

Summary
During gonorrheal infection, there is a heterogeneous population of Neisseria gonorrhoeae (Gc) varied in their expression of opacity-associated (Opa) proteins. While Opa proteins are important for bacterial attachment and invasion of epithelial cells, Opa+ Gc has a survival defect after exposure to neutrophils. Here, we use constitutively Opa- and OpaD+ Gc in strain background FA1090 to show that Opa+ Gc is more sensitive to killing inside adherent, chemokine-treated primary human neutrophils due to increased bacterial residence in mature, degradative phagolysosomes that contain primary and secondary granule antimicrobial content. Although Opa+ Gc stimulates a potent oxidative burst, neutrophil killing of Opa+ Gc was instead attributable to non-oxidative components, particularly neutrophil proteases and the bactericidal/permeability-increasing protein. Blocking interaction of Opa+ Gc with carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) or inhibiting Src family kinase signaling, which is downstream of CEACAM activation, enhanced the survival of Opa+ Gc in neutrophils. Src family kinase signaling was required for fusion of Gc phagosomes with primary granules to generate mature phagolysosomes. Conversely, ectopic activation of Src family kinases or coinfection with Opa+ Gc resulted in decreased survival of Opa- Gc in neutrophils. From these results, we conclude that Opa protein expression is an important modulator of Gc survival characteristics in neutrophils by influencing phagosome dynamics and thus bacterial exposure to neutrophils’ full antimicrobial arsenal.
Keywords: Neisseria gonorrhoeae, neutrophil, polymorphonuclear leukocyte, Opa proteins, CEACAM, Src-family kinase, granules, serine protease
Introduction
Gonorrhea is a major global health problem, with 106 million cases estimated worldwide each year (World Health Organization, 2012). Infection with the causative agent of gonorrhea, Neisseria gonorrhoeae (Gc), promotes the local influx of neutrophils (polymorphonuclear leukocytes, or PMNs), resulting in the purulent exudate characteristic of gonorrheal infection. PMNs are professional phagocytes and a key component of the innate immune response responsible for controlling bacterial infection. PMN antimicrobial mechanisms include generation of reactive oxygen species (ROS) via NADPH oxidase, release of antimicrobial enzymes and peptides, and production of neutrophil extracellular traps (NETs) (Borregaard et al., 2007;Papayannopoulos et al., 2009;Borregaard, 2010). Despite the abundance of PMNs during infection, viable Gc can be cultured from patient exudates and primary human PMNs infected ex vivo with Gc (Wiesner et al., 1980;Simons et al., 2005;Criss et al., 2009;Johnson et al., 2013b). These observations suggest that Gc has developed multiple mechanisms to avoid and defend against PMN mediated killing.
Gc interactions with human cells are modulated by surface-exposed components, including the opacity-associated (Opa) proteins. Gc strains possess approximately 11 opa genes (Connell et al., 1990;Bhat et al., 1991;Dempsey et al., 1991). Slipped strand mispairing at pentameric repeats on the 5’ end of opa genes causes opa genes to shift in or out of frame (Stern et al., 1986;Murphy et al., 1989), resulting in, a mixed population of bacteria expressing no Opa proteins, one Opa protein, or multiple Opa proteins in in vitro culture and during infection. Opa proteins are 25-30 kDa proteins which form an eight-stranded β-barrel in the Gc outer membrane, with four extracellular loops. The β-barrel is highly conserved, in contrast to the extracellular loops, which contain semivariable and hypervariable regions. The variability in these extracellular loops determines receptor specificity (Swanson, 1978;Malorny et al., 1998;Vandeputte-Rutten et al., 2003;de Jonge et al., 2003). Opa proteins can bind to carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) and/or heparan sulfate proteoglycans (HSPGs) on host cells (Sadarangani et al., 2011). Opa proteins that bind CEACAMs specifically interact with the N-terminal immunoglobulin fold of the receptor (Bos et al., 1998). In the human CEA family there are 12 proteins in the CEACAM subgroup, but to date, Opa proteins have been shown to bind only CEACAM1, CEACAM3, CEACAM5, and CEACAM6 (Sadarangani et al., 2011).
Human PMNs express three of the CEACAMs known to bind Opa proteins: CEACAM1, CEACAM3, and CEACAM6 (Gray-Owen et al., 2006). Binding of Opa proteins to CEACAMs on PMNs is sufficient to drive bacterial internalization in the absence of antibody or complement opsonization (Gray-Owen et al., 1997a;Gray-Owen et al., 1997b;Chen et al., 1996;Virji et al., 1996). Different CEACAMs engage different signaling pathways to lead to Gc internalization. The cytoplasmic regions of CEACAM1 and CEACAM3 have immunoreceptor tyrosine-based inhibition motifs and immunoreceptor tyrosine-based activation motifs, respectively. CEACAM1 activation results in recruitment of the phosphatase SHP-1, while CEACAM3 activation recruits kinases such as Syk and Src family kinases, which stimulate proinflammatory signaling cascades (McCaw et al., 2003;Hauck et al., 1998;Hauck et al., 1999;Sintsova et al., 2014). In contrast, CEACAM5 and CEACAM6 are glycosylphosphatidylinositol-anchored and the signaling is largely unknown (McCaw et al., 2004;Sadarangani et al., 2011). Sarantis et al. recently reported that CEACAMs can signal cooperatively in a cell; in transfected mouse PMNs, engagement of CEACAM1 and CEACAM6 results in signaling via CEACAM3 (Sarantis et al., 2012). Although Opa protein expression was thought to be required for PMN phagocytosis of unopsonized Gc, we have reported that adherent, chemokine-primed PMNs can internalize Opa- Gc, albeit with slower kinetics than Opa-CEACAM driven internalization (Criss et al., 2009;Ball et al., 2013;Smirnov et al., 2014).
In addition to mediating adherence and internalization, Gc expressing CEACAM-binding Opa proteins are also more potent activators of PMNs. Opa+ Gc stimulates human PMN NADPH oxidase complex assembly in order to generate reactive oxygen species (ROS). In contrast, Opa- Gc not only fails to induce an oxidative burst, but also suppresses the oxidative burst induced by other stimuli (Rest et al., 1982;Virji et al., 1986;Fischer et al., 1988;Criss et al., 2008;Ball et al., 2013;Smirnov et al., 2014). Infection with Opa+ Gc also increases the exocytosis of PMN primary and secondary granules (Sarantis et al., 2007;Sintsova et al., 2014). Together, these results would suggest there is a disadvantage to Gc expressing CEACAM-binding Opa proteins during infection. In support of this notion, Opa+ Gc have decreased survival compared to Opa- Gc in human PMNs (Rest et al., 1982;Rest et al., 1985;Virji et al., 1986;Fischer et al., 1988;Rest et al., 1989;Sarantis et al., 2007;Criss et al., 2009;Ball et al., 2013). However, the precise mechanism leading to this survival defect is not yet defined.
There are two nonexclusive hypotheses to explain the observed survival defect of Opa+ Gc in neutrophils. First, Opa proteins alter activation of PMN antimicrobial responses. Second, Opa proteins alter sensitivity to PMN antimicrobial components. In this work, we tested these two hypotheses in primary human PMNs, using Gc derivatives we recently engineered in the FA1090 strain background with in frame deletions of all opa genes (Opaless) and into which a constitutively expressed opaD allele was introduced (Ball et al., 2013). The OpaD protein engages CEACAM1 and CEACAM3 (Fulcher, 2004) (J. Martin, L. Ball, T. Solomon, A. Criss, and L. Columbus, manuscript in preparation). We found that that OpaD+ Gc has significantly reduced intracellular survival in PMNs due to its CEACAM-dependent trafficking into a mature, degradative phagolysosome and increased sensitivity to primary granule proteins. Exposure to non-oxidative antimicrobial components, predominantly proteases and the bactericidal/permeability increasing protein (BPI), in the phagolysosome compromises Opa+ Gc survival. These results establish a link between CEACAM ligation, Src family kinase signaling, and primary granule-phagosome fusion, resulting in enhanced PMN antigonococcal activity against Opa+ Gc.
