

ABSTRACT
Many pathogens use homologous recombination to vary surface antigens to avoid immune surveillance. Neisseria gonorrhoeae achieves this in part by changing the properties of its surface pili in a process called pilin antigenic variation (AV). Pilin AV occurs by high-frequency gene conversion reactions that transfer silent pilS sequences into the expressed pilE locus and requires the formation of an upstream guanine quartet (G4) DNA structure to initiate this process. The MutS and MutL proteins of the mismatch correction (MMC) system act to correct mismatches after replication and prevent homeologous (i.e., partially homologous) recombination, but MutS orthologs can also bind to G4 structures. A previous study showed that mutation of MutS resulted in a 3-fold increase in pilin AV, which could be due to the loss of MutS antirecombination properties or loss of G4 binding. We tested two site-directed separation-of-function MutS mutants that are both predicted to bind to G4s but are not able to perform MMC. Pilus phase variation assays and DNA sequence analysis of pilE variants produced in these mutants showed that all three mutS mutants and a mutL mutant had similar increased frequencies of pilin AV. Moreover, the mutS mutants all showed similar increased levels of pilin AV-dependent synthetic lethality. These results show that antirecombination by MMC is the reason for the effect that MutS has on pilin AV and is not due to pilE G4 binding by MutS.
IMPORTANCE Neisseria gonorrhoeae continually changes its outer surface proteins to avoid recognition by the immune system. N. gonorrhoeae alters the antigenicity of the pilus by directed recombination between partially homologous pilin copies in a process that requires a guanine quartet (G4) structure. The MutS protein of the mismatch correction (MMC) system prevents recombination between partially homologous sequences and can also bind to G4s. We confirmed that loss of MMC increases the frequency of pilin antigenic variation and that two MutS mutants that are predicted to separate the two different functions of MutS inhibit pilin variation similarly to a complete-loss-of-function mutant, suggesting that interaction of MutS with the G4 structure is not a major factor in this process.
INTRODUCTION
Neisseria gonorrhoeae is the sole causative agent of gonorrhea, the second most commonly reported sexually transmitted infection in the United States, with an estimated 800,000 new cases per year (1). N. gonorrhoeae extensively uses phase and antigenic variation to provide a reversible subpopulation of genetic variants that can be selected for during infection (2). The use of various surface antigens is one of the most effective strategies used by pathogens to evade immune surveillance. By changing outer surface components, N. gonorrhoeae can avoid recognition by the adaptive immune system, which can prolong a current infection and enable reinfection. These diversity generation mechanisms can also provide functional changes for N. gonorrhoeae.
It is estimated that there are over 100 phase-variable genes in the pathogenic species of Neisseria, with an average of 80 phase-variable genes per strain (3–5). Phase variation is the reversible change between different expression states of a gene (for example, “on” or “off”) and in Neisseria is mediated by the mispairing of polynucleotide repeats during replication (6). Phase variation in Neisseria alters the expression of many genes involved in virulence, including the pilus assembly factor PilC and genes encoding lipooligosaccharide biosynthetic enzymes and Opa attachment proteins, which use phase variation to achieve antigenic variation (reviewed in reference 7).
Pilin antigenic variation (AV) is a main system of genetic diversification used by Neisseria to change their type IV pili, long surface-exposed fibers involved in attachment, aggregation, DNA transformation, twitching motility, and protection from polymorphonuclear leukocyte (PMN) killing (8–11). Pilin AV differs from the major forms of phase variation in that the amino acid sequence of the protein is altered, rather than just its expression level, allowing for the production of multiple forms of the antigen. However, due to the many different pilin products produced, both pilus antigenic and phase variants can be produced by pilin AV (12), and pilus phase variation has been a major assay used as a surrogate measure of pilin AV (e.g., see references 13, 14, and 15).
