

Abstract
Cell-free systems provide a flexible platform for probing specific networks of biological reactions isolated from the complex resource sharing (e.g., global gene expression, cell division) encountered within living cells. However, such systems, used in conventional macro-scale bulk reactors, often fail to exhibit the dynamic behaviors and efficiencies characteristic of their living micro-scale counterparts. Understanding the impact of internal cell structure and scale on reaction dynamics is crucial to understanding complex gene networks. Here we report a microfabricated device that confines cell-free reactions in cellular scale volumes while allowing flexible characterization of the enclosed molecular system. This multilayered poly(dimethylsiloxane) (PDMS) device contains femtoliter-scale reaction chambers on an elastomeric membrane which can be actuated (open and closed). When actuated, the chambers confine Cell-Free Protein Synthesis (CFPS) reactions expressing a fluorescent protein, allowing for the visualization of the reaction kinetics over time using time-lapse fluorescent microscopy. Here we demonstrate how this device may be used to measure the noise structure of CFPS reactions in a manner that is directly analogous to those used to characterize cellular systems, thereby enabling the use of noise biology techniques used in cellular systems to characterize CFPS gene circuits and their interactions with the cell-free environment.
Keywords: Bioengineering, Issue 97, Cell-free, synthetic biology, microfluidics, noise biology, soft lithography, femtoliter volumes
Introduction
Cell-free systems offer a simplified and flexible platform for viewing biological reactions free from complicating factors such as fitness, division, and mutation that are unavoidable in the study of living cells. Such approaches have been employed to study cellular systems including the characterization of membrane proteins1, the probing of protein interactions2, and the exploration of fundamental aspects of translation3-7. Recently cell-free systems have begun to gain a foothold as viable platforms for synthetic biology8-10. The appeal of such approaches is that they free synthetic biology from the resource sharing and ‘extrinsic noise’ that affects reaction dynamics in living cells. However questions remain as to how the physical environment in which cell-free reactions are embedded affects the progression and outcome of the reaction. Cell-free reaction environments — particularly confined environments that approach cell-relevant volumes — remain poorly characterized. Cell-Free Protein Synthesis (CFPS) is conventionally thought of as being ‘scale-free,’ exhibiting equivalent kinetics across a range of microliter to liter-scale reaction volumes11. Nonetheless, confining reactions to cellular scale volumes has been shown to significantly affect protein expression rates12.
The stochastic nature of cell-free reactions — especially as these systems approach or even go below femtoliter volumes — may be of particular importance. Noise in gene expression is a property greatly influenced by confinement as small cell volumes and high densities of components force many of the important molecules to very low population levels — for example, Escherichia coli confines within a 1 fl volume as many as 4,300 different polypeptides under the inducible control of several hundred different promoters13. This inherent noise has been implicated as a central driving force in numerous biological processes including chemotaxis14, the HIV decision between active replication and latency15, the λ phage decision between lysis and lysogeny16,17, and the Bacillus subtillus decision between competence and sporulation17. Cell-free synthetic biology then provides both an opportunity to explore the stochastic properties of cellular gene circuits and networks, and manipulate these behaviors to achieve specific technological goals. While the noise behavior of cellular systems has been well-studied18-27, there has been little exploration of the fundamental noise behavior of cell-free systems8, particularly at the cellular scale.
Here we present a platform for the study of stochastic effects in cell-free synthetic biology. This microfabricated platform contains femtoliter-scale reaction chambers which may be quickly transitioned between open (free diffusion in and out of the chamber) and closed (reactants confined within the chamber) states. In the closed state, we confine Cell-Free Protein Synthesis (CFPS) reactants expressing a green fluorescent protein (GFP), and follow gene expression using time-lapse fluorescence microscopy28 (Figure 1). We characterize this cell-free environment by measuring the structure of the stochastic fluctuations in gene expression in a manner directly analogous to those used to characterize cells25. Non-microfabrication methods for confining cell-free reactions include vesicles and liposomes29-32, water-in-oil emulsions12, and porous media33. However, while these methods can provide control over the size distribution of the confined volumes34, microfabrication methods create highly replicable features with tightly specified dimensions, even on the nanoscale. Moreover, these rigid structures can be easily tracked over time without being susceptible to evaporation or changes in the external environment. Microfabricated container designs used in previous work8,35 cannot quickly seal the reaction chambers following reaction initiation, complicating the clear assignment of the time when the reaction was initiated (time zero). Using the method presented here, only 4-5 min are needed between initiation and visualization of the reaction on the device, thereby providing a well-defined “time zero”. The following protocols describe the methods for fabricating and testing this device, including optical lithography, device assembly, device testing, and methods for image analysis.
Protocol
1. Optical Lithography of Device Masters
Dehydrate clean silicon wafers on a hot plate at ~250 °C for at least 1 hr. NOTE: It is good practice to use more than one wafer when preparing a master, in case of user error.
Prepare photoresist aliquots. Prepare aliquots of both SU-8 2015 photoresist and a dilution of SU-8 2015 photoresist in 2:1 ratio using SU-8 thinner as diluent. NOTE: Approximately 1 ml of photoresist is needed for spin-coating one wafer.
