

Abstract
Stimulation of the aryl hydrocarbon receptor (AHR) by xenobiotics is known to affect epidermal differentiation and skin barrier formation. The physiological role of endogenous AHR signaling in keratinocyte differentiation is not known. We used murine and human skin models to address the hypothesis that AHR activation is required for normal keratinocyte differentiation. Using transcriptome analysis of Ahr-/- and Ahr+/+ murine keratinocytes, we found significant enrichment of differentially expressed genes linked to epidermal differentiation. Primary Ahr-/- keratinocytes showed a significant reduction in terminal differentiation gene and protein expression, similar to Ahr+/+ keratinocytes treated with AHR antagonists GNF351 and CH223191, or the selective AHR modulator (SAhRM), SGA360. In vitro keratinocyte differentiation led to increased AHR levels and subsequent nuclear translocation, followed by induced CYP1A1 gene expression. Monolayer cultured primary human keratinocytes treated with AHR antagonists also showed an impaired terminal differentiation program. Inactivation of AHR activity during human skin equivalent development severely impaired epidermal stratification, terminal differentiation protein expression and stratum corneum formation. As disturbed epidermal differentiation is a main feature of many skin diseases, pharmacological agents targeting AHR signaling or future identification of endogenous keratinocyte-derived AHR ligands should be considered as potential new drugs in dermatology.
Keywords: AHR, skin, epidermal differentiation, antagonists
Introduction
The Aryl Hydrocarbon Receptor (AHR) is a ligand activated transcription factor and member of the bHLH/PAS (basic Helix-Loop-Helix/Per-Arnt-Sim) family (Omiecinski et al., 2010; Shimizu et al., 2000). The AHR regulates drug metabolism, and environmental toxicants, such as 2,3,7,8--tetrachlorodibenzo-p-dioxin (TCDD) and polycyclic aromatic hydrocarbons, as well as plant polyphenols and tryptophan photoproducts, are primarily agonists, although some flavonoid antagonists have been described (Murray et al., 2010a). The AHR resides in a cytoplasmic multiprotein complex, which translocates to the nucleus upon agonist binding. There the receptor dissociates from HSP90 and dimerizes with the aryl hydrocarbon nuclear translocator (ARNT) to transactivate target genes primarily through dioxin response elements (DRE) in promoters of responsive genes (Omiecinski et al., 2010). In humans, TCDD toxicity causes chloracne, associated with epidermal hyperproliferation and hyperkeratinization (Poland et al., 1982), and increased expression of genes critical for formation of the cornified envelope (Greenlee et al., 1985; Loertscher et al., 2001; Sutter et al., 2009; Sutter et al., 2011). In cell culture, TCDD induces expression of genes in the epidermal differentiation complex, causing aberrant differentiation of keratinocytes (Geusau et al., 2005; Sutter et al., 2009; Sutter et al., 2011). 6-8 month old Ahr-/- mice exhibit alopecia associated with dystrophic and degenerating hair follicles, ulceration and regenerative hyperplasia (Fernandez-Salguero et al., 1997). We recently demonstrated in atopic dermatitis that coal tar therapy activates the AHR through an unknown agonist(s) and restores defective differentiation and barrier function (van den Bogaard et al., 2013). Together these data suggest that dysregulation of normal AHR function could be important in the pathogenesis of chronic skin diseases with aberrant epidermal differentiation.
In addition to pathological AHR agonists such as TCDD, other AHR ligands can act either as full antagonists or selective modulators of AHR function with overlapping but non-identical effects on AHR cellular activities in the cell (Boitano et al., 2010; Choi et al., 2012; Lahoti et al., 2013; Murray et al., 2010b; Smith et al., 2011). These too may have significant therapeutic potential in suppressing some or all of AHR driven pathways in human disease. However, despite this extensive characterization of the pathogenesis and molecular biology of cutaneous responses to TCDD (Chiaro et al., 2008a; Schroeder et al., 2010; Veldhoen et al., 2009) the role of the AHR in normal epidermal differentiation and homeostasis and the potential role of AHR ligands in therapy for cutaneous disease is poorly understood. Here we show that in vitro expression of differentiation genes and proteins is suppressed in Ahr-/- keratinocytes; that the AHR undergoes nuclear translocation during in vitro differentiation, and that AHR antagonists and selective modulators can block differentiation of human and mouse keratinocytes in monolayer culture and in human skin equivalents. These data underscore a significant physiological role of the AHR in normal epidermal differentiation.
Results
The AHR regulates epidermal differentiation, attachment and inflammatory cytokine gene expression
To identify AHR dependent genes we compared gene expression between Ahr+/+ and Ahr-/- keratinocytes cultured in 0.05 mM Ca2+ growth media using Affymetrix ST arrays and ArrayStar 11 Software (DNASTAR, Madison WI). We identified 391 genes whose expression was altered by at least 1.5 fold (p<.05) in Ahr-/- keratinocytes relative to Ahr+/+. We used DAVID Bioinformatics Software (Huang et al., 2009b) to identify functional annotation clusters within the group of differentially regulated genes, and consistent with initial analysis, the top functional annotation clusters were extracellular matrix and adhesion (Enrichment score 7.2, 5.4 p=7.3 × 10-7 and 9.7 × 10-8 respectively) and keratinocyte, epidermal cell differentiation (Enrichment Score 3.3, p=2.1 × 10-7). Of the top down regulated transcripts in Ahr-/- keratinocytes 22 were linked to epidermal differentiation, including those for structural proteins, proteins involved in the formation of the cornified envelope, proteases and protease inhibitors and the transcription factor Pou2f3 (Skin1) (Table S1). Thirteen of the top upregulated transcripts in Ahr-/- keratinocytes encoded proteins associated with extracellular matrix and adhesion (Table S2). Expression of genes linked to skin inflammatory diseases: Il33, Il36g and thymic stromal lymphopoietin (Tslp) was downregulated in Ahr-/- keratinocytes while expression of Il24 and Il18r was induced (Table S1 and S2).