Pilin AV is mediated by a gene conversion process that involves the nonreciprocal transfer of DNA from one of many silent donor pilin gene copies (pilS) to the recipient pilin expression (pilE) locus without the donor locus being changed (Fig. 1A and B). There are four to six pilS loci in each gonococcal isolate, with 19 silent copies located in six separate loci in strain FA1090 (16). These silent pilS copies share significant sequence similarity to the expressed pilE gene and act as reservoirs of variant genetic information. Silent copies lack a promoter and approximately 150 bp of the 5′ end of the 500-bp gene but have homology to conserved parts of the pilE gene (Fig. 1A).
FIG 1.
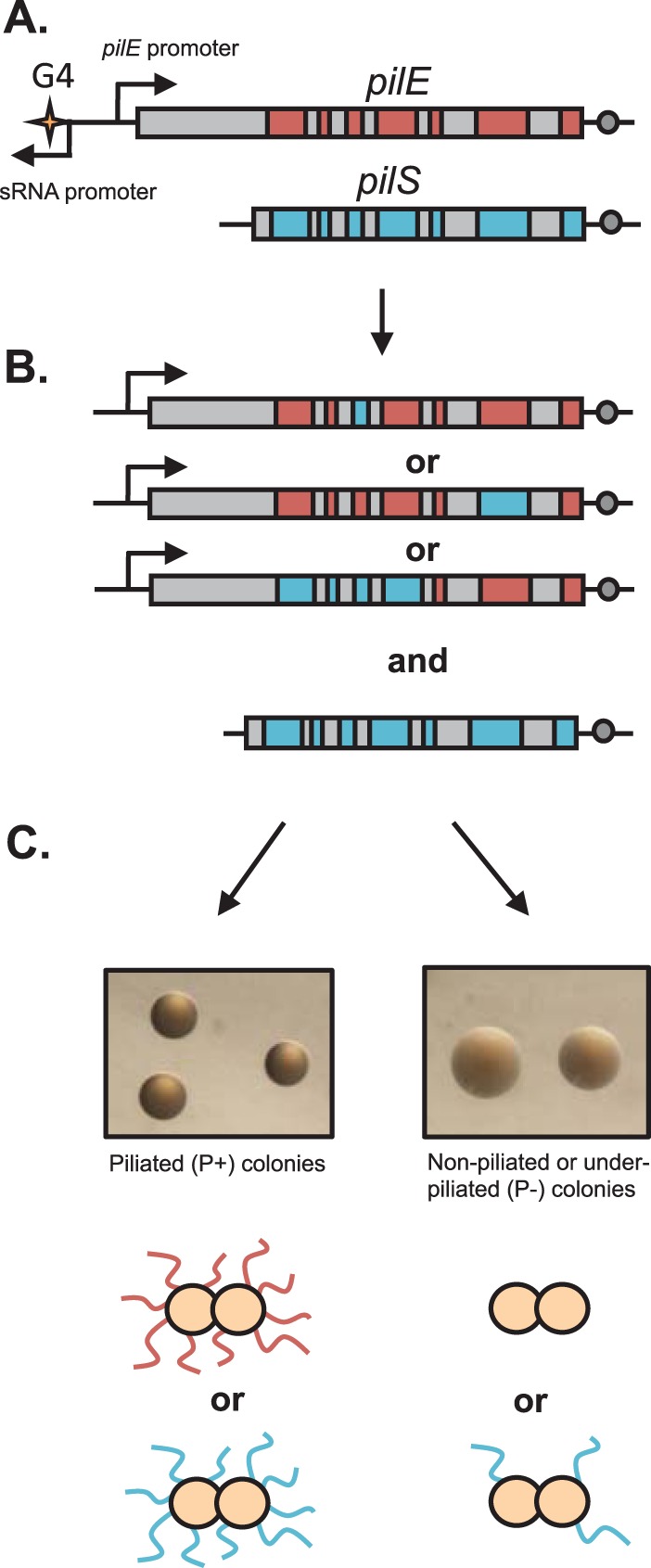
Pilin antigenic variation in N. gonorrhoeae. (A) Graphic representation of the pilE gene (red) and a pilS copy (blue) with homologous regions represented in gray. Bent arrows represent promoters, the G4 sequence is represented by a star, and the Sma/Cla region is represented by a circle. (B) Some of the many possible homeologous recombination products of pilin AV, demonstrating its segmental characteristics and the stability of the pilS locus. (C) Pilin AV can result in piliated gonococcal cells with a changed PilE amino acid sequence (P+) or nonpiliated or underpiliated (P−) gonococcal cells. The FA1090 recA6 Avd-1 and FA1090 recA6 ΔpilE strains, both grown in the presence of IPTG for 22 h, are shown as examples of P+ and P− colonies, respectively.