Prepare three mask patterns for producing these masters. For the Membrane Master, prepare two masks: one patterning the membrane channel and the other patterning the reaction chambers. For the Control Valve master, prepare only one mask pattern. NOTE: For more details on lithographic techniques, including mask patterning, see Ito and Okazaki, 2000.36 See Fowlkes and Collier, 2013 for a more detailed description of device design.37
- Prepare the Membrane Master
- Spin-coat 2:1 SU-8 2015 photoresist dilution on wafers at 1,000 rpm for 45 sec.
- Soft bake wafers at 95 °C for 2 min. Using a contact aligner, expose wafers with membrane channel pattern for 10 sec, and perform a post-exposure bake for 2 min at 95 °C.
- Develop wafers in SU-8 developer for 1 min, or until photoresist residue is removed. Rinse wafer with isopropanol, moving from top to bottom. Dry wafer with nitrogen, again moving from top to bottom. Bake wafers at 180 °C for 4 min.
- Spin-coat patterned wafers again with 2:1 SU-8 dilution at 2,000 rpm for 45 sec.
- Soft bake patterned wafers for 2 min at 95 °C. Using contact aligner, align patterned wafers with reaction chamber pattern, and expose for 10 sec. Perform post-exposure bake for 2 min at 95 °C.
- Develop wafers as described in step 1.4.3. After developing and drying wafers, bake wafers at 180 °C for 4 min. NOTE: The wafers may be developed in the same developer that was used in the previous step.
- Prepare the Control Valve Master
- Spin-coat undiluted SU-8 photoresist onto clean wafers at 2,000 rpm for 45 sec.
- Soft bake wafer at 95 °C for 6 min. Using a contact aligner, expose wafers with control valve pattern for 10 sec. Perform a post-exposure bake at 95 °C for 6 min.
- Develop wafers in SU-8 Developer for 2 min, or until residue is removed. Rinse with isopropanol, moving from top to bottom. Dry wafer with nitrogen and bake at 180 °C for 4 min.
2. PDMS Device Fabrication
- Silanize all masters with ~0.2 ml trimethylchlorosilane via vapor deposition.
- Quickly enclose the master in an airtight container at RT with a few drops of the silanizing agent. NOTE: Other silanizing protocols may be acceptable.38 If performed properly, the PDMS will be easy to remove.
- Mix a commercial poly(dimethylsiloxane) (PDMS) base and curing agent in different ratios for both the membrane and control valve layers of the device, as has been demonstrated in similar multilayer valve designs39. Use 20:1 and 5:1 ratios of base:curing agent for the membrane and control valve molds, respectively.
- For the membrane mold, mix 10 g of base with 0.5 g of curing agent. NOTE: This volume will be spin-coated onto the membrane master.
- For the control valve mold, mix the base and curing agent in a 5:1 ratio. The amount of PDMS necessary to mold the control valve will depend on the container used to hold the control valve master; fill the container such that the master is coated with ~1 cm of PDMS.
Thoroughly mix both PDMS preparations, and de-gas them in a vacuum chamber until no air bubbles are visible. Place the control valve master in a heat-resistant container, such as a glass dish. Carefully pour 5:1 ratio PDMS over the master, and de-gas the container a second time.
While the control valve PDMS container is being de-gassed, spin-coat the 20:1 ratio PDMS on the membrane master by carefully pouring the PDMS mixture onto the membrane master to minimize air bubble formation, then spin-coating the master at 1,000 rpm for 45 sec.
Partially cure both masters in an oven at 80 °C for 6 min for the membrane master and 15 min for the control valve master. NOTE: When partially cured, the PDMS should hold its form, but the material will be slightly tacky. If PDMS is not yet cured, bake again in increments of a few minutes at a time until the material holds its form when pressed.
Cut rectangular PDMS molds from control valve master, peeling the molds away gently. Punch inlet holes through the molded component using a 0.75 mm hole punch. NOTE: The hole may be cleaned by inserting a 23 gauge blunt tip needle, and the mold exterior may be cleaned with cellophane tape, if necessary.
Using an optical microscope to locate the reaction chambers on the membrane master, align the control valve mold component with the features of the reaction chamber membrane and place the control valve component directly on top of the membrane master. Orient the control valve inlet to the bottom left corner of the device, and ensure that the reaction chambers and channel of the membrane master are visible inside the rectangular control valve.
Bake the aligned mold components at 80 °C for 2 hr. NOTE: The membrane and control valve molds will now be sealed together, and manipulated as one mold.
Cut the layered PDMS mold away from the membrane master, peeling the mold away from the master very gently so as not to perforate the membrane.
Punch inlet and outlet holes for the cell extract input using a 0.75 mm hole punch. Punch holes through both layers, and clean them in the same way as described in step 2.6.
- Using an inductively-coupled plasma cleaner, plasma treat both the mold (membrane side up) and a No. 0 glass coverslip at 10.5 W for 20 sec. Immediately remove the coverslip and mold from the plasma cleaner and layer the components, membrane side towards the glass, attempting to minimize air pockets between the glass and the mold. Do not press directly on the membrane input channel, or the membrane may anneal to the glass, making it difficult to fill the channel with reactants.
- Take special care when handling the assembled devices to avoid breaking the glass layer. Use thin glass coverslips as the device must be imaged through the glass coverslip using high magnification oil-immersion objectives — if the glass is too thick, the device features may not be visible.
