 (MutLα-deficient) and HCT116 + chromosome 3 (MutLα-proficient) cells were obtained from Richard Boland (27) and were cultured in McCoy's 5a medium (Gibco) supplemented with 10% bovine calf serum (Gibco). The medium for the chromosome 3-complemented cell line was supplemented with 400 μg/ml G418. LoVo (MutSα-deficient) cells were grown in DMEM (Gibco) supplemented with 10% bovine calf serum. All media additionally contained 1% penicillin/streptomycin.
Nuclear Extracts of Human Cells
Nuclei were isolated as described previously (28), resuspended in ⅓ of their packed volume in cold extraction buffer (25 mm HEPES-KOH, pH 7.5, 292 mm sucrose, 1 mm PMSF, 0.5 mm DTT, 1 μg/ml leupeptin), and transferred to a small beaker fitted with a magnetic stirrer bar. NaCl was added dropwise to a final concentration of 150 mm, and extraction continued for 1 h at 4 °C. The nuclei were pelleted by centrifugation at 14,500 × g for 20 min at 4 °C in a tabletop centrifuge. The supernatant was transferred and dialyzed 2 × 1 h at 4 °C against 1 liter of cold dialysis buffer (25 mm HEPES-KOH, pH 7.5, 50 mm KCl, 0.1 mm EDTA, 231 mm sucrose, 1 mm PMSF, 2 mm DTT, 1 μg/ml leupeptin). The dialyzed extract was clarified by centrifugation at 20,000 × g for 15 min at 4 °C. The supernatant was aliquoted, snap frozen in liquid nitrogen, and stored at −80 °C. The protein concentration was determined with the Bradford assay, and the salt concentration was measured using a conductivity meter.
X. laevis Egg Extracts
S phase extract was prepared as described previously (29). Briefly, eggs were dejellied, activated with calcium ionophore (Sigma-Aldrich), rinsed with S buffer (50 mm HEPES-KOH, pH 7.5, 50 mm KCl, 2.5 mm MgCl2, 250 mm sucrose), transferred to 2-ml Eppendorf tubes, and crushed by centrifugation for 12 min at 13,200 rpm. The cytoplasmic layer was removed and after addition of CytB (Sigma-Aldrich) cleared by centrifugation for 25 min at 70,000 rpm (Sorvall TL55 swinging bucket rotor). The extract was supplemented with 250 μg/μl cycloheximide, 25 mm phosphocreatine, and 10 μg/ml creatine phosphokinase before use.
Western Blotting and Antibodies
Whole cell extracts of the cell lines were prepared using 2× Laemmli buffer (120 mm Tris-HCl, pH 6.8, 4% SDS, 20% glycerol). Upon determination of the protein concentration by the Lowry assay, proteins were separated by SDS-PAGE. Western blot analyses were carried out using standard procedures. The following antibodies and dilutions were used: MYH (mouse monoclonal, Abcam, ab55551), 1:333; OGG1 (rabbit monoclonal, Abcam, ab124741),1:10,000; MSH6 (mouse monoclonal, BD Transduction Laboratories, 610919), 1:1000; MSH2 (mouse monoclonal, Calbiochem, NA-27), 1:500; MLH1 (mouse monoclonal, BD Transduction Laboratories, 554073), 1:500; and TF II H (rabbit polyclonal, Santa Cruz Biotechnology, sc-293), 1:1000. The anti-MTH1 antibody was a generous gift from Yusaku Nakabeppu and was used at a dilution of 1:250. Horseradish peroxidase (HRP)-coupled anti-mouse and anti-rabbit secondary antibodies (GE Healthcare) were used at a dilution of 1:5000.
Substrate Generation
The detailed procedure was described previously (30). Briefly, hetero- and homoduplexes were constructed by primer extension on single-stranded phagemid templates. The templates differed in the position of the Nt.BstNBI site, which was situated at nucleotide 2850 either in the viral (top) or complementary (bottom) strand. Incubation of the substrates with the nickase yielded substrates in which MMR occurred either 5′ to 3′ or 3′ to 5′, respectively. The mismatches were located within a SalI and/or AclI restriction site, which was restored upon repair. The desired closed-circular heteroduplex substrates were purified on cesium chloride gradients.
Primers
The GO- or AO-containing primers were obtained from Eurogentec (Seraing, Belgium). All other primers were obtained from Microsynth (Balgach, Switzerland). The SalI (GTCGAC) and AclI (AACGTT) restriction sites are highlighted in bold and gray, respectively. PvuI restriction sites (CGATCG) are italicized, and mispaired residues are underlined. Primer sequences correspond to the outer strand sequence of the substrate. The substrates and the primer/template combinations are listed in Table 1.
TABLE 1.
Oligonucleotide primers used to generate the MMR substrates
The SalI (GTCGAC) and AclI (AACGTT) restriction sites are highlighted in bold and gray, respectively. Pvul restriction sites (CGATCG) are italicized, and mispaired residues are underlined.

a Template was pRichi-2850topAclI.
b Template was pRichi-2850botAclI.
c Template was pRichi-2850topSalI.
Prenicking of Substrates
MYH + APE1
200 ng of the substrates were incubated with 10 ng of purified recombinant MYH-GST and 10 units of APE1 (New England Biolabs) in 1× MMR buffer (20 mm Tris-HCl, pH 7.6, 40 mm KCl, 5 mm MgCl2, 1 mm glutathione, 50 μg/ml BSA, 0.1 mm dNTPs) for 4 h at 37 °C. The reaction was stopped by heat inactivation, and 50 ng of the prenicked substrate were analyzed on a 1% GelRed-stained agarose gel for nicking efficiency. The remaining 100 ng of the prenicked substrate were used in a MMR assay.
OGG1
1 μg of GO/C-G/T substrate was incubated with 0.8 μg of purified recombinant OGG1 in 20 mm Tris-HCl, pH 8, 1 mm DTT, 1 mm EDTA, and 0.1 mg/ml BSA for 2.5 h at 37 °C and subsequently purified on a MinElute Spin column (Qiagen). 100 ng of the prenicked substrate were used in a MMR assay.
Nt.BstNBI
Closed-circular DNA substrates (100 ng) were incubated with 1 unit of Nt.BstNBI (New England Biolabs) according to the recommendations of the manufacturer. Subsequently, they were purified on a MinElute Spin column and used in MMR assays.
In Vitro MYH Nicking Assay
100 ng of closed-circular GO/A, AO/G, AO/C, or homoduplex substrate were incubated with 10 ng of purified recombinant MYH-GST, 10 units of APE1, and 1.5 mm ATP in 1× MMR buffer in a total volume of 10 μl. For time course experiments, 5-μl aliquots were withdrawn at the indicated time points. The reactions were stopped by the addition of 2 μl of 6× loading dye (37.5 mg/ml Ficoll 400, 23% glycerol, 0.03% bromphenol blue) and subsequent heat inactivation. The samples were separated on 1% agarose gels and visualized with GelRed. The nicking efficiency was quantified from the ratio of the amount of open-circular DNA product to the total amount of DNA (closed-circular + open-circular).
In Vitro MMR Assay in Nuclear Extracts of Human Cells
The MMR assays were described previously (7, 30, 31). Unless otherwise specified, the reactions were carried out with 100 ng of substrate and 100 μg of nuclear cell extract in a total volume of 25 μl in a buffer containing 20 mm Tris-HCl, pH 7.6, 5 mm MgCl2, 1 mm glutathione, 50 μg/ml BSA, 0.1 mm dNTPs, 1.5 mm ATP, and 80 mm KCl (in experiments involving MYH-addressed substrates) or 110 mm KCl (in experiments involving OGG1-addressed substrates). 1.8 pmol of purified MutLα or MutSα were added where indicated. The reactions were incubated at 37 °C for 1 h. The reaction was stopped by adding an equal volume of STOP solution (50% Proteinase K, 1 mm EDTA, 3% SDS) and subsequent incubation for 1 h at 50 °C. Substrates were purified on MinElute spin columns and subjected to SalI/DraI or AclI restriction digestion, and the DNA fragments were separated on 1% agarose gels stained with GelRed. The percentage of mismatch repair was quantified from the ratio of the repaired bands (c + d) to the total amount of DNA (c + d + (c + d)) using ImageQuantTL. The band intensities were corrected according to their respective DNA fragment sizes. Some assays were supplemented with 2 μCi of [α-32P]dATP. To monitor incorporation of the radiolabeled nucleotide, the agarose gels were vacuum-dried, exposed to a Phosphor Screen, and scanned with a Typhoon Scanner (FLA 9500, GE Healthcare).
