

Abstract
Skeletal muscle specification and morphogenesis during early development are critical for normal physiology. In addition to mediating locomotion, skeletal muscle is a secretory organ that contributes to metabolic homeostasis. Muscle is a highly adaptable tissue, as evidenced by the ability to increase muscle cell size and/or number in response to weight bearing exercise. Conversely, muscle wasting can occur during aging (sarcopenia), cancer (cancer cachexia), extended hospital stays (disuse atrophy), and in many genetic diseases collectively known as the muscular dystrophies and myopathies. It is therefore of great interest to understand the cellular and molecular mechanisms that mediate skeletal muscle development and adaptation. Muscle morphogenesis transforms short muscle precursor cells into long, multinucleate myotubes that anchor to tendons via the myotendinous junction. This process requires carefully orchestrated interactions between cells and their extracellular matrix microenvironment. These interactions are dynamic, allowing muscle cells to sense biophysical, structural, organizational, and/or signaling changes within their microenvironment and respond appropriately. In many musculoskeletal diseases, these cell adhesion interactions are disrupted to such a degree that normal cellular adaptive responses are not sufficient to compensate for accumulating damage. Thus, one major focus of current research is to identify the cell adhesion mechanisms that drive muscle morphogenesis, with the hope that understanding how muscle cell adhesion promotes the intrinsic adaptability of muscle tissue during development may provide insight into potential therapeutic approaches for muscle diseases. Our objectives in this review are to highlight recent studies suggesting conserved roles for cell-extracellular matrix adhesion in vertebrate muscle morphogenesis and cellular adaptive responses in animal models of muscle diseases.
Keywords: skeletal muscle, cell adhesion, extracellular matrix, morphogenesis, cellular adaptation, muscular dystrophy
INTRODUCTION
Proper muscle specification and morphogenesis during development are critical for adult muscle physiology. In addition, active homeostasis maintains muscle function despite the normal wear and tear that occurs with activity. Skeletal muscle is a remarkably regenerative and adaptable tissue, with muscle hypertrophy (increase in cell size) and/or hyperplasia (increase in cell number), occurring in response to exercise or injury. These adaptive cellular behaviors can be triggered by stimuli (physical or chemical) both inside the cell and in the microenvironment surrounding cells. A key fundamental question is how do cells detect changes in their microenvironment, integrate and transduce these inputs, and respond with discrete outputs? It is likely that that cell-extracellular matrix (ECM) adhesion complexes play a major role in this process because: (1) cell-ECM adhesion complexes provide a physical link between the cytoskeleton and the microenvironment, (2) bidirectional signaling between cells and their microenvironment occurs through cell-ECM adhesion complexes, and (3) cell-ECM adhesion complexes are dynamic. As cellular morphogenetic or adaptive responses require the perception and integration of multiple signaling inputs, cell-ECM adhesions may function as a link between signaling inputs and cellular behaviors. Here, we will focus on recent progress in understanding how cell-ECM adhesion mediates cellular responses during muscle morphogenesis and disease.
To highlight dynamic roles for cell-ECM adhesions in physiological and pathological processes in skeletal muscle tissue, we will focus on two major themes – connecting signaling/specification to normal muscle morphogenesis via cell-ECM adhesion, and connecting cell-ECM adhesion to cellular adaptation in muscle diseases. Excellent recent reviews have extensively documented what is known about cell-ECM and cell-cell adhesion during muscle development and disease (Borycki, 2013; Lund and Cornelison, 2013; Thorsteinsdóttir et al., 2011); thus, we will mainly focus on newer information through the lens of the innate adaptability of muscle tissue. We will begin by providing a brief introduction to proteins involved in muscle cell-ECM adhesion and then discuss roles for cell-ECM adhesion components in multiple stages of muscle morphogenesis, homeostasis, and disease.
CELL ADHESION AND THE MUSCLE EXTRACELLULAR MATRIX
Tissue morphogenesis and homeostasis require carefully orchestrated interactions between cells and their ECM microenvironment. A tissue’s characteristics result from the properties and proportions of its ECM proteins. Thus, the stiffness of bones and the elasticity of (young) skin result from the biophysical properties of their constituent ECMs. Within the last two decades it has become clear that the ECM also modulates cellular signaling by multiple mechanisms. One way that the ECM modulates cellular signaling is by sequestering signaling molecules within the matrix (Droguett et al., 2006). Cell-ECM interactions can also modify cellular responses to signaling. For example, ECM receptors act as non-canonical co-receptors that impart specificity to FGF signaling (Polanska et al., 2009). ECM receptors are also both modulated and activated by Notch and MAP-kinase signaling (Karsan, 2008; Yee et al., 2008). Thus, the “ECM interactome” is ubiquitous and may play a central role in integrating cellular responses to multiple signaling pathways as well as mechanical stimuli. As morphogenesis and adaptation require the perception and integration of multiple signaling inputs, cell-ECM adhesions may function to ‘translate’ signaling inputs into discrete cellular responses.
The muscle extracellular matrix
The structure of mature mammalian muscle ECM has recently been described in detail (Gillies and Lieber, 2011). In brief, ECM is found at all spatial levels of muscle: muscle fibers are surrounded by basement membranes (BMs), ECM surrounds bundles of fibers, and whole muscles are encased in ECM (Fig. 1). The bridges between muscle and other tissues, the NMJs and the MTJs, are also rich in ECM.
Figure 1.

The muscle ECM. A: Simplified cartoon of proteins involved in muscle cell adhesion to the ECM. Multiple transmembrane receptor complexes indirectly link the intracellular cytoskeleton to the ECM. The BM attaches to the collagen-rich interstitial matrix. B: Larger size scale view of the organization of muscle ECM. Not only are individual muscle fibers encased by ECM, but there are also specialized matrices that define the neuromuscular and myotendinous junctions (NMJ and MTJ, respectively). C: A 24 hpf zebrafish embryo stained with phalloidin (red) to visualize actin and an antibody against laminin-111 (green). Side view, anterior left, dorsal top. Note the long muscle cells (red arrow) that connect to laminin-111 at the myotome boundaries (green arrow). These boundaries will generate the MTJ.
Why is it important to understand how the muscle ECM develops and is maintained? One dramatic demonstration of muscle ECM function comes from comparing natural fiber bundles with groups of fibers dissected away from ECM and tied together. The groups (without native ECM) bore the same stress as single muscle fibers whereas the naturally occurring bundles (with ECM) had a significantly higher modulus (Lieber and Ward, 2013; Meyer and Lieber, 2011). These data strongly argue that the muscle ECM bears much of the passive load. This would in turn suggest that many of the clinical symptoms involving range of motion and stiffness are derived from changes to the ECM (Gillies and Lieber, 2011). Therefore, elucidating how the muscle ECM develops normally and discovering manipulations that can improve the muscle ECM in pathological conditions will likely inform efforts to maintain muscle health and prevent muscle diseases.
Although assembly of the ECM during development is an especially interesting phenomenon because the ECM has to continue to function despite being remodeled and reorganized as animals grow and change, this process is not well understood. We and others have shown that the ECM changes during muscle development and regeneration in both zebrafish and mouse (Bajanca et al., 2004; Bentzinger et al., 2013; Costa et al., 2008; Deries et al., 2012; Marques and Thorsteinsdóttir, 2013; Snow and Henry, 2009). These dynamic changes allow the muscle ECM to be tuned to the morphogenetic or adaptive responses that generate and maintain this amazing structure. Expression of many cell-ECM adhesion proteins during muscle development is similar across vertebrate species. Here, we will highlight how studies that exploit the advantages of different vertebrate systems provide complementary insight into ECM modulation of muscle development and homeostasis.
Extracellular matrix components
The ECM is a meshwork of secreted proteins that are assembled into a complex network (Frantz et al., 2010) (Fig. 1). There are many different proteins in the ECM, only the most prominent of which (proteoglycans and fibrous glycoproteins) will be introduced here. Proteoglycans are heavily glycosylated ECM proteins which, due to their one or more glycosaminoglycan chains, absorb water to generate a hydrous gel that resists compressive forces (Frantz et al., 2010). The glycosaminoglycan chains that covalently or non-covalently associate with core proteins to form proteoglycans are hyaluronan, heparin sulfate, chondroitin sulfate, dermatan sulfate, and keratan sulfate. Specific proteoglycan molecules include Aggrecan, Decorin, Perlecan, Agrin, and Syndecans. Proteoglycans play roles in regulating both collagen fibrillogenesis and the movement/activity of signaling molecules through the ECM (Yoon and Halper, 2005).
The fibrous glycoproteins Fibronectin (Fn) and laminin will be the main focus of this review, although roles for other fibrous glycoproteins in muscle development and disease will be mentioned as well. Other fibrous glycoproteins include collagens, Vitronectin, Tenascin, Thrombospondins, Entactin, Nephronectin, and Fibrinogen. Collagens, which are the most abundant protein in the human body, comprise 1–2% of skeletal muscle tissue (Sikorski, 2001) and maintain the structural integrity of tissues. There are 28 types of collagens in vertebrates and over 40 different collagen-encoding genes in humans. Different collagen types (fibrillar, globular) comprise different structures. The tensile strength of the ECM primarily depends upon the alignment and density of collagens (Kjaer, 2004). Collagen proteins undergo many post-translational modifications, including hydroxylation, glycosylation, assembly into triple helixes, proteolytic cleavages, and crosslinking.
Fn is an interstitial matrix protein that is perhaps most renowned for its functions during branching morphogenesis and cell migration (Davidson et al., 2006; Jiang et al., 2000; Larsen et al., 2006; Matsui et al., 2007; Roman, 1997; Roman et al., 1991; Sakai et al., 2003; Trinh and Stainier, 2004). Fn also plays a critical role in muscle development and repair (Bentzinger et al., 2013; Snow et al., 2008b). There are 2 forms of Fn: (1) plasma Fn which circulates in the blood and is globular, and (2) cellular Fn which is assembled into fibrils that in turn polymerize to form a fibrillar matrix. Fn fibril formation is regulated on many levels, including dimerization and Integrin binding, as well as stretch-mediated uncovering of a cryptic self-binding site (Leiss et al., 2008). The ability of fibrillar Fn to be stretched up to fourfold by living cells may be particularly relevant for dynamic cell movements that occur during development (Erickson, 2002).
Laminin is necessary for the assembly of the BM (Hohenester and Yurchenco, 2013). BMs are specialized parts of the ECM that serve a myriad of functions, playing both structural and signaling/organizational roles. BMs interface with epithelial and endothelial tissues, surround muscle cells and Schwann cells, and act as barriers among other functions (Durbeej 2010). As BMs were first described in muscle tissue over a hundred and fifty years ago (Bowman, 1840), it is not surprising that the field of muscle research has contributed immeasurably to our understanding of laminin biology. Laminins are cross-shaped heterotrimeric proteins comprised of an alpha, beta, and gamma chain. Five alpha, four beta, and three gamma chains (Miner and Yurchenco, 2004) assemble in different combinations to generate the 16 known laminins (Aumailley et al., 2005) whose names reflect their composition (e.g. laminin-332 is laminin alpha3beta3gamma2). One way in which laminin function is tailored to particular tissues is through tissue specific expression of different laminin chains. In addition, there are multiple splice variants of laminin chains and laminin is post-translationally glycosylated. The laminin network in BMs is attached to the muscle cell by interacting with transmembrane laminin receptors. Agrin and Perlecan also link laminin to the cell surface. A meshwork of type IV collagen is linked to the laminin network by Entactin and heparin sulfate proteoglycans (HSPGs). The BM is then linked to the fibrous interstitial matrix that is mainly composed of collagens and proteoglycans (Sanes, 2003; 1982).
Comprehensive reviews describing structures and functions for these and other ECM molecules are highly recommended for interested readers (Halper and Kjaer, 2013; Hohenester and Yurchenco, 2013; Mienaltowski and Birk, 2014; Schwarzbauer and DeSimone, 2011; Tzu and Marinkovich, 2008; Yurchenco, 2011).
Extracellular matrix receptors
The ECM is indirectly linked to the intracellular cytoskeleton via transmembrane receptor complexes (Campbell and Humphries, 2011) (Fig. 1). The extracellular domain of these receptors binds to ECM proteins. The short cytoplasmic domain is the hub for assembly of large protein complexes that link to the cytoskeleton. These receptor complexes are signaling hubs that relay signals bidirectionally (Bissell et al., 1982). Signaling from the ECM modifies intracellular signaling, gene expression, and cell morphology; and signals from within the cell modify the molecular composition and structure of the ECM. Cell-ECM adhesion complexes also aid in resisting stress experienced during muscle contraction (Petrof et al., 1993).
Transmembrane Integrin heterodimers function as receptors for many of the ECM glycoproteins described above, such as collagens, Fn, and laminins (Campbell and Humphries, 2011). Integrins consist of an alpha and a beta subunit (e.g. Integrin alpha7beta1) and play key roles in nearly every cellular process. Integrin receptors can be promiscuous: certain Integrin heterodimers can bind multiple ECM proteins, and multiple Integrin heterodimers can bind certain ECM proteins. Integrin receptors for collagens include Integrins alpha1, 2, 10, or 11 dimerized with Integrin beta1. Integrin receptors for Fn include Integrins alpha5beta1, alphaIIbbeta3, and alphaVbeta1, 3, 5, 6, or 8. Laminin-binding Integrin receptors include Integrins alpha3, 6, or 7beta1 and alpha6beta4 (Srichai and Zent, 2010). The many different isoforms of Integrin receptors and ECM ligands and their tissue- and temporally-restricted expression imparts specificity to Integrin-ECM interactions and signaling. The conformation of Integrin heterodimers adds another layer of regulation. Integrins can exist in three conformational states: bent (inactive), primed (active), and ligand bound (Askari et al., 2009). Integrin conformational changes can be induced by interactions involving their cytoplasmic domain (inside-out activation) or extracellular domain (outside-in activation).