We compared expression of representative epidermal differentiation genes in Ahr+/+ and Ahr-/- keratinocytes cultured in proliferation medium or 24 h after switching to differentiation medium (0.12 mM Ca2+). In the absence of the AHR, both basal and induced expression levels of Krt1, Lor, Ivl, Dsc1 and the transcription factor Pou2f3 were significantly reduced (Figure 1a). Induction of differentiation with elevated calcium also increased expression of the well-characterized AHR target gene Cyp1a1 in Ahr+/+ keratinocytes, but was blocked in Ahr-/- keratinocytes (Figure 1a). As expected, TCDD caused significant induction of Cyp1a1 in Ahr+/+ but not Ahr-/- keratinocytes (data not shown). Similarly, both Il33 and Il36γ were significantly repressed in Ahr-/- keratinocytes relative to Ahr+/+ in cells cultured in proliferation media (Figure 1S).
Figure 1. Downregulation of differentiation gene expression in Ahr-/- mouse keratinocytes.
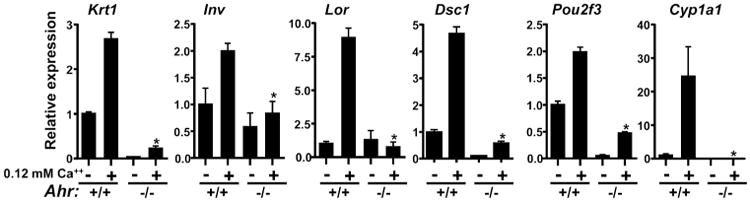
(a) Expression of indicated genes during calcium induced differentiation of primary Ahr+/+ and Ahr-/- keratinocytes was determined by quantitative PCR from triplicate cultures, repeated twice. Expression was normalized to Gapdh. * significantly different from Ahr +/+ p<.05. Krt1, Keratin 1; Lor, Loricrin; Ivl, Involucrin; Dsc1, Desmocollin 1; Pou2f3, POU Class 2 Homeobox 3, Cyp1a1, Cytochrome P450 1A1.
AHR antagonists and selective modulators block epidermal differentiation in monolayer culture
AHR ligands that act as either full antagonists or selective modulators have been identified. GNF351 is a full antagonist of DRE and non-DRE AHR function, interacts directly with the AHR ligand binding pocket and competes with a well-characterized photoaffinity AHR ligand for binding to the AHR, with an IC50 of 62 nM (Smith et al., 2011). GNF351 blocks AHR target gene induction by TCDD but has no agonist activity for either DRE dependent or independent functions of the AHR. SGA360 is a selective modulator (SAhRM) of the AHR as it blocks TCDD induced DRE-mediated AHR activity, but has agonist-like activity for non-DRE mediated AHR functions (Patel et al., 2009b; Tanos et al., 2012). We treated Ahr+/+ and Ahr-/-keratinocytes with TCDD or Ahr+/+ differentiating keratinocytes with GNF351 or SGA360, and measured changes in gene expression by quantitative RT-PCR. Consistent with its known effects, TCDD induced expression of genes involved in epidermal differentiation under proliferation conditions (Sutter et al., 2011), which was blocked in Ahr-/- keratinocytes (Figure 2a). Both GNF351 and SGA360 suppressed expression of early (Krt1, Pou2f3) and late (Ivl, Lor, Dsc1) differentiation genes in both basal and differentiation culture conditions. Similarly GNF351 completely blocked induction of Cyp1a1, in differentiating keratinocytes supporting the concept that the AHR becomes activated during normal epidermal differentiation (Figure 2a). In addition, both Il33 and Il36g were significantly downregulated in Ahr+/+ keratinocytes treated with either GNF351 or SGA360 (Figure S1). To test whether similar effects occurred in human keratinocytes, we treated differentiating human primary keratinocytes with the AHR antagonists GNF351 and CH223191 and observed a comparable reduction in expression of CYP1A1, FLG, hornerin (HRNR), and LOR relative to the untreated control differentiating keratinocyte cultures. There was a trend towards induced epidermal differentiation with the AHR agonist indirubin but this was not statistically significant (Figure 2b). FICZ (6-Formylindolo(3,2-b)carbazole) an AHR agonist generated in the skin from tryptophan by UV light (Fritsche et al., 2007) also induced expression of some but not all differentiation genes in human keratinocytes (Figure S2). There was minimal toxicity of these AHR ligands in mouse or human keratinocytes (Figure S3). Immunoblot analysis confirmed that genetic or pharmacological inactivation of AHR blocked induction of keratin 10 and loricrin protein expression in mouse (Figure 2c) and involucrin, loricrin and pro-filaggrin in human keratinocytes (Figure 2d).
Figure 2. AHR antagonists and selective modulators suppress epidermal differentiation in monolayer culture.

(a) Effect of TCDD (10 nM); GNF351 (500 nM) and SGA360 (10 μM) on gene expression in proliferating (0.05 mM CaCl2) or differentiating (0.12 mM CaCl2) primary mouse keratinocytes (triplicate, repeated twice) * p<.05 compared to vehicle control (b) Effect of GNF351 (500 nM); CH223191 (CH, 5 μM) and indirubin (IR, 50 nM) on differentiation gene expression in primary human keratinocytes. (2 separate experiments, total of n=5 donors). (c) Immunoblot analysis showing effect of Ahr ablation, GNF351 or SGA360 on differentiation induced expression of keratin 10 and loricrin in primary mouse keratinocytes. (d) Immunoblot analysis showing effect of GNF351 and CH223191 (CH) on pro-filaggrin (FLG), involucrin (IVL) and loricrin (LOR) in monolayer cultured primary human keratinocytes.