Finally, cure the completed devices at 80 °C for 2 hr.
3. Experimental Setup for Cell-free Protein Synthesis Reaction
Hydrate a device by boiling it in deionized water for 1 hr. NOTE: Device should have a cloudy appearance when completely hydrated. Device may also be left O/N in sterile water at RT in order to hydrate it.
Using an inverted microscope with an incubation chamber, set the ambient temperature to 30 °C. NOTE: This temperature was chosen to optimize expression of GFP with a T7 promoter, so optimal temperatures for other reactions may vary40.
Mount device to microscope stage holder with cellophane tape and wrap edges of device with wet tissue paper in order to maintain local hydration.
- Use two high precision closed-loop voltage-pressure transducers to modulate nitrogen gas pressure for control valve actuation and reagent input. NOTE: This protocol has only been tested with low-purity nitrogen, though other inert gases may be used.
- Connect the first transducer by 24-gauge PTFE tubing to a water reservoir held in a 4 ml glass vial with a septum lid. Connect the reservoir to the control valve inlet using a second tube terminated by a 23 gauge blunt tip needle. NOTE: Both tubes penetrate the reservoir septum with two sharp 23 gauge needles.
- Connect the second transducer by 24-gauge PTFE tubing connected to a male-to-male Luer-lok connector. Attach this to a Luer-lok 23 gauge needle connected by tubing with another 23 gauge blunt tip needle, which is assembled individually for each device. This needle connects to the membrane reaction channel; use it to flush water from the reaction channel and input reagents.
- Using a cell-free protein expression system, assemble the components for the CFPS reaction on ice, according to manufacturer’s instructions. Minimize the time spent holding CFPS reagents on ice and place the reaction into the device immediately after assembly. NOTE: This device has been used with a commercial E. coli extract protein expression kit and a plasmid constitutively expressing GFP. The total reaction volume was scaled to 25 µl — it may be possible to use an even lower volume for reactants, if desired. As CFPS reagents tend to be sensitive to freeze-thaw cycles, it may be helpful to make aliquots of the reagents at the appropriate volume prior to the experiment. Other reagents may be added to the reaction mixture, but the reaction must be fully assembled before being applied to the device.
- Assemble the reaction, adding the DNA input last. NOTE: Once assembled in an Eppendorf tube, the CFPS reaction will begin if not held on ice. Since the time taken to apply the reagents to the device and begin the experiment may vary, it is helpful to start a timer once the reaction is assembled and mixed — this will keep the timescale between experiments consistent, and aid in troubleshooting.
Using the tubing and needle connector described in Step 3.4.2, withdraw the assembled reaction into the tube using a 1 ml syringe. Insert the blunt tip needle into the reaction chamber inlet. Detach the needle connector from the syringe and attach it to the male-to-male connector used for the reaction chamber transducer.
Apply pressure (<10 psi) to the CFPS reactants to fill the channel. Remove the needle when the reaction is filled.
Insert the blunt tube from the other transducer into the control valve inlet. Do not pressurize the control valve yet.
Place the mounted device on the stage. Using brightfield imaging, locate the reaction chambers with a 100X oil-immersion objective.
Actuate the control valve by pressurizing the control valve transducer to 20 psi; a visible change in the membrane will be evident when the control valve is actuated. Focus on the bottoms of the reaction chambers.
Begin the image acquisition; growth in fluorescence will be visible in the interior and around the exterior of the reaction chambers, though it will likely not be evident in the early stages of the reaction. Capture images every 1-3 min until the reaction reaches a steady state fluorescence. If an automatically focusing stage is not available, briefly refocus each image prior to the images being taken. NOTE: While some photobleaching will occur, the effects on relative fluorescence due to photobleaching may be accounted for as long as the rate of photobleaching is known. This photobleaching rate may be estimated by exposing a fluorescent standard, such as a known concentration of GFP or a fluorophore mixture, to constant photobleaching over a period of time.
Record the time elapsed from the reaction assembly to the first image acquired. NOTE: This typically takes 4-5 min.
4. Image Analysis and Data Processing
- Using an image analysis software such as ImageJ, select the interior of the reaction chambers as an ROI. Acquire the mean fluorescence intensity value of the ROI for all images. NOTE: This is the raw fluorescence intensity trace.
- Perform this task in ImageJ using the Time Series Analyzer and ROI Manager plugins — Use Time Series Analyzer to choose regions of interest around the interior of each reaction chamber. Set “AutoROIProperties” to an area which corresponds to the interior of each reaction chamber, check “Add On Click”, and select each chamber. NOTE: This step may also be done using the ellipse tool to draw an ROI around the fluorescent chamber. This ROI size usually corresponds to a 30 x 30 pixel ellipse for a 10 µm diameter chamber viewed with a 100X objective.
- Highlight all ROIs in the ROI Manager. Use the “Multi Measure” function to determine the fluorescence intensity mean of each ROI through the entire image stack. NOTE: A plugin named StackReg may be used to align the image stack, if necessary.
After acquiring the raw fluorescence intensity traces for all chambers in an experiment, determine the deterministic component of the reaction by taking an inter-experimental average across all traces, and subtracting the average from individual raw traces. Use data analysis software such as IGOR or MS Excel for this analysis. NOTE: This provides noise traces for each reaction chamber.