There are also non-Integrin receptors for collagens and laminins (Bello et al., 2014). Additional laminin receptor complexes that are critical for skeletal muscle function are the Dystrophin- or Utrophin-glycoprotein complexes (DGC or UGC, respectively) (Carmignac and Durbeej, 2012). These complexes also link the ECM to the intracellular cytoskeleton. Both the DGC and UGC contain the transmembrane proteins beta-Dystroglycan, Sarcoglycans, and Sarcospan. Alpha-Dystroglycan binds to laminin. The distinguishing feature between these complexes is whether Dystrophin or Utrophin anchor the complexes to actin within the cell. The UGC is somewhat homologous to the DGC, but is spatially localized to the neuromuscular junction (NMJ) and myotendinous junction (MTJ) regions of the muscle cell membrane whereas the DGC is expressed throughout the membrane (Matsumura et al., 1992; Pearce et al., 1993; Tinsley et al., 1992; Zhao et al., 1992). In addition to differing spatial localization, the UGC and DGC differ with regards to anchoring specific cytoplasmic proteins to the membrane (Li et al., 2010).
Again, this is a brief overview of ECM receptors and detailed reviews on transmembrane receptor complexes for ECM ligands are recommended for additional reading (Campbell and Humphries, 2011; Hara and Campbell, 2014; Harburger and Calderwood, 2009).
CELL ADHESION TO THE ECM PLAYS A CRITICAL ROLE IN SEGMENTATION
The first step in muscle development is segmentation of the paraxial mesoderm flanking the notochord into somites, which give rise to most of musculature in vertebrates (Buckingham et al., 2003; Hollway and Currie, 2003). Somites in the amniotes and teleosts studied thus far consist of an outer epithelial layer that surrounds an inner mesenchymal cell mass (Gossler and Hrabĕ de Angelis, 1998; Kimmel et al., 1995). Somite formation proceeds in an anterior to posterior progression during development. Thus, anterior somites are older than posterior somites. This results in asynchronous muscle development as well, with anterior segments differentiating earlier than posterior segments.
As a discussion of how somites are patterned is outside the scope of this review, we will concentrate on somite boundary formation and maturation of somites into myotomes (groups of specified muscle fibers). Somites are transient structures. Shortly after somite formation in amniotes, the ventral aspect of the somite undergoes an epithelial to mesenchymal transition (EMT) to form the sclerotome that will give rise to axial cartilage and bones and the syndetome that will give rise to tendons (Deries et al., 2010; Tozer and Duprez, 2005). In contrast, the medial and dorsal aspect of the somite remains epithelial and is termed the dermomyotome. The dermomyotome will give rise to the primary myotome, to muscle progenitors that sustain embryonic and fetal growth, and to muscle stem cells that mediate homeostasis and repair in the adult (Relaix et al., 2005; Ben-Yair and Kalcheim, 2005; Kassar-Duchossy et al., 2005; Gros et al., 2005; Schienda et al., 2006). In zebrafish, the majority of somitic cells give rise to the primary myotome, with the sclerotome and syndetome elements occupying a small proportion of the somite (Kimmel et al., 1995; Morin-Kensicki and Eisen, 1997). Although zebrafish lack a dermomyotome per se, there is a population of somitic cells that gives rise to an external cell layer (ECL) that covers the myotome (Devoto et al., 2006; Siegel et al., 2013; Stellabotte and Devoto, 2007) (Fig. 2). The ECL is composed of mitotically active Pax7 expressing cells that contribute to muscle growth and function in a manner analogous to the amniote dermomyotome. Thus, although the relative proportions and exact morphology of these elements (sclerotome, syndetome, dermomyotome) differ between amniotes and teleosts, there is largely functional conservation of these somitic subdomains.
Figure 2.

Structure of the zebrafish and amniote myotomes. A: Top Panel - Muscle is the major constituent of the zebrafish myotome. Tendon progenitors and sclerotome are located medially. Most of the muscle cells are fast-twitch muscle. The most superficial muscle fibers are slow-twitch muscle fibers (gray). The external cell layer (red) is hypothesized to be somewhat equivalent to the amniote dermomyotome. Bottom panel - The ECM at the MTJ is superimposed upon a myotome. Laminin is expressed throughout the medial-lateral extent of the MTJ, but Fn is degraded medially to migrating slow-twitch fibers to end up primarily concentrated at the MTJ adjacent to slow-twitch fibers. B: Top Panel - Structure of the amniote myotome. The epithelial dermomyotome contains muscle progenitor cells that will sustain growth and will also give rise to satellite cells. The connective tissue progenitor region is termed the syndetome. Bottom panel - ECM of the amniote myotome. Note that the myotomal BM separates the sclerotome from the myotome. Fn is primarily concentrated at myotome boundaries.
There is remarkable conservation of roles for ECM during muscle development in amniotes and zebrafish despite the difference in somitic structure. In both amniotes and zebrafish, different regions of the myotome have distinct matrices (Deries et al., 2012; Snow and Henry, 2009) (Fig. 2). In amniotes, the dermomyotome and sclerotome are separated by a distinctive BM in addition to the BM and Fn-rich matrix present at segment boundaries (Anderson et al., 2007; Bajanca et al., 2004; 2006; Tosney et al., 1994). In zebrafish muscle tissue, ECM surrounds muscle fibers and concentrates at the boundaries between muscle segments. As muscle differentiates, the Fn-rich matrix becomes concentrated adjacent to slow-twitch fibers. This is in contrast to the laminin-rich BM that concentrates adjacent to both slow-twitch and fast-twitch muscle fibers. In teleosts, these ECM-rich areas between muscle segments will mature into MTJs, which are the functional equivalent of mammalian MTJs (Gemballa and Vogel, 2002). Next, we will focus on how cell-ECM adhesion guides the myriad of cell behaviors that generate functional muscle tissue.
Fn is the driving force for somite boundary formation
Multiple ECM proteins and their transmembrane receptors are expressed during segmentation and become concentrated at somite boundaries, raising the question of which of these proteins guide somite boundary formation. Transmembrane receptors expressed in muscle include the DGC, Integrin alpha7, Integrin alpha6, Integrin alpha5, and Integrin alphaV (Bajanca et al., 2004; Lunardi and Dente, 2002; Moreau et al., 2003; Parsons et al., 2002; Schofield et al., 1995; Song et al., 1992; Bajanca et al., 2006; Julich et al., 2005). ECM proteins include Fn, laminin, Perlecan, and Vitronectin (Crawford et al., 2003; Henry et al., 2001; Zoeller et al., 2008; Handler et al., 1997; Gullberg et al., 1995). Within the last decade, it has become clear that adhesion to Fn mediates somite boundary formation in mouse, chick, Xenopus, and zebrafish.
Fn null mouse embryos have a truncated anterior-posterior axis, blood vessel defects, neither notochord nor somites form (George et al., 1993; Georges-Labouesse et al., 1996), and while the paraxial mesoderm is specified there is a complete failure of somite condensation (Georges-Labouesse et al., 1996). Similar results are observed in mice where the Integrin binding motif in Fn is mutated (Girós et al., 2011). Fn is also required for segmentation in zebrafish: Fn-deficient zebrafish display dramatic disruption of somitogenesis and cardiac development (Julich et al., 2005; Koshida et al., 2005; Trinh and Stainier, 2004). Time-lapse analyses have shown that ectodermal Fn plays fundamental roles in somite epithelialization during chick development (Martins et al., 2009; Rifes et al., 2007). Finally, Integrin alpha5 is necessary for somite boundary formation in Xenopus (Kragtorp and Miller, 2007). Taken together, these data indicate that adhesion to Fn plays an important role in morphogenesis of somites, but do not elucidate the underlying molecular mechanisms.
Fn assembly at somite boundaries is triggered by inside-out Integrin signaling
One major question is how Fn is assembled at the somite boundary. Cells bind Fn through specific Integrin receptor proteins. Thus, it is necessary to determine which Integrin receptors are required for somite formation, and how Fn assembly is controlled in space and time. Progress in answering these questions was recently made by the Holley lab using the advantages of the zebrafish system (Fig. 3). Using high resolution time-lapse microscopy and bimolecular fluorescence complementation, Julich et al. (2009) observed that Integrin alpha5 clustered in the presomitic mesoderm prior to Fn polymerization. Integrin alpha5 and Integrin beta1 form heterodimers that clustered along nascent somite boundaries. They hypothesized that Integrin activation (inside-out signaling) might play a role in regulating Fn polymerization. A key result in support of this hypothesis was that a variant of Integrin alpha5, which was unable to bind Fn, was capable of binding Integrin beta1 and undergoing initial clustering at somite boundaries. However, in the absence of the ability to bind Fn, Integrin clustering was not maintained and Fn was not polymerized. One aspect of ECM polymerization that is not as readily understood, whether in somite boundaries or in other tissues, is how “available” the matrix is for assembly. Julich et al. (2009) investigated how available the ECM at somite boundaries was for assembly using genetic mosaic analysis. Donor cells that lacked Fn but expressed Integrin alpha5 were transplanted into host embryos that lacked Integrin alpha5 but expressed Fn. Borders of Fn matrix formed around the donor cells. This indicates that Fn in the host was readily available to be polymerized by Integrin alpha5 inside-out signaling in donor cells.
Figure 3.
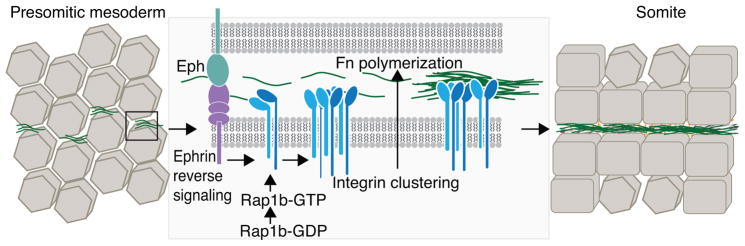
Ephrin reverse signaling and Rap1b contribute to Integrin clustering/activation, which results in Fn polymerization at the nascent somite boundary. Fn (green) is available for polymerization in the presomitic mesoderm but is not organized. Signaling through Ephrin receptors results in Integrin clustering (and presumably activation). Next, inside-out Integrin signaling potentiates Fn polymerization, leading to formation of intersomitic clefts. The alignment and enlargement of these clefts, along with epithelialization of the border cells, results in the formation of stable somite boundaries.
These experiments addressed some of the questions regarding somite boundary formation in vivo, but did not answer how Integrin alpha5 inside-out activation occurs or why Integrin alpha5 only polymerizes Fn at somite boundaries. Elegant experiments showed that reverse signaling through Ephrin receptors plays a major role in spatial regulation of Integrin activation (Julich et al., 2009). Additional experiments showed that a GTPase, Rap1b, acts downstream of Ephrin reverse signaling and contributes to Integrin inside-out activation and maintenance of Fn polymerization at somite boundaries (Lackner, 2013). Taken together, these studies clearly show that Integrin alpha5beta1 is a significant receptor for Fn matrix assembly at somite boundaries (Integrin alphaV also plays a role and is partially redundant with Integrin alpha5 (Dray et al., 2013; Yang et al., 1999)), Integrin alpha5 activation and clustering is the key prerequisite for Fn polymerization, and that Fn assembly by Integrin receptors involves Rap1b and is spatially regulated to somite boundaries by Eph/Ephrin signaling (Fig. 3).
Roles for Fn in adaptation when initial somite formation is disrupted
Although somite formation per se is not necessary for muscle specification, segmentation is critical for organized muscle development because segment boundaries halt elongating muscle fibers and therefore pattern muscle fiber length (Henry et al., 2005). Remarkably, some zebrafish with mutations that disrupt somite formation are viable and survive to adulthood (van Eeden et al., 1996). These fish are viable because segmentation partially recovers and patterns the imperfect formation of segmental elements such as vertebrae and skeletal muscle. What are the cellular and molecular adaptations that allow this recovery of segmentation? How does muscle properly organize in the absence of somite boundary formation? One clue, as mentioned above, is that slow-twitch fibers in zebrafish are specified medially and migrate laterally. Slow-twitch fibers elongate to irregular lengths in the absence of somite formation but still migrate through the presumptive fast-twitch muscle domain. As the length of fast-twitch fibers correlates with slow-twitch fibers in segmentation mutants, it was hypothesized that slow-twitch fibers would be necessary for segmental recovery. Indeed, treatment of segmentation mutants with cyclopamine, to inhibit slow-twitch muscle specification via disrupting Hedgehog (Hh) signaling, obliterated segmental recovery (Henry et al., 2005). Therefore, slow-twitch fibers instruct the recovery of segmentation in segmentation mutants, but the molecular mechanisms are not known. What is known is that Fn plays a fundamentally critical role in this recovery because in the absence of Fn, fast-twitch fibers are not patterned by the migrating slow-twitch fibers and they are significantly less organized (Snow et al., 2008b). Thus, as has been hypothesized in amniotes (Bajanca et al., 2004), Fn plays a major role in limiting/guiding muscle patterning. These data show that Fn plays a significant role in cellular adaptation to generate MTJs in the absence of initial segmentation.
CELL ADHESION TO THE ECM MEDIATES VERTEBRATE MUSCLE MORPHOGENESIS
Muscle morphogenesis begins after segmentation. During muscle development, initial cell movements result in the formation of the primary myotome and a population of self-renewing muscle satellite cells (Siegel et al., 2013; Scaal and Wiegreffe, 2006). Adhesion to the ECM plays an important role in regulating both muscle specification and morphogenesis. Musculoskeletal differentiation then involves multiple morphogenetic processes that require adhesion to the ECM. In brief, initially short muscle precursor cells will elongate, attach to the ECM (Gros et al., 2004; Snow et al., 2008a), fuse to generate multinucleate myotubes (Sieiro-Mosti et al., 2014), and contractile myofibrils will be assembled (Sanger et al., 2009) The BM that surrounds muscle cells will be secreted and assembled, as will the BM at the MTJ. In this next section, we will describe the large-scale cell movements that generate the myotome in chick, mouse, and zebrafish to provide a framework for later discussion of how cell adhesion to the ECM regulates multiple steps of muscle morphogenesis.