Pilin AV is dependent on a 16-nucleotide (nt) guanine-rich sequence located ∼350 bp upstream of the pilE promoter (Fig. 1A) that has been shown to form a G4 (guanine quartet) structure in vitro (13). G4 structures are composed of G-rich repeats that form quartets with a monovalent ion using Hoogstein bonds, and the quartets stack to form stable alternative DNA structures (17). G4 structures have key roles in diverse processes in eukaryotic cells, such as telomere maintenance, gene regulation, immunoglobulin class switching, DNA replication, translational control, and packaging of retroviral DNA (reviewed in references 17 and 18). A significant percentage of putative G4 sequences are predicted to form G4 structures in bacterial and eukaryotic promoters (18, 19), indicating a regulatory function. It is likely that G4 structures also have pleotropic roles in bacterial cells, but determination of the role of these G4 structures in prokaryotic biology has lagged behind studies with eukaryotic cells. Other than N. gonorrhoeae, functional roles for G4s have only been suggested for Escherichia coli, Deinococcus radiodurans, and Clostridium difficile (20–22).
Pilin AV in N. gonorrhoeae also requires the transcription of a small noncoding RNA (sRNA) that initiates within the pilE G4 motif (23) (Fig. 1A). The current model for pilin AV proposes that transcription of the pilE G4 sRNA melts the duplex and that the RNA-DNA hybrid formed between the pilE G4 RNA and the C-rich complement allows the formation of the G4 structure on the opposite strand. Since there are a few G4 structures that rely on a protein chaperone to facilitate structure formation (e.g., nucleolin at the G4 of the c-myc oncogene [24]), it is possible that there are one or more proteins that are involved in initiation of pilin AV through interactions with the G4.
One candidate protein for pilE G4 binding is the MutS protein of the DNA mismatch correction (MMC) system, which uses the MutS and MutL proteins to avoid mutations and to preserve replication fidelity in many bacteria and eukaryotes (reviewed in references 25 and 26). During MMC, MutS recognizes mismatches, particularly the pairing between G and T, and alternative perturbations of the DNA duplex, such as a G4 structure or mismatched heteroduplex DNA (27). MutL then binds to mismatch-bound MutS and other downstream MMC proteins (28). In E. coli, the parental strand is distinguished from the newly synthesized strand via differential methylation, and the process is referred to as methyl-directed mismatch repair (26). MutH cuts the DNA at unmethylated GATC sites during the window of time after replication but before Dam methylase has yet to act on the newly synthesized strand. Like many bacterial species, N. gonorrhoeae lacks a MutH homologue, and it is proposed that the weak endonuclease activity of MutL is sufficient to cleave the DNA (29). The nature of strand differentiation is unknown but is likely based on nicks in the newly synthesized strand, such as at the ends of Okazaki fragments, as proposed for eukaryotes (30). The UvrD helicase unwinds the processed DNA, while various exonucleases digest the single-stranded tails of the incorrect strand. Disruption to MMC in E. coli results in a strong mutator phenotype, with primarily single-nucleotide changes in the progeny (31). MMC mutations in N. gonorrhoeae result in a modest increase in spontaneous mutations and an increase in pilus phase variation, mediated mainly by increases in the phase variation of pilC expression (32).
In addition to its role in MMC during replication, MutS also acts to limit recombination between homeologous (partially homologous) DNA molecules. In vitro, the E. coli MutS (EcMutS) protein will prevent RecA from performing branch migration on mismatched homologues during strand exchange (33, 34) and addition of MutS from thermostable bacteria to PCRs can inhibit the formation of nonspecific banding by blocking the polymerase action on primer mispairings (35). The neisserial pilS copies can be considered homeologous to pilE, and it is likely that the increased frequencies of pilin AV in MMC-deficient gonococci result from loss of disruption of mismatched heteroduplex DNA (32).