Analyze the gene expression noise from these reaction chambers using the same methods used to analyze gene expression noise derived from cells25.
Representative Results
The distinct advantage of this microfabricated platform is in the application of the controllable elastomeric “control valve” which is independently actuated in order to confine CFPS reactions (Figure 2A). When the device is actuated, the membrane chambers are pressed against the glass slide to confine fluorescent reagents into an array of reaction chambers (Figure 2C). In order to verify that the chambers reliably confine the reaction through the duration of the experiment, a basic FRAP (Fluorescence Recovery After Photobleaching) test was conducted37. A fluorophore (AF 555) was applied to the device, and the control valve was actuated; using the shutter aperture of the microscope, a single well confining the fluorophore was isolated and photobleached individually (Figure 2D). The chosen well became dark and did not recover in brightness until the control valve was depressurized 20 min later, releasing the chamber from the glass. This test verifies that these reaction chambers remain well-sealed for the duration of the experiment.
In optimal conditions, a CFPS reaction expressing an easily visualized protein (such as GFP or Luciferase) expresses detectable protein within a few minutes of being applied to this device. Over the lifetime of the reaction, protein synthesis in the interior and exterior of the reaction chambers is imaged and quantified by measuring units of fluorescence intensity within each chamber (Figure 3A). Fluorescence intensity, corresponding to protein concentration, may be mapped over time for each reaction chamber (Figure 3D).
Gene expression is an inherently stochastic process that introduces fluctuations (noise) at every molecular step (synthesis, degradation, protein-DNA binding, etc.)20. One branch of noise biology focuses on the probative value of gene circuit noise41 . Expression in cell-free systems will have extrinsic noise effects that arise from interactions between the molecular machinery of expression and the surfaces that define the boundaries of the reaction vessels. These extrinsic effects will likely become more pronounced as cell-free reactions are confined into even smaller reaction chambers. The ability to perform time-lapse imaging of multiple confined CFPS reactions then enables the careful analysis of noise structure (magnitude and dynamics) in confined cell-free systems in a way directly analogous to methods that have been reported for cellular systems25. Figure 3C and 3D show the time courses of constitutive GFP expression from a T7 promoter in a standard 384-well microplate with a well volume of 15 µl, compared to in PDMS reaction chambers 10 µm in diameter, corresponding to volumes of only about 300 fl, about seven orders of magnitude less. The variability in protein expression rates in the 10 µm reaction chambers is much higher than in the well-plate measurements, approaching those seen in cells.
Multiplexed reactions performed on the device exhibit similar kinetics to CFPS reactions performed in bulk on a microplate reader (Figure 3B), where there is a swift increase in fluorescence which plateaus, often assumed to be caused by resource limitation within the reaction volume42,43. This deterministic growth behavior, though fluctuating, is generally consistent across all reaction chambers, and between experiments — by averaging traces between chambers across experiments, the deterministic trend may be subtracted from trace values, leaving only the noise components of the reaction (Figure 4A). Figure 4B shows the GFP expression noise after removal of the deterministic, transient component (top), and the autocorrelation of the noise (bottom), while Figure 4A shows the corresponding traces in the 10 µm reaction chambers. The distribution in the half-times of the autocorrelation traces gives the frequency dependence of the noise while the zero lag time of the autocorrelation traces gives the magnitudes of the noise, as the variance.
 Figure 1. Cell-Free Protein Synthesis reactants are confined in femtoliter scale reaction chambers for the purpose of measuring gene expression noise. Reactants from a commercial cell-free protein expression system are used to constitutively express GFP inside confined PDMS reaction chambers. An array of these chambers may be visualized with time-lapse fluorescence microscopy in order to characterize protein expression and gene expression noise. The fluorescence intensity of each reaction chamber over time may be plotted as an individual trace. Please click here to view a larger version of this figure.
Figure 1. Cell-Free Protein Synthesis reactants are confined in femtoliter scale reaction chambers for the purpose of measuring gene expression noise. Reactants from a commercial cell-free protein expression system are used to constitutively express GFP inside confined PDMS reaction chambers. An array of these chambers may be visualized with time-lapse fluorescence microscopy in order to characterize protein expression and gene expression noise. The fluorescence intensity of each reaction chamber over time may be plotted as an individual trace. Please click here to view a larger version of this figure.
