

Abstract
Bardet–Biedl syndrome is a rare ciliopathy characterized by retinal dystrophy, obesity, intellectual disability, polydactyly, hypogonadism and renal impairment. Patients are at high risk of cardiovascular disease. Mutations in BBS1 and BBS10 account for more than half of those with molecular confirmation of the diagnosis. To elucidate genotype–phenotype correlations with respect to cardiovascular risk indicators 50 patients with mutations in BBS1 were compared with 19 patients harbouring BBS10 mutations. All patients had truncating, missense or compound missense/truncating mutations. The effect of genotype and mutation type was analysed. C-reactive protein was higher in those with mutations in BBS10 and homozygous truncating mutations (p = 0.013 and p = 0.002, respectively). Patients with mutations in BBS10 had higher levels of C peptide than those with mutations in BBS1 (p = 0.043). Triglyceride levels were significantly elevated in patients with homozygous truncating mutations (p = 0.048). Gamma glutamyl transferase was higher in patients with homozygous truncating mutations (p = 0.007) and heterozygous missense and truncating mutations (p = 0.002) than those with homozygous missense mutations. The results are compared with clinical cardiovascular risk factors. Patients with missense mutations in BBS1 have lower biochemical cardiovascular disease markers compared with patients with BBS10 and other BBS1 mutations. This could contribute to stratification of the clinical service.
Keywords: Bardet–Biedl syndrome, cardiovascular morbidity, genotype–phenotype correlation, mutation type
Bardet–Biedl syndrome (BBS) is a pleiotropic autosomal recessive ciliopathy characterized by retinal dystrophy, post-axial polydactyly, obesity, learning difficulties, hypogonadism and renal dysfunction (1, 2). There is a high prevalence of cardiovascular, endocrine and renal disorders among patients with BBS (3). In order to optimize the clinical management, it is imperative to identify patients who are most at risk of disease-associated morbidity and mortality.
Since the clinical criteria for a diagnosis were proposed, 19 genes (BBS1–BBS19) have been discovered (4, 5). BBS genes code for proteins that localize to the basal body of the cilium. Mutations lead to defective cilia accounting in part for the variable effects observed in BBS. A clinical diagnosis can be confirmed by sequencing the known disease-causing genes in 80% of patients (authors' own unpublished data).
The majority of pathogenic mutations are found in BBS1 and BBS10 accounting for 23.2% and 20%, respectively in populations of northern European descent (4). The commonest mutation is M390R found in 82.5% of a cohort of British patients with BBS1 mutations (4). The frameshift mutation C91LfsX5 is prevalent in patient populations with BBS10 mutations.
Variable phenotypic expressivity is a hallmark of BBS, however, even among patients with the same genotype, interfamilial and intrafamilial phenotypic variability is common. Mutations in other BBS genes may modify the phenotype, accounting for this variability (6). Several studies have attempted to identify a genotype–phenotype correlation in BBS (7–10). These have primarily focused on physical features and have been limited by small sample sizes or participants from the same kindred.
This is the first study to explore the correlation between genotype, mutation type and morbidity in BBS. We examine indicators of cardiovascular, metabolic and renal morbidity in a large cohort of patients with BBS and compare the two most commonly mutated genes: BBS1 and BBS10.
Methods
Patients
Two hundred and thirty nine patients attending the national Bardet-Biedl Syndrome clinics in London and Birmingham were assessed for height, weight, blood pressure, BBS mutation analysis, full blood count, renal function, liver function, inflammatory markers, endocrine and lipid profile. Information on cardiovascular risk factors was collected retrospectively from patient notes. The following clinical parameters were ascertained: (i) hypertension (defined as a blood pressure over 140/90 or normotensive requiring antihypertensive medication), (ii) hypercholesterolaemia requiring hypolipidaemic agents, (iii) diabetes mellitus requiring hypoglycaemic medication, (iv) structural renal abnormalities and/or dialysis or renal transplant, and (v) structural cardiac abnormalities. Patients were predominantly of Caucasian origin. Referrals were made primarily via the British national patient support group and clinical geneticists in the United Kingdom.