AHR antagonists suppress epidermal differentiation and stratum corneum thickness in human skin equivalents
To further examine the effect of AHR antagonists on epidermal differentiation we generated epidermal skin equivalents using human primary keratinocytes cultured on plastic inert filters. We tested the effect of antagonists added at different time points during generation of the human skin equivalents. When the keratinocytes were in submerged culture (proliferation/attachment phase) or when monolayers were initially brought to the air-liquid interface, addition of GNF351 or CH223191 substantially suppressed the stratification process and formation of the stratum corneum (Figure 3a). Expression of late differentiation markers involucrin and filaggrin was strongly reduced, but the early differentiation marker keratin 10 was less affected (Figure 3b). Addition of antagonists during the last phase of air-liquid interface culture (from day 4 or 7 onwards) resulted in thinning of the stratum corneum but did not affect involucrin or filaggrin expression (Figure 3b). However, when skin equivalents were generated using de-epidermized dermis, treatment with GNF351 4 days after transfer to air-liquid interface reduced the expression and number of cell layers expressing loricrin and filaggrin, while expression of keratin 10 was delayed (Figure 4). Since AHR antagonists were added during the proliferation phase of the skin equivalent development (submerged culture), we tested if they affected keratinocyte proliferation. There was a significant reduction in the percentage of Ki67 positive cells and cell number after treating proliferating monolayer cultures of human keratinocytes with AHR antagonists for 48 h (Figure 5, S4). In contrast, skin equivalents generated on inert filters and treated with GNF351 during the submerged phase or at day one of transfer to the air-liquid interphase had more Ki67 positive basal cells at the end of the skin equivalent development compared to untreated cultures (Figure 3b).
Figure 3. Epidermal stratification defects and reduced stratum corneum thickness caused by AHR inactivation.

Human skin equivalents (epidermis-only) were generated on plastic inert filters. At indicated time points (arrows) during skin equivalent development (each block represents one day of culture), AHR antagonists were added to the culture medium. All skin equivalents were harvested at day 10 of air-liquid interface culture. (a) Hematoxylin and Eosin staining of skin equivalents treated with GNF351 (500 nM) or CH223191 (CH) (5 μM). (b) Immunohistochemical staining of Keratin 10 (KRT10, early differentiation), filaggrin (FLG, terminal differentiation), involucrin (IVL, terminal differentiation) and Ki67 (proliferation) of skin equivalents treated with GNF351 as depicted in 2A. (n=2 keratinocyte donors). Scale bar = 100 μm.
Figure 4. Reduced terminal differentiation protein expression caused by AHR inactivation.
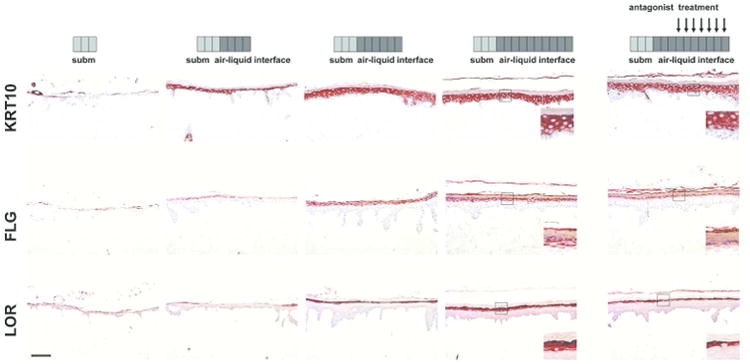
Human skin equivalents were generated using de-epidermised dermis and expression of keratin 10 (KRT10), filaggrin (FLG) and loricrin (LOR) was followed in time by harvesting the skin equivalents directly after submerged culture, and after 4, 6 and 10 days of air-liquid interface culture (each block represents one day of culture). Treatment with GNF351 (500 nM, arrows) was initiated at day 6 and sustained until day 10 of air-liquid interface culture. Magnification inlays show epidermal differentiation protein expression affected by AHR inactivation. (n= 2 keratinocytes donors). Scale bar = 100 μm.
Figure 5. AHR antagonists suppress human keratinocyte proliferation.

Monolayer cultures of human primary keratinocytes (n=3 keratinocyte donors, * p<0.05) were treated with AHR antagonists (GNF: GNF351, 500 nM; CH: CH223191, 5μM; SR1, 500 nM) for 48h during the proliferation stage of the culture. (a) Quantification of Ki67 positive cells and (b) total cell count of antagonist treated keratinocytes as compared to untreated cells.
Increased AHR nuclear localization during epidermal differentiation in vitro
To confirm whether the observed AHR dependence of differentiation gene expression was associated with AHR nuclear translocation, we isolated nuclear and cytoplasmic protein from primary mouse keratinocytes at specific time points after induction of differentiation with elevated medium calcium. Under basal proliferating conditions AHR protein expression was detected in the cytosolic extracts, but not in nuclear extracts (Figure 6a). Six hours after induction of differentiation there was an increase in nuclear AHR reaching a maximum at 12 h that was sustained through 48 h. AHR levels in cytosolic extracts also increased 12 h after induction of differentiation and this was sustained through 48 h (Figure 6a). ARNT levels retained in the nucleus also increased during differentiation, while slightly reduced levels were detected in Ahr-/- keratinocytes (Figure 6a). Treatment with GNF351 for 24 h enhanced nuclear AHR levels under differentiation conditions, while SGA360 increased cytosolic AHR levels and prevented nuclear retention. In proliferating keratinocytes, AHR agonists such as TCDD and indolo[3,2-b]carbazole (ICZ) caused rapid nuclear localization followed by significant loss of the protein from both the nuclear and cytoplasmic compartments, as expected, most likely due to proteosomal degradation (Davarinos et al., 1999; Ikuta et al., 2000; Ikuta et al., 1998). GNF351 caused a slower and sustained increase in nuclear AHR and cytoplasmic AHR, while SGA360 completely blocked the presence of AHR retained in nuclear extracts and increased cytoplasmic AHR levels.
Figure 6. Nuclear localization of AHR during epidermal differentiation.