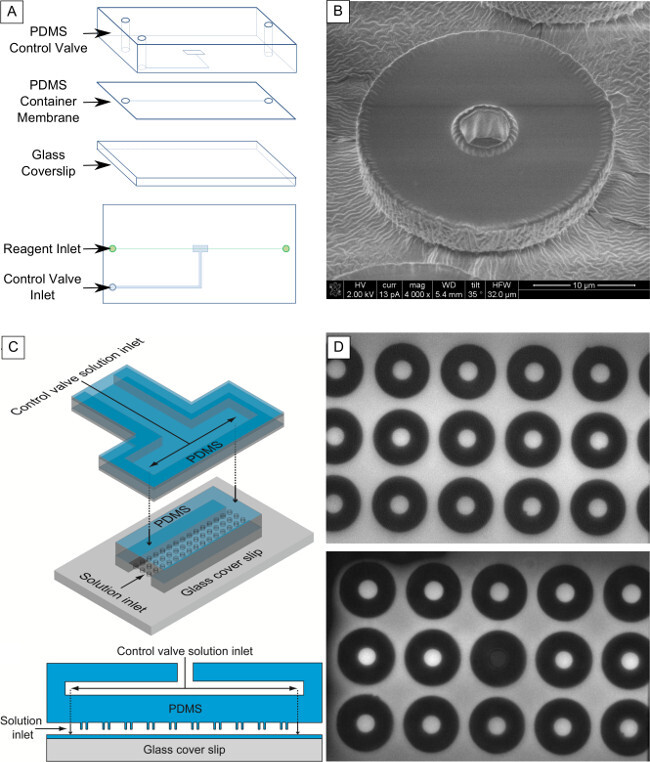 Figure 2. Fabrication of two-layer microfluidic device with sealable femtoliter-scale chambers. (A) Layout and exploded view of device layers. The device is composed of two PDMS layers and a glass coverslip. The PDMS membrane, sealed between the glass and control valve layers, holds the reaction chambers. (B) SEM image of PDMS reaction chamber. The interior diameter is 10 µm. (C) Schematic of input channels in device. Cell-Free Protein Synthesis (CFPS) reagents are flown through the reaction channel. Water is pressurized in the control valve to compress the reaction chambers against the glass slide, sealing the chambers37. Reproduced from Ref. 37 with permission from The Royal Society of Chemistry. (D) Fluorescence Recovery After Photobleaching (FRAP) test on a single well using FITC indicates chamber is well-sealed against external environment. The fluorophore was captured in the chambers (upper image) and a single well was photobleached (lower image). No fluorescence recovery was seen in the photobleached chamber until the control valve was released. Please click here to view a larger version of this figure.
Figure 2. Fabrication of two-layer microfluidic device with sealable femtoliter-scale chambers. (A) Layout and exploded view of device layers. The device is composed of two PDMS layers and a glass coverslip. The PDMS membrane, sealed between the glass and control valve layers, holds the reaction chambers. (B) SEM image of PDMS reaction chamber. The interior diameter is 10 µm. (C) Schematic of input channels in device. Cell-Free Protein Synthesis (CFPS) reagents are flown through the reaction channel. Water is pressurized in the control valve to compress the reaction chambers against the glass slide, sealing the chambers37. Reproduced from Ref. 37 with permission from The Royal Society of Chemistry. (D) Fluorescence Recovery After Photobleaching (FRAP) test on a single well using FITC indicates chamber is well-sealed against external environment. The fluorophore was captured in the chambers (upper image) and a single well was photobleached (lower image). No fluorescence recovery was seen in the photobleached chamber until the control valve was released. Please click here to view a larger version of this figure.
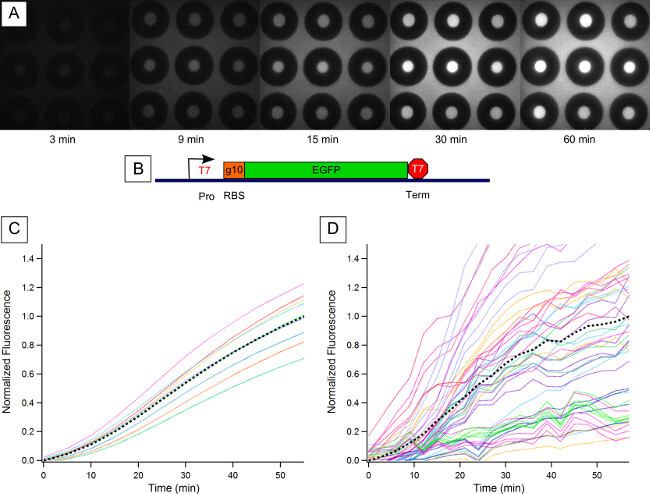 Figure 3. EGFP Expression in Confined Cell-Free Reaction. (A) Fluorescence images of sealed reaction chambers at chosen time points in the reaction. Protein production can be seen both inside the reaction chambers and outside the chambers in the main channel. (B) EGFP was cloned into a Pet3a vector, providing a T7 polymerase promoter and terminator and a strong ribosome binding site (RBS). (C) Normalized fluorescence measurements of constitutive expression of EGFP in a bulk cell-free reaction performed in a microplate reader. CFPS reactions usually produce protein quickly before slowing to a 'steady state' fluorescence — this is associated with resource limitation43. Black dashes indicate the average trace. (D) Normalized fluorescence of 51 raw fluorescence intensity traces read from 51 reaction chambers over several experiments. Black dashes indicate the average trace over several experiments, which illustrate the deterministic component of the protein expression. Please click here to view a larger version of this figure.
Figure 3. EGFP Expression in Confined Cell-Free Reaction. (A) Fluorescence images of sealed reaction chambers at chosen time points in the reaction. Protein production can be seen both inside the reaction chambers and outside the chambers in the main channel. (B) EGFP was cloned into a Pet3a vector, providing a T7 polymerase promoter and terminator and a strong ribosome binding site (RBS). (C) Normalized fluorescence measurements of constitutive expression of EGFP in a bulk cell-free reaction performed in a microplate reader. CFPS reactions usually produce protein quickly before slowing to a 'steady state' fluorescence — this is associated with resource limitation43. Black dashes indicate the average trace. (D) Normalized fluorescence of 51 raw fluorescence intensity traces read from 51 reaction chambers over several experiments. Black dashes indicate the average trace over several experiments, which illustrate the deterministic component of the protein expression. Please click here to view a larger version of this figure.