In Vitro MMR Assay in X. laevis Egg Extracts
Briefly, the reaction mixture containing 150 ng of substrate, 26 μl of S phase extract, and 2 μCi of [α-32P]dATP in a total volume of 30 μl was incubated at 23 °C for 45 min. To inhibit replication, 500 nm geminin and 40 μg/ml p27 were added. The reaction was stopped by the addition of 70 μl of STOP solution (76 mm EDTA, 1.5% SDS) and 40 μg of RNase (Sigma-Aldrich) and incubated for 30 min at 37 °C. Subsequently, 200 μg of Proteinase K (AppliChem) were added, and incubation continued at 37 °C overnight. The substrates were purified on MinElute Spin columns and subjected to SalI/DraI or AclI restriction digestion in the presence of RNase. The digested DNA was cleaned up again and analyzed on a 1% agarose gel stained with GelRed.
Immunodepletions
20 μl of Protein A/G PLUS-agarose beads (sc-2003, Santa Cruz Biotechnology) were washed twice with 750 μl of binding buffer (30 mm HEPES-KOH, pH 7.6, 7 mm MgCl2) and spun down at 2700 × g for 2 min at 4 °C. The beads were resuspended in binding buffer, and 1 μg anti-MYH or anti-MSH6 antibody was added. The mixture was then incubated for 3 h at 4 °C, the beads were washed three times with 750 μl of binding buffer, and subsequently used to immunodeplete 100 μg of nuclear cell extracts for 30 min at 4 °C. MMR assays were performed immediately after depletion. Mock-depleted nuclear cell extracts were obtained by incubation with beads only.
MMR- and/or BER-dependent Incorporation Assays
This assay was used to test MMR or BER activities. It was performed similarly to the MMR assay except for the following modifications. To track MMR-, MYH-, or OGG1-dependent nucleotide incorporation, the reactions were supplemented with 2 μCi of [α-32P]dATP, [α-32P]dCTP, or [α-32P]dGTP, respectively. Finally, the substrates were digested with NotI/BsaI and analyzed on a GelRed-stained 1% agarose gel. Repair tracts of up to 330 bp in Nt.BstNBI-nicked substrates, up to 29 bp in the GO/A substrates, and up to 22 bp in the GO/C substrates are seen in the 1808-bp fragment a. Longer repair tracts appear in the 1387-bp fragment b (see Fig. 1A). Although MMR-dependent [32P]dAMP incorporation gave rise to a strong radioactive signal in both bands, MYH-dependent [32P]dCMP or OGG1-dependent [32P]dGMP incorporation occurred only in DNA fragment a. Quantification of [32P]dCMP or [32P]dGMP incorporation in MYH- or OGG1-induced BER was determined from the ratio between the band intensities of fragment a on the autoradiographs and on the GelRed-stained agarose gels.
FIGURE 1.
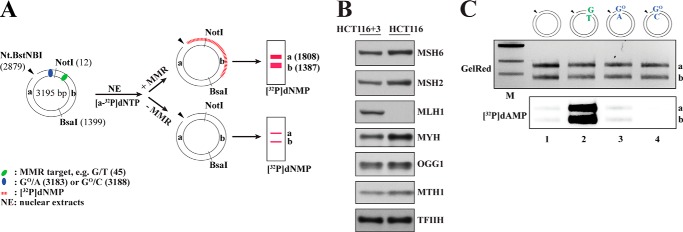
GO-containing mispairs are poor substrates for mismatch repair in human cell extracts. A, schematic representation of the MMR-dependent nucleotide incorporation assay using 5′-nicked substrates. In the absence of an MMR target or in MMR-deficient extracts (−MMR), very little repair-mediated [32P]dNMP incorporation takes place. In the presence of an MMR target in MMR-proficient extracts (+MMR), MMR-mediated [32P]dNMP incorporation can be detected upon NotI/BsaI digestion of the phagemid DNA in both the 1808-bp fragment a and 1387-bp fragment b. Positions of the modified base pairs/mispairs in the plasmid are as follows: GO/A, 3183; GO/C, 3188; G/T, 45. B, Western blot analysis of HCT116 (MMR-deficient) and HCT116 + chromosome 3 (MMR-proficient) nuclear cell extracts to verify the presence and relative abundance of MMR (MSH6, MSH2, and MLH1) and BER (MYH, OGG1, and MTH1) proteins. The absence of MutLα in HCT116 cells is well documented (27). Where indicated, the extracts were complemented with the respective purified recombinant proteins during the MMR assays. C, GO/A and GO/C base pairs are not efficient substrates for MMR. 5′-Nicked substrates containing either no mismatch (lane 1), a G/T (lane 2) or GO/A (lane 3) mismatch, or a GO/C base pair (lane 4) 306–363 nucleotides away from the nick were incubated with HCT116 nuclear cell extracts supplemented with purified recombinant MutLα and [α-32P]dATP. Upon NotI/BsaI restriction digestion, GelRed-stained DNA served as the DNA loading control, whereas the autoradiograph visualized MMR-dependent repair tracts in fragments a and b by means of [32P]dAMP incorporation. M, molecular size marker (1-kb ladder; New England Biolabs).
BER Assay
To determine AO/G to C/G or AO/C to G/C repair by MYH-dependent BER, the substrates were incubated with the extracts as described for the MMR assay. After the reaction, 50% of the purified, eluted substrate was digested with SalI/DraI (MMR assay), whereas the other half was used for PvuI digestion (BER assay).
RESULTS
GO/A and GO/C Mispairs Are Not Addressed by Canonical MMR
BER and MMR are mechanistically distinct biochemical pathways. They differ principally in their substrate specificity and in repair patch size. The specificity of BER is dictated by the enzymes that initiate the process, DNA glycosylases, which recognize and excise a limited number of damaged or modified bases (32). The repair process is thus initiated at the site of the base modification and involves the replacement of one (short patch BER) (33) or only 2–6 (long patch BER) nucleotides (34, 35). Canonical MMR addresses non-Watson-Crick base pairs that arise during replication, but in contrast to BER, the excision process initiates at a site distal to the mispair at a strand discontinuity that marks the nascent DNA strand. The repair tracts can thus be several hundred nucleotides long (36, 37). Because we wished to study the potential interplay of BER and MMR in the processing of substrates that could conceivably be addressed by both repair systems, we first needed to learn whether both repair pathways were active in our cell extracts and to establish experimental conditions that would allow us to differentiate between the two processes (Figs. 1A, 2B, and 5F). We first tested whether MMR was active in our cell extracts. To this end, we generated covalently closed circular phagemid substrates containing a single G/T, GO/C, or GO/A base pair or a control substrate containing a G/C at the same site. By subsequent nicking of the substrates at their Nt.Bst.NBI sites, we generated an initiation site for MMR (Fig. 1A). We then incubated the nicked substrates with extracts of MutLα-deficient HCT116 human cells (Fig. 1B) that were supplemented with purified recombinant MutLα and [α-32P]dATP. Following recovery of the phagemids, restriction digestion with NotI and BsaI, and separation of the fragments on agarose gels, we anticipated that long patch repair events (MMR) would give rise to heteroduplexes labeled in both fragments a and b (Fig. 1A).
FIGURE 2.