Myotome morphogenesis: a paradigm for impacts of ECM on cellular morphogenesis and differentiation
The dermomyotome in chick and mouse contains multipotent progenitor cells that proliferate and express Pax3 and Pax7 (Ben-Yair et al., 2003; Buckingham and Relaix, 2007). Adhesion to laminin in the ECM via Integrin alpha6beta1 is critical to prevent precocious differentiation in the dermomyotome (Bajanca et al., 2006). Presumptive muscle cells then translocate into the myotome and begin to differentiate. Time-lapse studies in chick embryos have elucidated the spatial regulation of entry into the myotome (Gros et al., 2005). The first myocytes translocate from the dorsomedial lip of the dermomyotome into the presumptive myotome. These cells then elongate bidirectionally to span the anterior-posterior length of the somite. Next, cells from the caudal border of the dermomyotome, followed by cells from the rostral border, translocate to the nascent myotome and begin to form elongated muscle cells. Finally, cells from the ventral lateral lip of the dermomyotome contribute to the myotome. The central region of the dermomyotome will give rise to satellite cells and also support growth (Gros et al., 2005). This process results in the formation of the primary myotome, which is comprised of postmitotic, mononucleate muscle cells.
In zebrafish, the functional equivalent of the dermomyotome (the ECL) is derived from anterior border cells (the cells on the anterior border of the somite) that also express Pax3 and Pax7 (Devoto et al., 2006; Siegel et al., 2013). These progenitor cells migrate laterally soon after somite boundary formation to form an extremely thin layer of cells that covers the myotome. An additional difference in myotome structure between amniotes and zebrafish is that slow-twitch and fast-twitch muscle fibers are distinguished much earlier in zebrafish and are spatially segregated in the zebrafish myotome (Devoto et al., 1996). Slow-twitch muscle progenitors are initially the most medial muscle cells, but shortly after somite formation they undergo a dramatic lateral migration through the presumptive fast-twitch muscle domain to become the most superficial layer of muscle (Devoto et al., 1996). Slow-twitch fiber migration is necessary and sufficient to trigger elongation of the fast-twitch fibers (Henry and Amacher, 2004). The terminal ends of muscle fibers attach to somite boundaries, which then become myotome boundaries and give rise to the MTJ.
The spatial segregation of fast- and slow-twitch muscle fibers in zebrafish has facilitated understanding of fiber type specification. In zebrafish, superficial slow-twitch fibers (SSFs), muscle pioneer fibers (MPs), and medial fast-twitch fibers (MFFs) are specified early in muscle development by Hh, BMP, and FGF signaling (Du et al., 1997; Hirsinger et al., 2004; Nguyen-Chi et al., 2012; Wolff et al., 2003). Engrailed expression in response to high levels of Hh ligand secreted from the notochord normally patterns MPs and MFFs. Recent experiments have shown that the ECM functions upstream of fiber specification. In the context of muscle fiber specification in zebrafish, it was recently shown that laminin is required for normal engrailed expression. Engrailed is greatly reduced in lamc1 mutant embryos. Surprisingly, this phenotype was not due to a lack of Hh signal transduction, but rather ectopic activation of BMP signaling which, in this context, antagonizes Hh signaling (Maurya et al., 2011). BMP ligand availability in the zebrafish muscle ECM is regulated by laminins via HSPGs (Dolez et al., 2011). The addition or removal of sulfate groups on the glycosaminoglycan side chains of HSPGs appears to impact the ability of HSPGs to regulate extracellular signaling ligands and muscle cell fate in zebrafish. Similar to lamc1 mutants, zebrafish deficient for Sulf1 (an enzyme that removes sulfate groups from HSPG side chains) have reduced muscle engrailed expression, normal Hh signal transduction, and increased BMP signaling (Meyers et al., 2013). An earlier report found that zebrafish deficient for hs6st, an enzyme that adds sulfate groups to HSPG glycosaminoglycans, have an expanded domain of engrailed expression in muscle despite normal Hh signaling (Bink et al., 2003). While levels of BMP signaling were not investigated in these embryos, one can hypothesize that BMP signaling would be reduced. Altogether, these data show that interactions between different ECM proteins and the post-translation modification of ECM components regulate cell signaling and muscle cell fate.
Cell biology of muscle fusion
One key process involved in generating multinucleate muscle fibers is muscle fusion. Studies in Drosophila have identified a number of proteins required for myoblast fusion (Beckett and Baylies, 2007; Chen and Olson, 2004). The fact that the primary myotome in amniotes and zebrafish is mononucleate (Gros et al., 2004; Waterman, 1969) indicates that myoblast fusion does not mediate the earliest stages of muscle morphogenesis in vertebrates. This fact was confirmed with live imaging studies in both chick and zebrafish (Gros et al., 2004; Snow et al., 2008a). Although use of the Drosophila system has provided tremendous insight into fusion mechanisms during fly muscle development, it is generally unclear these whether mechanisms are conserved in vertebrates (Powell and Wright, 2012). In addition, the cell biology underlying fusion (i.e. what populations of muscle cells fuse) is not well understood. Two excellent studies (Sieiro-Mosti et al., 2014; Powell and Wright, 2011) have provided some insight into cellular and molecular aspects of vertebrate muscle cell fusion.
Identifying the cellular mechanisms underlying muscle fusion is critical for understanding muscle development and regeneration. Electroporation of fluorescent proteins into different regions of the amniote dermomyotome – the dorsal medial lip, dorsal dermomyotome, or anterior/posterior borders of the dermomyotome – allowed quantification of fusion with regards to muscle cell length and volume. This study showed cells in the primary myotome typically fuse with themselves and resident progenitors generally fuse with each other, but that the primary myotome cells do not fuse with resident muscle progenitors in trunk muscle. Limb muscle fusion differed from trunk muscle fusion in two ways. In limb muscle, addition of myonuclei correlated with an increase in muscle cell length. In contrast, trunk muscle cells remained the length of one segment regardless of the number of nuclei. The speed of fusion was also different in the two populations, with limb muscles fusing dramatically faster (Sieiro-Mosti et al., 2014). Thus, this study provides a conceptual framework for future molecular studies.
Another excellent study identified, for the first time, mutants with specific defects in muscle fusion. Given that the initial myotome contains mononucleate muscle cells, any putative fusion mutant would be expected to have long muscle cells that are mononucleate. In other words, fusion would not be predicted to be required for muscle elongation. What was not known was whether fusion was required for sarcomerogenesis or what molecules mediate fusion. Powell and Wright identified two vertebrate specific cell surface receptors required for fast-twitch muscle fusion in zebrafish: Jamb and Jamc (Powell and Wright, 2011). These proteins are deuterostome specific immunoglobulin superfamily cell surface proteins with two extracellular immunoglobulin superfamily domains, a transmembrane domain, and a short cytoplasmic domain (Ebnet et al., 2004). Although fast-twitch fibers are specified and appear morphologically normal in jamb or jamc mutants, fusion does not occur (Fig. 4). Biochemical and genetic mosaic analyses demonstrated that Jamb and Jamc interact and this interaction is required for fusion. Although mechanisms downstream of Jamb and Jamc were not identified, this landmark study has set the bar for rigorous analysis of molecules required specifically for vertebrate muscle fusion and not other aspects of muscle morphogenesis. It will be interesting in the future to determine if these genes are also required for muscle fusion in mammals.
Figure 4.

Cartoon of muscle morphogenesis in chick and zebrafish embryos. A: Developing zebrafish myotome. Slow-twitch fibers (gray) have migrated partway through the fast-twitch domain. Fast-twitch muscle cells (dark blue) medial to the slow-twitch fibers are long. Fast-twitch cells (medium blue) directly intermingled with migrating slow-twitch fibers are elongating. Lateral to the slow-twitch fibers are short muscle precursor cells (light blue) extending protrusions in all directions and the external cell layer (red). B: Developing chick myotome. Note that short myoblasts extend protrusions in all directions, but once elongation has begun protrusions are extended only in the direction of elongation. C: More detailed view of the initial stages of muscle fiber morphogenesis that are similar in amniotes and zebrafish. Short precursor cells extend protrusions in all directions. Once cells begin elongating, they do so in an oriented fashion. Adhesion to laminin is required for oriented myocyte elongation in both species, WNT11 is required for oriented elongation in chick. Boundary capture involves the cessation of muscle cell elongation and regulates muscle cell length. Fn regulates myotomal muscle cell length in both zebrafish and chick, and laminin has been shown to play a role in boundary capture in the zebrafish system. D: Genes required for muscle fusion in zebrafish. To date, the only proteins whose disruption leads to a specific defect in fusion (with all other aspects of muscle morphogenesis appearing normal) are jamb and jamc. Once fusion has occurred, myofibrillogenesis, growth, innervation, and homeostasis all require adhesion to the ECM.
Muscle cell elongation requires adhesion to laminin
The above studies elucidated cell movements that generate the myotome but not how short muscle precursor cells elongate to the segment boundaries. Determining the cellular basis of primary myotome morphogenesis is necessary for a more precise understanding of the questions that need to be resolved at a molecular level. Live imaging studies in the chick and zebrafish models have highlighted a remarkable degree of conservation of the cellular mechanisms underlying initial fiber elongation. These complementary studies have identified discrete morphogenetic steps mediating muscle fiber morphogenesis and thus provide an excellent framework for molecular analyses of muscle morphogenesis.
Initially short precursor cells extend short filopodia-like protrusions in all directions in both model systems. Once cells begin to elongate they do so in a directed fashion, with protrusions extended in the direction of elongation (Fig. 4). In zebrafish, this phase of intercalation/elongation occurs via a repetitive two-step process of protrusion extension and filling (Snow et al., 2008a). In chick, polarized lamellipodia drive myocyte elongation (Gros et al., 2004). One interpretation of the fact that short muscle cells extend protrusions in all directions is that these cells are “searching” for the appropriate direction in which to elongate. Thus some questions are what are the molecules that guide oriented elongation? What are the signaling pathways that mediate the switch to oriented elongation? How do elongating myocytes gain traction for elongation?
The morphology of elongating muscle cells is remarkably similar to that of migrating cells. Cell migration requires cycles of membrane extension, adhesion to the ECM, cell body translocation, and detachment from the ECM (Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996; Murray and Oster, 1984). The difference between elongating muscle cells and migrating cells is that the “tail” of elongating muscle cells does not detach and retract back to the cell body. Thus, it is quite reasonable to hypothesize that cell adhesion to the ECM is required for traction forces necessary for muscle cell elongation, but which ECM proteins are required? Given that Fn is renowned for its role in neural crest cell migration, it is somewhat surprising that Fn is not required for muscle cell elongation in zebrafish (Snow et al., 2008b). In contrast, adhesion to laminin is necessary for muscle cell elongation in both mouse and zebrafish. Muscle cell elongation is disrupted in mouse explants incubated with an antibody against the laminin receptor Integrin alpha6beta1 (Bajanca et al., 2006). Fast-twitch muscle cell elongation is delayed in zebrafish lamb1 or lamc1 mutant embryos (Snow et al., 2008a). These data clearly indicate that adhesion to laminin is necessary for muscle fiber elongation in zebrafish and mouse but the mechanisms are not known. Are cells unable to exhibit traction to elongate initially? Are the tips of elongating cells unable to stably adhere to the matrix and thus retract? What occurs downstream of laminin binding to promote muscle cell elongation? These questions need to be answered in order to obtain a complete understanding of mechanisms guiding muscle fiber morphogenesis.
Observations of older embryos showed that although adhesion to laminin is required for efficient elongation of muscle precursor cells, muscle cells eventually recover and elongate in zebrafish lamb1 or lamc1 mutant embryos (Snow et al., 2008a) (it is not known if elongation recovers in the mouse). This recovery demonstrates how critical the dimension of time is when collecting and interpreting results in developing embryos, and implies the existence of a latent compensatory mechanism for fast-twitch muscle morphogenesis. What is this secondary mechanism? Hh signaling is known to be involved because fast-twitch muscle cells in lamc1 mutant embryos incubated in cyclopamine do not elongate, even after several days of development (Peterson and Henry, 2010). Hh signaling is known to act both directly and indirectly in the zebrafish myotome (Feng et al., 2006; Henry and Amacher, 2004). In this context, it was shown that Hh signaling acts indirectly through the specification of slow-twitch fibers (Peterson and Henry, 2010). In summary, although laminin is required for efficient elongation of fast-twitch muscle cells, elongation recovers in laminin mutant embryos. The recovery mechanism involves the cell population that triggers fast-twitch muscle cell elongation initially – the slow-twitch muscle fibers. Clearly, there is much to be learned but understanding these latent “back-up” mechanisms of morphogenesis could be important to provide insight into cellular adaptation, the evolution of development, and potential alternative therapeutic approaches for muscle diseases.
Some inroads have been made into understanding how muscle cells elongate in an oriented fashion. The myotome is highly organized because elongated myotubes are aligned with each other and oriented in the anterior-posterior axis. How is this order controlled? What are the cues that guide oriented elongation of short muscle precursor cells? Adhesion to laminin via Integrin alpha6beta1 is necessary for fiber organization in the mouse and zebrafish (Bajanca et al., 2006; Goody et al., 2012) but the mechanism of how laminin contributes to myotome order is not clear. Recent data using head and neck squamous carcinoma cells shows that cellular traction forces guide invadopodia and ECM degradation (Jerrell and Parekh, 2014). Thus, it is possible that efficient elongation, and the resulting deformations in the ECM, guide muscle cell elongation and myotome organization. An excellent study showed that WNT11 acts as a directional cue to guide muscle cell elongation in chick embryos: WNT11 is both necessary and sufficient for oriented elongation (Gros et al., 2009) (Fig. 4). The mechanism of WNT11 action is not known and it is not clear whether WNT11 and laminin adhesion pathways interact. The only known interaction between WNT11 and laminin is that WNT11 transcription is significantly downregulated in kidneys of lamc1 mutant embryos (Yang et al., 2011). It will be quite interesting in the future to clarify mechanisms of oriented elongation, whether these mechanisms are conserved, and whether laminin and WNT11 interact in oriented muscle cell elongation.