Recently, the E. coli MutS protein has been shown to bind to G4 structures more strongly than to its canonical G-T mismatch in vitro (36), strengthening MutS as a reasonable candidate protein for binding to the pilE G4. In this study, we tested the idea that the role of MutS in pilin AV is dependent primarily on MMC by constructing two different mutS separation-of-function mutants (Fig. 2) that disrupt MMC but should retain G4 binding. Both separation-of-function mutants showed increased levels of spontaneous mutation but no decrease in pilin AV relative to a mutS null mutation. These results demonstrate that MutS interaction at the G4 structure is not important for pilin AV.
FIG 2.

The mutS locus of N. gonorrhoeae. (A) Schematic representation of the mutS locus in N. gonorrhoeae strain FA1090 with the Kanr marker used to introduce the site-directed mutations. Open reading frames are indicated by arrows drawn in the direction of transcription. Gene or locus names are written either inside the arrow or directly underneath. The lines between genes represent intergenic regions (Igr). Stars represent the locations of the F32A and S661A amino acid substitutions. Drawings are to scale. (B) Alignment of N. gonorrhoeae (Neigo) MutS and E. coli (Ecoli) MutS amino acid sequences. Identical and related residues are shown in the center line, and the conserved phenylalanine and serine residues (in red) are indicated with an asterisk.
MATERIALS AND METHODS
Bacteria and growth conditions.
The strains used in this study were derivatives of N. gonorrhoeae FA1090 isolates, with all strains containing an IPTG (isopropyl-β-d-thiogalactopyranoside)-regulatable recA6 allele (37) to prevent antigenic variation of the pilE gene in the absence of inducer. The detailed description of all strains is outlined in Table 1. Strains were incubated for 22 h on GCB medium with Kellogg's supplements (38) at 37°C with 5% CO2. When required, IPTG (Diagnostic Chemicals, Ltd.) was added to 1 mM, and the final concentrations of antibiotics were 50 μg/ml kanamycin, 0.5 to 5 μg/ml chloramphenicol, 2.5 μg/ml erythromycin, 0.2 μg/ml tetracycline, and 70 ng/ml rifampin.
TABLE 1.
List of N. gonorrhoeae strains used
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| FA1090 | Background strain | J. Cannon |
| Phase-variable parent | FA1090 recA6::Tetr | 37 |
| Phase-variable ΔmutS mutant | FA1090 recA6 mutS::Ermr | 32 |
| Phase-variable Kanr parent | FA1090 recA6 igr1584::Kanr | This study |
| Phase-variable mutS-F32A mutant | FA1090 recA6 mutS-F32A igr1584::Kanr | This study |
| Phase-variable ΔmutL mutant | FA1090 recA6 ΔmutL | 32 |
| Tn9/G4mtAvd-1 | FA1090 recA6 Avd-1 | 13 |
| Phase-variable ΔpilE mutant | FA1090 recA6 ΔpilE | A. Criss |
| FA7458-1A | FA1090 A23a ΔpilC2 pilC1PL | 47 |
| Parent | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat | This study |
| Kanr parent | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat igr1584::Kanr | This study |
| ΔmutS mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS::Ermr | This study |
| Kanr ΔmutS mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS::Ermr igr1584::Kanr | This study |
| mutS-F32A mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS-F32A igr1584::Kanr | This study |
| mutS-S661A mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS-S661A igr1584::Kanr | This study |
| Complemented parent | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS-NICS | This study |
| ΔmutL mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat ΔmutL | This study |
| ΔmutS ΔmutL mutant | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS::Ermr ΔmutL | This study |
| Complemented ΔmutS strain | FA1090 A23a ΔpilC2 pilC1PL recA6 RS1::cat mutS::Ermr mutS-NICS | This study |
| Phase-variable ruvB recG mutant | FA1090 ruvB::Ermr recG::Kanr recA6 | 49 |
| Phase-variable ruvB mutant | FA1090 ruvB::Ermr recA6 | 49 |
| ruvB mutant | FA1090 ruvB::Ermr recA6 pilC1PL | This study |
| ruvB recG mutant | FA1090 ruvB::Ermr recG::Kanr recA6 pilC1PL | This study |
| ruvB recG ΔmutS mutant | FA1090 ruvB::Ermr recG::Kanr recA6 pilC1PL mutS::Ermr | This study |
| ruvB recG mutS-F32A mutant | FA1090 ruvB::Ermr recG::Kanr recA6 pilC1PL mutS-F32A igr1584::Kanr | This study |
Bacterial transformations.