MYH is active in HCT116 extracts and addresses GO/A but not AO/G or AO/C mispairs. A, nicking assay. The closed-circular homoduplex or the GO/A, AO/G, or AO/C substrates were incubated with recombinant, purified MYH-GST and APE1 for the indicated times and analyzed by agarose gel electrophoresis. The mobility of the closed-circular (cc), open-circular (oc), and linear (l) DNA molecules is indicated on the left. The quantification shows the percentage of open-circular substrate compared with the total amount of DNA (open-circular + closed-circular). Only the GO/A substrate was cleaved with appreciable efficiency. B, schematic representation of the MYH-dependent nucleotide incorporation assay. Short patch BER should replace the mispaired A with a radiolabeled C (asterisk). This should result in detectable radioactivity in band a of the NotI/BsaI digest. C, BER-dependent incorporation assay. The supercoiled homoduplex (lanes 1–3), GO/A (lanes 4–6), and AO/G (lanes 7–9) substrates were incubated with HCT116 extracts for the indicated time points. The reactions were supplemented with [α-32P]dCTP. As visualized in the autoradiograph, [32P]dCMP incorporation into fragment a of a NotI/BsaI-digested phagemid, indicative of short patch repair, was detected above background only in the GO/A substrate. D, the supercoiled homoduplex (lanes 1–3) and AO/C (lanes 4–6) substrates were incubated with the extracts for the indicated time points in reactions supplemented with [α-32P]dGTP. Only low levels of radioactive nucleotide were incorporated into fragment a of a NotI/BsaI-digested AO/C phagemid, indicative of inefficient BER. M, molecular size marker.
FIGURE 5.

A GO/C base pair does not act as an initiation site for MMR in nuclear extracts of human cells. A, schematic representation of the GO/C-G/T substrate used in the in vitro MMR assay. The circular heteroduplex substrate carries a GO/C base pair 54 nucleotides from a G/T mismatch in the recognition site of AclI endonuclease. The positions of two further AclI cleavage sites and the Nt.BstNBI site where a nick can be introduced selectively into the outer strand are indicated. In the absence of repair, digestion of the phagemid with AclI gives rise to fragments of 2824 (fragments c + d) and 373 bp (fragment e). Repair of the G/T mismatch to A/T regenerates a third AclI restriction site such that the phagemid DNA is cleaved into three fragments of 1518 (fragment c), 1306 (fragment d), and 373 bp (fragment e). B, substrates G/T (lanes 1 and 2), nicked G/T (lanes 3 and 4), GO/C-G/T (lanes 5 and 6), and GO/C-A/T (lanes 7 and 8) were incubated with extracts of HCT116 cells supplemented (+) or not (−) with purified recombinant MutLα and analyzed on a GelRed-stained agarose gel. The autoradiograph visualizes [32P]dAMP incorporation into the different substrate fragments. The indicated MMR efficiencies (%) were estimated by ImageQuant from scans of GelRed-stained agarose gels and represent an average of three independent experiments. Error bars represent S.D. Only background levels of repair were detected in the GO/C-G/T substrate (lane 6). C, OGG1-generated DNA termini at GO/C sites act as MMR initiation sites. The G/T phagemid substrate was preincubated with Nt.BstNBI (lane 2), and the GO/C-G/T phagemid was preincubated with recombinant, purified OGG1 (lane 4) to generate nicked, open-circular substrates (see E). The substrates were then purified and incubated with LoVo nuclear cell extracts supplemented with purified MutSα (lanes 1–4). The closed-circular G/T (lane 1) and GO/C-G/T (lane 3) phagemid substrates served as controls. The OGG1 prenicked GO/C-G/T substrate (lane 4) was at least as efficiently repaired as the positive control, nicked G/T (lane 2). D, Western blot analysis of the relative abundance of MMR (MSH6, MSH2, and MLH1) and BER (MYH, OGG1, and MTH1) proteins in nuclear extracts of HCT116 and LoVo cells. The absence of MutLα in HCT116 cells and of MutSα in LoVo cells is evident. Where required, these extracts were complemented with the purified recombinant proteins. E, the GO/C-G/T phagemid substrate was preincubated with purified recombinant OGG1 (lane 2) to give rise to an open-circular (oc) substrate, which migrates in the 1% agarose gel in the same position as the Nt.BstNBI-nicked G/T substrate (lane 1). These prenicked substrates were purified and used in the MMR assay shown in C. F, schematic representation of the OGG1-dependent nucleotide incorporation assay. Short patch BER should replace the oxidized G with a radiolabeled G (asterisk). This should result in detectable radioactivity in band a of the NotI/BsaI digest. G, absence of OGG1 activity in human nuclear cell extracts. Upper panel, homoduplex (lanes 1–3) or GO/C (lanes 4–6) substrate was incubated with LoVo extracts supplemented with [α-32P]dGTP for the indicated times. As seen in the autoradiograph, both substrates incorporated similar amounts of [32P]dGMP that were close to background. H, Western blot showing the amount of OGG1 in 100 μg of LoVo nuclear extract used in one MMR reaction (lane 1) and the amount of recombinant purified OGG1-GST required to supplement the reaction (lane 2). I, a GO/C base pair activates MMR in LoVo extracts supplemented with purified, recombinant OGG1-GST. The nicked G/T (lane 1) and the GO/C-G/T (lanes 2–3) substrates were incubated with LoVo nuclear cell extracts and purified MutSα supplemented (+) or not (−) with an amount of purified recombinant (Rec.) OGG1-GST shown in H, lane 2. H and I show representative images and quantifications of three independent experiments. M, molecular size marker.
As shown in Fig. 1C, intense radiolabeling was seen only in the bona fide MMR substrate G/T (lane 2). That both fragments a and b were labeled with similar intensity indicates that the repair patch spanned at least the distance of 361 nucleotides between the nick and the mispair. Only background amounts of [32P]dAMP were detected in fragments a and b of the control homoduplex, GO/A, or GO/C substrates (lanes 1, 3, and 4), which confirmed that the GO/C and GO/A pairs failed to activate nick-dependent long patch excision and resynthesis characteristic of MMR (38, 39) and exemplified in lane 2.
MYH-dependent BER in Human Nuclear Cell Extracts
Having shown that MMR was active in the extracts, but that it did not address the GO/A and GO/C substrates, we wanted to see whether they were processed by BER. GO/A repair should be initiated by MYH, which should remove the mispaired adenine. The resulting apurinic site should then be incised by APE1. We therefore asked whether incubation of the supercoiled GO/A substrate with purified, recombinant MYH-GST and APE1 gave rise to nicked circular molecules. As shown in Fig. 2A (lanes 3–6), the GO/A substrate was efficiently converted to the open-circular form upon incubation of the plasmid with the recombinant proteins.
MYH has been reported to excise (in addition to adenine from mispairs with GO) also the oxidation product of adenine, AO, from mispairs with guanine or cytosine (23, 24). We therefore included the covalently closed AO/G and AO/C substrates in this assay. As shown in Fig. 2A, only limited nicking was detected on the AO/G (lanes 7–10) and AO/C (lanes 11–14) substrates with only 42 and 38% of the plasmids having been nicked after 8 h, respectively. This was only slightly above the nonspecific nicking levels observed on the homoduplex (26%).
We next set out to test BER activity in the extracts. MYH-initiated BER of GO/A should give rise to GO/C. Thus, upon incubation of the covalently closed GO/A substrate with extracts supplemented with [α-32P]dCTP, specific incorporation of [32P]dCMP into fragment a of the substrate (Fig. 2B) should be detectable. This was indeed the case (Fig. 2C, lanes 4–6). As in the case of the purified enzymes, after incubation of the AO/G (Fig. 2C, lanes 7–9) or AO/C (Fig. 2D, lanes 4–6) substrate with [α-32P]dCTP- or [α-32P]dGTP-supplemented HCT116 extracts, respectively, the amount of radionucleotide incorporated into the AO-containing fragment a of the two substrates was only slightly greater than that detected in the homoduplex substrate.
MYH-dependent Mismatch Repair in Human Nuclear Cell Extracts
Having obtained preliminary evidence that both MYH-dependent BER and MMR were active in the extracts, we next wanted to learn whether there was a cross-talk between these processes. To address this question, we deployed a phagemid substrate containing a G/T mismatch within the unique SalI restriction site (G/T; covalently closed or nicked with Nt.BstNBI 376 nucleotides 3′ from the mispaired T), a phagemid containing a single GO/A mispair in addition to the G/T (GO/A-G/T), and a control phagemid (GO/A-G/C). Upon incubation with the cell extracts, the recovered phagemids were digested with SalI and DraI to give rise to the pattern of bands shown in Fig. 3A. Successful repair of the G/T mismatch to G/C regenerates the SalI site, and the plasmid is cut into four instead of three fragments (the smallest DraI fragment runs out of the gel and is therefore not seen). This process should be dependent on the presence of a nick in the strand containing the mispaired T as canonical MMR requires a strand discontinuity for initiation of the EXO1-dependent excision step (40, 41).