Results
Expression of CEACAM-binding Opa proteins reduces the survival of Gc inside PMNs
To begin to understand the mechanism of increased sensitivity of Opa+ Gc to PMNs, we measured the viability of intracellular and extracellular Gc after infection of adherent, IL-8 primed human PMNs, using Baclight viability dyes (Johnson et al., 2013b;Criss et al., 2009;Johnson et al., 2013a). We found that Opa+ Gc had a significant reduction in intracellular survival in PMNs compared to Opa- Gc. While Opa+ Gc also exhibited a reduction in extracellular survival compared to Opa- Gc, this difference was slight compared with the decrease in intracellular survival (Figure 1A–C). Treatment of PMNs with cytochalasin D to inhibit phagocytosis prior to infection with Opa- or Opa+ Gc significantly rescued Opa+ Gc survival to levels observed for Opa- Gc, supporting the importance of intracellular antimicrobial mechanisms to the decreased survival of Opa+ Gc (Figure 1D). We also observed a significant increase in the percentage of neutrophils associated with Opa+ Gc (Figure 1E), as well as an increase in the number of Opa+ Gc per neutrophil (Figure 1F), compared to Opa- bacteria. From these data we conclude that the observed survival defect of Opa+ Gc in PMNs is primarily intracellular and enhanced by the increased association of Opa+ Gc with PMNs, and our subsequent experiments focused on the mechanism underlying this survival defect.
Figure 1.

Reduced survival of Opa+ Gc after exposure to PMNs. PMNs were infected with Opa- (A) or Opa+ (B) Gc for 1 hr. Viable Gc (green) and nonviable Gc (red) were discriminated using Baclight viability dyes SYTO9 and propidium iodide, and extracellular Gc were labeled with soybean lectin (blue). Extracellular viable Gc appear teal, intracellular nonviable Gc appear red, and intracellular viable Gc appear green. Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. Scale bar, 5 μm. The percent of viable extracellular and intracellular Gc are reported in C. D. Opa- and Opa+ Gc were exposed to PMNs that were left untreated or treated with cytochalasin D (CytoD). Bacterial survival was calculated as CFU enumerated from PMN lysates at each time point, divided by the CFU present at 0 min. E, F. PMNs were exposed to Opa- or Opa+ Gc as in Figure 1A. In E, the percentage of PMNs with associated bacteria was calculated and presented as the average from three independent experiments. In F, the number of Gc per PMN was determined for each PMN with associated bacteria for three independent experiments (n = 210 PMNs with Opa- Gc, 253 PMNs with Opa+ Gc). Asterisks indicate P < 0.05 by Student’s two-tailed t test.
To determine if the survival defect of OpaD-expressing Gc in PMNs was dependent on CEACAM engagement, we incubated Opa+ Gc with the N-terminal domain of CEACAM1 (N-CEACAM1) prior to infection of PMNs. Incubation with N-CEACAM1 reduced the PMN oxidative burst in response to Opa+ Gc in a dose- dependent manner, demonstrating that soluble N-CEACAM1 blocks Opa-mediated interactions with neutrophils (Supplemental Figure 1). Preincubation with N-CEACAM1 significantly increased the intracellular viability of Opa+ Gc (Figure 2A–C). Opa+ Gc preincubated with N-CEACAM1 was still internalized by PMNs, although not as effectively as bacteria without N-CEACAM1, and was similar to the percent internalization we reported for Opa- Gc (Johnson et al., 2013b), (Supplemental Figure 1). From these data, we conclude that the observed internal survival defect of Opa+ Gc in PMNs is mediated by Opa engagement of CEACAM(s), and blocking this interaction increases the intracellular survival of Opa+ Gc.
Figure 2.
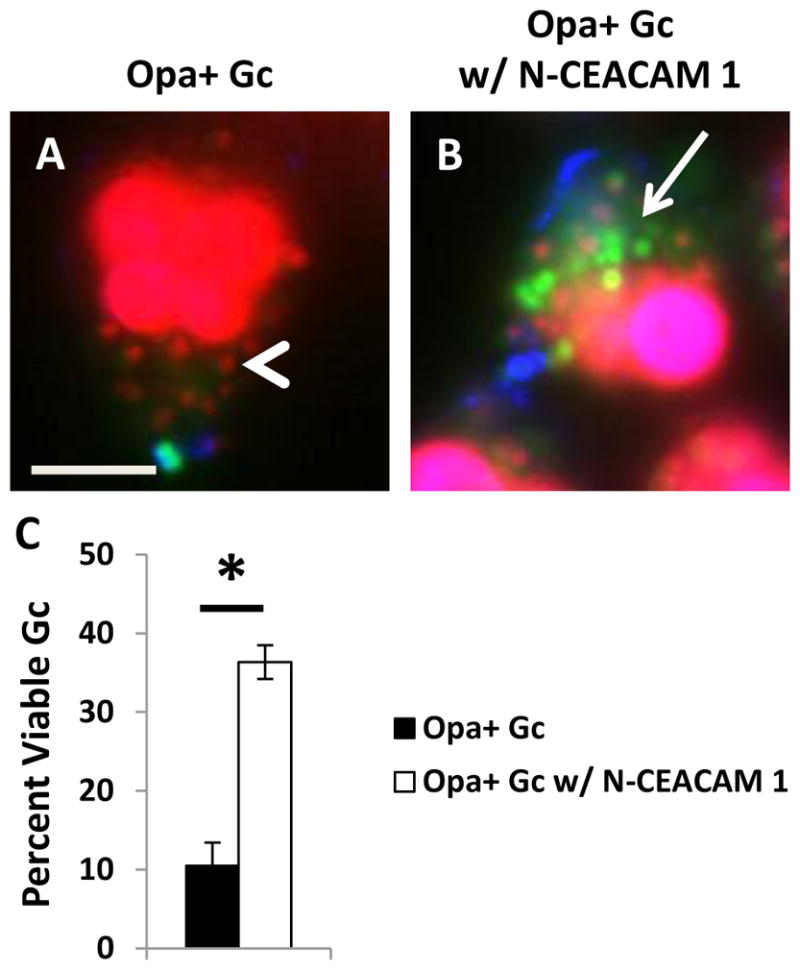
Survival of Opa+ Gc in PMNs can be rescued by preincubating the bacteria with soluble N-terminal domain of CEACAM1 (N-CEACAM1). PMNs were infected with Opa+ (A) or Opa+ Gc incubated with N-CEACAM 1 (B). Viability of individual Gc was assessed as in Figure 1A–B. Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. Scale bar, 5 μm. The percent of viable intracellular Gc are reported in C. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Opa+ Gc predominantly resides in mature phagolysosomes, in which the bacteria are exposed to PMN non-oxidative antigonococcal components
We previously showed that phagosome maturity affects Gc viability in PMNs (Johnson et al., 2013b). In order to assess if Opa protein expression influences maturation of Gc phagosomes in PMNs, adherent, IL-8 primed PMNs were infected with Opa+ or Opa- Gc for 1 hr, and phagosome composition was determined by immunofluorescence, using antibodies against components of primary (neutrophil elastase) and secondary (lactoferrin) granules. A phagosome was considered positive for a granule class if the fluorescent signal for the granule marker of interest surrounded > 50% of the bacterial circumference. While both Opa+ and Opa- Gc phagosomes were positive for lactoferrin (Figure 3A and 3C), there was a significant increase in the percent of phagosomes positive for neutrophil elastase in PMNs infected with Opa+ Gc (Figure 3B and 3D). Thus Opa protein expression directs Gc to a mature phagolysosome which has fused with both primary and secondary granules, while Opa- Gc predominantly resides in immature, primary granule-negative phagosomes.
Figure 3.

Phagosomes containing Opa+ Gc are significantly more mature than those containing Opa- Gc. PMNs infected with Opa- or Opa+ Gc for 1 hr were stained with antibodies against the secondary granule protein lactoferrin (A) or the primary granule protein neutrophil elastase (B), which appear green. External Gc appear red/blue and internal Gc appear blue. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. Scale bar, 5 μm. The percent of Opa+ and Opa- Gc phagosomes positive for lactoferrin or neutrophil elastase are reported in C and D, respectively. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Given that Opa expression continually varies in vivo, we sought to determine how Opa+ and Opa- Gc affect each others’ survival in coinfected PMNs. Compared with Opa- Gc alone, we observed a significant decrease in Opa- Gc survival during coinfection with Opa+ Gc (Figure 4D). Notably, the presence of Opa- Gc did not rescue the survival of Opa+ Gc in PMNs (Figure 4D). These results suggest that during coinfection, the presence of Opa+ Gc enhances PMN killing of Opa- Gc, perhaps by affecting the maturity of Opa- Gc phagosomes. To test this possibility, we evaluated phagosome maturity in coinfected PMNs, using antibodies directed against the primary granule protein neutrophil elastase. The percent of Opa- Gc phagosomes positive for neutrophil elastase was significantly increased in PMNs coinfected with Opa+ Gc (Figure 4A) compared to PMNs infected with Opa- Gc only (Figure 4B, quantified in Figure 4C). We observed no difference in the percent of Opa+ Gc phagosomes positive for neutrophil elastase in coinfected or singly infected PMNs (Figure 4A–C). Together, these results demonstrate that during coinfection with Opa+ and Opa- Gc, the presence of Opa+ Gc is sufficient to alter maturation of Opa- Gc phagosomes and thereby decrease Opa- Gc survival in PMNs.