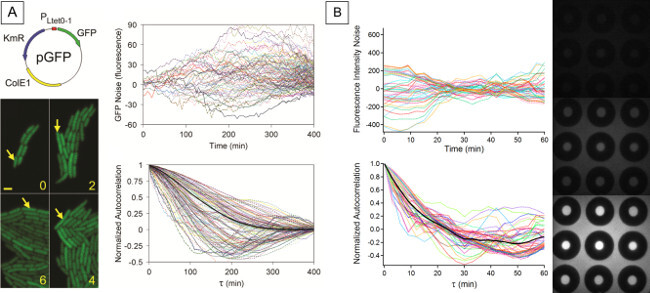 Figure 4. Individual Noise Traces and Noise Autocorrelation of a Cellular and Cell-Free System. (A) From Austin et al., 2006. Noise in GFP expression (top) and normalized autocorrelation functions (bottom) acquired from tracking GFP production in living bacteria25. Reprinted by permission from Macmillan Publishers Ltd: [Nature] 25 (Vol. 439), copyright (2006). (B) Noise in GFP expression (top) and normalized autocorrelation functions (bottom) acquired from GFP production in cell-free system, tracked in microfluidic device reaction chambers. Please click here to view a larger version of this figure.
Figure 4. Individual Noise Traces and Noise Autocorrelation of a Cellular and Cell-Free System. (A) From Austin et al., 2006. Noise in GFP expression (top) and normalized autocorrelation functions (bottom) acquired from tracking GFP production in living bacteria25. Reprinted by permission from Macmillan Publishers Ltd: [Nature] 25 (Vol. 439), copyright (2006). (B) Noise in GFP expression (top) and normalized autocorrelation functions (bottom) acquired from GFP production in cell-free system, tracked in microfluidic device reaction chambers. Please click here to view a larger version of this figure.
Discussion
Gene expression in cells is inherently noisy due to small cellular volumes and low copy numbers of important reactants. Noise biology often focuses on the sources, processing, and biological consequences of fluctuations in the populations, concentrations, positions, or states of molecules that control gene circuits and networks44. The vast majority of this work has been performed in cellular systems, which has the advantage of viewing the noise of a gene circuit within the natural context of the genetic networks within the cell. However, cell-free systems allow the characterization of the intrinsic fluctuations of an individual gene circuit without the confounding extrinsic effects18 that cannot be avoided in cellular systems. Analysis of noise can offer important physical insights into how genetic circuits are structured and how they function, and has been used in cellular systems to characterize negative25 and positive27 autoregulation, extrinsic and intrinsic contributions to expression noise18 , and transcriptional bursting45,46. Here we describe the study of a cell-free expression system in microfluidic devices that enable the simultaneous control of reactor size and reaction initiation times, in order to better understand the roles that confinement and crowding47,48 have on intrinsic protein expression noise without the complications associated with living cells.
The key enabling feature of the design is the integration of arrays of femtoliter-volume (micron-scale) reaction chambers used for confining the reactants of a cell-free protein expression system, with an elastomeric “control valve” membrane in PDMS that traps the reactants at a well-defined, “time zero” for reaction initiation (Figure 1). This control allows the kinetics of the reactions involved in protein synthesis to be followed in real time with high precision. As such, it is important to manage cell-free reactants so that inter-experimental variability is minimized as much as possible. This control allows us to evaluate noise structure of cell-free genetic circuits in a manner that is analogous to techniques previously used to evaluate gene expression in living cells.
As reactants used in CFPS systems can be sensitive to freeze-thaw cycles, it is important to keep the reactants cold and minimize the time the reactants spend thawing on ice. It is good practice to periodically test the expression of the CFPS system in bulk in order to identify changes in expression levels over time — this may be done in a 10-15 µl reaction in an Eppendorf tube, or in a device like a microplate reader, which performs multiple reads over time to capture reaction kinetics. Noting the age and thaw times of the reactants for every experiment will help when troubleshooting low expression levels. Furthermore, when assembling CFPS reagents, it is important to note that the reaction will begin once it is fully assembled and removed from the ice. In order to maintain a consistent “time zero”, it is helpful to record the time following the initiation of the CFPS reaction after the final addition of the DNA input, and to apply the reaction as quickly as possible to the incubated device. This process should take about 4-5 min, and fluorescence should not yet be visible within the reaction chambers. This control assures that the time available to visualize the growth portion of the reaction curve is maximized.
Before running CFPS reactions on the device, it is advisable to run quality-control tests to verify there is no leakage from the chambers. A FRAP test can be performed (as in Figure 2D) by applying a fluorophore to the device and exposing an individual well until the well is completely bleached. If the chambers are well-sealed, no recovery should be visible inside the well — there should be a stark contrast between the walls of the compartment and the interior and exterior spaces. If fluorescence recovery is apparent or the walls of the reaction chamber are not well defined, the pressure on the control valve should be increased or the device should be checked for leakage or delamination from the glass slide.