(a) Immunoblot of AHR and ARNT following induction of differentiation with elevated calcium medium. (b) Immunoblot of cytoplasmic and nuclear AHR in primary mouse keratinocytes induced to differentiate in presence of GNF351 or SGA360. (c) Immunoblot showing time course of nuclear and cytoplasmic AHR levels in response to AHR ligands in primary mouse keratinocytes cultured in proliferation media.
Discussion
The AHR is the mediator of TCDD toxicity in the skin and other tissues. TCDD causes induction of keratinocyte terminal differentiation in vitro and accelerated skin barrier function in utero (Loertscher et al., 2001; Muenyi et al., 2014; Sutter et al., 2011) suggesting that AHR activation by exogenous ligands can cause pathological dysregulation of keratinocyte differentiation. In contrast, recent studies point to the beneficial effects of AHR activation in skin and other tissues and its potential as a therapeutic target (DiMeglio et al., 2014; Qiu et al., 2012). However the role of the AHR in normal skin physiology is poorly understood. Here we show that normal epidermal differentiation is regulated by AHR signaling in both murine and human keratinocytes: Ahr-/- mouse keratinocytes have defects in differentiation gene expression; AHR antagonists and SAhRMs suppress differentiation gene expression in monolayer culture of human and mouse keratinocytes and epidermal differentiation and stratification in human skin equivalent models. These data suggest a physiological role for the AHR during epidermal differentiation and stratification amenable to manipulation by pharmacological AHR antagonists or SAhRMs.
Our results are in concordance with earlier reports of elevated CYP1A1 enzyme levels in differentiated keratinocytes in absence (Sadek et al., 1994) or presence of xenobiotics (Reiners, Jr. et al., 1992) and the response to TCDD was higher in differentiated keratinocytes (Swanson 2004; Wanner et al., 1995). Additionally, retinoic acid, which interferes with epidermal differentiation, suppresses AHR induction during keratinocyte differentiation (Wanner et al., 1995). In contrast, lentiviral knockdown of the AHR in 3D skin equivalents had no effect on epidermal morphology (Forrester et al., 2014), but this may be due to insufficient AHR inactivation (40% knockdown) compared to the AHR antagonists used here. The ability of AHR antagonists to suppress proliferation in human keratinocyte monolayers is consistent with cell cycle arrest and reduced proliferation caused by AHR knockdown in HaCaT cells (Kalmes et al., 2011). Since the AHR can activate the EGFR/ERK pathway through c-Src (Fritsche et al., 2007; Sutter et al., 2009), it is possible that AHR antagonist and SAhRM-induced growth inhibition is indirect. However, this is unlikely since keratinocyte cultures were supplemented with EGF. The increased numbers of Ki-67 positive cells in skin equivalents treated with AHR antagonist at early stages of skin equivalent development is most likely due to some type of feedback response to the disturbed differentiation or retention of cells with a basal cell phenotype due to suppressed differentiation.
We observed increased levels of nuclear AHR in mouse keratinocytes following induction of differentiation. Translocation and activation of AHR is unlikely to be due to the indirect mechanism reported by Wincent and colleagues (Wincent et al., 2012), whereby inhibition of CYP1A1 increases culture media levels of the agonist FICZ, as differentiation induces CYP1A1 enzyme activity (Jones et al., 1997; Reiners, Jr. et al., 1990). We hypothesize rather that keratinocyte differentiation generates endogenous AHR ligands, which drive translocation and activation. It is possible that differentiation-induced prostaglandin synthesis could mediate the observed nuclear translocation of the AHR, since prostaglandins and other arachidonic acid metabolites can act as AhR agonists (Chiaro et al., 2008b; Seidel et al., 2001) and increased Cox-2 expression is associated with epidermal differentiation in vitro and in vivo (Cameron et al., 1990; Evans et al., 1993; Leong et al., 1996; Xu et al., 2008), although this remains to be determined directly. Surprisingly, GNF351 treatment in differentiation and basal culture conditions also increased nuclear AHR levels after 24 h, while SGA360 blocked AHR retention in the nucleus. This is in direct contrast to TCDD and ICZ, AHR agonists which cause rapid nuclear translocation, followed by loss of receptor as previously reported, suggesting that AHR nucleocytoplasmic shuttling and degradation induced by differentiation and AHR antagonists, are distinct from that driven by exogenous agonists (Davarinos et al., 1999; Ikuta et al., 2000; Ikuta et al., 1998). Our results also show distinct mechanisms of action of the pure antagonist GNF351 and the SAhRM SGA360, as GNF351 appears to allow AHR nuclear translocation but prevent AHR mediated gene expression while SGA360 blocks AHR nuclear translocation. Similar distinctions have been made for regulation of inflammatory gene expression by SAhRMs, which is DRE-independent and involves inhibition of AHR interaction with pro-inflammatory pathways although pure antagonists also block nuclear retention of the AHR in other cell types (DiNatale et al., 2010; Patel et al., 2009a; Smith et al., 2011; Tanos et al., 2012). Computational analysis has indicated the presence of DRE in promoters for many terminal differentiation genes, and functional DREs have been identified in the human FLG gene (Sutter et al., 2011). Our expression results reveal that keratinocyte differentiation gene expression is suppressed upon ablation or pharmacological inhibition of AHR activity. In addition, nuclear localization of the AHR during differentiation suggests that endogenous AHR ligands drive translocation and binding of AHR/ARNT complexes to DREs present in the promoter region of differentiation genes, although this remains to be directly determined through ChIP and ChIP seq. Interestingly, ablation of ARNT in mouse skin or knockdown in human keratinocytes induces expression of a number of epidermal differentiation genes (Geng et al., 2006; Robertson et al., 2012), although Flg and Lor are reduced in Arnt-/- skin (Geng et al., 2006), similar to our findings with Ahr-/-keratinocytes. However, this aberrant induction of differentiation gene expression may be indirect through downregulation of the EGFR ligand amphiregulin in the absence of ARNT (Robertson et al., 2012). It is possible that some effects of AHR ablation or antagonism on differentiation could be indirect, through downregulation of the transcription factor Pou2f3 (Skn-1a) critical for epidermal proliferation and differentiation (Andersen et al., 1997; Takemoto et al., 2010).