Figure 4.

Coinfection with Opa- and Opa+ Gc increases primary granule fusion with Opa- Gc phagosomes and decreases survival of Opa- Gc in neutrophils. A. PMNs were infected either with Opa- or Opa+ Gc for 1 h. Maturity of Gc phagosomes was measured by immunofluorescence for the primary granule protein neutrophil elastase (NE) which appears green. External Gc appear red/blue and internal Gc appear blue. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. B. PMNs were coinfected with CFSE-labeled Opa+ Gc and unstained Opa- Gc. Maturity of Gc phagosomes was measured by immunofluorescence for the primary granule protein neutrophil elastase, which appears red. External Opa+ Gc appear purple/green and external Opa- Gc appear purple/blue. Internal Opa+ Gc appear green and internal Opa- Gc appear blue. Arrowheads indicate Opa+ Gc phagosomes positive for elastase and triangles indicate Opa- Gc phagosomes positive for NEelastase The percent of phagosomes positive for neutrophil elastase is reported in C. Asterisks indicate P < 0.05 by Student’s two-tailed t test. Scale bar, 5 μm. D. PMNs were infected with Opa-, Opa+ Gc, or a 1:1 mix of Opa- and Opa+ Gc. Gc CFU were enumerated over time as described in Figure 1D. Opa- and Opa+ Gc were differentiated by their colony opacity. Mix Opa- and Mix Opa+ Gc indicate the percent change in translucent and opaque CFU measured in the coinfection, relative to the CFU of each at 0 min. Asterisks indicate P < 0.01 by Student’s two tailed t test between Opa- and Opa+ Gc at 30 and 60 min. Hash indicates P < 0.01 by Student’s two tailed t test between Opa- and Mix Opa- Gc at 60 min.
Mature PMN phagolysosomes contain a full array of non-oxidative antimicrobial proteins and peptides, as well as the NADPH oxidase holoenzyme to generate ROS. We sought to determine the contribution of these oxidative and non-oxidative components to the reduced survival of Opa+ Gc inside PMNs. FA1090 Gc expressing OpaD induces a strong oxidative burst in PMNs, while no ROS is detectable from PMNs exposed to Opa- Gc (Supplemental Figure 1A) (Rest et al., 1982;Virji et al., 1986;Fischer et al., 1988;Ball et al., 2013;Smirnov et al., 2014). In order to directly test the role of ROS in killing Opa+ Gc by PMNs, PMNs were treated with diphenyleneiodonium (DPI) to inhibit production of ROS or were left untreated prior to infection with Opa+ Gc. Treatment with DPI did not rescue Opa+ Gc survival in PMNs (Figure 5). Thus, ROS is dispensable for PMN- mediated killing of Gc, even under conditions (OpaD expression) where the bacteria stimulate a strong oxidative response.
Figure 5.

Increased killing of Opa+ Gc by PMNs is independent of the PMN oxidative burst. PMNs were pretreated with 10 μM DPI and infected with Opa- or Opa+ Gc. CFU of Gc were enumerated from PMN lysates as described in Figure 1D. Asterisks indicate P < 0.05 for Opa+ Gc vs Opa- Gc at matched time points by Student’s two tailed t test.
Non-oxidative components of PMNs include antimicrobial peptides such as LL-37 and defensins, BPI, and primary granule proteases such as cathepsin G, proteinase 3, and neutrophil elastase. We previously reported that PMN proteases contribute to the killing of Gc localizing to mature phagolysosomes (Johnson et al., 2013b). Therefore, we hypothesized that the increased intracellular killing of Opa+ Gc was due to increased bacterial exposure to these proteases. To test this hypothesis, PMNs were incubated with a protease inhibitor cocktail prior to infection with Opa- or Opa+ Gc. Protease inhibition significantly enhanced the percent of live Opa- and Opa+ Gc inside PMNs by Baclight viability stain (Figure 6A–E). Similarly, inhibition of PMN proteases significantly increased the number of Opa+ Gc recovered from PMNs by colony count (Figure 6F). These findings reinforce our previous observations that PMN proteases have antigonococcal activity. Thus Opa+ Gc resides in a mature PMN phagolysosome with primary and secondary granule characteristics, and exposure to primary granule proteases in this compartment is responsible for the observed survival defect of Opa+ Gc inside PMNs.
Figure 6.

Protease inhibition increases the survival of Gc inside PMNs, regardless of bacterial Opa status. PMNs were treated with protease inhibitors or left untreated prior to infection with Opa+ or Opa- Gc. In A-D, bacterial viability was determined using Baclight viability dyes as in Figure 1A-B. The percent of viable Opa- and Opa+ Gc is reported in E. In F, bacterial survival was measured by CFU enumeration from PMN lysates as in Figure 1D. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
In addition to residence in a mature phagolysosome, intrinsic susceptibility of Opa- or Opa+ Gc could contribute to the observed differences in survival after exposure to PMNs. To examine this possibility, survival of Opa- and Opa+ Gc was measured after bacterial exposure to LL-37, cathepsin G, defensin, and BPI. While exposure to LL-37 and cathepsin G resulted in dose-dependent killing of both Opa- and Opa+ Gc, there was no significant difference in their susceptibility to these antimicrobial components (Figure 7A and 7B). In contrast, Opa+ Gc displayed significantly increased sensitivity to BPI at concentrations under 3 μg/ml (Figure 7C). Both Opa- and Opa+ Gc were resistant to human β-defensin-2 at 200 μg/ml (data not shown). We conclude that increased susceptibility of Opa+ Gc to PMN killing is due to residence in mature phagolysosomes, where the bacteria are exposed to antimicrobial components, as well as increased sensitivity to at least one of these antimicrobial components, BPI.
Figure 7.

Expression of Opa proteins by Gc affects bacterial sensitivity to BPI. Gc were incubated with increasing concentrations of cathepsin G (A), LL-37 (B), or BPI (C) for 45 min. While survival of each strain was significantly reduced at each concentration of cathepsin G and LL-37 relative to untreated Gc, there was no statistically significant difference in Opa+ vs Opa- Gc sensitivity at matched concentrations of these proteins. For matched concentrations of BPI, single asterisk indicates P < 0.05 and double asterisks indicate P < 0.005 by Student’s two-tailed t test.
Src-family kinase signaling is required for PMNs to direct Gc into mature phagolysosomes, where they are killed
Degranulation and induction of the oxidative burst in PMNs involves signaling via Syk kinase, phosphoinositide 3-kinase (PI3K), the small GTPase Rac, and Src family kinases (Sengelov et al., 1993;Mohn et al., 1995;Fensome et al., 1996;Werner, 2004;Abdel-Latif et al., 2005;Van Ziffle et al., 2009). These signaling components are activated upon engagement of CEACAM3 (reviewed in (Buntru et al., 2012). Therefore, we tested the contribution of these signaling components to the antimicrobial activity of PMNs against Opa+ Gc, using inhibitors specific for each enzyme. The inhibitors were used at concentrations and under conditions that significantly reduced the PMN oxidative burst in response to infection with Opa+ Gc (Supplemental Figure 2A). Adherent, IL-8 treated PMNs were treated with each of the inhibitors prior to infection with Opa+ Gc, and bacterial survival was evaluated using Baclight viability dyes. Only PMN treatment with the Src family kinase member inhibitor PP1 significantly increased the viability of Opa+ Gc inside PMNs relative to control, untreated cells (Figure 8A–F). None of the inhibitors significantly affected the efficiency of Gc internalization by adherent, IL-8 primed PMNs (Supplemental Figure 2B-C). These results suggest that signaling via Src family kinase members is critical to the ability of PMNs to kill internalized Opa+ Gc.
Figure 8.