Mutation screening
Eighty four of the 239 patients had two known pathogenic mutations in BBS genes. Of these, 73 harboured two mutations in BBS1 or BBS10. Mutation analysis was primarily undertaken through targeted sequencing of the four most common mutations: M390R in BBS1, and Y24X and R275X in BBS2 and C91LfsX5 in BBS10. Where only one mutation was found, full sequencing of the relevant gene was performed to identify a second mutation.
Statistical analysis
Analysis was targeted to BBS1 and BBS10 patients with truncating and/or missense mutations to allow for adequate sample sizes.
We applied a two pronged approach to statistical analysis. Mann–Whitney U test, Kruskal–Wallis test and analysis of variance (anova) were performed as appropriate to identify associations between genes (BBS1 vs BBS10) and mutation types (homozygous truncating mutations, heterozygous missense and truncating mutations or homozygous missense mutations). Multivariable linear regression analysis was applied to variables which were statistically significant on univariable analysis and/or known indicators of metabolic, renal and cardiovascular disease. Statistical analyses were carried out using spss version 21.0 (SPSS Inc., Chicago IL).
Results
Distribution of patients
Fifty two patients harboured two mutations in BBS1 and 21 harboured two mutations in BBS10. This included nine pairs and two sets of three siblings. DNA results were classified according to gene and mutation. Mutation type was further classified according to the predicted severity. Most patients had either homozygous missense mutations, homozygous truncating mutations or a heterozygous missense and truncating mutations. Two patients with BBS1 mutations and two patients with BBS10 mutations harboured splice site mutations and were excluded from the statistical analysis. Figure 1 demonstrates the distribution of mutation types in the BBS1 and BBS10 genotypes analysed in this study, illustrating the higher proportions of missense mutations in BBS1 and truncating mutations in BBS10.
Fig 1.
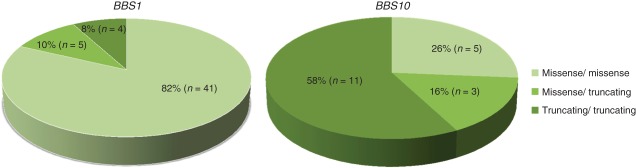
Distribution of mutation type in patients with BBS1 and BBS10 included in the analysis.
Of the remaining 69 patients seen in the clinic with a mutation in BBS1 or BBS10 the mean age was 28.25 (SD: 14.41, range: 0–59). The mean (SD) age of patients with a BBS1 mutation was 30.5 (15.6) years. In contrast the mean age (SD) of patients with a mutation in BBS10 was 22.32 (8.95) years (p = 0.034). Figure 2 illustrates the age distribution of patients with BBS1 and BBS10 included in this study. Twenty four (48%) patients with a mutation in BBS1 were female and 26 (52%) were male. Twelve (63.2%) patients with mutations in BBS10 were female and seven (36.8%) were male.
Fig 2.
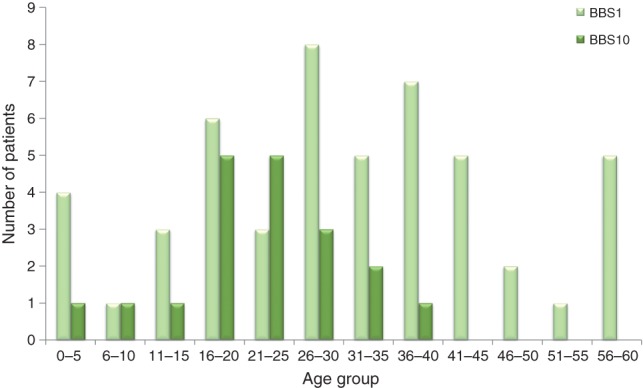
Age distribution of patients with mutations in BBS1 and BBS10 included in the analysis.
Clinical parameters
Genotype–phenotype associations were tested for all clinical variables. Univariable analysis comparing patients with BBS1 vs BBS10 demonstrated a statistically significant difference in age, height, C-reactive protein (CRP), c-peptide, triglycerides, potassium, and albumin–creatinine ratio (Table 1). Comparison of mutation types revealed a statistically significant difference in high density lipoprotein (HDL) cholesterol and gamma glutamyl transferase (GGT) (Table 2).
Table 1.