Boundary capture and regulation of fiber length: roles for Fn and laminin
A critical aspect of muscle morphogenesis is the cessation of muscle fiber elongation. The mechanisms that instruct elongating muscle cells to stop elongating are fundamental for normal muscle morphogenesis. Axial skeletal muscle fibers stop elongating when they hit the anterior and posterior segment boundaries (Henry et al., 2005). This process is called boundary capture. Boundary capture is mediated by laminin signaling in the zebrafish model (Snow et al., 2008a). Data from both zebrafish and mouse models suggest that Fn may play a role in ensuring proper length of axial skeletal muscle fibers (Snow et al., 2008b; Bajanca et al., 2004). Here, we will briefly discuss evidence for Fn in muscle cell-ECM interactions at segment boundaries. Next, we will discuss roles for laminin polymerization in boundary capture in zebrafish and myotome organization in mouse.
In mouse, elongating myocytes express the Fn receptors Integrin alpha4beta1 and alpha5beta1. These myocytes appear to attach to the intersegmental ECM that is rich in Fn (Bajanca et al., 2004). These data suggest that Fn plays a role in boundary capture. Unfortunately, disrupted mesoderm development in Fn null mice precludes analysis of how Fn contributes to muscle organization (Georges-Labouesse et al., 1996). Data from zebrafish using morpholinos to reduce but not abolish Fn also suggest that Fn is critical to reinforce segment boundaries. In embryos with reduced Fn, fast-twitch fibers are not patterned by migrating slow-twitch fibers and they are significantly less organized (Snow et al., 2008b). Exciting recent data suggest that a tetraspanin acts upstream of Integrin alpha5 in regulating fiber length. The transmembrane protein Tm4sf5 is best known as a promoter of EMT via enhancing activation of the cell-ECM adhesion protein focal adhesion kinase (FAK) (Jung et al., 2012). Tm4sf5 is expressed during muscle development in zebrafish (Choi et al., 2014). Tm4sf5 morphants display abnormal segment boundaries in addition to fewer and disorganized muscle fibers. Injection of Integrin alpha5 rescued embryos deficient for Tm4sf5, indicating that this protein functions upstream of Integrin alpha5 (Choi et al., 2014). One critical function of Fn is to regulate deposition of type I and III collagens in the ECM (Sottile and Hocking, 2002; Velling et al., 2002). Interestingly, it has recently been shown that the ability of Fn to bind to collagen is critical for Fn-stimulated cell migration during wound healing (Sottile et al., 2007). Although this study was in vitro, the dynamic interaction between Fn-dependent collagen deposition and Fn-dependent cell migration highlights the need for integration of multiple ECM components when studying developmental processes. Taken together, these studies indicate that Fn plays a conserved role in limiting/guiding muscle patterning; although Fn may act directly in mammals and indirectly through slow-twitch fibers and/or collagen deposition in zebrafish.
Laminin also plays a conserved role in regulating myotome organization. In mice, laminin is thought to act as a barrier that keeps myogenic cells inside the myotome. When laminin polymerization is compromised, myocytes disperse to the sclerotome region (Bajanca et al., 2006). In lamb1 or lamc1 mutant zebrafish, some fast-twitch muscle cells do not stop elongating at segment boundaries and instead extend into the adjacent myotome (Snow et al., 2008a). A dense network of polymerized laminin could function as a physical barrier or, alternatively, laminin binding may result in cell autonomous signaling that retains cells within their myotomes. To discriminate between these possibilities, genetic mosaic experiments were performed where wild-type muscle cells were transplanted into laminin gamma1-deficient embryos. The rationale was that wild-type cells might be able to secrete small amounts of laminin that would not act as a physical barrier (too little laminin), but could play a role in signaling that would facilitate boundary capture. Indeed, transplanted cells were significantly less likely to cross MTJs, indicating that boundary capture is a cell autonomous process (Snow et al., 2008a). Thus, it is likely that intracellular signaling downstream of laminin binding plays a major role in boundary capture and therefore muscle shape. In the future, it will be important to identify events downstream of laminin binding.
Laminin polymerization
One major focus of recent research, given the importance of laminin in muscle development and homeostasis, has been to understand how laminin polymerization and BM assembly are mediated in vivo. Translated laminin gene products (one alpha, one beta, and one gamma) assemble into cross-shaped laminin proteins with the alpha subunit constituting the long arm of the cross and the beta and gamma subunits forming the short arms of the cross. The short arms of laminin bind to each other, resulting in self-assembly (Colognato and Yurchenco, 2000; LeBleu et al., 2007). Assembled laminin proteins can then go on to polymerize with other laminin proteins and other ECM molecules to form specialized BMs. Roles for laminin-111 in triggering BM assembly in mouse muscle were shown by providing exogenous laminin-111 to Shh−/−;Gli3−/− embryos that do not express laminin-111. These elegant experiments clearly demonstrated that laminin-111 is necessary and sufficient to initiate myotomal BM assembly (Anderson et al., 2009). In addition to self assembly, laminin polymerization can be guided by cellular receptors for laminin that nucleate and organize laminin polymerization (Colognato and Yurchenco, 2000; LeBleu et al., 2007; Thorsteinsdóttir, 1992). Important questions that remain are which laminin receptors play roles in laminin polymerization and what mechanisms are involved.
The location of the myotomal BM differs in amniotes and teleosts. Whereas the somite boundary gives rise to the MTJ in teleosts, the myotomal BM in amniotes is assembled de novo (Anderson et al., 2009; 2007; Deries et al., 2012). Despite these differences, laminin receptors appear to potentiate polymerization in amniotes and teleosts. Correlative data implicate the laminin receptor Dystroglycan (Dag1) because Dag1 expression increases when laminin polymerizes in both mouse and zebrafish (Anderson et al., 2007; Snow and Henry, 2009). In addition, there are functional data showing conserved roles for the laminin receptor Integrin alpha6beta1 in laminin polymerization. Blocking Integrin alpha6beta1 interactions with laminin results in a patchy BM in mouse myotomes (Bajanca et al., 2006). Laminin polymerization at the MTJ is also less organized in zebrafish deficient for Integrin alpha6 or the Integrin beta1 binding protein Nrk2b (Goody et al., 2012; 2010). While Integrin alpha6beta1 appears to be the main laminin receptor mediating MTJ BM polymerization, Integrin alpha7 and Dag1 were also found to play minor roles in zebrafish MTJ morphogenesis (Goody et al., 2012). Taken together, these data indicate that multiple laminin receptor complexes are required for normal laminin polymerization at the MTJ during muscle development.
In zebrafish, failure to polymerize laminin at the MTJ can result in abnormally long muscle fibers that span the length of multiple muscle segments (referred to as boundary crossings). Similar to inside-out signaling through Integrin alpha5beta1 promoting Fn matrix polymerization during segmentation, inside-out signaling through Integrin alpha6beta1 appears to be one way of potentiating laminin polymerization at the MTJ BM and limiting muscle fiber length. Overexpression of the intracellular Integrin binding protein Paxillin rescued boundary crossings in Nrk2b-deficient zebrafish and laminin polymerization in Dag1 deficient zebrafish, but could not rescue these muscle phenotypes without the presence of Integrin alpha6 (Goody et al., 2012; 2010). Boundary crossings were also observed in zebrafish deficient for the extracellular BM glycoprotein Periostin (Kudo et al., 2004). It is not known exactly how Periostin limits muscle fiber length in zebrafish, but neither segmentation nor boundary capture phenotypes were observed suggesting that Periostin likely limits fiber length by contributing to MTJ development.
ECM molecules are required to maintain MTJ integrity during muscle tissue stress
A final step of muscle morphogenesis that must be continued throughout life is maintenance of MTJ structure despite repeated cycles of contraction and relaxation, injury and repair. Multiple genes encoding ECM molecules (lama2, tsp4b, and col22a1) have been shown to be necessary for MTJ homeostasis in zebrafish. Not surprisingly, these genes are expressed during later stages of muscle morphogenesis and their expression becomes restricted to the MTJ (Charvet et al., 2013; Hall et al., 2007; Subramanian and Schilling, 2014; Sztal et al., 2012). Zebrafish mutants or morphants deficient for laminin alpha2, thrombospondin 4b or collagen 22a1 proteins have mild muscle defects; however, when muscle contraction is stimulated (either mechanically or electrically), the muscle phenotype becomes severe with many muscle fibers retracting from the MTJ (Charvet et al., 2013; Hall et al., 2007; Subramanian and Schilling, 2014). Unlike the case in mouse and zebrafish models with dystrophin mutations, the muscle fibers didn’t rupture, they were found to retract from MTJ intact (Charvet et al., 2013; Gupta et al., 2012; Hall et al., 2007; Subramanian and Schilling, 2014). Therefore, rather than stabilizing the sarcolemma, these ECM proteins play a role in muscle health by maintaining integrity of muscle cell adhesion to the ECM at the MTJ.
THE EXTRACELLULAR MATRIX, CELLULAR ADAPTATION, AND MUSCLE DISEASE
We have focused on roles for ECM molecules in early muscle morphogenesis. Changes in the muscle ECM also play major roles – both positive and negative – later in life during muscle aging and disease. We will now address our second theme by discussing how muscle cell-ECM interactions regulate cellular adaptive responses in homeostasis and disease. One well-known (and frequently sought after) example of cellular adaptation in normal physiology is hypertrophy of skeletal muscle cells in response to weight bearing exercise (McDonagh and Davies, 1984). A far less desired, pathological cellular adaptation is resistance of cancer cells to therapy (reviewed in Cojoc et al., 2014); such as cancer cells using adaptive evasion to exit mitosis without proper cell division in response to drugs that induce mitotic arrest (Díaz-Martínez et al., 2014). Cellular adaptations are becoming a focus of biomedical research because, if better understood, innate cellular adaptation mechanisms could be augmented or inhibited for therapeutic benefit. As musculoskeletal health plays a major role in system health (e.g. posture, strength, mobility, metabolism) and pioneering work on cellular adaptation was conducted in skeletal muscle tissue, we believe muscle is a prime fertile ground for understanding the contribution of cellular adaptation to system health in normal and disease states. In our opinion, whether it be for improving physique or conquering cancer, understanding the mechanisms underlying cellular adaptation is one of the next major frontiers in systems biology. In the sections to follow, we will review roles for three ECM glycoproteins in either physiologically beneficial or deleterious skeletal muscle adaptations to disease.
Adhesion to laminin plays a major role in cellular adaptation during muscle disease
In skeletal muscle, adhesion of muscle fibers to the BM is necessary for muscle development, regeneration, and homeostasis. This requirement for muscle-BM adhesion is highlighted by the genetic basis for many muscular dystrophies; these are progressive, debilitating diseases without cures. Mutations in genes encoding muscle ECM components or proteins in ECM receptor complexes can lead to congenital muscular dystrophies (CMDs). For example, mutations in the human lama2 gene result in Merosin-deficient congenital muscular dystrophy (MDC1A), a common CMD (Helbling-Leclerc et al., 1995). Perhaps the most well known of the dystrophies is Duchenne muscular dystrophy (DMD), caused by mutations in the dystrophin gene, resulting in loss of muscle cell/ECM connection (Hoffman et al., 1987). Because disruption of muscle cell adhesion to the matrix can lead to muscle diseases, a major focus of current research is to identify approaches that enhance adhesion of muscle fibers to their ECM.
As mentioned above, there are multiple receptor complexes for laminin (Integrin alpha6beta1, Integrin alpha7beta1, the DGC, and the UGC). Generation of double mutants and overexpression studies have shown that although these receptor complexes have unique roles; they exhibit some functional overlap and can act synergistically. It is possible that understanding the intrinsic compensatory functions of these laminin receptor complexes would allow development of therapeutics that improve muscle pathology. Compensatory adhesion approaches towards developing therapies to improve muscle structure and function have included: (1) increasing the expression/functionality of an uncompromised adhesion complex (e.g. the UGC), and (2) augmenting the BM. Readers interested in other approaches toward therapeutics that are outside the scope of this review (exon skipping, stem cell therapies) are referred to excellent recent reviews on alternate therapeutic avenues (Aartsma-Rus, 2012; Meregalli et al., 2013). Here, we will discuss exciting data regarding the remarkable ability of muscle cells to adapt when one mode of cell adhesion to laminin is disrupted, and the potential for this intrinsic adaptation to be exploited for therapeutic purposes.
In some mouse models for CMDs, there are intrinsic adaptive cellular responses that upregulate an uncompromised receptor complex. This upregulation can lead to an improved BM. This was first discovered when it was noticed that the mdx mouse model of DMD had reduced muscle pathology compared to boys with DMD (Tanabe et al., 1986). It was hypothesized that upregulation of the UGC and Integrin receptors could be compensating for the disrupted DGC. Indeed, phenotypes are more severe in mdx/Itga7 (Guo et al., 2006; Rooney et al., 2006), mdx/utr (Deconinck et al., 1997; Grady et al., 1997), and utr/Itga7 (Welser et al., 2009) double mutants. Critically, the background strain of mouse on which the mdx mutation was placed had a central role in the ability of the muscle to maintain itself (Fukada et al., 2010), with the DBA/2J strain exhibiting greatly decreased satellite cell renewal potential. Exactly what the genetic and molecular mechanisms of this defect are in DBA/2J compared to the original, more adaptive C57BL/10 strain remain to be determined and may provide key insight into the ability of muscle to compensate. Similar results have been observed in the zebrafish model. Muscle pathology is also exacerbated in zebrafish deficient for Integrin alpha7 and Dag1, Integrin-linked kinase and Dag1, or Integrin-linked kinase and Dystrophin compared to knockdown or mutation of a single gene (Goody et al., 2012; Sztal et al., 2012). These data from two different vertebrates provide validation for the approach of potentiating the activity of compensatory adhesion complexes to ameliorate muscle diseases in humans. Indeed, approaches for upregulation of the UGC and/or Integrin alpha7beta1 expression are being pursued. One interesting approach is Sarcospan overexpression because Sarcospan appears to have a pleiotropic beneficial effect in dystrophic muscle by both upregulating DGC/Integrin expression and membrane trafficking (Marshall and Crosbie-Watson, 2013). Similarly, upregulating Utrophin is a promising potential therapy for DMD (Phase I clinical trials) (Fairclough et al., 2013).