Inhibition of Src family kinase signaling increases Opa+ Gc survival in PMNs. Untreated PMNs (A) or PMNs treated with Syk kinase inhibitor piceatannol (B), Src family kinase inhibitor PP1 (C), Rac inhibitor EHT1864 (D), or phosphoinositide-3-kinase inhibitor LY294002 (E) were infected with Opa+ Gc for 1 hr. The viability of individual intracellular Opa+ Gc was assessed as in Figure 1A–B and is reported in F. Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. Scale bar, 5 μm. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Src family kinases, specifically Hck, have been shown to associate with PMN primary granules and to be recruited to mature phagolysosomes in activated PMNs (Mohn et al., 1995;Mocsai et al., 2000;Fumagalli et al., 2007). Given the increased residence of Opa+ Gc in mature PMN phagosomes, we hypothesized that Src family kinase signaling is required for primary granule fusion with the Gc phagosome. Treatment of PMNs with PP1 significantly reduced the fraction of Opa+ Gc residing in neutrophil elastase-positive phagolysosomes (Figure 9A and 9C). Moreover, we observed increased recruitment of Hck to Opa+ Gc phagosomes in PMNs (Figure 9B and 9D). In contrast, PMN treatment with the Syk kinase inhibitor piceatannol or the PI3K inhibitor LY294002 did not affect Opa+ Gc residence in mature phagolysosomes (Figure 9A and 9C, and Supplemental Figure 3). Therefore, we conclude that in adherent, IL-8 primed human PMNs, Src family kinase signaling, particularly Hck, is required for phagosomes to fuse with PMN primary granules and become mature degradative phagolysosomes that are capable of killing Opa+ Gc.
Figure 9.

Src family kinase activity is crucial for Opa+ Gc phagosome fusion with primary granules. A, PMNs were treated with piceatannol or PP1 or were left untreated, then infected with Opa+ Gc for 30 min. Immunofluorescence was conducted to discriminate external Gc (red/blue) and internal Gc (blue). PMNs were stained with antibodies against the primary granule protein neutrophil elastase, which appears green. B. PMNs were infected with Opa- or Opa+ Gc (blue) for 1 hr. PMNs were stained with an antibody against Hck, which appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins or Hck, while arrows indicate phagosomes negative for granule proteins or Hck. Scale bar, 5 μm. The percent of phagosomes positive for neutrophil elastase and Hck is reported in C and D, respectively. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Given that approximately 40% of intracellular Opa- Gc reside in mature PMN phagolysosomes (Figure 3D) (Johnson et al., 2013b) and we observed Hck recruitment to Opa- Gc phagosomes (Figure 9B), we examined the contribution of Src family kinase signaling to survival of Opa- Gc in PMNs. Strikingly, the viability of intracellular Opa- Gc was also increased in PMNs treated with PP1, as we observed for Opa+ Gc (Figure 10A–C). In order to determine if activation of Src family kinase signaling is sufficient to decrease Opa- Gc survival in PMNs, PMNs were treated with a synthetic activator of Src family kinase members prior to infection with Opa- Gc. Treatment with the Src family kinase activator significantly reduced the survival of Opa- Gc in PMNs (Figure 10D-F). From these results we conclude that the relative level of Src family kinase signaling modulates primary granule fusion with bacterial phagosomes and thus influences Gc survival in PMNs.
Figure 10.

Activation of Src family kinases decreases Opa- Gc survival in PMNs. (A–C) PMNs were left untreated (A) or treated with PP1 (B) prior to infection with Opa- Gc for 1 hr. Intracellular bacterial viability was assessed as in Figure 1A–B and is reported in C. (D–F) Untreated PMNs (D) and PMNs treated with a Src family kinase activator (E) were infected with Opa- Gc for 1 hr. The viability of individual bacteria was measured as above and reported in F. Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. Scale bar, 5 μm. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Discussion
Opa+ Gc that engages CEACAMs is more sensitive to killing by PMNs compared to Opa- Gc, but the mechanisms underlying this difference were poorly understood. Using constitutively Opa- and Opa+ Gc, we show that the decreased survival of Opa+ Gc after PMN challenge is largely due to their increased residence in mature phagolysosomes, where they are susceptible to serine proteases, BPI, and other antimicrobial components of primary and secondary granules. Although CEACAM ligation also stimulates signaling events in PMNs that lead to ROS production, ROS do not contribute to the increased killing of Opa+ Gc. Our work supports a model in which Opa+ Gc that interact with CEACAMs on the PMN surface triggers Src family kinase-dependent signaling events, which promote internalization of the bacteria into mature, degradative phagolysosomes that contain components with antigonococcal activity (Figure 11).
Figure 11.

Model depicting mechanisms which contribute to differential survival of Opa- and Opa+ Gc in PMNs. Opa- Gc is internalized by a currently unknown mechanism, and a fraction of the bacteria initially resides in immature phagosomes that do not fuse with primary granules. Avoidance of the non-oxidative antimicrobial components found in PMN primary granules enhances Opa- Gc survival inside PMNs. In contrast, Opa+ Gc engages CEACAM receptors, which stimulates Src family kinase (green oval) signaling. As a result, Opa+ Gc resides predominantly in mature phagolysosomes that fuse with primary (1°, yellow circle) and secondary granules (2°, purple rectangle), which reduces bacterial intracellular survival. Survival of Opa+ Gc in PMNs is rescued when Opa protein engagement of CEACAMs is blocked. Under these circumstances, Opa+ Gc is internalized by an alternative mechanism akin to Opa- Gc. These bacteria reside in immature phagosomes, avoid exposure to non-oxidative antimicrobial components in primary granules, and thus survive intracellularly in PMNs.
PMNs kill bacteria intracellularly by directing their phagosomes to fuse with antimicrobial granules (Borregaard, 2010). Our results indicate that in contrast to most Opa- Gc phagosomes, which remain immature, Opa+ Gc phagosomes fuse with both secondary and primary granules, exposing the bacteria to oxidative and non-oxidative antimicrobial components. Secondary granules contain the NADPH oxidase subunits gp91phox and p22phox, and the primary granule enzyme myeloperoxidase uses NADPH oxidase-derived hydrogen peroxide to generate hypochlorous acid (Borregaard et al., 2007;Borregaard, 2010). Although Opa+ Gc induces a potent oxidative burst in PMNs, and NADPH oxidase holoenzyme assembles on Opa+ Gc phagosomes (Rest et al., 1982;Virji et al., 1986;Fischer et al., 1988;Criss et al., 2008;Ball et al., 2013;Smirnov et al., 2014), ROS does not contribute to the intracellular survival defect of Opa+ Gc. Our finding is in agreement with several studies demonstrating that PMN killing of Gc is primarily via non-oxidative granule components (Rest et al., 1982;Seib et al., 2005;Criss et al., 2008;Criss et al., 2009;Wu et al., 2009). These components include the primary granule proteases cathepsin G, neutrophil elastase, and proteinase 3, as well as BPI, lysozyme, LL-37, and defensins,(Borregaard et al., 2007); LL-37, cathepsin G, and BPI have been shown to have antigonococcal activity (Casey et al., 1985;Shafer et al., 1986;Shafer et al., 1998). We found that protease activity is particularly important for mediating killing of Opa+ Gc inside PMNs. This is in agreement with our previous report that the fraction of Opa- Gc in primary granule-positive, mature phagolysosomes are killed by exposure to PMN proteases (Johnson et al., 2013b). PMN serine proteases have direct antigonococcal activity, cleave the propeptide hCAP-18 to produce the active LL-37 antimicrobial peptide and enhance release of BPI from the primary granule matrix, all of which may contribute to the survival defect of Opa+ Gc (Sorensen et al., 2001;Edwards, 2005;Rest et al., 1981;Shafer et al., 1986;Shafer et al., 1987;Shafer et al., 1990). While Sintsova et al. recently reported that Opa+ Gc is not more sensitive to killing by PMNs (Sintsova et al., 2014), in that study infected PMNs were treated with protease inhibitors prior to cell lysis and CFU enumeration. Our results suggest that any survival defect of Opa+ Gc in PMNs would be masked by treating the cells with protease inhibitors, which may explain the discrepancy between these two reports. Our data supports a model in which Gc residence within or avoidance of a degradative phagolysosome is a key factor in determining Gc survival in PMNs, with a two-fold difference in phagosome maturity between Opa- and Opa+ Gc being sufficient to significantly affect intracellular Gc viability.