Genotype–phenotype correlation: univariable comparison of statistically significant parameters. Patients with BBS1 vs BBS10. Only statistically significant results are included. The full analysis is available in Tables S1, Supporting information
| BBS 1 |
BBS 10 |
||||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | p-Valuea | |
| Anthropomorphic measurements | |||||
| Age at clinic | 30.5 | −15.48 | 22.32 | −8.95 | 0.034 |
| Height (cm) | 163.92 | −28.17 | 162.93 | −9.31 | 0.031 |
| Inflammatory markers | |||||
| CRP (mg/l) | 5.69 | −2.74 | 9.53 | −7.12 | 0.04 |
| Endocrine profile | |||||
| C peptide (ng/ml) | 1295.88 | −740.13 | 2333.3 | −1501.27 | 0.014 |
| Lipid profile | |||||
| Triglycerides (mmol/l) | 1.5 | −0.73 | 1.98 | −0.94 | 0.049 |
| Renal profile | |||||
| Potassium (mmol/l) | 4.16 | −0.48 | 4.38 | −0.42 | 0.015 |
| Albumin/creatinine ratio | 7.1 | −22.39 | 5.3 | −11.23 | 0.032 |
ANOVA, analysis of variance; BBS, Bardet–Biedl syndrome, CRP, C-reactive protein.
p-Value obtained from anova test or Mann–Whitney U test.
Table 2.
Mutation type-phenotype comparison: univariable comparison of statistically significant parameters. Homozygous missense; heterozygous truncating and missense and homozygous truncating mutations. Only statistically significant results are included. The full analysis is available in Table S2
| Missense/missense |
Missense/truncating |
Truncating/truncating |
|||||
|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | p-Valuea | |
| Lipid profile | |||||||
| HDL cholesterol (mmol/l) | 1.28 | −0.25 | 1.09 | −0.12 | 1.1 | −0.23 | 0.022 |
| Liver profile | |||||||
| Gamma glutamyl transferase (U/l) | 29.21 | −16.22 | 70.33 | −10.02 | 62.75 | −40.01 | 0.027 |
HDL, high-density lipoprotein.
p-value obtained from anova test or Kruskal–Wallis test.
We applied multivariable analysis to selected variables based on association with cardiovascular risk and controlled for confounding factors. Statistically significant results are displayed in Tables 3 and 4 and discussed here.
Table 3.
Genotype–phenotype comparison: multivariable comparison of selected parameters found to be statistically significant. BBS1 vs BBS10. Only statistically significant results are included in this table. The full analysis is available in Table S3
| β estimate | 95.0% CI | p-Valuea | |
|---|---|---|---|
| CRP (mg/l) | |||
| Genotype | |||
| BBS1 | Reference | — | — |
| BBS10 | 4.08 | (0.90, 7.25) | 0.013 |
| Age | 0.06 | (−0.05, 0.18) | 0.295 |
| BMI | 0.1 | (−0.08, 0.29) | 0.266 |
| C peptide (ng/ml) | |||
| Genotype | |||
| BBS1 | Reference | — | — |
| BBS10 | 942.94 | (32.26, 1853.61) | 0.043 |
| BMI | −4.86 | (−67.84, 58.12) | 0.876 |
| Blood glucose | 92.08 | (−71.24, 255.41) | 0.258 |
BBS, Bardet–Biedl syndrome; BMI, body mass index; CI, confidence interval; CRP, C-reactive protein. Significant values are highlighted in bold.
p-value obtained from linear regression model.
Table 4.
Mutation type-phenotype comparison: multivariable comparison of selected parameters found to be statistically significant. Homozygous missense; heterozygous truncating and missense and homozygous truncating. Only statistically significant results are included in this table. The full analysis is available as Table S4
| β estimate | 95.0% CI | p-Valuea | |
|---|---|---|---|
| CRP (mg/l) | |||
| Mutation type | |||
| Missense/missense | Reference | — | — |
| Missense/truncating | −0.65 | (−4.42, 3.12) | 0.729 |
| Truncating/truncating | 5.33 | (1.99, 8.68) | 0.002 |
| Age | 0.06 | (−0.05, 0.17) | 0.272 |
| BMI | 0.14 | (−0.03, 0.31) | 0.11 |
| Triglycerides (mmol/l) | |||
| Mutation type | |||
| Missense/missense | Reference | — | — |
| Missense/truncating | 0 | (−0.67, 0.67) | 0.996 |
| Truncating/truncating | 0.56 | (0.01, 1.11) | 0.048 |
| Gender | |||
| Female | Reference | — | — |
| Male | 0.52 | (0.07, 0.98) | 0.026 |
| BMI | 0.03 | (0.00, 0.06) | 0.05 |
| Age | −0.01 | (−0.03, 0.01) | 0.452 |
| Gamma glutamyl transferase (U/l) | |||
| Mutation type | |||
| Missense/missense | Reference | — | — |
| Missense/truncating | 44.22 | (17.90, 70.54) | 0.002 |
| Truncating/truncating | 29.32 | (8.72, 49.91) | 0.007 |
| Gender | |||
| Female | Reference | — | — |
| Male | 17.94 | (4.12, 31.76) | 0.013 |
| BMI | 1.05 | (0.15,1.95) | 0.025 |
| Age | 0.24 | (−0.28, 0.76) | 0.349 |
Significant values are highlighted in bold.