Gonococcal genomic DNA was isolated from the donor strain by swabbing a half plate of confluent lawn growth into 1 ml GCBL (1.5% peptone protease no. 3 [Difco], 0.4% K2HPO4 [Fisher], 0.1% KH2PO4 [Fisher], 0.1% NaCl [Fisher]) and washed once in 1× phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4). The pellet was resuspended in 180 μl buffer ATL from the QIAmp DNA minikit (Qiagen), and total DNA was extracted following the manufacturer's instructions. The recipient strain was transformed by plating a small patch of cells on an IPTG plate using 3 to 5 starting colonies. Ten microliters of supplemented GCBL with 10 mM MgSO4 was combined with 10 μl chromosomal or plasmid DNA, spotted on the lawn, and grown for 22 h. The growth containing the spot was swabbed into GCBL and spread onto a GCB plate containing the antibiotic whose resistance marker was linked to the donor DNA. Colonies were streaked twice on antibiotic plates, and 5 to 6 candidates were saved for confirmation of the mutation (by the size of locus-specific PCR products) and for the identity of pilE (direct sequencing). All mutS alleles were sequenced (using the first 14 primers listed in Table 2) at the Northwestern University Genomics Core Facility. For the ΔmutL and pilC1PL (i.e., phase-locked pilC1) mutations without antibiotic resistance markers, 960 colonies were collected in 96 pools to check the transformants by PCR. Pools with correctly sized bands were streaked out, with 10 colonies subsequently rechecked.
TABLE 2.
List of primers used
| Primer | Sequence |
|---|---|
| 1585_mutS_for | ATGCAAGCTTCAGTACCTTGGAAACGGC |
| 1584_mutS_rev | ATGCCGATCGATTCCGGAACCCTGTTGG |
| MutS_for_int_EcoRI | AACCAACCGCATCGTTGC |
| MutS_rev_int_EcoRI | ATGCCGTCTGAAGTTGCG |
| MutS_intfor | TTGGACAGCAAAGAACACGC |
| MutSfor_N_His | GCATACATATGTCCAAATCCGCCGTTTCC |
| MutS_intrev | AAGATTTGATCGACAGGCCC |
| MutSrev_N_His | CGTCCGAAGCTTAAGATACGGATTTGCACAAAT |
| Irg4-7 | AGCACGTACTCGGATGGAAG |
| Irg4-7RC | CTTCCATCCGAGTACGTGCT |
| mutS_int_endF | CCCAAAAGCATTTGAACGG |
| MutS_int-f13 | AAACAACTCTTTGACGGCG |
| MutS_int-r14 | CGTATTGGCTTTCCAGCG |
| PILRBS | GGCTTTCCCCTTTCAATTAGGAG |
| SP3A | CCGGAACGGACGACCCCG |
| SmaClaI | TTGCAAACCCTTAAAAGACAAGC |
| CONSTF2 | TACCAAGACTACACCGCCCG |
| Cat_intF | AATACCACGACGATTTCCG |
| Cat_intR | GGTATTCACTCCAGAGCG |
| pilCupstream | TAGGCGGTTAAGTTGTTGGGAAAG |
| pilCdownstream | CCATCTTTGGCGGTACCCTCGCTG |
| Ngo055up | TATGTTCCAACACGCAGGG |
| pilC_int | CCCAACCAAGGATAATCCG |
| pilCPLfor | GGCGGAGGTGGCGGGGCC |
| SDM_MutSfor | GTTTTACCGTATGGGTGATGCCTACGAGCTGTTTTTGGATG |
| SDM_MutSrev | CATCCAAAAACAGCTCGTAGGCATCACCCATACGGTAAAAC |
| MutS-T1981G-for | CGCCTCCAACCGCGCCACCTTCATGGT |
| MutS-T1981G-rev | ACCATGAAGGTGGCGCGGTTGGAGGCG |
| ermCout1 | CAATTCTTATCTCTTTTCAATAGC |
| ermCout2 | GAAGTAATAAAGTTTTGACTGTG |
| RuvBfor2 | TGCCGTCTGAAACGCGCCG |
| RuvBrev2 | CAAACGTCTGATAACAATGCCG |
| recGfor2 | CCAACAACAGCAGGAAGCCG |