Other innovative approaches have been to consider providing different isoforms of laminin. In MDC1A, caused by mutations in lama2, there is a partial increase in laminin alpha4 synthesis and a decrease in amounts of two laminin receptors, Integrin alpha7beta1 and the DGC (Bentzinger et al., 2005; Kortesmaa et al., 2000; Moll et al., 2001; Patton et al., 1997; Ringelmann et al., 1999; Vachon et al., 1997). Unfortunately, increased laminin alpha4 does not fully compensate for the lack of laminin alpha2 because laminin alpha4 doesn’t self-polymerize or bind to the DGC or Integrin alpha7beta1 with high affinity (Kortesmaa et al., 2000; Talts et al., 2000). As mentioned above, laminin-111 is the major developmental isoform of laminin. Remarkably, laminin-111 protein therapy significantly improves pathology of mouse models for MDC1A, DMD, and congenital myopathy with Integrin alpha7 deficiency (Rooney et al., 2009a; 2009b; 2012). Thus, laminin-111 protein therapy may be a viable approach, although effective delivery of laminin-111 to muscle tissue in large mammals has not yet been demonstrated. Striking data indicate that some dystrophic muscle cells can generate their own ectopic BM and maintain viability. Zebrafish softy mutants are homozygous viable despite being identified as part of the dystrophic class of mutants in one of the first large-scale zebrafish forward genetic screens (Granato et al., 1996). The softy phenotype is caused by a mutation in the lamb2 gene. In softy mutants, muscle fibers detach from the BM at the MTJ but their sarcolemma remains intact. What is remarkable is that some of the fibers do not detach fully, but rather remain striated and attach to an ectopic BM. As these ectopic fiber terminations are unique in the zebrafish dystrophy models, and softy mutants are unique in that they are homozygous viable, it is hypothesized that these ectopic fiber terminations function to stabilize the damaged myotome (Jacoby et al., 2009). This could allow more robust regeneration or slow down degeneration. Certainly, understanding the mechanisms underlying the recovery from early and severe muscle degeneration is critical because it may provide novel insight into therapeutic approaches.
An alternate approach is to identify small molecules that potentiate muscle cell adhesion to laminin either through hypothesis-driven testing of chemicals or large-scale chemical screening. Zebrafish models of muscular dystrophies are being used to pursue both of these approaches. The Nrk2b pathway that mediates laminin-111 polymerization during development ameliorates muscle damage in Integrin alpha7 or Dag1-deficient zebrafish (Fig. 5) (Goody et al., 2012). Chemical screens have shown phosphodiesterase inhibitors (Kawahara et al., 2011), upregulation of heme oxygenase 1 (Kawahara et al., 2014), and selective serotonin reuptake inhibitors (Waugh et al., 2014) reduce disease severity in the zebrafish model of DMD. Thus, understanding and being able to manipulate the intrinsic cellular adaptive responses that occur in diseased muscle tissue, whether they be between laminin receptors, ECM components, or metabolic/hormone signaling pathways, could be of benefit to the many people with genetic or acquired muscle diseases.
Figure 5.
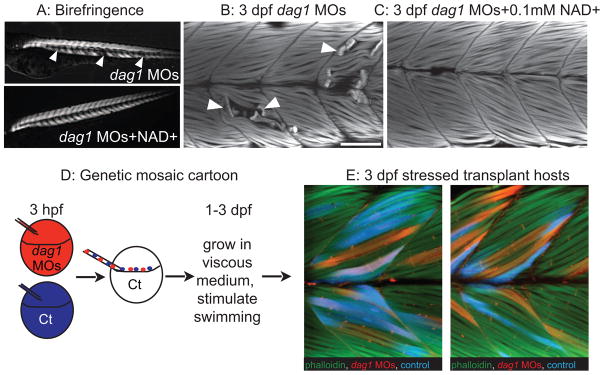
An organized ECM microenvironment rescues fiber resiliency in Dag1-deficient cells (from Goody et al., 2012). Anterior left, dorsal top, side-mounted, 3 dpf embryos. A: Polarized light microscopy shows loss of birefringence in dag1 morphant myotomes (white arrowheads). Birefringence is rescued in NAD+-supplemented dag1 morphants. B: 3 dpf embryos stained with phalloidin (white). Fiber detachment is readily observed in dag1 morphants (arrowheads). C: NAD+ supplementation reduces fiber detachment in dag1 morphants. D: Genetic mosaic cartoon depicting transplantation of fluorescent dextran-labeled dag1 morphant (red) and control (blue) cells into unlabeled, control hosts. Embryos were stressed (frequently stimulated to swim in a viscous medium), and reared to 3 dpf. E: Transplanted control cells (blue) and dag1 morphant cells (red) remain attached to MTJs, even when hosts are stressed. This suggests that a normal host ECM microenvironment is sufficient for resiliency of dag1 morphant cells and supports that NAD+ functions via augmentation of the ECM microenvironment.
The dark side of Fn: fibrosis
Fn plays a key role in the stem cell niche during muscle regeneration, being both necessary and sufficient for satellite cell expansion (Bentzinger et al., 2013). This role for Fn in generating a supportive ECM microenvironment is reminiscent of how Fn upregulation by bone marrow-derived hematopoietic progenitor cells generates a permissive premetastatic niche and instructs organ-specific tumor spread (Kaplan et al., 2005; 2006). However, despite playing critical roles in muscle development and regeneration, Fn may also contribute to pathology in aging and diseased muscle. Abnormal ECM deposition, including Fn deposition, is observed in fibrotic tissue in both cardiac and skeletal muscle (Bertolotto et al., 1983; Booth et al., 2012; Li et al., 2008). Fn is frequently assessed as a marker of fibrosis and disrupted Fn has been observed in humans with various muscular dystrophies (Martin et al., 2014; Higashi et al., 2005; Martoni et al., 2013; Sáenz et al., 2008). The replacement of contractile muscle tissue by Fn-rich fibrotic material leads to diminished organ function.
Fibrosis can be thought of as an aberrant wound healing response (Mann et al., 2011). The inflammatory state induced in response to an acute tissue injury activates and recruits cells (e.g. macrophages, fibroblasts) that clean up debris and synthesize new cytoskeletal and ECM components (e.g. alpha-smooth muscle actin, Vimentin, collagens, Fn). This phase is followed by resolution of inflammation, degradation of the temporary ECM, and restoration of normal cellular and tissue architecture and function. Chronic muscle damage, in contrast, is often associated with chronic inflammation that results in aberrant deposition of ECM. While Fn can promote muscle repair, there seems to be a limited time, place, and duration of Fn expression that is beneficial in muscle regeneration. In Itga7 mutant mice, Integrin alpha5 and Fn remain concentrated around muscle fibers and at MTJs before muscle wasting is observed (Nawrotzki et al., 2003). In lama2 mutant mice Fn was the most upregulated of the ECM components assayed early in disease progression (Mehuron et al., 2014). The increase in Fn in these animal models of muscular dystrophy was suggested to be an early marker and pathomechanism in these diseases. It is clear that understanding how Fn expression is dynamically regulated will be critical to promote proper muscle repair even when damage is chronic. Proinflammatory cytokines and growth factors are known to regulate Fn expression in muscle tissue: Connective Tissue Growth Factor is sufficient to increase Fn in wild-type mouse muscle (Morales et al., 2011) and Transforming Growth Factor-beta is sufficient to increase Fn protein in cultured muscle cells (Li et al., 2004). Proteoglycans in the ECM of skeletal muscle likely play important roles in regulating muscle tissue fibrosis because they can bind proinflammatory cytokines and growth factors and modulate their signaling (reviewed in Brandan and Gutierrez, 2013). Fn upregulation is not only a result of the proinflammatory state, but alternative splicing of Fn can itself activate inflammatory signaling cascades and induce production of proinflammatory cytokines (Kelsh et al., 2014). This results in a positive feedback loop between inflammation and Fn expression. Therefore, interrupting the cycle between inflammation-induced Fn expression and Fn-induced inflammatory signaling could be a fruitful approach to prevent the fibrosis associated with muscle diseases and aging.
Do collagens compensate for each other?
Cellular adaptations in response to disruption of many ECM molecules remain largely unexplored; such is the case for the collagen family of genes. Type IV collagen is expressed in BMs and collagen 22a1 is required for MTJ homeostasis in zebrafish muscle, but it is mutations in type VI collagen genes that cause the second most common form of congenital muscular dystrophy: Ullrich congenital muscular dystrophy (UCMD). Mutations in type VI collagen genes can also cause a less severe muscular dystrophy called Bethlam myopathy. Mice with col6a1 mutations have yielded important pathological and therapeutic insights for UCMD and Bethlam myopathy (e.g. identifying mitochondrial disruptions (Irwin et al., 2003) and roles for autophagy in muscle homeostasis (Grumati et al., 2010)); however, these mice failed to recapitulate the severity of UCMD. This could potentially be due to the genetics of the background strain used as discussed above for the mdx mouse model of DMD. Recently, zebrafish models of UCMD and Bethlam myopathy were generated using morpholinos to block the incorporation of exon 9 or 13 of col6a1, respectively (Telfer et al., 2010). Injection of the morpholino targeting exon 9 produced early onset disruptions to muscle architecture, mitochondria, and motility in zebrafish. Mitochondria morphology and embryo motility could be rescued by a drug inhibiting the mitochondrial permeability transition pore (Telfer et al., 2010), similar to what was seen in col6a1 mutant mice (Irwin et al., 2003). Despite these important insights into collagen function, the cellular adaptive responses to collagen disruptions are just beginning to be uncovered in multiple tissues. Is there any functional redundancy or the potential for compensation between various collagen molecules in the muscle ECM? Could muscle ECM molecules regulating collagen processes be manipulated to improve disease symptoms? There does not appear to be compensation between type XV and XVIII collagens, despite similar structures and tissue expression, because double mutant mice do not display any novel, major defects compared to single mutants (Ylikärppä et al., 2003). In contrast, double knock out of two proteoglycans (Decorin and Biglycan) that each regulate collagen fibril formation by competing for the same binding site in type I collagen (Pogany et al., 1994) resulted in more severe bone (Young et al., 2002) and corneal (Zhang et al., 2009) phenotypes in mice than knock out of either alone, suggesting redundancy and compensation between Decorin and Biglycan in collagen regulation in these tissues. Finally, in col4a3 mutant mice modeling Alport Syndrome, which affects the kidney glomerular BM, expression of col4a1, col4a2, lama1, and lama5 either persist or are upregulated in the glomerular BM (Abrahamson et al., 2007). Whether there is compensation between different collagen molecules in muscle tissue or whether collagen disruptions can be compensated for by changes to other muscle ECM proteins or receptors in animal models of UCMD or Bethlam myopathy remain to be tested and could provide important therapeutic insights for these muscle diseases and potentially other muscle pathologies as well.
CONCLUSIONS
It has become increasingly clear that signaling pathways mediating embryonic development also can regulate homeostasis and aging. Cell-ECM adhesion interacts with these pathways throughout the lifespan. We believe muscle is a robust and strategic tissue for interrogating these interactions and that these interactions contain tremendous potential for therapeutic discoveries. As we begin to understand the foundations of adhesion-dependent adaptive cellular responses, we will be exploring the innate ability of tissue to maintain homeostasis and repair its own damage, rather than treating downstream symptoms. The promise of regenerative medicine will likely require a clear and mechanistic understanding of how cell-ECM interactions potentiate robust cellular adaptation to stresses so that these intrinsic responses can be exploited for therapeutic purposes.
Table 1.
ECM related cellular adaptive responses in diseases.
| Gene/gene product disrupted | Cellular adaptive response demonstrated | Reference |
|---|---|---|
| mdx/utr, mdx/itga7, or utr/itga7 double knock out mice | Compensation between Dystrophin, Utrophin, and Integrin alpha7 | Guo et al., 2006; Rooney et al., 2006; Deconinck et al., 1997; Grady et al., 1997; Welser et al., 2009 |
| Dystroglycan/Integrin alpha7, Dystroglycan/Integrin-linked kinase, or Dystrophin/Integrin-linked kinase double knockdown in zebrafish | Compensation between Dystroglycan or Dystrophin and Integrin alpha7 and/or Integrin linked kinase | Goody et al., 2012; Sztal et al., 2012 |
| mdx mice | Sarcospan overexpression sufficient to increase DGC, UGC, and Integrin alph7beta1 sarcolemma expression | Marshall and Crosbie-Watson, 2013 |
| Human DMD | Increased Utrophin is beneficial | Fairclough et al., 2013 |
| lama2 | Partial compensation by increased laminin alpha4 | Kortesmaa et al., 2010; Talts et al., 2010 |
| lama2, mdx, or itga7 mutant mice | Laminin-111 protein therapy beneficial | Rooney et al., 2009a; 2009b; 2012 |
| lamb2 mutant zebrafish | Fibers form and attach to ectopic BMs | Jacoby et al., 2009 |
| Integrin alpha7 or Dystroglycan deficient zebrafish | Exogenous NAD+ supplementation enhances basement membrane | Goody et al., 2012 |
| dmd mutant zebrafish | Phosphodiesterase inhibitors, upregulation of heme oxygenase 1, or selective serotonin reuptake inhibitors beneficial | Kawahara et al., 2011; 2014; Waugh et al., 2014 |
| itga7 mutant mice | Integrin alpha5 and Fn are upregulated | Nawrotzki et al., 2003 |
| lama2 mutant mice | Fn is upregulated | Mehuron et al., 2014 |
| Decorin/Biglycan double knock out mice | Compensation between Decorin and Biglycan in collagen fibrillogenesis in bone and cornea | Young et al., 2002; Zhang et al., 2009 |
| col4a3 mutant mice | Upregulation of col4a1, col4a2, lama1, and lama5 | Abrahamson et al., 2007 |
Highlights.