p-Value obtained from linear regression model.
Inflammatory markers
A statistically significant difference in CRP is observed. Patients with a mutation in BBS10 or homozygous truncating mutations have a significantly higher CRP (p = 0.013 and p = 0.002, respectively). Analysis of white cell count and other blood count parameters did not reach statistical significance.
C peptide
C peptide levels were significantly higher in patients with BBS10 compared with BBS1 mutations (p = 0.043).
Lipid profile
Triglycerides levels were significantly higher in patients with homozygous truncating mutations (p = 0.048) than those with other mutation types.
Liver function
Multivariable analysis demonstrated significantly higher GGT in patients with homozygous truncating or heterozygous missense and truncating mutations than those with homozygous missense mutations (p = 0.007 and p = 0.002, respectively).
Clinical cardiovascular risk factors
Sixty seven patients had a full lipid profile, of whom 14 had hypercholesterolaemia. Hypertension was identified in 23 of 65 patients. Fifteen of 69 patients had a diagnosis of diabetes mellitus. Five of 69 patients had the results of an echocardiogram documented. Of these, one had an innocent murmur, two had a ventricular septal defect, one had an atrioventricular septal defect and one patient had aortic valve stenosis. All of these patients had two missense mutations in BBS1. Twenty nine patients had renal ultrasounds. Sixteen of these had abnormal results ranging from benign structural malformations to sonographic evidence of chronic renal failure. Two patients had renal transplants and one was on dialysis, all of whom had two truncating mutations in BBS10. Figure 3a,b illustrate the prevalence of the clinical cardiovascular risk factors. Cardiac abnormalities are not included in these diagrams as only five patients had documented echocardiograms and we therefore perceive the results not be representative of the group as a whole.
Fig 3.
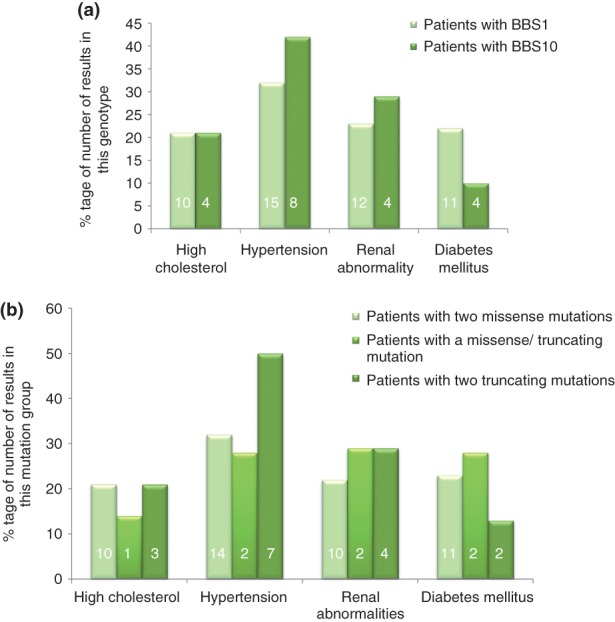
Prevalence of cardiovascular risk factors by genotype (a) and mutation type (b). The prevalence is illustrated as a percentage to compensate for disproportionate population sizes. Absolute numbers are given in each column.