This protocol has been tested with CFPS reagents from a commercial E. coli cell-free protein expression kit (scaled to 25 µl), though other robust CFPS systems may be used. It is possible to use volumes much lower than 25 µl when applying reactions to the device, which may be helpful when reagent cost is a limiting factor in experiments. Once reactants are added to the device and the reaction chambers are sealed, it is not possible to add reactants to the solution without de-actuating the control valve — thus this device is not suitable for reactions which require the addition of reagents during the course of the reaction. This device is also not optimized for observing CFPS reactions which may run longer than 3 hr — the effects of dehydration and drying of the device after this time period have not been evaluated. If longer reaction times are desired, these effects may be mitigated by sealing the device to prevent evaporation, changing the incubation temperature, or by using a humidity chamber. Modifications to the device design, such as nanoporous structures in the chamber walls49,50 or the inclusion of a porous membrane layer, represent a few methods which could allow reagent exchange and thus lengthen reaction timescales.
Microfabricated reaction compartments of uniform volume are valuable for maintaining consistent dimensions across experiments and highly suitable for investigation into “side reactions” with the compartment walls. Unlike methods using non-microfabricated techniques, these reactions must be evaluated in small numbers, and do not provide dimensional flexibility during experiments. However, the controllable design for these reaction chambers is highly suitable for time-lapse microscopy, and may be an illuminating complement to a high-throughput method of confinement.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. Mitch Doktycz, Dr. Jennifer Morrel-Falvey, Dr. Amber Bible, and Dr. Brandon Razooky for helpful advice and conversations, and acknowledge Dr. Sukanya Iyer for constructing the Pet3a-EGFP plasmid used in the gene expression tests. We acknowledge support from the Center for Nanophase Materials Sciences, which is sponsored by the Scientific User Facilities Division, Office of Science, U.S. Department of Energy. SEN and PMC acknowledge support from Bredesen Center Fellowships at the University of Tennessee, Knoxville. This research was performed at Oak Ridge National Laboratory (ORNL). ORNL is managed by UT-Battelle, LLC, for the U.S. Department of Energy.
References
- Klammt C, et al. High level cell-free expression and specific labeling of integral membrane proteins. European Journal of Biochemistry. 2004;271:568–580. doi: 10.1111/j.1432-1033.2003.03959.x. [DOI] [PubMed] [Google Scholar]
- Wong RW, Blobel G. Cohesin subunit SMC1 associates with mitotic microtubules at the spindle pole. Proceedings of the National Academy of Sciences. 2008;105:15441–15445. doi: 10.1073/pnas.0807660105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederholtmeyer H, Stepanova V, Maerkl SJ. Implementation of cell-free biological networks at steady state. Proceedings of the National Academy of Sciences. 2013;110:15985–15990. doi: 10.1073/pnas.1311166110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jardine P, Noireaux V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS synthetic biology. 2012;1:408–413. doi: 10.1021/sb300049p. [DOI] [PubMed] [Google Scholar]
- Algire MA, et al. Development and characterization of a reconstituted yeast translation initiation system. RNA. 2002;8:382–397. doi: 10.1017/s1355838202029527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka N, Najita L, Franzusoff A, Sarnow P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Molecular and cellular biology. 1994;14:7322–7330. doi: 10.1128/mcb.14.11.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZZ, et al. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. Journal of visualized experiments: JoVE. 2013. p. e50762. [DOI] [PMC free article] [PubMed]
- Karig DK, Jung S-Y, Srijanto B, Collier CP, Simpson ML. Probing cell-free gene expression noise in femtoliter volumes. ACS synthetic biology. 2013;2:497–505. doi: 10.1021/sb400028c. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS synthetic biology. 2012;1:29–41. doi: 10.1021/sb200016s. [DOI] [PubMed] [Google Scholar]
- Karzbrun E, Tayar AM, Noireaux V, Bar-Ziv RH. Programmable on-chip DNA compartments as artificial cells. Science. 2014;345:829–832. doi: 10.1126/science.1255550. [DOI] [PubMed] [Google Scholar]
- Zawada JF, et al. Microscale to manufacturing scale-up of cell-free cytokine production - a new approach for shortening protein production development timelines. Biotechnology and bioengineering. 2011;108:1570–1578. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Yanagisawa M, Sato YT, Fujiwara K, Yoshikawa K. Cell-Sized confinement in microspheres accelerates the reaction of gene expression. Scientific reports. 2012;2:283. doi: 10.1038/srep00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson ML, Sayler GS, Fleming JT, Applegate B. Whole-cell biocomputing. Trends in biotechnology. 2001;19:317–323. doi: 10.1016/s0167-7799(01)01691-2. [DOI] [PubMed] [Google Scholar]