FIGURE 3.

A single GO/A base pair in a DNA heteroduplex can act as an initiation site for MMR in human nuclear cell extracts. A, schematic representation of the GO/A-G/T substrate and the in vitro MMR assay. The substrate carries a GO/A mispair 57 nucleotides away from a G/T mismatch, which is located in a SalI recognition site. The presence of the mismatch makes the phagemid refractory to cleavage with the enzyme such that incubation with SalI and DraI yields fragments c + d (2484 bp) and e (692 bp). (The third, 19-bp-long fragment is not detectable on 1% agarose gels stained with GelRed.) The presence of a nick in the inner strand, introduced either by Nt.BstNBI or through BER-catalyzed incision of the A strand results in a repair of the G/T mismatch to G/C in human nuclear cell extracts that regenerates the SalI site. Upon incubation with SalI and DraI, the repaired phagemid gives rise to fragments c (1328 bp), d (1156 bp), and e (692 bp). B, GO/A mispairs serve as initiation sites for MMR. The MMR assay shows the efficiency of repair of a G/T mismatch in closed-circular G/T (lane 1), nicked G/T (lanes 2 and 3), closed-circular GO/A-G/T (lanes 4 and 5), and closed-circular GO/A-G/C (lanes 6 and 7) substrates that were incubated with nuclear extracts of MutLα-deficient HCT116 cells supplemented with purified MutLα (+) where indicated. The autoradiograph shows MMR-dependent [32P]dAMP incorporation into the different substrate fragments. C, G/T repair in the GO/A-G/T substrate is dependent on MYH. The closed-circular G/T (lanes 1–3), nicked G/T (lanes 4–6), and closed-circular GO/A-G/T (lanes 7–9) substrates were incubated with MutLα-supplemented extracts of HCT116 cells, which were either mock-depleted (lanes 1, 4, and 7), MYH-depleted (lanes 2, 5, and 8), or MYH-depleted and supplemented with recombinant MYH-GST (lanes 3, 6, and 9). The efficiency of the repair reactions shown in B and C was estimated by ImageQuant from scans of GelRed-stained agarose gels. The indicated MMR efficiencies (%) represent an average of three independent experiments. Error bars represent S.D. D, Western blot showing MYH immunodepletion efficiency of HCT116 nuclear cell extracts and the amount of recombinant MYH-GST used to supplement the depleted extracts. E, MYH/APE1-generated DNA termini at GO/A sites are recognized by MMR. Upper panel, GO/A-G/T phagemid (lane 3) was incubated with purified recombinant MYH-GST and APE1 (lane 3), and the G/T substrate either closed-circular (lane 1) or prenicked with Nt.BstNBI (lane 2) was mock-incubated (lanes 1 and 2) under the same experimental conditions. Lower panel, the pretreated substrates were purified and incubated with HCT116 nuclear extracts supplemented with MutLα. Subsequently, they were digested with SalI/DraI, and repair efficiencies were analyzed on a GelRed-stained agarose gel and quantified. The G/T mispair in the MYH/APE1-pretreated GO/A-G/T substrate was efficiently repaired. The panel shows a representative image of two independent experiments. F, schematic representation of a BER assay using phagemid substrates containing AO mispaired with guanine or cytosine. The AO residue is located within a PvuI restriction site, which makes the phagemid refractory to cleavage with this enzyme. AO/G to C/G or AO/C to G/C repair by BER restores the PvuI site. Incubation with the enzyme gives rise to 1846-bp fragment f and 1351-bp fragment g. Positions are as follows: AO/G, 3060; AO/C, 3057. G, AO/G and AO/C mispairs do not serve as MMR initiation sites. The indicated substrates were incubated with MMR-deficient HCT116 extracts supplemented (+) or not (−) with purified MutLα. Upper panel, the efficiency of G/T to G/C (lanes 1–3) or T/G to C/G (lanes 4 and 5) repair mediated by MMR was estimated by SalI/DraI restriction digestion of the following recovered substrates: closed-circular GO/A-G/T (lane 1), closed-circular G/T (lane 2), nicked G/T (lane 3), closed-circular AO/G-T/G (lane 4), and closed-circular AO/C-T/G (lane 5). Lower panel, an aliquot of the recovered phagemids was digested with PvuI, which detects MYH-dependent BER of AO to C in the AO/G substrate or of AO to G in the AO/C substrate. The nicked G/T substrate (lane 3) served as the positive control for PvuI digestion. AO/G-T/G and AO/C-T/G substrates that were not incubated with the extracts (lanes 6 and 7) contain defective PvuI sites and thus served as negative controls. MMR and BER efficiencies (%) are indicated below the panels. M, molecular size marker.
When the above substrates were incubated with MMR-deficient HCT116 nuclear cell extracts supplemented with [α-32P]dATP, only background levels of repair and [32P]dAMP incorporation were detected in the bona fide MMR substrate, nicked G/T (Fig. 3B, lane 2). However, the same substrate was repaired with high efficiency when the extracts were supplemented with purified recombinant MutLα (lane 3), particularly when compared with the unnicked control G/T plasmid (lane 1), which was processed to a limited extent by non-canonical MMR (42). Strikingly, although only low levels of [32P]dAMP incorporation were detected in this system when the closed-circular GO/A-G/C control substrate was used (lanes 6 and 7), around 60% of the covalently closed GO/A-G/T phagemid were repaired in the extract supplemented with recombinant MutLα (lane 5). This result implied that A opposite GO can serve as a cryptic strand discrimination signal for MMR.
We postulated that the MMR machinery might be hijacking intermediates of GO/A processing for EXO1 loading. We therefore set out to confirm that the above described phenomenon was dependent on MYH by immunodepleting it from the extract (Fig. 3C) prior to incubation with the substrates. This reduced MMR efficiency on the GO/A-G/T substrate from 52 (lane 7) to ∼20% (lane 8). The latter level was comparable with the efficiency of non-canonical MMR on the covalently closed G/T substrates (∼30%) (lanes 1–3). That the observed inhibition was due to MYH depletion was confirmed by complementation of the depleted extracts with purified, recombinant MYH-GST (Fig. 3D), which restored the MMR efficiency on the GO/A-G/T substrate to 43% (Fig. 3C, lane 9). As anticipated, MMR efficiencies on the G/T (lanes 1–3) and nicked G/T (lanes 4–6) substrates were unaffected by the amount of MYH-GST in the extracts.
To confirm that processing of GO/A intermediates by the BER machinery indeed generates DNA termini that can be utilized by EXO1 in the strand degradation step of MMR, we preincubated the closed-circular GO/A-G/T substrate with purified, recombinant MYH-GST and APE1 (Fig. 3E, upper panel, lane 3), which cleaves the sugar-phosphate backbone at the MYH-generated abasic site. We then incubated the MYH/APE1-nicked substrate with HCT116 extracts supplemented with MutLα. As shown in Fig. 3E (lower panel), the MYH/APE1-generated nick in the GO/A-G/T substrate (lane 3) was efficiently recognized by MMR factors. That the latter substrate was even more efficiently repaired than the Nt.BstNBI-nicked G/T phagemid (lane 2) is most likely indicative of the shorter distance between the nick and the mispair in the GO/A-G/T substrate (54 nucleotides) as compared with the Nt.BstNBI-nicked G/T phagemid (361 nucleotides).