We also observed upregulation of a significant number of genes encoding extracellular matrix and cell attachment proteins in Ahr-/- keratinocytes relative to Ahr+/+, but it is not known if this represents secondary changes due to long term ablation of AHR function, possible function of AHR as a repressor of gene expression, or reprogramming of primary keratinocyte epithelial phenotype. In Ahr-/- keratinocytes we found a significant downregulation of three cytokine genes that are important in different inflammatory skin diseases Il33, Il36g and Tslp (Balato et al., 2012; Carrier et al., 2011; Hueber et al., 2011; Larson et al., 2010; Shigeno et al., 2009; Tortola et al., 2012). Interestingly, suppression of Il33 and Il36g expression by GNF351 and SGA360 suggests that AHR antagonists and selective modulators may be useful therapeutics for regulating skin inflammation. The observed upregulation of the proinflammatory cytokine IL24 (Kumari et al., 2013; Kunz et al., 2006) in Ahr-/- keratinocytes indicate that this may be more complex within a treatment setting.
Similar to our recent finding that AHR activation mediated by coal tar restores disturbed epidermal differentiation and improved skin barrier function in atopic dermatitis (van den Bogaard et al., 2013), this study opens an additional avenue for the development of AHR antagonists or selective modulators that can regulate AHR signaling in keratinocytes to modulate epidermal differentiation and inflammation.
Materials and Methods
Isolation and culture of primary keratinocytes
Primary keratinocytes were isolated from newborn Ahr+/+ and Ahr-/- mice, PCR genotyped (Schmidt et al., 1996) and cultured as described in 0.05 mM CaCl2 medium (Markell et al., 2011). Cells were treated with 10 nM TCDD, 500 nM ICZ, 200 nM GNF351 and 10 μM SGA360 or DMSO for the indicated times in either 0.05 mM or 0.12 mM CaCl2 containing media. All animal studies were done using protocols approved by the Penn State University IACUC. Human keratinocytes were isolated from abdominal skin (Rheinwald et al., 1975) in accordance with the Declaration of Helsinki principles, approval by Radboud University Medical Center and written informed patient consent. For submerged culture, keratinocytes were cultured in KGM (Lonza Ltd, Slough, UK) and differentiated by growth factor depletion (Van Ruissen et al., 1996). Cells were treated with 50 nM indirubin (IR), 500 nM GNF351, 5 μM CH223191, 500 nM SR1 0.1 and 1 μM FICZ at indicated time points or DMSO as vehicle control.
Microarray Analysis
RNA isolated from 4 independent pooled Ahr+/+ primary keratinocyte cultures and 3 Ahr-/- cultures were reverse transcribed and hybridized to Affymetrix Mouse Gene ST 2.0 arrays in the Penn State Genomics Core Facility according to the manufacturers protocol. Arrays were scanned using a GeneChip Scanner 3000 7G and analyzed using ArrayStar 11 Software (DNASTAR, Madison WI) with RMA background correction and quantile normalization. Mean log2 signal was used to compare gene expression between groups and significantly different genes identified using a 1.5-fold cut off and p value <.05 using a one sided equal variance Student t test. Functional annotation clustering with genes identified as differentially expressed using ArrayStar11 was conducted using DAVID Bioinformatics Software (Huang et al., 2009a; Huang et al., 2009b). GEO accession number:GSE62490.
Human skin equivalent development
Human primary keratinocytes were seeded onto plastic inert filters (ThinCerts, Greiner Bio-one, Breda, The Netherlands) in CnT-PR medium (CELLnTEC, Bern, Switzerland). After 48h, cells were switched to CnT-PR-3D barrier (CELLnTEC) for 24h, and then cultured at the air-liquid interface for 10 days. Human skin equivalents using de-epidermised dermis were generated as described previously (van den Bogaard et al., 2012). Skin equivalents were treated, as indicated, with 500 nM GNF351 or 5 μM CH223191.
Immunostaining
Formalin-fixed paraffin-embedded skin equivalents were stained with hematoxylin and eosin (H&E) (Sigma) or processed for immunohistochemistry. Human keratinocytes on glass coverslips were treated twice over 48h with AHR antagonists (GNF351, 500 nM; CH223191, 5 μM; SR1, 500 nM) fixed with 4% paraformaldehyde, permeabilized with 1% Triton-X/PBS and stained for Ki-67. Nuclei were counterstained with DAPI.
RNA and q-PCR
Total RNA was isolated from keratinocytes using Ribozol and quantitative RT-PCR (qPCR) was performed as described, in triplicate (Markell et al., 2011) and normalized to Gapdh (mouse) or RPLP0 (human). Intron spanning primer sequences for analyzed genes were obtained using Primer 3 (Rozen et al., 2000) software with Genebank sequence information.
Western Blot Analysis
Proteins were isolated from mouse keratinocytes as described (Cheng et al., 1990; Hogan et al., 2013), and from human keratinocytes with RIPA buffer. Separated proteins were detected by ECL (Markell et al., 2011).
Statistical Analysis
For gene expression statistical significance was determined between genotypes or between treatment and control using a Student t test and GraphPad Prism4 with significance determined as a p value <0.05.
Supplementary Material
Figure S1. AhR regulates Il33 and Il36g in mouse keratinocytes. Quantitative Rt-PCR analysis of Il33 and Il36g expression primary cultures of Ahr +/+ and Ahr -/- keratinocytes or in Ahr +/+ keratinocytes treated with 200 nM GNF351 or 10 μM SGA360 for 24h or DMSO in proliferation media (0.05 mM CaCl2). Expression was normalized to that of Gapdh and levels in vehicle treated Ahr +/+ cells set as 1. Each histogram the average expression of three independent cultures, representative of two independent experiments. * = significantly different from Ahr +/+ p<.05.