- Korobkova E, Emonet T, Vilar JM, Shimizu TS, Cluzel P. From molecular noise to behavioural variability in a single bacterium. Nature. 2004;428:574–578. doi: 10.1038/nature02404. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage λ-infected Escherichia coli cells. Genetics. 1998;149:1633–1648. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowitz MB. Tunability and noise dependence in differentiation dynamics. Science. 2007;315:1716–1719. doi: 10.1126/science.1137455. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- McAdams HH, Arkin A. Stochastic mechanisms in gene expression. Proceedings of the National Academy of Sciences. 1997;94:814–819. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nature Reviews Genetics. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Blake WJ, Kærn M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Rao CV, Wolf DM, Arkin AP. Control, exploitation and tolerance of intracellular noise. Nature. 2002;420:231–237. doi: 10.1038/nature01258. [DOI] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- Austin D, et al. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- Simpson ML, Cox CD, Sayler GS. Frequency domain analysis of noise in autoregulated gene circuits. Proceedings of the National Academy of Sciences. 2003;100:4551–4556. doi: 10.1073/pnas.0736140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nature genetics. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- Locke JC, Elowitz MB. Using movies to analyse gene circuit dynamics in single cells. Nature Reviews Microbiology. 2009;7:383–392. doi: 10.1038/nrmicro2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourian Z, Danelon C. Linking genotype and phenotype in protein synthesizing liposomes with external supply of resources. ACS synthetic biology. 2013;2:186–193. doi: 10.1021/sb300125z. [DOI] [PubMed] [Google Scholar]
- Pereira de Souza T, Stano P, Luisi PL. The minimal size of liposome-based model cells brings about a remarkably enhanced entrapment and protein synthesis. ChemBioChem. 2009;10:1056–1063. doi: 10.1002/cbic.200800810. [DOI] [PubMed] [Google Scholar]
- Nomura SiM, et al. Gene expression within cell-sized lipid vesicles. ChemBioChem. 2003;4:1172–1175. doi: 10.1002/cbic.200300630. [DOI] [PubMed] [Google Scholar]
- Noireaux V, Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proceedings of the national academy of sciences of the United States of America. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N, Um SH, Funabashi H, Xu J, Luo D. A cell-free protein-producing gel. Nature. 2009;8:432–437. doi: 10.1038/nmat2419. [DOI] [PubMed] [Google Scholar]
- Swaay D, deMello A. Microfluidic methods for forming liposomes. Lab on a Chip. 2013;13:752–767. doi: 10.1039/c2lc41121k. [DOI] [PubMed] [Google Scholar]
- Okano T, Matsuura T, Kazuta Y, Suzuki H, Yomo T. Cell-free protein synthesis from a single copy of DNA in a glass microchamber. Lab on a Chip. 2012;12:2704–2711. doi: 10.1039/c2lc40098g. [DOI] [PubMed] [Google Scholar]
- Ito T, Okazaki S. Pushing the limits of lithography. Nature. 2000;406:1027–1031. doi: 10.1038/35023233. [DOI] [PubMed] [Google Scholar]
- Fowlkes JD, Collier CP. Single-molecule mobility in confined and crowded femtolitre chambers. Lab on a Chip. 2013;13:877–885. doi: 10.1039/c2lc40907k. [DOI] [PubMed] [Google Scholar]
- Herold KE, Rasooly A. Lab on a Chip Technology: Biomolecular separation and analysis. Vol. 2. Horizon Scientific Press; 2009. [Google Scholar]
- Unger MA, Chou H-P, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Karig DK, Iyer S, Simpson ML, Doktycz MJ. Expression optimization and synthetic gene networks in cell-free systems. Nucleic acids research. 2012;40:3763–3774. doi: 10.1093/nar/gkr1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CD, McCollum JM, Allen MS, Dar RD, Simpson ML. Using noise to probe and characterize gene circuits. Proceedings of the National Academy of Sciences. 2008;105:10809–10814. doi: 10.1073/pnas.0804829105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal-Gaskins D, Noireaux V, Murray RM. American Control Conference (ACC), 2013. 2013. pp. 1531–1536.
- Jewett MC, Swartz JR. Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm) Biotechnology and bioengineering. 2004;87:465–471. doi: 10.1002/bit.20139. [DOI] [PubMed] [Google Scholar]
- Simpson ML, et al. Noise in biological circuits. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009;1:214–225. doi: 10.1002/wnan.22. [DOI] [PubMed] [Google Scholar]
- Dar RD, et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proceedings of the National Academy of Sciences. 2012;109:17454–17459. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So L-h, et al. General properties of transcriptional time series in Escherichia coli. Nature genetics. 2011;43:554–560. doi: 10.1038/ng.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C, Saurabh S, Bruchez MP, Schwartz R, LeDuc P. Molecular crowding shapes gene expression in synthetic cellular nanosystems. Nat Nano. 2013;8:602–608. doi: 10.1038/nnano.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, et al. Enhanced transcription rates in membrane-free protocells formed by coacervation of cell lysate. Proceedings of the National Academy of Sciences. 2013;110:11692–11697. doi: 10.1073/pnas.1222321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer ST, Siuti P, Choi C-K, Thomas DK, Doktycz MJ. Development and fabrication of nanoporous silicon-based bioreactors within a microfluidic chip. Lab on a Chip. 2010;10:1174–1181. doi: 10.1039/b921592a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti P, Retterer ST, Doktycz MJ. Continuous protein production in nanoporous, picolitre volume containers. Lab Chip. 2011;11:3523–3529. doi: 10.1039/c1lc20462a. [DOI] [PubMed] [Google Scholar]