| RECGREV | GTCCTGATTTTTGTTAATCCACT |
| KAN FP-1 | ACCTACAACAAAGCTCTCATCAACC |
| LACPFOR | GAGCGGATAACAATTTCACA |
| GCRecA-Rev2 | CAAAGCCGAAGAAACCGC |
F7458-1A was transformed to pilE variant 1-81-S2 (2) by selecting for a chloramphenicol resistance cassette in the upstream RS1 region (pUSS2 5-13 with Tn9 excised by NruI and SalI and replaced by cat amplified with primers containing the same restriction sites).
Site-directed mutagenesis.
The mutS gene is flanked in N. gonorrhoeae FA1090 by intergenic region 1584 (igr1584) and igr1585 (Fig. 2A). Primers 1585_mutS_for (with a HindIII site) and MutS_rev_int_EcoRI were designed to amplify the region between igr1585 and the 5′ end of the mutS gene just beyond the naturally occurring EcoRI site at 448 nt (1.15 kb). Primers 1584_mutS_rev (with the PvuI site) and MutS_for_int_EcoRI were designed to amplify the mutS gene downstream of the EcoRI site and igr1584 (2.8 kb). Both PCR fragments were ligated separately into pCR-Blunt-II (Invitrogen). The pBlunt-1584 construct, containing the majority of the mutS gene, had difficulty growing in E. coli cells. pBlunt-1585 was changed to mutS-F32A (TTT-GCC) with primers SDM_MutSfor and SDM_MutSrev using the QuikChange II XL site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. Amino acid substitutions were chosen using commonly used codons in N. gonorrhoeae FA1090 from http://www.kazusa.or.jp. The 1585-mutS fragment was excised using EcoRI and HindIII and ligated into similarly double-digested pBR322 (ATCC 37017). pBlunt-1584 was digested with NdeI and blunted with T4 polymerase. The 1.25-kb Kanr cassette was excised from pBSL86 (ATCC 87129) with SmaI and ligated codirectionally with the mutS gene in pBlunt-1584. The 4-kb PvuI-EcoRI fragment containing Kanr in igr1584 along with the 3′ end of mutS was excised and cloned into both pBR322 and pBR322 with the mutS-F32A. The mutS-S661A (T1981G) mutant was created using the QuikChange kit on pET15b (Novagen) containing the mutS gene. Both mutS-F32A and mutS-S661A constructs were transformed by selecting for kanamycin resistance. Sequencing was confirmed along the entire mutS region for intermediate and final products.
Construction of mutS complements.
Complementation with the wild-type mutS gene was performed using the neisserial insertional complementation system (NICS) (39), where the gene of interest is placed in the N. gonorrhoeae chromosome between the lctP and aspC genes. ΔmutS was transformed with the chloramphenicol-resistant plasmid pGCC5MutS (32) using 5 μg/ml chloramphenicol. The mutS+ parent was transformed using the erythromycin-resistant plasmid pGCC2MutS, created by excising the SalI-PmeI mutS fragment from pGCC5MutS and ligating it into similarly digested pGCC2. The entirety of both mutS genes (at the normal locus and at the external NICS locus) was confirmed by sequencing along the length of the mutS region.