We extended the in vitro MMR assays also to substrates containing an AO/G or an AO/C pair in the vicinity of a T/G mismatch. These substrates differed from those described above inasmuch as the AO residues were located within a PvuI site (see “Experimental Procedures”). Successful repair of AO/G to C/G or of AO/C to G/C by BER would restore the restriction site such that PvuI digestion of the recovered DNA would allow quantification of BER activity at the AO sites (Fig. 3F). As anticipated from the absence of detectable BER activity on these substrates, incubation of the AO/G-T/G and AO/C-T/G substrates with MMR-proficient HCT116 extracts supplemented with recombinant MutLα yielded only 15 and 12% PvuI-sensitive products, respectively (Fig. 3G, lower panel, lanes 4 and 5), whereas the G/T phagemid was completely digested under identical conditions (lane 3). These results were reflected in MMR efficiency determined from the same assays by digestion of the recovered phagemids with SalI/DraI (Fig. 3G, upper panel). Repair of the T/G mismatch in the AO/G-T/G and AO/C-T/G substrates was very inefficient (20 and 18%, respectively), whereas the nicked G/T (lane 3) and the covalently closed GO/A-G/T (lane 1) substrates were efficiently repaired (93 and 59%, respectively). Taken together, these data show that AO/G or AO/C pairs are inefficiently processed by MYH in cell extracts and therefore fail to act as entry sites for mismatch-activated excision.
GO/A Mispairs Act as MMR Initiation Sites also in X. laevis Egg Extracts
Because GO/A mispairs arise during replication, MYH should be able to act on newly replicated DNA, and indeed, available experimental evidence shows that this glycosylase is more abundant during S phase (43). In an attempt to learn whether MYH is also more efficient during S phase, we decided to make use of X. laevis egg extracts, which are enriched in S phase proteins (44).
In the first experiment, we wanted to learn whether the extracts were proficient in MYH-initiated BER. We decided to make use of the assay described above (Fig. 2B) in which the homoduplex or GO/A closed-circular substrates were incubated with X. laevis egg extracts supplemented with [α-32P]dCTP. Digestion of the recovered phagemid DNA with NotI/BsaI revealed a 3-fold greater incorporation of [32P]dCMP into the GO-containing fragment of the GO/A substrate than into the corresponding fragment of the homoduplex phagemid (Fig. 4A). This demonstrated that MYH-dependent BER was active in the X. laevis extracts.
FIGURE 4.
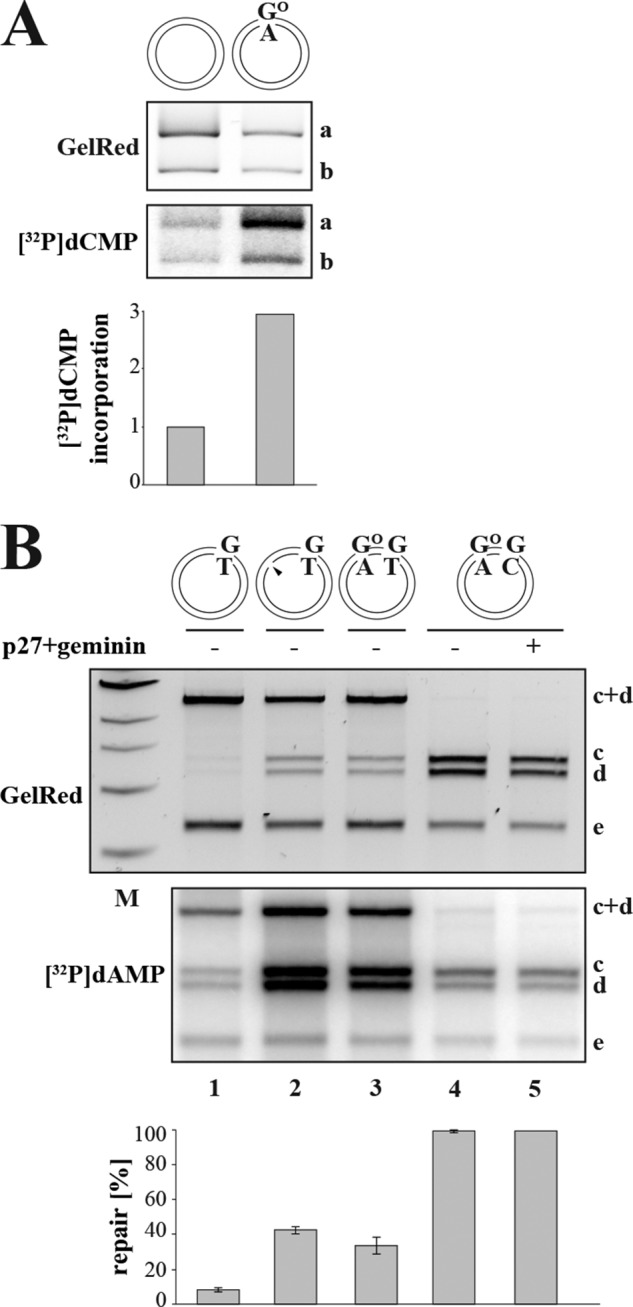
Efficiency of MMR initiation at GO/A sites in X. laevis egg extracts. A, MYH is active in X. laevis eggs. Closed-circular homoduplex or GO/A substrate was incubated with X. laevis egg extracts supplemented with [α-32P]dCTP and subsequently digested with NotI/BsaI (see Fig. 1A). Although only background levels of [32P]dCMP were incorporated into the homoduplex substrate (lane 1), substantial incorporation into the 1808-bp fragment a of the GO/A substrate was detected (lane 2), which is indicative of base excision repair. For quantification, [32P]dCMP incorporation into the homoduplex substrate was set to 1. The panel shows a representative image of two independent experiments. B, GO/A serves as an initiation site for MMR in X. laevis egg extracts. G/T (lane 1), nicked G/T (lane 2), GO/A-G/T (lane 3), or GO/A-G/C (lanes 4–5) substrate was incubated with X. laevis egg extracts. Upon SalI/DraI digestion of the recovered phagemids, MMR efficiencies were estimated from the GelRed-stained agarose gels, whereas MMR-dependent DNA synthesis was visualized by [32P]dAMP incorporation seen in the autoradiograph. A control reaction (lane 5) contained the replication inhibitors p27 and geminin; because [32P]dAMP incorporation was similar in the presence and absence of these inhibitors (cf. lanes 4 and 5), the plasmid was apparently not replicated in the extracts (52). The indicated MMR efficiencies (%) represent an average of three independent experiments. Error bars represent S.D. M, molecular size marker.
We also had to show that the extracts supported nick-directed MMR because an earlier report indicated that mismatch processing in X. laevis oocyte extracts was efficient but not nick-directed (45). We therefore incubated the covalently closed and the nicked isoforms of the G/T phagemid with the extracts supplemented with [α-32P]dATP. As shown in Fig. 4B, we were able to detect nick-directed MMR on the G/T substrate (lane 2), whereas the covalently closed phagemid was only inefficiently processed (lane 1). Under these conditions, the covalently closed GO/A-G/T substrate (lane 3) was almost as efficiently repaired as the nicked G/T phagemid (lane 2). Because extracts of X. laevis eggs support plasmid replication under certain conditions, which would result in considerable [32P]dAMP incorporation and conversion of 50% of the G/T mismatches to G/C in the absence of MMR, we carried out a control experiment in extracts supplemented with the DNA replication inhibitors p27 and geminin. No detectable differences between the levels of radionucleotide incorporation in the presence and absence of these inhibitors were observed, indicating that the plasmid did not replicate in the assay (cf. lanes 4 and 5).
OGG1-dependent Mismatch Repair Is Inefficient in Human Nuclear Cell and X. laevis Egg Extracts
Assuming that MTH1 hydrolyzes oxidized dGTP in the nucleotide precursor pool with a 100% efficiency, all GO residues in oxidized DNA should be in the template strand following replication irrespective of whether they are paired with A or C. As discussed above, MYH-initiated BER of GO/A mispairs arising during replication would be directed to the A strand that is also the nascent DNA strand. In contrast, BER-mediated repair of GO/C pairs arising during replication would have the opposite effect on MMR because OGG1-dependent GO/C repair would introduce breaks in the template DNA strand and thus provide MMR with an incorrect strand bias.