Figure S2. Regulation of differentiation gene expression by FICZ. Human primary keratinocytes (n=3 donors) were cultured with FICZ at the indicated concentrations and harvested after 3, 6 and 24 h. Gene expression was normalized using the housekeeping gene, RPLP0 and expression of vehicle control was set to 1, indicated by the dotted line. * p<0.05; ** p<0.01; *** p<0.001, compared to vehicle control.
Figure S3. AhR ligands have minimal toxicity in human and mouse keratinocytes. Triplicate cultures of a) Human, or b) mouse keratinocytes were treated with inidicated compounds for 24 h after which viability was determined using the LDH Cytotoxicity Assay (Roche). GNF351: 500 nM (human), 200 nM (mouse); CH223191: 5 uM; SR1: 500 nM; TCDD: 10 nM; FICZ: 1 μM; SGA360 10 μM; ICZ, 500 nM.
Figure S4. AhR antagonists inhibit human keratinocyte proliferation in monolayer culture. Human keratinocytes were seeded on coverslips and cultured for three days until colonies formed. Cells were treated with AHR antagonists (GNF351, 500 nM; CH223191 (CH), 5 μM; SR1, 500 nM) for 48h and then were fixed and stained for Ki67 and counterstained with DAPI to visualize nuclei.
Table S1. Genes Significantly Downregulated in Ahr -/- Mouse Keratinocytes
Table S2. Genes Significantly Upregulated in Ahr -/- Mouse Keratinocytes
Acknowledgments
This work was supported by a grant from the Ter Meulen Foundation (KNAW), The Netherlands and the Radboud University Medical Center, The Netherlands (to EB and JS), and The National Institutes of Health 1R21ES020922 (ABG and GHP) and RO1ES19964 (GHP). The authors would like to acknowledge Kyle Breech for excellent technical assistance.
Grant Support: Ter Meulen Foundation (KNAW), The Netherlands and the Radboud University Medical Center, The Netherlands (to EB and JS), and The National Institutes of Health 1R21ES020922 (ABG) and RO1ES19964 and RO1ES004869 (GHP)
Abbreviations
- AHR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- SAhRM
selective aryl hydrocarbon receptor modulator
- bHLH/PAS
basic Helix-Loop-Helix/Per-Arnt-Sim
- DRE
dioxin response element
- TCDD
2,3,7,8-Tetrachloro-p-dibenzodioxin
- ICZ
indolo[3,2-b]carbazole
- FICZ
6-Formylindolo(3,2-b)carbazole
Footnotes
Conflict of Interest: The authors have no conflict of interest to declare.
See Supplemental Material & Methods for mice, chemicals, antibodies and primer sequences.
References
- Andersen B, Weinberg WC, Rennekampff O, et al. Functions of the POU domain genes Skn-1a/i and Tst-1/Oct-6/SCIP in epidermal differentiation. Genes Dev. 1997;11:1873–84. doi: 10.1101/gad.11.14.1873. [DOI] [PubMed] [Google Scholar]
- Balato A, Lembo S, Mattii M, et al. IL-33 is secreted by psoriatic keratinocytes and induces pro-inflammatory cytokines via keratinocyte and mast cell activation. Exp Dermatol. 2012;21:892–4. doi: 10.1111/exd.12027. [DOI] [PubMed] [Google Scholar]
- Boitano AE, Wang J, Romeo R, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–8. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron GS, Baldwin JK, Jasheway DW, et al. Arachidonic acid metabolism varies with the state of differentiation in density gradient-separated mouse epidermal cells. J Invest Dermatol. 1990;94:292–6. doi: 10.1111/1523-1747.ep12874434. [DOI] [PubMed] [Google Scholar]
- Carrier Y, Ma HL, Ramon HE, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–37. doi: 10.1038/jid.2011.234. [DOI] [PubMed] [Google Scholar]
- Cheng C, Kilkenny AE, Roop D, et al. The v-ras oncogene inhibits the expression of differentiation markers and facilitates expression of cytokeratins 8 and 18 in mouse keratinocytes. Mol Carcinog. 1990;3:363–73. doi: 10.1002/mc.2940030608. [DOI] [PubMed] [Google Scholar]
- Chiaro CR, Morales JL, Prabhu KS, et al. Leukotriene A4 metabolites are endogenous ligands for the Ah receptor. Biochemistry. 2008a;47:8445–55. doi: 10.1021/bi800712f. [DOI] [PubMed] [Google Scholar]
- Chiaro CR, Patel RD, Perdew GH. 12(R)-Hydroxy-5(Z),8(Z),10(E),14(Z)-eicosatetraenoic acid [12(R)-HETE], an arachidonic acid derivative, is an activator of the aryl hydrocarbon receptor. Mol Pharmacol. 2008b;74:1649–56. doi: 10.1124/mol.108.049379. [DOI] [PubMed] [Google Scholar]
- Choi EY, Lee H, Dingle RW, et al. Development of novel CH223191-based antagonists of the aryl hydrocarbon receptor. Mol Pharmacol. 2012;81:3–11. doi: 10.1124/mol.111.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem. 1999;274:28708–15. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- DiMeglio P, Duarte JH, Ahlfors H, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity. 2014;40:989–1001. doi: 10.1016/j.immuni.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNatale BC, Schroeder JC, Perdew GH. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol Carcinog. 2010 doi: 10.1002/mc.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CB, Pillai S, Goldyne ME. Endogenous prostaglandin E2 modulates calcium-induced differentiation in human skin keratinocytes. Prostaglandins Leukot Essent Fatty Acids. 1993;49:777–81. doi: 10.1016/0952-3278(93)90025-r. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Ward JM, Sundberg JP, et al. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol. 1997;34:605–14. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- Forrester AR, Elias MS, Woodward EL, et al. Induction of a chloracne phenotype in an epidermal equivalent model by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is dependent on aryl hydrocarbon receptor activation and is not reproduced by aryl hydrocarbon receptor knock down. J Dermatol Sci. 2014;73:10–22. doi: 10.1016/j.jdermsci.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche E, Schafer C, Calles C, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A. 2007;104:8851–6. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S, Mezentsev A, Kalachikov S, et al. Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J Cell Sci. 2006;119:4901–12. doi: 10.1242/jcs.03282. [DOI] [PubMed] [Google Scholar]