Mutator assay.
The phase-locked parent, ΔmutS, mutS-F32A, and mutS-S661A strains were heavily streaked on GCB solid medium (10 colonies per half a plate) and were incubated for 22 h. The lawns were swabbed into 4 ml GCBL with 500 μl spread on GCB Rif plates, and serial dilutions of the suspension were plated on GCB plates to measure the total CFU per milliliter. The titer plates were counted after overnight incubation, while the Rif plates were counted after 42 h. Both large and small Rifr colonies formed, both of which were able to be propagated on GCB Rif plates.
PDCMC assays.
For the pilus-dependent colony morphology change (PDCMC) assays, strains were revived from frozen stock to GCB plates and grown for 22 h. One colony was picked with a filter disk and dispersed into 500 μl GCBL. After dilution of 1 μl into 500 μl GCBL, 30 μl was spread onto a fresh IPTG plate. At 22 h, colonies were examined under a stereomicroscope, and 10 were selected that were entirely piliated (P+). The number of nonpiliated or underpiliated (P−) blebs was counted every 2 h, with each bleb receiving a score of 1, until 4 or more appeared, which was scored as a maximum of 4. The assay was repeated 5 to 7 times, and all colony scores were considered for the Student t test. The standard error of the mean (SEM) was provided for the average of each 10-colony PDCMC repeat.
Traditional sequencing assay.
After growth on GCB IPTG for 22 h, seven progenitor colonies of the phase-variable Kanr parent and the phase-variable mutS-F32A strain were propagated on GCB without IPTG to prevent variation of the pilE sequence. The pilE sequences from 28 to 32 piliated (P+) colonies were amplified, sequenced, and analyzed for variants differing from the starting pilE sequence (pilin variant 1-81-S2 [2]) using AlignX (Vector NTI software; Invitrogen).
Next-generation sequencing assay and analysis.
Approximately 300 colonies per strain were grown for 22 h on GCB IPTG plates and pooled for total DNA extraction. The 500-bp pilE sequence was amplified with primers CONSTF2 and SmaClaI on 10 to 20 ng of chromosomal DNA with unique multiplex identification tags encoded into the primers. The samples were sent for next-generation sequencing at the Roche 454 Sequencing Center in Branford, CT, with 13,207 reads for the parent strain and 5,037 for the ΔmutL derivative, 18,756 for the ΔmutS derivative, 1,686 for the mutS-F32A derivative, and 6,118 for the mutS-S661A derivative. The percentages of variants and silent copy identities were determined using an in-house-generated Perl program that matched sequences from silent copies to different parts of each read. The frequency was calculated as the percentage of variant pilE reads over total pilE sequences. Statistics were calculated using the prop.test function in R on the number of variant reads per total reads for each strain. The MMC mutants showed a statistically significant difference of P < 0.0001 between the mutants and the parent.
Viability assay.
Strains were streaked onto GCB plates from frozen stocks and grown for 22 h. One colony was picked and diluted in GCBL so that ∼200 colonies were evenly spread onto plain GCB and GCB-IPTG plates. At 22 h, 3 to 4 colonies were individually picked with a filter disk and dispersed into 500 μl GCBL. Serial dilutions were plated onto GCB plates, and the resulting titer was counted. The assay was performed 4 to 6 times, and the SEM was taken from each of the averaged titers.
Detection of pilE deletion events.
Nonpiliated colonies that arose after growth on IPTG-containing medium were lysed and used as the templates for PCR with primers PILRBS and SP3A. Samples that yielded a 630-bp band contained an intact pilE region, blank lanes were presumed to contain a deletion, and a larger product indicated an L-pilin (extra-long pilin variant) duplication. The template DNA for all blank lanes was amplified with pilC primers (either PilCdownstream/pilCPLfor or Ngo055up/pilC_int) to confirm the presence of DNA in the reaction. Over 200 colonies were analyzed for the parent, ΔmutL, ΔmutS, and mutS-F32A strains, while 25 colonies were analyzed for the ruvB, ruvB recG, ruvB recG ΔmutS, and ruvB recG mutS-F32A strains.