Our results suggest that the Opa+ Gc survival defect in PMNs is dependent on signaling events downstream of CEACAM engagement, which is in agreement with numerous studies reporting PMN phagocytosis and activation downstream of CEACAM-binding Opa+ bacteria (Chen et al., 2001;McCaw et al., 2003;Sarantis et al., 2007;Sarantis et al., 2012;Sintsova et al., 2014). While CEACAM1 and CEACAM6 signaling do not result in substantial PMN activation, binding to CEACAM1 or CEACAM6 results in cooperative signaling via CEACAM3, which is sufficient to stimulate PMN antimicrobial processes such as the oxidative burst and granule exocytosis (Sarantis et al., 2012). Opa interaction with CEACAM3 stimulates signaling via Syk, PI3K, Rac GTPase, and Src family kinases(Lacy, 2006;Sadarangani et al., 2011;Hauck et al., 1998;Sintsova et al., 2014). In our infection model, we identified a role for Src family kinases in intracellular killing of Opa+ Gc by PMNs. PMNs express the Src family kinases Hck, Fgr, and Lyn; we specifically focused on Hck, which associates with primary granules and mature phagolysosomes in activated PMNs (Mohn et al., 1995;Mocsai et al., 2000;Fumagalli et al., 2007) and associates with Opa+ Gc in JOSK-M phagocytes (Hauck et al., 1998). We found that primary granule-positive Opa+ Gc phagolysosomes are enriched for Hck, and inhibiting Src family kinase activity decreases primary granule fusion with Opa+ Gc phagosomes and increases intracellular Gc survival. These findings support a model in which Hck activation downstream of CEACAMs promotes primary granule-phagosome fusion and creation of mature, degradative phagolysosomes. In agreement with this model, pathogenic mycobacteria can survive inside PMNs by reducing phagosome-primary granule fusion, which was associated with reduced activation and association of Hck with the phagosome membrane (N’Diaye et al., 1998;Astarie-Dequeker et al., 2000;Cougoule et al., 2002).
In our infection model, Syk, phosphoinositide-3-kinase, and Rac GTPase did not significantly contribute to phagosomal maturation or the killing of Opa+ Gc inside PMNs. This was especially surprising because Sarantis et al. previously reported that Syk is required for activation of PMN antimicrobial processes and killing of Gc (Sarantis et al., 2007). There are at least three reasons that could explain the discrepancy in these findings. First, our experimental system uses adherent, IL-8 primed primary human PMNs, which may bypass or negate the requirement for these signaling components. Second, Syk signaling was previously implicated in PMN granule exocytosis in response to Opa+ Gc, but was not examined for granule fusion with phagosomes. Third, the link between Syk signaling and PMN bactericidal activity was established using recombinant Escherichia coli expressing Opa proteins, and bacterial species may differ in their Syk-dependent effects on PMN activation and/or susceptibility to the PMN antimicrobial activities initiated by Syk signaling. Additional studies are required to fully dissect the signaling pathways required for PMN mediated killing of Opa+ Gc.
In order to make unambiguous observations about survival of Opa- and Opa+ Gc, we used an Opa- Gc isolate in which all 11 opa genes are deleted and Opa+ Gc that constitutively express OpaD. However, Gc has the ability to phase vary the expression of each Opa protein independently, such that during infection, the Gc population contains bacteria expressing anywhere from zero to multiple Opa proteins (Stern et al., 1986;Murphy et al., 1989). We observed that coinfection of PMNs with Opa- and Opa+ Gc resulted in increased primary granule fusion with Opa- Gc phagosomes and reduced survival of Opa- Gc in PMNs. However, Opa- Gc did not reduce the maturity of Opa+ Gc phagolysosomes. We speculate that the activating signals (e.g., Src family kinases) initiated by Opa-CEACAM ligation can affect the overall activation state of the PMN, leading to enhanced fusion between primary granules and Opa- Gc phagosomes. Although the mechanism by which Opa- Gc is internalized by adherent, IL-8 treated PMNs is currently undefined, these results would suggest that Opa- Gc does not send an inhibitory signal to interfere with CEACAM-mediated activation of Src family kinases. We predict that the ratio of Opa- and Opa+ Gc associated with each PMN thus influences the efficiency of the cells’ phagocytic killing machinery.
Selection for Opa+ Gc occurs in naturally and experimentally infected individuals and in the female mouse genital tract (James et al., 1978;Jerse, 1999;Cole et al., 2010;Hobbs et al., 2011). However, Opa+ Gc is more readily killed by PMNs (Rest et al., 1982;Rest et al., 1985;Virji et al., 1986;Fischer et al., 1988;Rest et al., 1989;Sarantis et al., 2007;Criss et al., 2009;Ball et al., 2013). Therefore, Opa protein expression would be both advantageous and disadvantageous during gonorrheal infection. Advantages of Opa protein expression include increased colonization and invasion of epithelial cells, and resistance to proteases found in cervical secretions (Makino et al., 1991;Kupsch et al., 1993;Wang et al., 1998;Jerse, 1999;Simms et al., 2006). As presented here, one major disadvantage is that Opa+ Gc stimulates PMN antimicrobial responses, leading to residence in a degradative phagolysosome and decreased bacterial survival. Therefore, in the immediate context of PMNs, Opa- Gc has a survival advantage. However, interactions between Opa+ Gc and PMNs may enhance Gc pathogenesis in two ways. First, the PMN oxidative burst and degranulation results in tissue damage, which may increase bacterial colonization of deeper layers of a stratified epithelium or even transmigration across epithelial cells to promote dissemination (Zen et al., 2003). Second, stimulation of the PMN oxidative burst may serve as a signal for Gc to upregulate genes to defend against PMN killing. In support of this hypothesis, Gc upregulates the expression of proteins such as RecN and Ngo1686/Mpg in response to oxidative stress, which provide resistance to PMN killing (Stohl et al., 2005;Criss et al., 2009;Stohl et al., 2013). Based on our current knowledge, we propose that a balance of Opa+ Gc and Opa- Gc, as well as the ability of Gc to vary expression of Opa proteins, play critical roles for the continual adaptation of Gc to its obligate human hosts, to enhance bacterial persistence and the progression of gonorrheal disease.
Experimental Procedures
Bacterial strains and growth conditions
The Gc used in this study are in the FA1090 background and constitutively express the pilin variant 1-81-S2 due to mutation of the guanine quartet sequence upstream of pilE (Cahoon et al., 2009). The majority of experiments were conducted using Opaless and OpaDnv (Ball et al., 2013), referred to here as “Opa-“ and “Opa+” Gc respectively. Experiments involving DPI used ΔopaBEGK Gc that were predominantly Opa- or phase ON for expression of OpaD (Ball et al., 2013), as confirmed by immunoblotting with the 4B12 pan-Opa antibody (data not shown). Piliated Gc was routinely grown on Gonococcal Medium Base (Difco) plus Kellogg’s supplements (Kellogg et al., 1963) for 20 hr at 37°C in 5% CO2. Gc was grown to exponential phase via successive rounds of bacterial growth in rich liquid medium (GCBL) and used to infect PMNs as previously described (Criss et al., 2008). Gc was labeled with 5 μg ml−1 carboxyfluorescein diacetate succinimidyl ester (CFSE) for 20 min at 37°C. In order to coat the Gc surface with the N-terminal Ig domain of CEACAM 1 (see below), 2×108 Gc were incubated with 6 μg ml−1 N-CEACAM1 at 37°C for 1 hr in 1 ml rich liquid medium.
N-domain CEACAM1 Purification
Escherichia coli strain MC1061 transformed with pGEX-2V plasmid containing the N-terminal 107 codons of human CEACAM1 (N-CEACAM1) was a kind gift of Alena Fedarovich (Medical University of South Carolina). A tobacco etch virus protease (TEV) cleavage domain was introduced between GST and N-CEACAM1. GST-TEV-N-CEACAM1 was purified from IPTG-induced E. coli by passage of the bacterial lysate over a glutathione column. The GST tag was then cleaved using TEV, and N-CEACAM1 was purified from GST and TEV using a Sephacryl S-200 gel-filtration column. Further details of N-CEACAM1 expression and purification are forthcoming (J. Martin, L. Ball, T. Solomon, A. Criss, and L. Columbus, manuscript in preparation).
PMN isolation
Venous blood was collected from healthy human subjects after receiving written informed consent. All human subject research was conducted in accordance with a protocol approved by the University of Virginia Institutional Review Board for Health Sciences Research. PMNs were isolated as previously described (Stohl et al., 2005) and resuspended at 1 × 107 cells per ml in Dulbecco’s PBS (without calcium and magnesium; Thermo Scientific) containing 0.1% dextrose. PMNs were kept on ice until use within 2 hr of preparation. PMN morphology was assessed by phase-contrast microscopy and indicated the PMN preparations contained approximately 95% PMNs.