Discussion
To our knowledge this is the only study comparing cardiovascular risk factors for patients with BBS1 and BBS10 – the two most commonly mutated genes in patients from Europe and North America. It is the first published study to compare the BBS phenotype according to mutation type. Several studies have suggested that there is little evidence of a genotype–phenotype correlation in BBS and proposed that this may be because BBS proteins contribute to a common molecular pathway (4). However, there is an emerging evidence of some genotype–phenotype correlations (7, 10). Feuillian et al. (10) reported that patients with BBS1 had lower insulin resistance compared with patients with BBS10. Observations from the British nationally commissioned clinic suggest that patients with two mutations in BBS10 are often more severely affected than those with BBS1 although there is considerable variation.
CRP increases with obesity and inflammation. Chronically raised CRP indicates a higher risk of cardiovascular morbidity (11). This study shows that patients with BBS10 genotypes and/or two truncating mutations have a significantly worse CRP value. As CRP is a physiological marker of inflammation and infection, it is notable that there was no statistically significant difference in white cell count or weight between patients with different genes or mutation types. This suggests that patients with missense mutations in BBS1 may be at lower risk of cardiovascular disease than patients with BBS10 or other mutations in BBS1.
C peptide is used as a marker of insulin resistance but has in recent years been recognized as an independent bioactive peptide exerting effects on microvascular function, correlating with macrovascular complications and cardiovascular death (12). Our results demonstrate that patients with mutations in BBS10 have significantly higher levels of C-peptide indicating insulin resistance, supporting the suggestion that they are at higher risk of cardiovascular disease than patients with mutations in BBS1.
Raised triglycerides are associated with an increased risk of cardiovascular disease (13). Our results demonstrate that patients with homozygous truncating mutations are more likely to have raised triglycerides than patients with other mutation types.
We demonstrated a statistically significant increase in GGT in patients with homozygous truncating mutations and heterozygous truncating and missense mutations compared with patients with homozygous missense mutations. Although used as a marker of chronic liver disease, GGT correlates with cardiovascular diseases and is an independent marker of cardiovascular risk (14) making it a potentially powerful tool in the risk stratification of patients with BBS.
The prevalence of clinical cardiovascular risk factors according to genotype and mutation type do not reveal a clear pattern. This may be because the patients in this study are young (mean age 28.25) and the natural progression of the disease has not yet unfolded, or because the patients with BBS1 mutations are significantly older than the patients with mutations in BBS10. Alternatively, it may reflect a true lack of genotype–phenotype correlation.
Although analysis of renal parameters did not reveal any statistically significant differences, it is noteworthy that the patient on dialysis and both patients who had received renal transplants had two truncating mutations in BBS10. This is in keeping with previous studies suggesting that the renal phenotype may be more severe in patients with mutations in BBS10 (3).
The high prevalence of hypertension (Figure 3a,b) among patients with mutations in BBS10 and two truncating mutations (42% and 50%) respectively is striking considering only one person in these groups is older than 31. This may represent the early development of a severe cardiovascular phenotype or a statistical error due to sample sizes (19 and 14, respectively).
Conclusion
Cardiovascular disease is a major cause of death and morbidity, and significant resources are allocated to primary prevention in the general population with the aim of reducing the overall disease burden. The same principles should apply to special groups within the population such as patients with BBS where there is an opportunity to practice personalized medicine as the genotype of many patients is already known. This study indicates that patients with missense mutations in BBS1 may be at lower risk of cardiovascular disease than patients with homozygous truncating mutations and mutations in BBS10. In practice most patients with BBS10 mutations harbour the common homozygous truncating mutation and it is possible that the resulting truncated protein product or a hypomorphic effect of the common missense mutation M390R in BBS1, rather than the affected gene, determines the phenotypic effect. Larger studies could clarify this, and longitudinal research will determine the clinical effect these risk factors have on cardiovascular morbidity.
Acknowledgments
E. F. is funded by an Academic Clinical Fellowship. P. L. B. is a Wellcome trust Senior Research Fellow and NIHR investigator funded by the Great Ormond Street Hospital Children's charity. The national Bardet–Biedl Syndrome clinic is funded by the NHS Highly Specialised Services (NHS England). The authors wish to thank patients and colleagues and in particular the Laurence–Moon–Bardet–Biedl syndrome (LMBBS) patient group for their ongoing support.