- Geusau A, Khorchide M, Mildner M, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs differentiation of normal human epidermal keratinocytes in a skin equivalent model. J Invest Dermatol. 2005;124:275–7. doi: 10.1111/j.0022-202X.2004.23541.x. [DOI] [PubMed] [Google Scholar]
- Greenlee WF, Dold KM, Osborne R. Actions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on human epidermal keratinocytes in culture. In Vitro Cell Dev Biol. 1985;21:509–12. doi: 10.1007/BF02620843. [DOI] [PubMed] [Google Scholar]
- Hogan KA, Ravindran A, Podolsky MA, et al. The TGFbeta1 pathway is required for NFkappaB dependent gene expression in mouse keratinocytes. Cytokine. 2013;64:652–9. doi: 10.1016/j.cyto.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang dW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang dW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hueber AJ, ves-Filho JC, Asquith DL, et al. IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur J Immunol. 2011;41:2229–37. doi: 10.1002/eji.201041360. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Eguchi H, Tachibana T, et al. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem. 1998;273:2895–904. doi: 10.1074/jbc.273.5.2895. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Tachibana T, Watanabe J, et al. Nucleocytoplasmic shuttling of the aryl hydrocarbon receptor. J Biochem. 2000;127:503–9. doi: 10.1093/oxfordjournals.jbchem.a022633. [DOI] [PubMed] [Google Scholar]
- Jones CL, Reiners JJ., Jr Differentiation status of cultured murine keratinocytes modulates induction of genes responsive to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch Biochem Biophys. 1997;347:163–73. doi: 10.1006/abbi.1997.0350. [DOI] [PubMed] [Google Scholar]
- Kalmes M, Hennen J, Clemens J, et al. Impact of aryl hydrocarbon receptor (AhR) knockdown on cell cycle progression in human HaCaT keratinocytes. Biol Chem. 2011;392:643–51. doi: 10.1515/BC.2011.067. [DOI] [PubMed] [Google Scholar]
- Kumari S, Bonnet MC, Ulvmar MH, et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity. 2013;39:899–911. doi: 10.1016/j.immuni.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Kunz S, Wolk K, Witte E, et al. Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp Dermatol. 2006;15:991–1004. doi: 10.1111/j.1600-0625.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- Lahoti TS, John K, Hughes JM, et al. Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2012-202639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson RP, Zimmerli SC, Comeau MR, et al. Dibutyl phthalate-induced thymic stromal lymphopoietin is required for Th2 contact hypersensitivity responses. J Immunol. 2010;184:2974–84. doi: 10.4049/jimmunol.0803478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong J, Hughes-Fulford M, Rakhlin N, et al. Cyclooxygenases in human and mouse skin and cultured human keratinocytes: association of COX-2 expression with human keratinocyte differentiation. Exp Cell Res. 1996;224:79–87. doi: 10.1006/excr.1996.0113. [DOI] [PubMed] [Google Scholar]
- Loertscher JA, Sattler CA, len-Hoffmann BL. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol Appl Pharmacol. 2001;175:121–9. doi: 10.1006/taap.2001.9202. [DOI] [PubMed] [Google Scholar]
- Markell LM, Masiuk KE, Blazanin N, et al. Pharmacologic Inhibition of ALK5 Causes Selective Induction of Terminal Differentiation in Mouse Keratinocytes Expressing Oncogenic HRAS. Mol Cancer Res. 2011 doi: 10.1158/1541-7786.MCR-11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenyi CS, Carrion SL, Jones LA, et al. Effects of in Utero Exposure of C57BL/6J Mice to 2,3,7,8-Tetrachlorodibenzo-p-dioxin on Epidermal Permeability Barrier Development and Function. Environ Health Perspect. 2014;122:1052–8. doi: 10.1289/ehp.1308045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Flaveny CA, DiNatale BC, et al. Antagonism of aryl hydrocarbon receptor signaling by 6,2′,4′-trimethoxyflavone. J Pharmacol Exp Ther. 2010a;332:135–44. doi: 10.1124/jpet.109.158261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Krishnegowda G, DiNatale BC, et al. Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem Res Toxicol. 2010b;23:955–66. doi: 10.1021/tx100045h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omiecinski CJ, Vanden Heuvel JP, Perdew GH, et al. Xenobiotic Metabolism, Disposition and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol Sci. 2010 doi: 10.1093/toxsci/kfq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RD, Murray IA, Flaveny CA, et al. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab Invest. 2009a;89:695–707. doi: 10.1038/labinvest.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RD, Murray IA, Flaveny CA, et al. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab Invest. 2009b;89:695–707. doi: 10.1038/labinvest.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–54. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Qiu J, Heller JJ, Guo X, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners JJ, Jr, Cantu AR, Pavone A. Modulation of constitutive cytochrome P-450 expression in vivo and in vitro in murine keratinocytes as a function of differentiation and extracellular Ca2+ concentration. Proc Natl Acad Sci U S A. 1990;87:1825–9. doi: 10.1073/pnas.87.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners JJ, Jr, Cantu AR, Thai G, et al. Differential expression of basal and hydrocarbon-induced cytochrome P-450 monooxygenase and quinone reductase activities in subpopulations of murine epidermal cells differing in their stages of differentiation. Drug Metab Dispos. 1992;20:360–6. [PubMed] [Google Scholar]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinising colonies from single cells. Cell. 1975;6:331–44. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Robertson ED, Weir L, Romanowska M, et al. ARNT controls the expression of epidermal differentiation genes through. J Cell Sci. 2012;125:3320–32. doi: 10.1242/jcs.095125. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sadek CM, len-Hoffmann BL. Cytochrome P450IA1 is rapidly induced in normal human keratinocytes in the absence of xenobiotics. J Biol Chem. 1994;269:16067–74. [PubMed] [Google Scholar]