Gc survival in adherent, IL-8 treated PMNs
CFU enumeration
PMNs were diluted into RPMI (Mediatech) containing 10% fetal bovine serum (Thermo Scientific) and 10 nM IL-8 (R&D Systems). 106 PMNs were added to tissue culture-treated plastic coverslips (Sarstedt) for 1 hr at 37°C, 5% CO2.to promote PMN attachment PMNs were synchronously infected with viable exponential-phase Gc at a multiplicity of infection of 1–5 bacterial colony forming units (CFU) per PMN, CFU were enumerated over time from saponin lysates of PMNs as described in (Criss et al., 2009).
Bacterial viability dyes
IL-8 treated PMNs in RPMI were allowed to attach to human serum-coated glass coverslips for 1 hr at 37°C, 5% CO2 as previously described (Johnson et al., 2013b). PMNs were infected with Gc as above, and detection of viable and nonviable Gc associated with PMNs as previously described, using Life Technologies Baclight Viability Dyes (Life Technologies) on saponin-permeabilized PMNs (Johnson et al., 2013a). Images were acquired within 30 min of exposure to the viability dyes. A minimum of 100 bacteria were analyzed for each experiment.
Immunofluorescence
PMNs infected for indicated times with Opa- or Opa+ Gc were fixed with 4% paraformaldehyde (PFA) in PBS for 15 min. External and internal Gc were distinguished using a polyclonal rabbit anti-Gc antibody with and without permeabilization, as previously described (Criss et al., 2009). For experiments evaluating recruitment of PMN secondary granules, Gc was labeled prior to infection with CFSE to stain the total Gc population as previously described (Johnson et al., 2013b) and external Gc were detected using the polyclonal rabbit anti-Gc antibody as described above. Cells were then permeabilized using a 1:1 ratio of acetone and methanol. Post permeabilization, PMN secondary granules were recognized using a polyclonal rabbit anti-lactoferrin antibody (MP Biomedicals) and PMN primary granules were recognized using monoclonal antibodies against neutrophil elastase (AHN-10) (Millipore).
For experiments evaluating recruitment of Hck, Gc was labeled prior to infection with CFSE to stain the total Gc population as previously described above. Intracellular Gc were identified by staining for PMN secondary granules using monoclonal antibodies against p22phox (44.1) and gp91phox (54.1) (both Santa Cruz Biotechnology) as previously described (Johnson et al., 2013b). Staining for Hck was conducted using a polyclonal rabbit anti-Hck antibody (Santa Cruz Biotechnology). Cells were incubated with Alexa Fluor-coupled goat anti-rabbit or goat anti-mouse antibodies (Life Technologies) following primary antibody incubation. Coverslips were mounted using Flouromount G (Southern Biotech) with 2.5 mg ml−1 propyl gallate (Acros Organics).
For coinfection experiments, PMNs were infected with unlabeled Opa- Gc and CFSE-labeled Opa+ Gc as described above. After 1 h, PMNs were fixed with 4% PFA in PBS for 15 min. External and internal Gc were distinguished using a polyclonal rabbit anti-Gc antibody with and without permeabilization, as previously described (Criss et al., 2009). PMN primary granules were recognized using monoclonal antibodies against neutrophil elastase as described above. Cells were incubated with Alexa Fluor-coupled goat anti-rabbit or goat anti-mouse antibodies (Life Technologies) following primary antibody incubation.
For each experiment, 50–200 intracellular bacteria were analyzed for enrichment of PMN granule proteins. Bacterial phagosomes were classified as positive for PMN granule proteins if antibody staining surrounded 50% of the bacterial circumference.
Image acquisition
Images were acquired on a Nikon Eclipse E800 with Hamamatsu Orca-ER digital camera using Openlab software and processed in Adobe Photoshop CS5. Images showing staining for Hck were acquired on an LSM-700 Confocal microscope using Zen software and processed in Adobe Photoshop CS5. For consistency, images depicting total bacteria, including bacteria labeled with CFSE, were false colored blue.
PMN oxidative burst
Generation of reactive oxygen species from PMNs exposed to Opa+ Gc was measured by luminol-dependent chemiluminescence as previously described (Criss et al., 2008;Ball et al., 2013) using a Victor 3 multilabel plate reader (Perkin-Elmer). Luminescence was measured in 1s intervals every 3 min for 60 min.
PMN Treatments
Cytochalasin D
PMNs were treated with 10 μg ml−1 cytochalasin D (CytoD; Sigma Aldrich) for 10 min prior to addition of Gc and for the duration of the infection.
DPI
PMNs were treated with 10 μM DPI (Sigma Aldrich) for 15 min prior to addition of Gc and for the duration of the infection.
Protease Inhibitors
PMNs were incubated with 1X Protease Inhibitor Cocktail Set V (Calbiochem) for 15 min prior to infection with Gc and for the duration of the infection, as previously described (Johnson et al., 2013b).
Signaling Inhibitors
PMNs were incubated at 37°C for 30 min prior to infection with 30 μM PP1 (Adipogen) or 10 μM LY294002. PMNs were incubated at 37°C for 20 min prior to infection with 50 μM piceatannol (Calbiochem) or 68.8 μM EHT1864 (Santa Cruz Biotechnology). Inhibitors were present for the duration of the infection.
Src family activator
PMNs were incubated at 37°C for 15 min prior to infection with 0.5 mg ml−1 Src family activator peptide (Santa Cruz Biotechnology) and was present for the duration of the infection. The Src family activator activates Src-family members by binding to the SH2 domain.
Antimicrobial Protein Susceptibility
Gc was incubated with the following proteins, and bacterial survival was calculated relative to the survival of untreated Gc (set to 100%) as previously described (Ball et al., 2013).
LL-37
Gc was incubated with the indicated concentrations of LL-37 (gift from William Shafer) in 0.5X GCBL for 45 min at 37 °C.
Cathepsin G
Cathepsin G (MP Biomedicals) was dialyzed for 12 hr at 4°C in distilled water using SpectraPor dialysis membrane MWCO 3,500 (Spectrum Labs) to remove acetate. Gc was incubated with the indicated concentrations of cathepsin G in 0.1X GCBL for 60 min at 37 °C.
BPI
Gc was incubated with the indicated concentrations of recombinant, full-length BPI (Novatein Biosciences; in 0.01% acetic acid) in 0.1x GCBL for 45 min at 37 °C.
Defensin
Gc was incubated with 0.2 mg/ml human β defensin-2 (Phoenix Pharmaceuticals; in 10 mM phosphate buffer, pH 7.4) in 0.2x GCBL for 45 min at 37 °C.
Statistics
Experimental values are expressed as a mean ± standard error of the mean for three replicate experiments (unless otherwise noted), performed with different donors’ PMNs for each assay. A Student’s two-tailed t-test was used to determine significance for each assay. A p value of less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Kodi Ravichandran for providing EHT1864, James Casanova for providing PP1, Ann Sutherland for providing LY294002, Jony Kipnis for providing the goat anti-rabbit Alexa-350 coupled antibody, William Shafer for providing LL-37, Alena Fedarovich for the N-CEACAM1 construct, and members of the Criss lab for consenting human subjects and for assistance with venipuncture. This work was supported by NIH R00 TW008042, R01 AI097312, and University of Virginia Research and Development Seed Award to A.K.C. and NIH R01 GM087828 and Cottrell Scholar Award (Research Corporation for the Advancement of Science) to L.C. M.B.J. was supported in part by NIH T32 AI007046, and J.N.M. was supported in part by NIH T32 GM008136.
Footnotes
The authors have no conflicts of interest to declare.