Cell-ECM adhesions mediate morphogenesis, cell fate, and adaptation in muscle
Cell-ECM adhesion complexes are highly dynamic during muscle development
Disrupted cell-ECM adhesion occurs in many pathological states
Augmenting specific cell-ECM adhesions holds therapeutic promise for muscle diseases
Acknowledgments
The authors would like to thank Ryan Cowan for help with the artistic renderings presented in the figures.
FUNDING
This work was supported by in part by NIH grant 5RO3HD077545 to CAH and grant number #1-FY14-284 from the March of Dimes Foundation to CAH.
ABBREVIATIONS
- ECM
extracellular matrix
- Fn
Fibronectin
- BM
basement membrane
- DGC
Dystrophin-glycoprotein complex
- UGC
Utrophin-glycoprotein complex
- NMJ
Neuromuscular junction
- MTJ
myotendinous junction
- HSPG
heparin sulfate proteoglycan
- EMT
epithelial to mesenchymal transition
- ECL
external cell layer
- Hh
Hedgehog
- SSF
superficial slow-twitch fiber
- MP
muscle pioneer fiber
- MFF
medial fast-twitch fiber
- FAK
focal adhesion kinase
- Dag1
dystroglycan
- Nrk2b
nicotinamide riboside kinase 2b
- CMD
congenital muscular dystrophy
- DMD
Duchenne muscular dystrophy
- MDC1A
Merosin-deficient congenital muscular dystrophy
- UCMD
Ullrich congenital muscular dystrophy
- hpf
hours post fertilization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aartsma-Rus A. Overview on DMD exon skipping. Methods Mol Biol. 2012;867:97–116. doi: 10.1007/978-1-61779-767-5_7. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, Isom K, Roach E. Laminin compensation in collagen α3 (IV) knockout (Alport) glomeruli contributes to permeability defects. JASN. 2007;18:2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- Anderson C, Thorsteinsdóttir S, Borycki AG. Sonic hedgehog-dependent synthesis of laminin alpha1 controls basement membrane assembly in the myotome. Development. 2009;136:3495–3504. doi: 10.1242/dev.036087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C, Winder SJ, Borycki A-G. Dystroglycan protein distribution coincides with basement membranes and muscle differentiation during mouse embryogenesis. Dev Dyn. 2007;236:2627–2635. doi: 10.1002/dvdy.21259. [DOI] [PubMed] [Google Scholar]
- Askari JA, Buckley PA, Mould AP, Humphries MJ. Linking integrin conformation to function. J Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JCR, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, Mark von der K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Bajanca F, Luz M, Duxson MJ, Thorsteinsdóttir S. Integrins in the mouse myotome: developmental changes and differences between the epaxial and hypaxial lineage. Dev Dyn. 2004;231:402–415. doi: 10.1002/dvdy.20136. [DOI] [PubMed] [Google Scholar]
- Bajanca F, Luz M, Raymond K, Martins GG, Sonnenberg A, Tajbakhsh S, Buckingham M, Thorsteinsdóttir S. Integrin alpha6beta1-laminin interactions regulate early myotome formation in the mouse embryo. Development. 2006;133:1635–1644. doi: 10.1242/dev.02336. [DOI] [PubMed] [Google Scholar]
- Beckett K, Baylies MK. 3D analysis of founder cell and fusion competent myoblast arrangements outlines a new model of myoblast fusion. Dev Biol. 2007;309:113–125. doi: 10.1016/j.ydbio.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello V, Moreau N, Sirour C, Hidalgo M, Buisson N, Darribère T. The dystroglycan: Nestled in an adhesome during embryonic development. Dev Biol. 2014 doi: 10.1016/j.ydbio.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Ben-Yair R, Kahane N, Kalcheim C. Coherent development of dermomyotome and dermis from the entire mediolateral extent of the dorsal somite. Development. 2003;130:4325–4336. doi: 10.1242/dev.00667. [DOI] [PubMed] [Google Scholar]
- Ben-Yair R, Kalcheim C. Lineage analysis of the avian dermomyotome sheet reveals the existence of single cells with both dermal and muscle progenitor fates. Development. 2005;132:689–701. doi: 10.1242/dev.01617. [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. Faseb J. 2005;19:934–942. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX, von Maltzahn J, Soleimani VD, Yin H, Rudnicki MA. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell. 2013;12:75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotto A, Palmucci L, Doriguzzi C, Mongini T, Gagnor E, Del Rosso M, Tarone G. Laminin and fibronectin distribution in normal and pathological human muscle. J Neurol Sci. 1983;60:377–382. doi: 10.1016/0022-510x(83)90148-x. [DOI] [PubMed] [Google Scholar]
- Bink RJ, Habuchi H, Lele Z, Dolk E, Joore J. Heparan sulfate 6-O-sulfotransferase is essential for muscle development in zebrafish. Journal of Biological Chemistry. 2003;278:31118–31127. doi: 10.1074/jbc.M213124200. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? Journal of theoretical biology. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, White ES, Bishop DK. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol. 2012;226:609–618. doi: 10.1002/path.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borycki AG. The myotomal basement membrane: insight into laminin-111 function and its control by Sonic hedgehog signaling. Cell Adh Migr. 2013;7:72–81. doi: 10.4161/cam.23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman W. On the minute structure and movements of voluntary muscle. Philosophical Transactions of the Royal Society of London. 1840;130:457–501. [Google Scholar]
- Brandan E, Gutierrez J. Role of proteoglycans in the regulation of the skeletal muscle fibrotic response. FEBS J. 2013;280:4109–4117. doi: 10.1111/febs.12278. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Relaix F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol. 2007;23:645–673. doi: 10.1146/annurev.cellbio.23.090506.123438. [DOI] [PubMed] [Google Scholar]
- Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harbor Perspectives in Biology. 2011 doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignac V, Durbeej M. Cell-matrix interactions in muscle disease. J Pathol. 2012;226:200–218. doi: 10.1002/path.3020. [DOI] [PubMed] [Google Scholar]
- Charvet B, Guiraud A, Malbouyres M, Zwolanek D, Guillon E, Bretaud S, Monnot C, Schulze J, Bader HL, Allard B, Koch M, Ruggiero F. Knockdown of col22a1 gene in zebrafish induces a muscular dystrophy by disruption of the myotendinous junction. Development. 2013;140:4602–4613. doi: 10.1242/dev.096024. [DOI] [PubMed] [Google Scholar]
- Chen EH, Olson EN. Towards a molecular pathway for myoblast fusion in Drosophila. Trends Cell Biol. 2004;14:452–460. doi: 10.1016/j.tcb.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Choi Y-J, Kim HH, Kim J-G, Kim H-J, Kang M, Lee M-S, Ryu J, Song HE, Nam SH, Lee D, Kim K-W, Lee JW. TM4SF5 suppression disturbs integrin α5-related signalling and muscle development in zebrafish. Biochem J. 2014;462:89–101. doi: 10.1042/BJ20140177. [DOI] [PubMed] [Google Scholar]
- Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin Cancer Biol. 2014 doi: 10.1016/j.semcancer.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Costa ML, Escaleira RC, Jazenko F, Mermelstein CS. Cell adhesion in zebrafish myogenesis: distribution of intermediate filaments, microfilaments, intracellular adhesion structures and extracellular matrix. Cell Motil Cytoskeleton. 2008;65:801–815. doi: 10.1002/cm.20301. [DOI] [PubMed] [Google Scholar]
- Crawford BD, Henry CA, Clason TA, Becker AL, Hille MB. Activity and distribution of paxillin, focal adhesion kinase, and cadherin indicate cooperative roles during zebrafish morphogenesis. Mol Biol Cell. 2003;14:3065–3081. doi: 10.1091/mbc.E02-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson LA, Marsden M, Keller R, DeSimone DW. Integrin alpha5beta1 and fibronectin regulate polarized cell protrusions required for Xenopus convergence and extension. Curr Biol. 2006;16:833–844. doi: 10.1016/j.cub.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Deries M, Gonçalves AB, Vaz R, Martins GG, Rodrigues G, Thorsteinsdóttir S. Extracellular matrix remodeling accompanies axial muscle development and morphogenesis in the mouse. Dev Dyn. 2012;241:350–364. doi: 10.1002/dvdy.23703. [DOI] [PubMed] [Google Scholar]
- Deries M, Schweitzer R, Duxson MJ. Developmental fate of the mammalian myotome. Dev Dyn. 2010;239:2898–2910. doi: 10.1002/dvdy.22425. [DOI] [PubMed] [Google Scholar]
- Devoto SH, Melancon E, Eisen JS, Westerfield M. Identification of separate slow and fast muscle precursor cells in vivo, prior to somite formation. Development. 1996;122:3371–3380. doi: 10.1242/dev.122.11.3371. [DOI] [PubMed] [Google Scholar]
- Devoto SH, Stoiber W, Hammond CL, Steinbacher P, Haslett JR, Barresi MJF, Patterson SE, Adiarte EG, Hughes SM. Generality of vertebrate developmental patterns: evidence for a dermomyotome in fish. Evol Dev. 2006;8:101–110. doi: 10.1111/j.1525-142X.2006.05079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Martínez LA, Karamysheva ZN, Warrington R, Li B, Wei S, Xie X-J, Roth MG, Yu H. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J. 2014;33:1960–1976. doi: 10.15252/embj.201487826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolez M, Nicolas JF, Hirsinger E. Laminins, via heparan sulfate proteoglycans, participate in zebrafish myotome morphogenesis by modulating the pattern of Bmp responsiveness. Development. 2011;138:97–106. doi: 10.1242/dev.053975. [DOI] [PubMed] [Google Scholar]
- Dray N, Lawton A, Nandi A, Julich D, Emonet T, Holley SA. Cell-fibronectin interactions propel vertebrate trunk elongation via tissue mechanics. Curr Biol. 2013;23:1335–1341. doi: 10.1016/j.cub.2013.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droguett R, Cabello-Verrugio C, Riquelme C, Brandan E. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol. 2006;25:332–341. doi: 10.1016/j.matbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Du SJ, Devoto SH, Westerfield M, Moon RT. Positive and negative regulation of muscle cell identity by members of the hedgehog and TGF-beta gene families. J Cell Biol. 1997;139:145–156. doi: 10.1083/jcb.139.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M. Laminins. Cell Tissue Research. 2010;339:259–268. doi: 10.1007/s00441-009-0838-2. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions? Journal of Cell Science. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- Erickson HP. Stretching fibronectin. J Muscle Res Cell M. 2002;23:575–580. doi: 10.1023/a:1023427026818. [DOI] [PubMed] [Google Scholar]
- Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nature Reviews Genetics. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- Feng X, Adiarte EG, Devoto S. Hedgehog acts directly on the zebrafish dermomyotome to promote myogenic differentiation. Dev Biol. 2006;300:736–746. doi: 10.1016/j.ydbio.2006.08.056. [DOI] [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. Journal of Cell Science. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada S-I, Morikawa D, Yamamoto Y, Yoshida T, Sumie N, Yamaguchi M, Ito T, Miyagoe-Suzuki Y, Takeda S, Tsujikawa K, Yamamoto H. Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol. 2010;176:2414–2424. doi: 10.2353/ajpath.2010.090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemballa S, Vogel F. Spatial arrangement of white muscle fibers and myoseptal tendons in fishes. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:1013–1037. doi: 10.1016/s1095-6433(02)00186-1. [DOI] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse EN, George EL, Rayburn H, Hynes RO. Mesodermal development in mouse embryos mutant for fibronectin. Dev Dyn. 1996;207:145–156. doi: 10.1002/(SICI)1097-0177(199610)207:2<145::AID-AJA3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. 2011;44:318–331. doi: 10.1002/mus.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girós A, Grgur K, Gossler A, Costell M. α5β1 integrin-mediated adhesion to fibronectin is required for axis elongation and somitogenesis in mice. PLoS ONE. 2011;6:e22002. doi: 10.1371/journal.pone.0022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody MF, Kelly MW, Lessard KN, Khalil A, Henry CA. Nrk2b-mediated NAD+ production regulates cell adhesion and is required for muscle morphogenesis in vivo: Nrk2b and NAD+ in muscle morphogenesis. Dev Biol. 2010;344:809–826. doi: 10.1016/j.ydbio.2010.05.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody MF, Kelly MW, Reynolds CJ, Khalil A, Crawford BD, Henry CA. NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. Plos Biol. 2012;10:e1001409. doi: 10.1371/journal.pbio.1001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossler A, Hrabĕ de Angelis M. Somitogenesis. Curr Top Dev Biol. 1998;38:225–287. [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Granato M, van Eeden FJ, Schach U, Trowe T, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nüsslein-Volhard C. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development. 1996;123:399–413. doi: 10.1242/dev.123.1.399. [DOI] [PubMed] [Google Scholar]
- Gros J, Manceau M, Thomé V, Marcelle C. A common somitic origin for embryonic muscle progenitors and satellite cells. Nature. 2005;435:954–958. doi: 10.1038/nature03572. [DOI] [PubMed] [Google Scholar]
- Gros J, Scaal M, Marcelle C. A two-step mechanism for myotome formation in chick. Dev Cell. 2004;6:875–882. doi: 10.1016/j.devcel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Gros J, Serralbo O, Marcelle C. WNT11 acts as a directional cue to organize the elongation of early muscle fibres. Nature. 2009;457:589–593. doi: 10.1038/nature07564. [DOI] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M, Bonaldo P. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–1320. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- Gullberg D, Sjoberg G, Velling T, Sejersen T. Analysis of fibronectin and vitronectin receptors on human fetal skeletal muscle cells upon differentiation. Exp Cell Res. 1995;220:112–123. doi: 10.1006/excr.1995.1297. [DOI] [PubMed] [Google Scholar]