We set out to test the above hypothesis by studying the effect on MMR efficiency and directionality of a GO/C pair situated in the vicinity of a G/T mismatch. We generated a GO/C-G/T substrate in which the GO was positioned 54 nucleotides 5′ from the mispaired G. OGG1-initiated BER of the GO would give rise to a break that should activate MMR to correct the G/T mismatch to A/T and thus regenerate an AclI site in the substrate. This repair bias should be identical to that introduced by a Nt.Bst.NBI-generated nick in the G/T substrate (Fig. 5A).
When we incubated the nicked G/T substrate or the covalently closed G/T, GO/C-G/T, or GO/C-A/T phagemid heteroduplexes with HCT116 nuclear cell extracts supplemented with [α-32P]dATP, no significant repair or [32P]dAMP incorporation was observed (Fig. 5B, lanes 1, 3, 5, and 7), but when the extract was supplemented with purified, recombinant MutLα, the Nt.BstNBI-nicked G/T substrate was repaired (lane 4). In contrast to what was observed with the GO/A-G/T substrate (Fig. 3B, lane 5), only background levels of repair and [32P]dAMP incorporation were detected with the GO/C-G/T substrate (Fig. 5B, lane 6) and with the control, covalently closed G/T phagemid (lane 2). Similar results were obtained also with MutSα-deficient LoVo extracts supplemented with purified recombinant MutSα (Fig. 5C, lanes 1–3). Given that both extracts contained the key MMR and BER factors as ascertained by Western blotting (Figs. 1B and 5D), these results at first suggested that GO processing failed to activate MMR on the GO/C-G/T substrate.
The above result could be explained in several ways. We first considered the possibility that the MMR machinery was unable to use the BER intermediates generated during GO processing as strand discrimination signals because OGG1 is a DNA glycosylase/lyase that generates a strand break on the 3′ side of the abasic site (46, 47). APE1-mediated cleavage at its 5′ side then generates a single nucleotide gap rather than just a break at the 5′ side of the abasic site as is the case for MYH/APE1. We therefore treated the covalently closed GO/C-G/T substrate with purified recombinant OGG1-GST fusion protein. As shown in Fig. 5E (lane 2), the substrate was as efficiently nicked as the G/T control with Nt.BstNBI (lane 1). When the OGG1-nicked phagemid was incubated with extracts of LoVo cells supplemented with purified recombinant MutSα, it was repaired with efficiency similar to that of the control, nicked G/T phagemid (Fig. 5C, cf. lanes 2 and 4). This shows that MMR can use OGG1-generated strand breaks as initiation sites.
The second possibility was that OGG1 in the tested cell extracts was inactive or that it was present in insufficient amounts. We therefore incubated the GO/C and homoduplex substrates with LoVo extracts supplemented with [α-32P]dGTP to test for OGG1-dependent BER, which should result in [32P]dGMP incorporation into fragment a (Fig. 5F). As shown in Fig. 5G, only background levels of the radiolabeled nucleotide were incorporated into fragment a of the phagemid heteroduplex, suggesting that GO/C repair was indeed inefficient despite the fact that the extracts contained readily detectable amounts of OGG1 (Figs. 1B and 5D). Similar results were obtained with 32P-labeled GO/C-containing oligomers, which were inefficiently processed in LoVo extracts, whereas processing of U/G-containing oligomers was very efficient. Moreover, purified, recombinant OGG1 was able to process the GO/C oligonucleotide substrate with ∼50% cleavage of the GO strand seen after only 10 min (data not shown).
To test whether the LoVo extracts contained an inhibitor of OGG1, we supplemented them with an amount of purified recombinant OGG1-GST that was comparable with that of the endogenous protein present in the nuclear extracts (Fig. 5H). Under these conditions, the GO/C-G/T substrate (Fig. 5I, lane 3) was repaired with efficiency similar to that of the nicked G/T phagemid (lane 1).
Importantly, the above described phenomenon was not limited to extracts of human cells as identical results were obtained with the X. laevis MMR system (Fig. 6). The reason underlying the low OGG1 activity in the extracts is currently unknown, but we were able to eliminate inappropriate salt concentration, lack of an activator protein in the cytoplasmic fraction, and short half-life of OGG1 in our assay as possible causes (data not shown).
FIGURE 6.
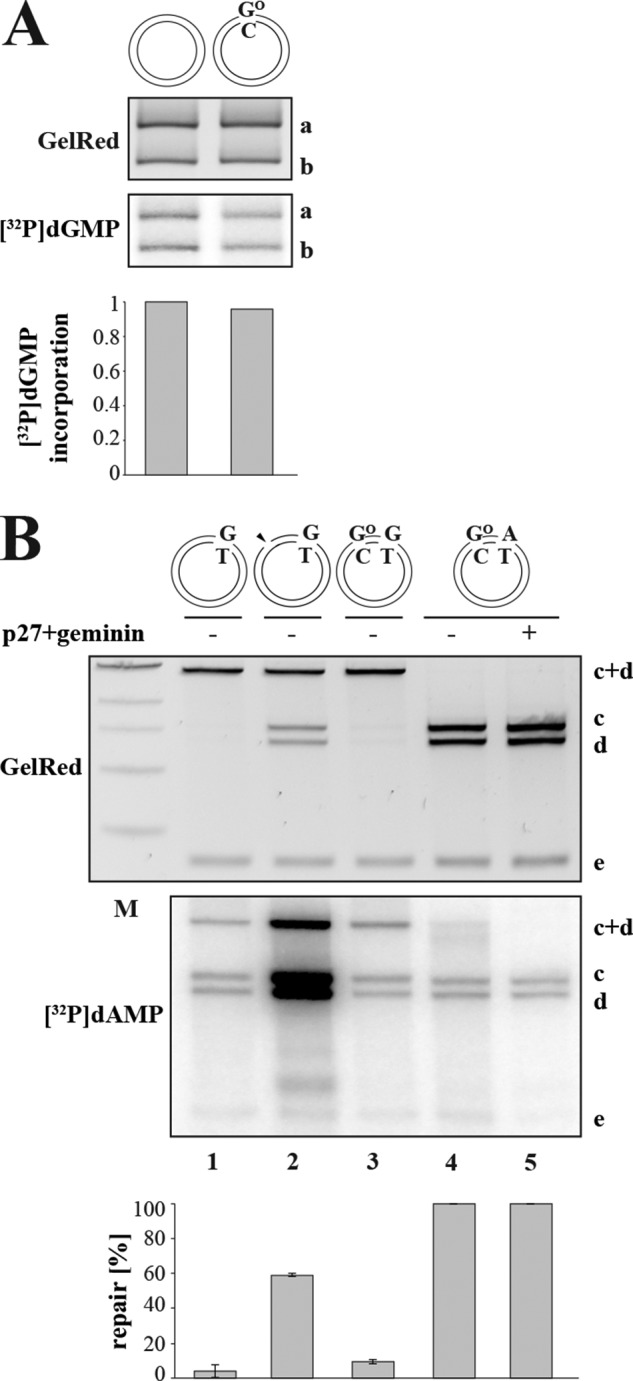
A GO/C base pair does not act as an initiation site for MMR in X. laevis egg extracts. A, BER-dependent incorporation assay. X. laevis egg extracts lack OGG1 activity as only background levels of [32P]dGMP were incorporated into fragment a of the control homoduplex (lane 1) and the GO/C (lane 2) substrates. OGG1-dependent BER efficiencies were quantified from the intensity ratio between band a in the autoradiograph and the GelRed-stained agarose gel using ImageQuant. The panel shows a representative image of two independent experiments. B, the MMR activity in X. laevis egg extracts was determined using closed-circular G/T (lane 1), nicked G/T (lanes 2), GO/C-G/T (lane 3), and GO/C-A/T (lanes 4 and 5) substrates. That similar background levels of [32P]dAMP were incorporated into the control GO/C-A/T substrate in the absence (lane 4) and presence (lane 5) of the replication inhibitors p27 and geminin demonstrates that the phagemids were not replicated under these conditions (52). The panel shows a representative image of three independent experiments. Their average quantitation is shown below. Error bars represent S.D. M, molecular size marker.