References
- Abdel-Latif D, Steward M, Lacy P. Neutrophil primary granule release and maximal superoxide generation depend on Rac2 in a common signalling pathway. Can J Physiol Pharmacol. 2005;83:69–75. doi: 10.1139/y04-123. [DOI] [PubMed] [Google Scholar]
- Astarie-Dequeker C, Nigou J, Puzo G, Maridonneau-Parini I. Lipoarabinomannans activate the protein tyrosine kinase Hck in human neutrophils. Infect Immun. 2000;68:4827–4830. doi: 10.1128/iai.68.8.4827-4830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LM, Criss AK. Constitutively Opa-Expressing and Opa-Deficient Neisseria gonorrhoeae Strains Differentially Stimulate and Survive Exposure to Human Neutrophils. J Bacteriol. 2013;195:2982–2990. doi: 10.1128/JB.00171-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KS, Gibbs CP, Barrera O, Morrison SG, Jahnig F, Stern A, et al. The opacity proteins of Neisseria gonorrhoeae strain MS11 are encoded by a family of 11 complete genes. Mol Microbiol. 1991;5:1889–1901. doi: 10.1111/j.1365-2958.1991.tb00813.x. [DOI] [PubMed] [Google Scholar]
- Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28:340–345. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Bos MP, Kuroki M, Krop-Watorek A, Hogan D, Belland RJ. CD66 receptor specificity exhibited by neisserial Opa variants is controlled by protein determinants in CD66 N-domains. Proc Natl Acad Sci U S A. 1998;95:9584–9589. doi: 10.1073/pnas.95.16.9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntru A, Roth A, Nyffenegger-Jann NJ, Hauck CR. HemITAM signaling by CEACAM3, a human granulocyte receptor recognizing bacterial pathogens. Arch Biochem Biophys. 2012;524:77–83. doi: 10.1016/j.abb.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Cahoon LA, Seifert HS. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SG, Shafer WM, Spitznagel JK. Anaerobiosis increases resistance of Neisseria gonorrhoeae to O2-independent antimicrobial proteins from human polymorphonuclear granulocytes. Infect Immun. 1985;47:401–407. doi: 10.1128/iai.47.2.401-407.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Bolland S, Chen I, Parker J, Pantelic M, Grunert F, Zimmermann W. The CGM1a (CEACAM3/CD66d)-mediated phagocytic pathway of Neisseria gonorrhoeae expressing opacity proteins is also the pathway to cell death. J Biol Chem. 2001;276:17413–17419. doi: 10.1074/jbc.M010609200. [DOI] [PubMed] [Google Scholar]
- Chen T, Gotschlich EC. CGM1a antigen of neutrophils, a receptor of gonococcal opacity proteins. Proc Natl Acad Sci U S A. 1996;93:14851–14856. doi: 10.1073/pnas.93.25.14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JG, Fulcher NB, Jerse AE. Opacity proteins increase Neisseria gonorrhoeae fitness in the female genital tract due to a factor under ovarian control. Infect Immun. 2010;78:1629–1641. doi: 10.1128/IAI.00996-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell TD, Shaffer D, Cannon JG. Characterization of the repertoire of hypervariable regions in the Protein II (opa) gene family of Neisseria gonorrhoeae. Mol Microbiol. 1990;4:439–449. doi: 10.1111/j.1365-2958.1990.tb00610.x. [DOI] [PubMed] [Google Scholar]
- Cougoule C, Constant P, Etienne G, Daffe M, Maridonneau-Parini I. Lack of fusion of azurophil granules with phagosomes during phagocytosis of Mycobacterium smegmatis by human neutrophils is not actively controlled by the bacterium. Infect Immun. 2002;70:1591–1598. doi: 10.1128/IAI.70.3.1591-1598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Katz BZ, Seifert HS. Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell Microbiol. 2009;11:1074–1087. doi: 10.1111/j.1462-5822.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Seifert HS. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol. 2008;10:2257–2270. doi: 10.1111/j.1462-5822.2008.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge MI, Hamstra HJ, van Alphen L, Dankert J, van der Ley P. Mapping the binding domains on meningococcal Opa proteins for CEACAM1 and CEA receptors. Mol Microbiol. 2003;50:1005–1015. doi: 10.1046/j.1365-2958.2003.03749.x. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Litaker W, Madhure A, Snodgrass TL, Cannon JG. Physical map of the chromosome of Neisseria gonorrhoeae FA1090 with locations of genetic markers, including opa and pil genes. J Bacteriol. 1991;173:5476–5486. doi: 10.1128/jb.173.17.5476-5486.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SW. Biochemistry and physiology of the neutrophil. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- Fensome A, Cunningham E, Prosser S, Tan SK, Swigart P, Thomas G, et al. ARF and PITP restore GTP gamma S-stimulated protein secretion from cytosol-depleted HL60 cells by promoting PIP2 synthesis. Curr Biol. 1996;6:730–738. doi: 10.1016/s0960-9822(09)00454-0. [DOI] [PubMed] [Google Scholar]
- Fischer SH, Rest RF. Gonococci possessing only certain P.II outer membrane proteins interact with human neutrophils. Infect Immun. 1988;56:1574–1579. doi: 10.1128/iai.56.6.1574-1579.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher NB. PhD thesis. Department of Microbiology and Immunology; Chapel Hill, NC: University of North Carolina; 2004. The role of Neisseria gonorrhoeae opacity proteins in host cell interactions and pathogenesis. [Google Scholar]
- Fumagalli L, Zhang H, Baruzzi A, Lowell CA, Berton G. The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol. 2007;178:3874–3885. doi: 10.4049/jimmunol.178.6.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- Gray-Owen SD, Dehio C, Haude A, Grunert F, Meyer TF. CD66 carcinoembryonic antigens mediate interactions between Opa-expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. EMBO J. 1997a;16:3435–3445. doi: 10.1093/emboj/16.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Owen SD, Lorenzen DR, Haude A, Meyer TF, Dehio C. Differential Opa specificities for CD66 receptors influence tissue interactions and cellular response to Neisseria gonorrhoeae. Mol Microbiol. 1997b;26:971–980. doi: 10.1046/j.1365-2958.1997.6342006.x. [DOI] [PubMed] [Google Scholar]
- Hauck CR, Gulbins E, Lang F, Meyer TF. Tyrosine phosphatase SHP-1 is involved in CD66-mediated phagocytosis of Opa52-expressing Neisseria gonorrhoeae. Infect Immun. 1999;67:5490–5494. doi: 10.1128/iai.67.10.5490-5494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck CR, Meyer TF, Lang F, Gulbins E. CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO J. 1998;17:443–454. doi: 10.1093/emboj/17.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs MM, Sparling PF, Cohen MS, Shafer WM, Deal CD, Jerse AE. Experimental Gonococcal Infection in Male Volunteers: Cumulative Experience with Neisseria gonorrhoeae Strains FA1090 and MS11mkC. Front Microbiol. 2011;2:123. doi: 10.3389/fmicb.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JF, Swanson J. Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect Immun. 1978;19:332–340. doi: 10.1128/iai.19.1.332-340.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerse AE. Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect Immun. 1999;67:5699–5708. doi: 10.1128/iai.67.11.5699-5708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Criss AK. Fluorescence microscopy methods for determining the viability of bacteria in association with mammalian cells. J Vis Exp. 2013a;79 doi: 10.3791/50729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Criss AK. Neisseria gonorrhoeae phagosomes delay fusion with primary granules to enhance bacterial survival inside human neutrophils. Cell Microbiol. 2013b;15:1323–1340. doi: 10.1111/cmi.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DS, Jr, Peacock WL, Jr, Deacon WE, Brown L, Pirkle DI. Neisseria gonorrhoeae. I. Virulence Genetically Linked to Clonal Variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupsch EM, Knepper B, Kuroki T, Heuer I, Meyer TF. Variable opacity (Opa) outer membrane proteins account for the cell tropisms displayed by Neisseria gonorrhoeae for human leukocytes and epithelial cells. EMBO J. 1993;12:641–650. doi: 10.1002/j.1460-2075.1993.tb05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol. 2006;2:98–108. doi: 10.1186/1710-1492-2-3-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S, van Putten JP, Meyer TF. Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J. 1991;10:1307–1315. doi: 10.1002/j.1460-2075.1991.tb07649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malorny B, Morelli G, Kusecek B, Kolberg J, Achtman M. Sequence diversity, predicted two-dimensional protein structure, and epitope mapping of neisserial Opa proteins. J Bacteriol. 1998;180:1323–1330. doi: 10.1128/jb.180.5.1323-1330.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaw SE, Liao EH, Gray-Owen SD. Engulfment of Neisseria gonorrhoeae: revealing distinct processes of bacterial entry by individual carcinoembryonic antigen-related cellular adhesion molecule family receptors. Infect Immun. 2004;72:2742–2752. doi: 10.1128/IAI.72.5.2742-2752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaw SE, Schneider J, Liao EH, Zimmermann W, Gray-Owen SD. Immunoreceptor tyrosine-based activation motif phosphorylation during engulfment of Neisseria gonorrhoeae by the neutrophil-restricted CEACAM3 (CD66d) receptor. Mol Microbiol. 2003;49:623–637. doi: 10.1046/j.1365-2958.2003.03591.x. [DOI] [PubMed] [Google Scholar]