- Guo C, Willem M, Werner A, Raivich G, Emerson M, Neyses L, Mayer U. Absence of alpha7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum Mol Genet. 2006;15:989–998. doi: 10.1093/hmg/ddl018. [DOI] [PubMed] [Google Scholar]
- Gupta VA, Kawahara G, Myers JA, Chen AT, Hall TE. A splice site mutation in laminin-α2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PLoS ONE. 2012 doi: 10.1371/journal.pone.0043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TE, Bryson-Richardson RJ, Berger S, Jacoby AS, Cole NJ, Hollway GE, Berger J, Currie PD. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. PNAS. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halper J, Kjaer M. Advances in Experimental Medicine and Biology, Advances in Experimental Medicine and Biology. Springer Netherlands; Dordrecht: 2013. Basic Components of Connective Tissues and Extracellular Matrix: Elastin, Fibrillin, Fibulins, Fibrinogen, Fibronectin, Laminin, Tenascins and Thrombospondins; pp. 31–47. [DOI] [PubMed] [Google Scholar]
- Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn. 1997;210:130–145. doi: 10.1002/(SICI)1097-0177(199710)210:2<130::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hara Y, Campbell KP. Dystroglycan: An Extracellular Matrix Receptor That Links to the Cytoskeleton. Glycoscience: Biology and Medicine. 2014:1245–1251. [Google Scholar]
- Harburger DS, Calderwood DA. Integrin signalling at a glance. Journal of Cell Science. 2009;122:159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FMS, Schwartz K, Fardeau M, Tryggvason K, Guicheney P. Mutations in the laminin alpha2–chain gene (LAMA2) cause merosin–deficient congenital muscular dystrophy. Nature Genetics. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Henry CA, Amacher SL. Zebrafish slow muscle cell migration induces a wave of fast muscle morphogenesis. Dev Cell. 2004;7:917–923. doi: 10.1016/j.devcel.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Henry CA, McNulty IM, Durst WA, Munchel SE, Amacher SL. Interactions between muscle fibers and segment boundaries in zebrafish. Dev Biol. 2005;287:346–360. doi: 10.1016/j.ydbio.2005.08.049. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J Cell Sci. 2001;114:1137–1144. doi: 10.1242/jcs.114.6.1137. [DOI] [PubMed] [Google Scholar]
- Higashi K, Higuchi I, Niiyama T, Uchida Y, Shiraishi T, Hashiguchi A, Saito A, Horikiri T, Suehara M, Arimura K, Osame M. Abnormal expression of proteoglycans in Ullrich’s disease with collagen VI deficiency. Muscle Nerve. 2005;33:120–126. doi: 10.1002/mus.20449. [DOI] [PubMed] [Google Scholar]
- Hirsinger E, Stellabotte F, Devoto S, Westerfield M. Hedgehog signaling is required for commitment but not initial induction of slow muscle precursors. Dev Biol. 2004;275:143–157. doi: 10.1016/j.ydbio.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Hoffman E, Brown R, Kunkel L. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adh Migr. 2013;7:56–63. doi: 10.4161/cam.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollway GE, Currie PD. Myotome meanderings. Cellular morphogenesis and the making of muscle. EMBO Rep. 2003;4:855–860. doi: 10.1038/sj.embor.embor920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- Jacoby AS, Busch-Nentwich E, Bryson-Richardson RJ, Hall TE, Berger J, Berger S, Sonntag C, Sachs C, Geisler R, Stemple DL, Currie PD. The zebrafish dystrophic mutant softy maintains muscle fibre viability despite basement membrane rupture and muscle detachment. Development. 2009;136:3367–3376. doi: 10.1242/dev.034561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerrell RJ, Parekh A. Cellular traction stresses mediate extracellular matrix degradation by invadopodia. Acta Biomater. 2014;10:1886–1896. doi: 10.1016/j.actbio.2013.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ST, Chuang WJ, Tang MJ. Role of fibronectin deposition in branching morphogenesis of Madin-Darby canine kidney cells. Kidney Int. 2000;57:1860–1867. doi: 10.1046/j.1523-1755.2000.00035.x. [DOI] [PubMed] [Google Scholar]
- Julich D, Geisler R, Holley SA, Tübingen Screen Consortium, 2005. Integrinalpha5 and delta/notch signaling have complementary spatiotemporal requirements during zebrafish somitogenesis. Dev Cell. 2000;8:575–586. doi: 10.1016/j.devcel.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Julich D, Mould AP, Koper E, Holley SA. Control of extracellular matrix assembly along tissue boundaries via Integrin and Eph/Ephrin signaling. Development. 2009;136:2913–2921. doi: 10.1242/dev.038935. [DOI] [PubMed] [Google Scholar]
- Jung O, Choi S, Jang SB, Lee SA, Lim ST, Choi YJ, Kim HJ, Kim DH, Kwak TK, Kim H, Kang M, Lee MS, Park SY, Ryu J, Jeong D, Cheong HK, Kim HJ, Park KH, Lee BJ, Schlaepfer DD, Lee JW. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. Journal of Cell Science. 2012;125:5960–5973. doi: 10.1242/jcs.100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner S, Schwendinger-Schreck J, Jülich D, Holley SA. Segmental Assembly of Fibronectin Matrix Requires rap1b and integrin α5. Dev Dyn. 2013;242:122–131. doi: 10.1002/dvdy.23909. [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Rafii S, Lyden D. Preparing the “Soil”: The Premetastatic Niche. Cancer Res. 2006;66:11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsan A. Notch and integrin affinity: a sticky situation. Sci Signal. 2008;1:e2. doi: 10.1126/stke.12pe2. [DOI] [PubMed] [Google Scholar]
- Kassar-Duchossoy L, Giacone E, Gayraud-Morel B, Jory A, Gomès D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 2005;19:1426–1431. doi: 10.1101/gad.345505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara G, Gasperini MJ, Myers JA, Widrick JJ, Eran A, Serafini PR, Alexander MS, Pletcher MT, Morris CA, Kunkel LM. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum Mol Genet. 2014;23:1869–1878. doi: 10.1093/hmg/ddt579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara G, Karpf JA, Myers JA, Alexander MS, Guyon JR, Kunkel LM. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proceedings of the National Academy of Sciences. 2011;108:5331–5336. doi: 10.1073/pnas.1102116108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PLoS ONE. 2014;9:e102974. doi: 10.1371/journal.pone.0102974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kjaer M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiological reviews. 2004;84:649–698. doi: 10.1152/physrev.00031.2003. [DOI] [PubMed] [Google Scholar]
- Kortesmaa J, Yurchenco P, Tryggvason K. Recombinant laminin-8 (alpha(4)beta(1)gamma(1)). Production, purification, and interactions with integrins. J Biol Chem. 2000;275:14853–14859. doi: 10.1074/jbc.275.20.14853. [DOI] [PubMed] [Google Scholar]
- Koshida S, Kishimoto Y, Ustumi H, Shimizu T, Furutani-Seiki M, Kondoh H, Takada S. Integrinalpha5-dependent fibronectin accumulation for maintenance of somite boundaries in zebrafish embryos. Dev Cell. 2005;8:587–598. doi: 10.1016/j.devcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Kragtorp KA, Miller JR. Integrin alpha5 is required for somite rotation and boundary formation in Xenopus. Dev Dyn. 2007;236:2713–2720. doi: 10.1002/dvdy.21280. [DOI] [PubMed] [Google Scholar]
- Kudo H, Amizuka N, Araki K, Inohaya K, Kudo A. Zebrafish periostin is required for the adhesion of muscle fiber bundles to the myoseptum and for the differentiation of muscle fibers. Dev Biol. 2004;267:473–487. doi: 10.1016/j.ydbio.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Larsen M, Wei C, Yamada KM. Cell and fibronectin dynamics during branching morphogenesis. Journal of Cell Science. 2006;119:3376–3384. doi: 10.1242/jcs.03079. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med. 2007;232:1121–1129. doi: 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- Leiss M, Beckmann K, Girós A, Costell M, Fässler R. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008;20:502–507. doi: 10.1016/j.ceb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Li D, Bareja A, Judge L, Yue Y, Lai Y, Fairclough R, Davies KE, Chamberlain JS, Duan D. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. Journal of Cell Science. 2010;123:2008–2013. doi: 10.1242/jcs.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J. Transforming Growth Factor-β1 Induces the Differentiation of Myogenic Cells into Fibrotic Cells in Injured Skeletal Muscle. Am J Pathol. 2004;164:1007–1019. doi: 10.1016/S0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZB, Kollias HD, Wagner KR. Myostatin Directly Regulates Skeletal Muscle Fibrosis. J Biol Chem. 2008;283:19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber RL, Ward SR. Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am J Physiol-Cell Ph. 2013;305:C241–52. doi: 10.1152/ajpcell.00173.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunardi A, Dente L. Molecular cloning and expression analysis of dystroglycan during Xenopus laevis embryogenesis. Gene Expr Patterns. 2002;2:45–50. doi: 10.1016/s0925-4773(02)00356-8. [DOI] [PubMed] [Google Scholar]
- Lund DK, Cornelison DDW. Enter the matrix: shape, signal and superhighway. FEBS J. 2013;280:4089–4099. doi: 10.1111/febs.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Muñoz-Cánoves P. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin FC, Hiller M, Spitali P, Oonk S, Dalebout H, Palmblad M, Chaouch A, Guglieri M, Straub V, Lochmüller H, Niks EH, Verschuuren JJGM, Aartsma-Rus A, Deelder AM, van der Burgt YEM, ‘t Hoen PAC. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics Clin Appl. 2014;8:269–278. doi: 10.1002/prca.201300072. [DOI] [PubMed] [Google Scholar]
- Marques L, Thorsteinsdóttir S. Dynamics of Akt activation during mouse embryo development: distinct subcellular patterns distinguish proliferating versus differentiating cells. Differentiation. 2013;86:48–56. doi: 10.1016/j.diff.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Marshall JL, Crosbie-Watson RH. Sarcospan: a small protein with large potential for Duchenne muscular dystrophy. Skelet Muscle. 2013;3:1. doi: 10.1186/2044-5040-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins GG, Rifes P, Amândio R, Rodrigues G, Palmeirim I, Thorsteinsdóttir S. Dynamic 3D cell rearrangements guided by a fibronectin matrix underlie somitogenesis. PLoS ONE. 2009;4:e7429. doi: 10.1371/journal.pone.0007429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martoni E, Petrini S, Trabanelli C, Sabatelli P, Urciuolo A, Selvatici R, D’Amico A, Falzarano S, Bertini E, Bonaldo P, Ferlini A, Gualandi F. Characterization of a rare case of Ullrich congenital muscular dystrophy due to truncating mutations within the COL6A1 gene C-terminal domain: a case report. BMC Med Genet. 2013;14:59. doi: 10.1186/1471-2350-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Raya A, Callol-Massot C, Kawakami Y, Oishi I, Rodriguez-Esteban C, Izpisúa Belmonte JC. miles-apart-Mediated regulation of cell-fibronectin interaction and myocardial migration in zebrafish. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S77–82. doi: 10.1038/ncpcardio0764. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- Maurya AK, Tan H, Souren M, Wang X, Wittbrodt J, Ingham PW. Integration of Hedgehog and BMP signalling by the engrailed2a gene in the zebrafish myotome. Development. 2011;138:755–765. doi: 10.1242/dev.062521. [DOI] [PubMed] [Google Scholar]
- McDonagh MJ, Davies CT. Adaptive response of mammalian skeletal muscle to exercise with high loads. Eur J Appl Physiol Occup Physiol. 1984;52:139–155. doi: 10.1007/BF00433384. [DOI] [PubMed] [Google Scholar]
- Mehuron T, Kumar A, Duarte L, Yamauchi J, Accorsi A, Girgenrath M. Dysregulation of matricellular proteins is an early signature of pathology in laminin-deficient muscular dystrophy. Skelet Muscle. 2014;4:14. doi: 10.1186/2044-5040-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli M, Farini A, Belicchi M, Parolini D, Cassinelli L, Razini P, Sitzia C, Torrente Y. Perspectives of stem cell therapy in Duchenne muscular dystrophy. FEBS J. 2013;280:4251–4262. doi: 10.1111/febs.12083. [DOI] [PubMed] [Google Scholar]
- Meyer GA, Lieber RL. Elucidation of extracellular matrix mechanics from muscle fibers and fiber bundles. J Biomech. 2011;44:771–773. doi: 10.1016/j.jbiomech.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JR, Planamento J, Ebrom P, Krulewitz N. Sulf1 modulates BMP signaling and is required for somite morphogenesis and development of the horizontal myoseptum. Dev biol. 2013;378:107–121. doi: 10.1016/j.ydbio.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Mienaltowski MJ, Birk DE. Structure, physiology, and biochemistry of collagens. Adv Exp Med Biol. 2014;802:5–29. doi: 10.1007/978-94-007-7893-1_2. [DOI] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Moll J, Barzaghi P, Lin S, Bezakova G, Lochmüller H, Engvall E, Müller U, Ruegg MA. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- Morales MG, Cabello-Verrugio C, Santander C, Cabrera D, Goldschmeding R, Brandan E. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J Pathol. 2011;225:490–501. doi: 10.1002/path.2952. [DOI] [PubMed] [Google Scholar]
- Moreau N, Alfandari D, Gaultier A, Cousin H, Darribere T. Cloning and expression patterns of dystroglycan during the early development of Xenopus laevis. Dev Genes Evol. 2003;213:355–359. doi: 10.1007/s00427-003-0328-6. [DOI] [PubMed] [Google Scholar]
- Morin-Kensicki EM, Eisen JS. Sclerotome development and peripheral nervous system segmentation in embryonic zebrafish. Development. 1997;124:159–167. doi: 10.1242/dev.124.1.159. [DOI] [PubMed] [Google Scholar]