DISCUSSION
The postreplicative mismatch repair system improves the fidelity of DNA replication by several orders of magnitude through removing from nascent DNA nucleotides that fail to form Watson-Crick base pairs. To fulfill this function, it has to satisfy two key criteria: it has to (i) recognize base-base mismatches and small insertion/deletion loops generated by the replicative polymerases during DNA synthesis and (ii) direct the repair process to the newly synthesized DNA strand. How the major mismatch recognition factor MutSα recognizes the different helical distortions that are caused by purine/purine, purine/pyrimidine, and pyrimidine/pyrimidine mispairs as well as by insertion/deletion loops is still poorly understood despite the fact that several structures of protein-DNA complexes exist (48). However, because MMR deficiency leads to transition, transversion, and frameshift mutations, MutSα clearly has broad substrate specificity. Although this characteristic of MutSα is beneficial as far as its role in the maintenance of replication fidelity is concerned, it might also be deleterious should MutSα bind to lesions that ought to be processed by other repair systems. One such example is mispairs containing O6-methylguanine; these lesions activate MMR, but because their processing does not lead to the removal of the modified nucleotide from the template strand, they trigger futile repair that eventually leads to cell death (1). Processing of GO/A mismatches arising through the incorporation of dAMP opposite GO in the template strand might have also been expected to trigger futile MMR, but the in vivo evidence for such a process is currently lacking, and in vitro data showing that GO/A mispairs are very poorly addressed by the MMR system in human cell extracts (this study and Ref. 39) would appear to argue against it. However, in vivo, the MMR system appears to remove from the nascent strand GO misincorporated opposite template A (49, 50) and to increase the efficiency of MYH-dependent BER at least in vitro (51). Thus, available experimental evidence suggests that MMR supports BER during oxidative DNA damage processing. In the present study, we asked whether the reverse might also be true, namely whether BER-dependent processing of GO affects MMR efficiency.
In our earlier studies, we showed that the MMR system could use breaks generated during the BER-mediated processing of uracil residues (7, 42) or during the RNase H2-mediated excision of ribonucleotides misincorporated into DNA during replication (9) as strand discrimination signals. We postulated that MMR might also use breaks generated during the BER-dependent processing of oxidative damage. However, the outcome of such interference would be positive only in the case that the breaks were generated in the nascent DNA strand. We show here that MYH-dependent processing of a GO/A mispair in extracts of human cells or of Xenopus laevis eggs directs MMR to the A strand (Figs. 3 and 4). In vivo, the ability of the MMR system to use breaks generated during the MYH-directed processing of GO/A mispairs arising through the incorporation of dAMP opposite GO would direct mismatch correction to the nascent strand and thus improve the fidelity of replication (Fig. 7, right panel). MYH-dependent BER and MMR would thus synergize during S phase as might have been anticipated from the physical interaction of some of the subunits of these pathways (51). It could be argued that the number of GO/A mispairs arising in the vicinity of a replication error might be too low to affect MMR efficiency. However, this number likely increases substantially when strand discontinuities arising during the processing of other “non-standard” nucleotides (uracils and thymines arising through deamination of cytosine and 5-methylcytosine, respectively; ribonucleotides; methylpurines; thymine glycols; etc.) are considered. Indeed, as we showed previously, ribonucleotide processing alone is sufficient to affect MMR efficiency in vivo (9).
FIGURE 7.

Schematic representation of the BER and MMR interplay in GO metabolism. Left panel, should dGOMP be incorporated into the nascent strand (red) opposite C, it could be removed by OGG1-dependent BER or by MMR without deleterious consequences. In contrast, should Atemplate/GOnascent mispairs arise during replication, MYH-mediated BER to C/GO would give rise to A to C mutations, whereas MMR would process the GO-containing strand and prevent mutagenesis. In cases where the oxidized base was in the vicinity of a replication error (e.g. a G/T mispair), BER intermediates of Ctemplate/GOnascent processing would direct MMR to the correct strand, whereas MYH-initiated BER of Atemplate/GOnascent would misdirect MMR to the parental template strand (black). However, the likelihood of incorporation of dGOMP into the nascent DNA strand is minimized by hydrolysis of dGOTP in the nucleotide pool by MTH1. Most GO residues in the DNA should therefore be in the template strand as shown in the right panel. Right panel, BER of GO/C pairs would lead to the removal of the oxidized base, but during S phase, the OGG1-generated strand break might result in replication fork collapse or in the misdirection of MMR to the template strand. Processing of GOtemplate/Anascent mispairs by MYH-dependent BER or by MMR would have no deleterious consequences. Moreover, in cases where the oxidized base was in the vicinity of a replication error (e.g. a G/T mispair), BER intermediates of GOtemplate/Anascent processing would direct MMR to the correct strand.
Breaks introduced into the nascent DNA strand by OGG1 during the processing of C/GO pairs arising through the incorporation of dGOMP opposite template C would also help improve MMR efficiency and replication fidelity. However, due to the sanitization of dNTP pools by MTH1, dGOTP concentrations should be extremely low such that dGOMP incorporation into nascent DNA during replication should be minimal (Fig. 7, left panel). As a result, most GO residues present in DNA during replication should be in the template strand. Incision of this strand by OGG1 behind the replication fork would misdirect MMR to the wrong strand (Fig. 7, right panel), and OGG1-catalyzed incision of DNA at GO/C sites in front of the replication fork could cause its collapse. It would therefore appear logical that GO/C processing should be completed prior to the onset of S phase and that OGG1 should be inactivated during this cell cycle stage. Interestingly, we found that GO/C processing in both in vitro systems was inefficient and that the presence of a GO/C pair in the heteroduplex substrate failed to activate MMR of the G/T mismatch (Figs. 5 and 6) despite the fact that OGG1 was present in considerable quantities (Figs. 1B and 5, D and H). Because addition of the recombinant polypeptide to the extracts resulted in GO-dependent G/T repair (Fig. 5I), we postulate that OGG1 present in the cell extracts is inactive either as a result of post-translational modifications or through complexation with an inhibitor. We are currently attempting to identify the underlying cause of this inhibition as well as carrying out a series of in vivo experiments that should show whether the observations described above correspond to the situation in living cells. Our current findings indicate that the repair of oxidative damage in vertebrate cells is highly regulated to prevent genomic instability, and future experiments should show how this regulation is mediated at the molecular level.
Acknowledgments
We are grateful to Mariela Artola-Borán for preparation of the phagemid substrates, Marie-Christine Weller for critical reading of the manuscript, Barbara van Loon for the gift of OGG1-GST and the bacterial MYH-GST expression vector, and to Yusaku Nakabeppu for the generous gift of the anti-MTH1 antibody.
This work was supported by Swiss National Science Foundation Grants 310030B-133123 and 31003A-149989 and European Research Council Grant “MIRIAM” 294537.
- MMR
- mismatch repair
- MYH
- MutY homolog
- GO
- 8-oxoguanine
- OGG1
- 8-oxoguanine DNA glycosylase
- BER
- base excision repair
- MTH1
- MutT homolog 1
- AO
- 2-hydroxyadenine
- APE1
- apurinic endonuclease 1
- TFIIH
- transcription factor II H.