- Mocsai A, Jakus Z, Vantus T, Berton G, Lowell CA, Ligeti E. Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol. 2000;164:4321–4331. doi: 10.4049/jimmunol.164.8.4321. [DOI] [PubMed] [Google Scholar]
- Mohn H, Le Cabec V, Fischer S, Maridonneau-Parini I. The src-family protein-tyrosine kinase p59hck is located on the secretory granules in human neutrophils and translocates towards the phagosome during cell activation. Biochem J. 1995;309 (Pt 2):657–665. doi: 10.1042/bj3090657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GL, Connell TD, Barritt DS, Koomey M, Cannon JG. Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell. 1989;56:539–547. doi: 10.1016/0092-8674(89)90577-1. [DOI] [PubMed] [Google Scholar]
- N’Diaye EN, Darzacq X, Astarie-Dequeker C, Daffe M, Calafat J, Maridonneau-Parini I. Fusion of azurophil granules with phagosomes and activation of the tyrosine kinase Hck are specifically inhibited during phagocytosis of mycobacteria by human neutrophils. J Immunol. 1998;161:4983–4991. [PubMed] [Google Scholar]
- Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Rest RF, Fischer SH, Ingham ZZ, Jones JF. Interactions of Neisseria gonorrhoeae with human neutrophils: effects of serum and gonococcal opacity on phagocyte killing and chemiluminescence. Infect Immun. 1982;36:737–744. doi: 10.1128/iai.36.2.737-744.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Lee N, Bowden C. Stimulation of human leukocytes by protein II+ gonococci is mediated by lectin-like gonococcal components. Infect Immun. 1985;50:116–122. doi: 10.1128/iai.50.1.116-122.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Pretzer E. Degradation of gonococcal outer membrane proteins by human neutrophil lysosomal proteases. Infect Immun. 1981;34:62–68. doi: 10.1128/iai.34.1.62-68.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Shafer WM. Interactions of Neisseria gonorrhoeae with human neutrophils. Clin Microbiol Rev. 1989;2(Suppl):S83–91. doi: 10.1128/cmr.2.suppl.s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadarangani M, Pollard AJ, Gray-Owen SD. Opa proteins & CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS Microbiol Rev. 2011 doi: 10.1111/j.1574-6976.2010.00260.x. [DOI] [PubMed] [Google Scholar]
- Sarantis H, Gray-Owen SD. The specific innate immune receptor CEACAM3 triggers neutrophil bactericidal activities via a Syk kinase-dependent pathway. Cell Microbiol. 2007;9:2167–2180. doi: 10.1111/j.1462-5822.2007.00947.x. [DOI] [PubMed] [Google Scholar]
- Sarantis H, Gray-Owen SD. Defining the roles of human carcinoembryonic antigen-related cellular adhesion molecules during neutrophil responses to Neisseria gonorrhoeae. Infect Immun. 2012;80:345–358. doi: 10.1128/IAI.05702-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Simons MP, Wu HJ, McEwan AG, Nauseef WM, Apicella MA, Jennings MP. Investigation of oxidative stress defenses of Neisseria gonorrhoeae by using a human polymorphonuclear leukocyte survival assay. Infect Immun. 2005;73:5269–5272. doi: 10.1128/IAI.73.8.5269-5272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengelov H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J Immunol. 1993;150:1535–1543. [PubMed] [Google Scholar]
- Shafer WM, Morse SA. Cleavage of the protein III and major iron-regulated protein of Neisseria gonorrhoeae by lysosomal cathepsin G. J Gen Microbiol. 1987;133:155–162. doi: 10.1099/00221287-133-1-155. [DOI] [PubMed] [Google Scholar]
- Shafer WM, Onunka VC, Jannoun M, Huthwaite LW. Molecular mechanism for the antigonococcal action of lysosomal cathepsin G. Mol Microbiol. 1990;4:1269–1277. doi: 10.1111/j.1365-2958.1990.tb00706.x. [DOI] [PubMed] [Google Scholar]
- Shafer WM, Onunka VC, Martin LE. Antigonococcal activity of human neutrophil cathepsin G. Infect Immun. 1986;54:184–188. doi: 10.1128/iai.54.1.184-188.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer WM, Qu X, Waring AJ, Lehrer RI. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci U S A. 1998;95:1829–1833. doi: 10.1073/pnas.95.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms AN, Jerse AE. In vivo selection for Neisseria gonorrhoeae opacity protein expression in the absence of human carcinoembryonic antigen cell adhesion molecules. Infect Immun. 2006;74:2965–2974. doi: 10.1128/IAI.74.5.2965-2974.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons MP, Nauseef WM, Apicella MA. Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect Immun. 2005;73:1971–1977. doi: 10.1128/IAI.73.4.1971-1977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintsova A, Sarantis H, Islam EA, Sun CX, Amin M, Chan CH, et al. Global Analysis of Neutrophil Responses to Neisseria gonorrhoeae Reveals a Self-Propagating Inflammatory Program. PLoS pathogens. 2014;10:e1004341. doi: 10.1371/journal.ppat.1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Daily KP, Criss AK. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression state of Neisseria gonorrhoeae. Infection and Immunity. 2014;82(3):1036–44. doi: 10.1128/IAI.00881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- Stern A, Brown M, Nickel P, Meyer TF. Opacity genes in Neisseria gonorrhoeae: control of phase and antigenic variation. Cell. 1986;47:61–71. doi: 10.1016/0092-8674(86)90366-1. [DOI] [PubMed] [Google Scholar]
- Stohl EA, Criss AK, Seifert HS. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol. 2005;58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Dale EM, Criss AK, Seifert HS. Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. MBio. 2013:4. doi: 10.1128/mBio.00399-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J. Studies on gonococcus infection. XIV. Cell wall protein differences among color/opacity colony variants of Neisseria gonorrhoeae. Infect Immun. 1978;21:292–302. doi: 10.1128/iai.21.1.292-302.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114:4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte-Rutten L, Bos MP, Tommassen J, Gros P. Crystal structure of Neisserial surface protein A (NspA), a conserved outer membrane protein with vaccine potential. J Biol Chem. 2003;278:24825–24830. doi: 10.1074/jbc.M302803200. [DOI] [PubMed] [Google Scholar]
- Virji M, Heckels JE. The effect of protein II and pili on the interaction of Neisseria gonorrhoeae with human polymorphonuclear leucocytes. J Gen Microbiol. 1986;132:503–512. doi: 10.1099/00221287-132-2-503. [DOI] [PubMed] [Google Scholar]
- Virji M, Makepeace K, Ferguson DJ, Watt SM. Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic neisseriae. Mol Microbiol. 1996;22:941–950. doi: 10.1046/j.1365-2958.1996.01551.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Gray-Owen SD, Knorre A, Meyer TF, Dehio C. Opa binding to cellular CD66 receptors mediates the transcellular traversal of Neisseria gonorrhoeae across polarized T84 epithelial cell monolayers. Mol Microbiol. 1998;30:657–671. doi: 10.1046/j.1365-2958.1998.01102.x. [DOI] [PubMed] [Google Scholar]
- Werner E. GTPases and reactive oxygen species: switches for killing and signaling. J Cell Sci. 2004;117:143–153. doi: 10.1242/jcs.00937. [DOI] [PubMed] [Google Scholar]
- Wiesner PJ, Thompson SE., 3rd Gonococcal diseases. Dis Mon. 1980;26:1–44. doi: 10.1016/s0011-5029(80)80002-2. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Global incidence and prevalence of selected curable sexually transmitted infections – 2008. Geneva, Switzerland: WHO, Department of Reproductive Health and Research; 2012. [Google Scholar]
- Wu H, Soler-Garcia AA, Jerse AE. A strain-specific catalase mutation and mutation of the metal-binding transporter gene mntC attenuate Neisseria gonorrhoeae in vivo but not by increasing susceptibility to oxidative killing by phagocytes. Infect Immun. 2009;77:1091–1102. doi: 10.1128/IAI.00825-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K, Parkos CA. Leukocyte-epithelial interactions. Curr Opin Cell Biol. 2003;15:557–564. doi: 10.1016/s0955-0674(03)00103-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.