Supporting Information
The following Supporting information is available for this article:
Table S1. Genotype–phenotype correlation: univariable comparison of clinical and laboratory parameters. Patients with BBS1 vs BBS10. Statistically significant results are highlighted in bold.
Table S2. Mutation type-phenotype comparison: univariable comparison of clinical and laboratory parameters. Homozygous missense, heterozygous truncating and missense and homozygous truncating mutations. Statistically significant results are highlighted in bold.
Table S3. Genotype–phenotype comparison: multivariable comparison of selected parameters associated with cardiovascular disease. BBS1 vs BBS10. Statistically significant results are highlighted in bold.
Table S4. Mutation type-phenotype comparison: multivariable comparison of selected parameters associated with cardiovascular disease. Homozygous missense, heterozygous truncating and missense and homozygous truncating. Statistically significant results are highlighted in bold.
References
- 1.Bardet G. On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity) 1920. Obes Res. 1995;3(4):387–399. doi: 10.1002/j.1550-8528.1995.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 2.Biedl A. A pair of siblings with adiposo-genital dystrophy 1922. Obes Res. 1995;3(4):404. doi: 10.1002/j.1550-8528.1995.tb00167.x. [DOI] [PubMed] [Google Scholar]
- 3.Imhoff O, Marion V, Stoetzel C, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6(1):22–29. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aldahmesh MA, Li Y, Alhashem A, Anazi S, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu044. Epub 12 February 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deveault C, Billingsley G, Duncan JL, et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat. 2011;32(6):610–619. doi: 10.1002/humu.21480. [DOI] [PubMed] [Google Scholar]
- 7.Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Fielding Hejtmacic J, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(7):901–907. doi: 10.1001/archophthalmol.2012.89. [DOI] [PubMed] [Google Scholar]
- 8.Carmi R, Elbedour K, Stone EM, Sheffield VC. Phenotypic differences among patients with Bardet-Biedl syndrome linked to three different chromosome loci. Am J Med Genet. 1995;59(2):199–203. doi: 10.1002/ajmg.1320590216. [DOI] [PubMed] [Google Scholar]
- 9.Hjortshøj TD, Grønskov K, Philp AR, et al. Bardet-Biedl syndrome in Denmark--report of 13 novel sequence variations in six genes. Hum Mutat. 2010;31(4):429–436. doi: 10.1002/humu.21204. [DOI] [PubMed] [Google Scholar]
- 10.Feuillan PP, Ng D, Han JC, Sapp JC, et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab. 2011;96(3):E528–E535. doi: 10.1210/jc.2010-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hribal ML, Fiorentino TV, Sesti G. Role of C reactive protein (CRP) in leptin resistance. Curr Pharm Des. 2014;20(4):609–615. doi: 10.2174/13816128113199990016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebherz C, Marx N. C-peptide and its career from innocent bystander to active player in diabetic atherogenesis. Curr Atheroscler Rep. 2013;15(7):339. doi: 10.1007/s11883-013-0339-3. [DOI] [PubMed] [Google Scholar]
- 13.Isaacs A, Willems SM, Bos D, et al. Risk scores of common genetic variants for lipid levels influence atherosclerosis and incident coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(9):2233–2239. doi: 10.1161/ATVBAHA.113.301236. [DOI] [PubMed] [Google Scholar]
- 14.Du G, Song Z, Zhang Q. Gamma-glutamyltransferase is associated with cardiovascular and all-cause mortality: A meta-analysis of prospective cohort studies. Prev Med. 2013;57(1):31–37. doi: 10.1016/j.ypmed.2013.03.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genotype–phenotype correlation: univariable comparison of clinical and laboratory parameters. Patients with BBS1 vs BBS10. Statistically significant results are highlighted in bold.
Table S2. Mutation type-phenotype comparison: univariable comparison of clinical and laboratory parameters. Homozygous missense, heterozygous truncating and missense and homozygous truncating mutations. Statistically significant results are highlighted in bold.
Table S3. Genotype–phenotype comparison: multivariable comparison of selected parameters associated with cardiovascular disease. BBS1 vs BBS10. Statistically significant results are highlighted in bold.
Table S4. Mutation type-phenotype comparison: multivariable comparison of selected parameters associated with cardiovascular disease. Homozygous missense, heterozygous truncating and missense and homozygous truncating. Statistically significant results are highlighted in bold.