- Murray JD, Oster GF. Cell traction models for generating pattern and form in morphogenesis. J Math Biol. 1984;19:265–279. doi: 10.1007/BF00277099. [DOI] [PubMed] [Google Scholar]
- Nawrotzki R, Willem M, Miosge N, Brinkmeier H, Mayer U. Defective integrin switch and matrix composition at alpha 7-deficient myotendinous junctions precede the onset of muscular dystrophy in mice. Hum Mol Genet. 2003;12:483–495. doi: 10.1093/hmg/ddg047. [DOI] [PubMed] [Google Scholar]
- Nguyen-Chi ME, Bryson-Richardson R, Sonntag C, Hall TE, Gibson A, Sztal T, Chua W, Schilling TF, Currie PD. Morphogenesis and Cell Fate Determination within the Adaxial Cell Equivalence Group of the Zebrafish Myotome. PLoS Genet. 2012;8:e1003014. doi: 10.1371/journal.pgen.1003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MJ, Campos I, Hirst EM, Stemple DL. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development. 2002;129:3505–3512. doi: 10.1242/dev.129.14.3505. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and Function of Laminins in the Neuromuscular System of Developing, Adult, and Mutant Mice. The Journal of Cell Biology. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce M, Blake DJ, Tinsley JM, Byth BC. The utrophin and dystrophin genes share similarities in genomic structure. Hum Mol Genet. 1993;2:1765–1772. doi: 10.1093/hmg/2.11.1765. [DOI] [PubMed] [Google Scholar]
- Peterson MT, Henry CA. Hedgehog signaling and laminin play unique and synergistic roles in muscle development. Dev Dyn. 2010;239:905–913. doi: 10.1002/dvdy.22204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. PNAS. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogány G, Hernandez DJ, Vogel KG. The in vitro interaction of proteoglycans with type I collagen is modulated by phosphate. Arch Biochem Biophys. 1994;313:102–11. doi: 10.1006/abbi.1994.1365. [DOI] [PubMed] [Google Scholar]
- Polanska UM, Fernig DG, Kinnunen T. Extracellular interactome of the FGF receptor-ligand system: complexities and the relative simplicity of the worm. Dev Dyn. 2009;238:277–293. doi: 10.1002/dvdy.21757. [DOI] [PubMed] [Google Scholar]
- Powell GT, Wright GJ. Jamb and jamc are essential for vertebrate myocyte fusion. Plos Biol. 2011 doi: 10.1371/journal.pbio.1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell GT, Wright GJ. Do muscle founder cells exist in vertebrates? Trends Cell Biol. 2012;22:391–396. doi: 10.1016/j.tcb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–53. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- Rifes P, Carvalho L, Lopes C, Andrade RP, Rodrigues G, Palmeirim I, Thorsteinsdóttir S. Redefining the role of ectoderm in somitogenesis: a player in the formation of the fibronectin matrix of presomitic mesoderm. Development. 2007;134:3155–3165. doi: 10.1242/dev.003665. [DOI] [PubMed] [Google Scholar]
- Ringelmann B, Röder C, Hallmann R, Maley M, Davies M, Grounds M, Sorokin L. Expression of Laminin α1, α2, α4, and α5 Chains, Fibronectin, and Tenascin-C in Skeletal Muscle of Dystrophic 129ReJdy/dyMice. Exp Cell Res. 1999;246:165–182. doi: 10.1006/excr.1998.4244. [DOI] [PubMed] [Google Scholar]
- Roman J. Fibronectin and fibronectin receptors in lung development. Exp Lung Res. 1997;23:147–159. doi: 10.3109/01902149709074027. [DOI] [PubMed] [Google Scholar]
- Roman J, Crouch EC, McDonald JA. Reagents that inhibit fibronectin matrix assembly of cultured cells also inhibit lung branching morphogenesis in vitro. Implications for lung development, injury, and repair. Chest. 1991;99:20S–21S. [PubMed] [Google Scholar]
- Rooney JE, Gurpur PB, Burkin DJ. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. PNAS. 2009a;106:7991–7996. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JE, Gurpur PB, Yablonka-Reuveni Z, Burkin DJ. Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am J Pathol. 2009b;174:256–264. doi: 10.2353/ajpath.2009.080522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JE, Knapp JR, Hodges BL, Wuebbles RD, Burkin DJ. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol. 2012;180:1593–1602. doi: 10.1016/j.ajpath.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ, Burkin DJ. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. Journal of Cell Science. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- Sakai T, Larsen M, Yamada KM. Fibronectin requirement in branching morphogenesis. Nature. 2003;423:876–881. doi: 10.1038/nature01712. [DOI] [PubMed] [Google Scholar]
- Sanes JR. Laminin, fibronectin, and collagen in synaptic and extrasynaptic portions of muscle fiber basement membrane. The Journal of Cell Biology. 1982;93:442–451. doi: 10.1083/jcb.93.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR. The Basement Membrane/Basal Lamina of Skeletal Muscle. J Biol Chem. 2003;278:12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- Sanger JW, Wang J, Holloway B, Du A, Sanger JM. Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motil Cytoskeleton. 2009;66:556–566. doi: 10.1002/cm.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz A, Azpitarte M, Armañanzas R, Leturcq F, Alzualde A, Inza I, García-Bragado F, De la Herran G, Corcuera J, Cabello A, Navarro C, De la Torre C, Gallardo E, Illa I, López de Munain A. Gene expression profiling in limb-girdle muscular dystrophy 2A. PLoS ONE. 2008;3:e3750. doi: 10.1371/journal.pone.0003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaal M, Wiegreffe C. Somite compartments in anamniotes. Anat Embryol (Berl) 2006;211:9–19. doi: 10.1007/s00429-006-0127-8. [DOI] [PubMed] [Google Scholar]
- Schienda J, Engleka KA, Jun S, Hansen MS, Epstein JA, Tabin CJ, Kunkel LM, Kardon G. Somitic origin of limb muscle satellite and side population cells. PNAS. 2006;103:945–950. doi: 10.1073/pnas.0510164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield JN, Górecki DC, Blake DJ, Davies K, Edwards YH. Dystroglycan mRNA expression during normal and mdx mouse embryogenesis: a comparison with utrophin and the apo-dystrophins. Dev Dyn. 1995;204:178–185. doi: 10.1002/aja.1002040208. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harbor Perspectives in Biology. 2011 doi: 10.1101/cshperspect.a005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel AL, Gurevich DB, Currie PD. A myogenic precursor cell that could contribute to regeneration in zebrafish and its similarity to the satellite cell. FEBS J. 2013;280:4074–4088. doi: 10.1111/febs.12300. [DOI] [PubMed] [Google Scholar]
- Sieiro-Mosti D, De La Celle M, Pelé M. A dynamic analysis of muscle fusion in the chick embryo. Development. 2014;141:3605–3611. doi: 10.1242/dev.114546. [DOI] [PubMed] [Google Scholar]
- Sikorski ZE. Chemical and Functional Properties of Food Proteins. Boca Raton: CRC Press; 2001. p. 242. [Google Scholar]
- Snow CJ, Goody M, Kelly MW, Oster EC, Jones R, Khalil A, Henry CA. Time-lapse analysis and mathematical characterization elucidate novel mechanisms underlying muscle morphogenesis. PLoS Genet. 2008a;4:e1000219. doi: 10.1371/journal.pgen.1000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CJ, Henry CA. Dynamic formation of microenvironments at the myotendinous junction correlates with muscle fiber morphogenesis in zebrafish. Gene Expr Patterns. 2009;9:37–42. doi: 10.1016/j.gep.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CJ, Peterson MT, Khalil A, Henry CA. Muscle development is disrupted in zebrafish embryos deficient for fibronectin. Dev Dyn. 2008b;237:2542–2553. doi: 10.1002/dvdy.21670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ. H36-alpha 7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J Cell Biol. 1992;117:643–657. doi: 10.1083/jcb.117.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol-Cell Ph. 2007;293:C1934–46. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- Srichai MB, Zent R. Integrin structure and function. Cell-Extracellular Matrix Interactions in Cancer. 2010 doi: 10.1007/978-1-4419-0814-8_2. [DOI] [Google Scholar]
- Stellabotte F, Devoto S. The teleost dermomyotome. Dev Dyn. 2007;236:2432–2443. doi: 10.1002/dvdy.21253. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Schilling TF. Thrombospondin-4 controls matrix assembly during development and repair of myotendinous junctions. eLife. 2014;3:e02372. doi: 10.7554/eLife.02372. http://dx.doi.org/10.7554/eLife.02372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztal TE, Sonntag C, Hall TE, Currie PD. Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum Mol Genet. 2012;21:4718–4731. doi: 10.1093/hmg/dds312. [DOI] [PubMed] [Google Scholar]
- Talts JF, Sasaki T, Miosge N, Göhring W, Mann K, Mayne R, Timpl R. Structural and functional analysis of the recombinant G domain of the laminin alpha4 chain and its proteolytic processing in tissues. J Biol Chem. 2000;275:35192–35199. doi: 10.1074/jbc.M003261200. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69:91–95. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- Telfer WR, Busta AS, Bonnemann CG, Feldman EL, Dowling JJ. Zebrafish models of collagen VI-related myopathies. Hum Mol Genet. 2010;19:2433–2444. doi: 10.1093/hmg/ddq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsteinsdóttir S. Basement membrane and fibronectin matrix are distinct entities in the developing mouse blastocyst. Anat Rec. 1992;232:141–149. doi: 10.1002/ar.1092320116. [DOI] [PubMed] [Google Scholar]
- Thorsteinsdóttir S, Deries M, Cachaço AS, Bajanca F. The extracellular matrix dimension of skeletal muscle development. Dev Biol. 2011;354:191–207. doi: 10.1016/j.ydbio.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- Tosney KW, Dehnbostel DB, Erickson CA. Neural crest cells prefer the myotome’s basal lamina over the sclerotome as a substratum. Dev Biol. 1994;163:389–406. doi: 10.1006/dbio.1994.1157. [DOI] [PubMed] [Google Scholar]
- Tozer S, Duprez D. Tendon and ligament: Development, repair and disease. Birth Defect Res C. 2005;75:226–236. doi: 10.1002/bdrc.20049. [DOI] [PubMed] [Google Scholar]
- Trinh LA, Stainier DYR. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell. 2004;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. The International Journal of Biochemistry & Cell Biology. 2008;40:199–214. doi: 10.1016/j.biocel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, Reed JC, Wewer UM, Engvall E. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997;100:1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeden FJ, Granato M, Schach U, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Warga RM, Allende ML, Weinberg ES, Nüsslein-Volhard C. Mutations affecting somite formation and patterning in the zebrafish, Danio rerio. Development. 1996;123:153–164. doi: 10.1242/dev.123.1.153. [DOI] [PubMed] [Google Scholar]
- Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- Waterman RE. Development of the lateral musculature in the teleost, Brachydanio rerio: a fine structural study. Am J Anat. 1969;125:457–493. doi: 10.1002/aja.1001250406. [DOI] [PubMed] [Google Scholar]
- Waugh TA, Horstick E, Hur J, Jackson SW, Davidson AE, Li X, Dowling JJ. Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum Mol Genet. 2014;23:4651–4662. doi: 10.1093/hmg/ddu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welser JV, Rooney JE, Cohen NC, Gurpur PB, Singer CA, Evans RA, Haines BA, Burkin DJ. Myotendinous Junction Defects and Reduced Force Transmission in Mice that Lack α7 Integrin and Utrophin. Am J Pathol. 2009;175:1545–1554. doi: 10.2353/ajpath.2009.090052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff C, Roy S, Ingham PW. Multiple muscle cell identities induced by distinct levels and timing of hedgehog activity in the zebrafish embryo. Curr Biol. 2003;13:1169–1181. doi: 10.1016/s0960-9822(03)00461-5. [DOI] [PubMed] [Google Scholar]
- Yang DH, McKee KK, Chen ZL, Mernaugh G, Strickland S, Zent R, Yurchenco PD. Renal collecting system growth and function depend upon embryonic γ1 laminin expression. Development. 2011;138:4535–4544. doi: 10.1242/dev.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Bader BL, Kreidberg JA, Ullman-Culleré M, Trevithick JE, Hynes RO. Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev Biol. 1999;215:264–277. doi: 10.1006/dbio.1999.9451. [DOI] [PubMed] [Google Scholar]
- Yee KL, Weaver VM, Hammer DA. Integrin-mediated signalling through the MAP-kinase pathway. IET Systems Biology. 2008;2:8–15. doi: 10.1049/iet-syb:20060058. [DOI] [PubMed] [Google Scholar]
- Ylikärppä R, Eklund L, Sormunen R, Muona A, Fukai N, Olsen BR, Pihlajaniemi T. Double knockout mice reveal a lack of major functional compensation between collagens XV and XVIII. Matrix Biol. 2003;22:443–448. doi: 10.1016/s0945-053x(03)00074-x. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Halper J. Tendon proteoglycans: biochemistry and function. J Musculoskelet Neuronal Interact. 2005;5:22–34. [PubMed] [Google Scholar]
- Young MF, Bi Y, Ameye L, Chen XD. Biglycan knockout mice: new models for musculoskeletal diseases. Glycoconjugate journal. 2002;19:257–262. doi: 10.1023/A:1025336114352. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harbor Perspectives in Biology. 2011;3 doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Chen S, Goldoni S, Calder BW. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. Journal of Biological Chemistry. 2009;284:8888–8897. doi: 10.1074/jbc.M806590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Yoshioka K, Miyatake M, Miike T. Dystrophin and a dystrophin-related protein in intrafusal muscle fibers, and neuromuscular and myotendinous junctions. Acta Neuropathol. 1992;84:141–146. doi: 10.1007/BF00311386. [DOI] [PubMed] [Google Scholar]
- Zoeller JJ, McQuillan A, Whitelock J, Ho SY, Iozzo RV. A central function for perlecan in skeletal muscle and cardiovascular development. J Cell Biol. 2008;181:381–394. doi: 10.1083/jcb.200708022. [DOI] [PMC free article] [PubMed] [Google Scholar]