REFERENCES
- 1. Jiricny J. (2006) The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 7, 335–346 [DOI] [PubMed] [Google Scholar]
- 2. Lahue R. S., Modrich P. (1988) Methyl-directed DNA mismatch repair in Escherichia coli. Mutat. Res. 198, 37–43 [DOI] [PubMed] [Google Scholar]
- 3. Modrich P., Lahue R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133 [DOI] [PubMed] [Google Scholar]
- 4. Peña-Diaz J., Jiricny J. (2010) PCNA and MutLα: partners in crime in triplet repeat expansion? Proc. Natl. Acad. Sci. U.S.A. 107, 16409–16410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pluciennik A., Dzantiev L., Iyer R. R., Constantin N., Kadyrov F. A., Modrich P. (2010) PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 107, 16066–16071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neuberger M. S., Di Noia J. M., Beale R. C., Williams G. T., Yang Z., Rada C. (2005) Somatic hypermutation at A·T pairs: polymerase error versus dUTP incorporation. Nat. Rev. Immunol. 5, 171–178 [DOI] [PubMed] [Google Scholar]
- 7. Schanz S., Castor D., Fischer F., Jiricny J. (2009) Interference of mismatch and base excision repair during the processing of adjacent U/G mispairs may play a key role in somatic hypermutation. Proc. Natl. Acad. Sci. U.S.A. 106, 5593–5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dianov G. L., Hübscher U. (2013) Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 41, 3483–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghodgaonkar M. M., Lazzaro F., Olivera-Pimentel M., Artola-Borán M., Cejka P., Reijns M. A., Jackson A. P., Plevani P., Muzi-Falconi M., Jiricny J. (2013) Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol. Cell 50, 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins A. R. (1999) Oxidative DNA damage, antioxidants, and cancer. BioEssays 21, 238–246 [DOI] [PubMed] [Google Scholar]
- 11. Gedik C. M., Collins A. (2005) Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 19, 82–84 [DOI] [PubMed] [Google Scholar]
- 12. Burrows C. J., Muller J. G. (1998) Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 98, 1109–1152 [DOI] [PubMed] [Google Scholar]
- 13. David S. S., O'Shea V. L., Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McAuley-Hecht K. E., Leonard G. A., Gibson N. J., Thomson J. B., Watson W. P., Hunter W. N., Brown T. (1994) Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry 33, 10266–10270 [DOI] [PubMed] [Google Scholar]
- 15. Shibutani S., Takeshita M., Grollman A. P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434 [DOI] [PubMed] [Google Scholar]
- 16. Avkin S., Livneh Z. (2002) Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutat. Res. 510, 81–90 [DOI] [PubMed] [Google Scholar]
- 17. Einolf H. J., Guengerich F. P. (2001) Fidelity of nucleotide insertion at 8-oxo-7,8-dihydroguanine by mammalian DNA polymerase δ. Steady-state and pre-steady-state kinetic analysis. J. Biol. Chem. 276, 3764–3771 [DOI] [PubMed] [Google Scholar]
- 18. Sakumi K., Furuichi M., Tsuzuki T., Kakuma T., Kawabata S., Maki H., Sekiguchi M. (1993) Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 268, 23524–23530 [PubMed] [Google Scholar]
- 19. Maki H., Sekiguchi M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355, 273–275 [DOI] [PubMed] [Google Scholar]
- 20. Mo J. Y., Maki H., Sekiguchi M. (1992) Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc. Natl. Acad. Sci. U.S.A. 89, 11021–11025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michaels M. L., Miller J. H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174, 6321–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fortini P., Pascucci B., Parlanti E., D'Errico M., Simonelli V., Dogliotti E. (2003) 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutat. Res. 531, 127–139 [DOI] [PubMed] [Google Scholar]
- 23. Ohtsubo T., Nishioka K., Imaiso Y., Iwai S., Shimokawa H., Oda H., Fujiwara T., Nakabeppu Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 28, 1355–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ushijima Y., Tominaga Y., Miura T., Tsuchimoto D., Sakumi K., Nakabeppu Y. (2005) A functional analysis of the DNA glycosylase activity of mouse MUTYH protein excising 2-hydroxyadenine opposite guanine in DNA. Nucleic Acids Res. 33, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Loon B., Hübscher U. (2009) An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase λ. Proc. Natl. Acad. Sci. U.S.A. 106, 18201–18206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dufner P., Marra G., Räschle M., Jiricny J. (2000) Mismatch recognition and DNA-dependent stimulation of the ATPase activity of hMutSα is abolished by a single mutation in the hMSH6 subunit. J. Biol. Chem. 275, 36550–36555 [DOI] [PubMed] [Google Scholar]
- 27. Koi M., Umar A., Chauhan D. P., Cherian S. P., Carethers J. M., Kunkel T. A., Boland C. R. (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 54, 4308–4312 [PubMed] [Google Scholar]
- 28. Iaccarino I., Marra G., Palombo F., Jiricny J. (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSα. EMBO J. 17, 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kubota Y., Takisawa H. (1993) Determination of initiation of DNA replication before and after nuclear formation in Xenopus egg cell free extracts. J. Cell Biol. 123, 1321–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baerenfaller K., Fischer F., Jiricny J. (2006) Characterization of the “mismatch repairosome” and its role in the processing of modified nucleosides in vitro. Methods Enzymol. 408, 285–303 [DOI] [PubMed] [Google Scholar]
- 31. Modrich P. (2006) Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jacobs A. L., Schär P. (2012) DNA glycosylases: in DNA repair and beyond. Chromosoma 121, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dianov G., Price A., Lindahl T. (1992) Generation of single-nucleotide repair patches following excision of uracil residues from DNA. Mol. Cell. Biol. 12, 1605–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L. S., Lane D. P., Abbondandolo A., Dogliotti E. (1996) Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 271, 9573–9578 [DOI] [PubMed] [Google Scholar]
- 35. Klungland A., Lindahl T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 16, 3341–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fang W. H., Modrich P. (1993) Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem. 268, 11838–11844 [PubMed] [Google Scholar]
- 37. Thomas D. C., Roberts J. D., Kunkel T. A. (1991) Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem. 266, 3744–3751 [PubMed] [Google Scholar]
- 38. Mazurek A., Berardini M., Fishel R. (2002) Activation of human MutS homologs by 8-oxo-guanine DNA damage. J. Biol. Chem. 277, 8260–8266 [DOI] [PubMed] [Google Scholar]
- 39. Larson E. D., Iams K., Drummond J. T. (2003) Strand-specific processing of 8-oxoguanine by the human mismatch repair pathway: inefficient removal of 8-oxoguanine paired with adenine or cytosine. DNA Repair 2, 1199–1210 [DOI] [PubMed] [Google Scholar]
- 40. Holmes J., Jr., Clark S., Modrich P. (1990) Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl. Acad. Sci. U.S.A. 87, 5837–5841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kadyrov F. A., Dzantiev L., Constantin N., Modrich P. (2006) Endonucleolytic function of MutLα in human mismatch repair. Cell 126, 297–308 [DOI] [PubMed] [Google Scholar]
- 42. Peña-Diaz J., Bregenhorn S., Ghodgaonkar M., Follonier C., Artola-Borán M., Castor D., Lopes M., Sartori A. A., Jiricny J. (2012) Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell 47, 669–680 [DOI] [PubMed] [Google Scholar]
- 43. Boldogh I., Milligan D., Lee M. S., Bassett H., Lloyd R. S., McCullough A. K. (2001) hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 29, 2802–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillespie P. J., Gambus A., Blow J. J. (2012) Preparation and use of Xenopus egg extracts to study DNA replication and chromatin associated proteins. Methods 57, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brooks P., Dohet C., Almouzni G., Méchali M., Radman M. (1989) Mismatch repair involving localized DNA synthesis in extracts of Xenopus eggs. Proc. Natl. Acad. Sci. U.S.A. 86, 4425–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nash H. M., Bruner S. D., Schärer O. D., Kawate T., Addona T. A., Spooner E., Lane W. S., Verdine G. L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol. 6, 968–980 [DOI] [PubMed] [Google Scholar]
- 47. Dherin C., Radicella J. P., Dizdaroglu M., Boiteux S. (1999) Excision of oxidatively damaged DNA bases by the human α-hOgg1 protein and the polymorphic α-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 27, 4001–4007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Warren J. J., Pohlhaus T. J., Changela A., Iyer R. R., Modrich P. L., Beese L. S. (2007) Structure of the human MutSα DNA lesion recognition complex. Mol. Cell 26, 579–592 [DOI] [PubMed] [Google Scholar]
- 49. Colussi C., Parlanti E., Degan P., Aquilina G., Barnes D., Macpherson P., Karran P., Crescenzi M., Dogliotti E., Bignami M. (2002) The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 12, 912–918 [DOI] [PubMed] [Google Scholar]
- 50. Russo M. T., Blasi M. F., Chiera F., Fortini P., Degan P., Macpherson P., Furuichi M., Nakabeppu Y., Karran P., Aquilina G., Bignami M. (2004) The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol. Cell. Biol. 24, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gu Y., Parker A., Wilson T. M., Bai H., Chang D. Y., Lu A. L. (2002) Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 277, 11135–11142 [DOI] [PubMed] [Google Scholar]
- 52. Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V. (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]