- Schmidt JV, Su GH, Reddy JK, et al. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A. 1996;93:6731–6. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JC, DiNatale BC, Murray IA, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49:393–400. doi: 10.1021/bi901786x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel SD, Winters GM, Rogers WJ, et al. Activation of the Ah receptor signaling pathway by prostaglandins. J Biochem Mol Toxicol. 2001;15:187–96. doi: 10.1002/jbt.16. [DOI] [PubMed] [Google Scholar]
- Shigeno T, Katakuse M, Fujita T, et al. Phthalate ester-induced thymic stromal lymphopoietin mediates allergic dermatitis in mice. Immunology. 2009;128:e849–e857. doi: 10.1111/j.1365-2567.2009.03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Nakatsuru Y, Ichinose M, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97:779–82. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Murray IA, Tanos R, et al. Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther. 2011;338:318–27. doi: 10.1124/jpet.110.178392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter CH, Bodreddigari S, Campion C, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol Sci. 2011;124:128–37. doi: 10.1093/toxsci/kfr205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter CH, Yin H, Li Y, et al. EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc Natl Acad Sci U S A. 2009;106:4266–71. doi: 10.1073/pnas.0900874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson HI. Cytochrome P450 expression in human keratinocytes: an aryl hydrocarbon receptor perspective. Chem Biol Interact. 2004;149:69–79. doi: 10.1016/j.cbi.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Takemoto H, Tamai K, Akasaka E, et al. Relation between the expression levels of the POU transcription factors Skn-1a and Skn-1n and keratinocyte differentiation. J Dermatol Sci. 2010;60:203–5. doi: 10.1016/j.jdermsci.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Tanos R, Patel RD, Murray IA, et al. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology. 2012;55:1994–2004. doi: 10.1002/hep.25571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122:3965–76. doi: 10.1172/JCI63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaard EH, Bergboer JG, Vonk-Bergers M, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest. 2013;123:917–27. doi: 10.1172/JCI65642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaard EH, Rodijk-Olthuis D, Jansen PA, et al. Rho kinase inhibitor Y-27632 prolongs the life span of adult human keratinocytes, enhances skin equivalent development, and facilitates lentiviral transduction. Tissue Eng Part A. 2012;18:1827–36. doi: 10.1089/ten.tea.2011.0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ruissen F, de Jongh GJ, Zeeuwen PL, et al. Induction of normal and psoriatic phenotypes in submerged keratinocyte cultures. J Cell Physiol. 1996;168:442–52. doi: 10.1002/(SICI)1097-4652(199608)168:2<442::AID-JCP23>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Christensen J, et al. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–9. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner R, Brommer S, Czarnetzki BM, et al. The differentiation-related upregulation of aryl hydrocarbon receptor transcript levels is suppressed by retinoic acid. Biochem Biophys Res Commun. 1995;209:706–11. doi: 10.1006/bbrc.1995.1556. [DOI] [PubMed] [Google Scholar]
- Wincent E, Bengtsson J, Mohammadi BA, et al. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2012;109:4479–84. doi: 10.1073/pnas.1118467109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yang L, Yang T, et al. Expression pattern of cyclooxygenase-2 in normal rat epidermis and pilosebaceous unit during hair cycle. Acta Histochem Cytochem. 2008;41:157–63. doi: 10.1267/ahc.08024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AhR regulates Il33 and Il36g in mouse keratinocytes. Quantitative Rt-PCR analysis of Il33 and Il36g expression primary cultures of Ahr +/+ and Ahr -/- keratinocytes or in Ahr +/+ keratinocytes treated with 200 nM GNF351 or 10 μM SGA360 for 24h or DMSO in proliferation media (0.05 mM CaCl2). Expression was normalized to that of Gapdh and levels in vehicle treated Ahr +/+ cells set as 1. Each histogram the average expression of three independent cultures, representative of two independent experiments. * = significantly different from Ahr +/+ p<.05.
Figure S2. Regulation of differentiation gene expression by FICZ. Human primary keratinocytes (n=3 donors) were cultured with FICZ at the indicated concentrations and harvested after 3, 6 and 24 h. Gene expression was normalized using the housekeeping gene, RPLP0 and expression of vehicle control was set to 1, indicated by the dotted line. * p<0.05; ** p<0.01; *** p<0.001, compared to vehicle control.
Figure S3. AhR ligands have minimal toxicity in human and mouse keratinocytes. Triplicate cultures of a) Human, or b) mouse keratinocytes were treated with inidicated compounds for 24 h after which viability was determined using the LDH Cytotoxicity Assay (Roche). GNF351: 500 nM (human), 200 nM (mouse); CH223191: 5 uM; SR1: 500 nM; TCDD: 10 nM; FICZ: 1 μM; SGA360 10 μM; ICZ, 500 nM.
Figure S4. AhR antagonists inhibit human keratinocyte proliferation in monolayer culture. Human keratinocytes were seeded on coverslips and cultured for three days until colonies formed. Cells were treated with AHR antagonists (GNF351, 500 nM; CH223191 (CH), 5 μM; SR1, 500 nM) for 48h and then were fixed and stained for Ki67 and counterstained with DAPI to visualize nuclei.
Table S1. Genes Significantly Downregulated in Ahr -/- Mouse Keratinocytes
Table S2. Genes Significantly Upregulated in Ahr -/- Mouse Keratinocytes