RESULTS
N. gonorrhoeae mutS-F32A and mutS-S661A mutants are MMC deficient.
The highly conserved phenylalanine residue at the N-terminal domain of MutS from E. coli, Thermus aquaticus, and Saccharomyces cerevisiae has been shown to be critical for the ability of MutS to recognize mismatches and small loops (40–42). An E. coli mutant carrying the mutS-F36A mutation is deficient in MMC (41), but the mutant protein can still bind to a G4 structure in vitro, at 70% of the wild-type level (36). In addition, mutation of the conserved E. coli mutS serine 668 to alanine (S668A) in the ATPase domain results in a mutant MutS with parental levels of DNA binding to mismatches (and presumably G4s) but is unable to perform MMC (43). The corresponding mutS-F32A and -S661A site-directed mutations were independently constructed and introduced into the mutS locus in N. gonorrhoeae FA1090 (Fig. 2A and B) to assay whether the gonococcal MutS (GcMutS) interaction with the pilE G4 might be important for regulating pilin AV.
To confirm the MMC-deficient phenotype of the site-directed mutS mutants, the frequency of spontaneous resistance to the antibiotic rifampin was measured for the ΔmutS, mutS-F32A, and mutS-S661A mutants. Mutator frequencies were calculated by determining the number of colonies that were rifampin resistant (Rifr) compared to the total number of CFU. The parental strain had a median mutation frequency of 3.3 × 10−7 Rifr CFU/total CFU. All three mutS mutations resulted in a 10-fold increase in the frequency of spontaneous rifampin resistance, with 2.5 × 10−6 Rifr CFU/total CFU for ΔmutS, 8.2 × 10−6 Rifr CFU/total CFU for mutS-F32A, and 4.0 × 10−6 Rifr CFU/total CFU for mutS-S661A (Fig. 3A). Similar increases in the spontaneous mutation frequency were obtained for nalidixic acid resistance (data not shown). The results show that both the F32A and S661A mutations each result in a mutator phenotype, similar to the ΔmutS mutation, and confirm the predicted loss of function resulting from the two site-directed mutations.
FIG 3.
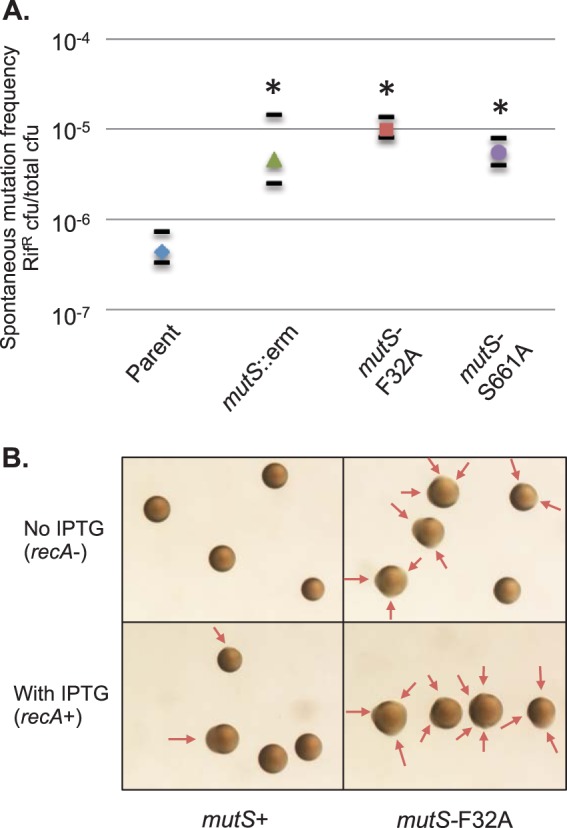
Phenotypes of N. gonorrhoeae mutS mutants. (A) Spontaneous resistance to rifampin of mutS mutants. The strains shown are the parent and isogenic ΔmutS (mutS::erm), mutS-F32A, and mutS-S661A derivatives. The frequency was calculated by the number of Rifr colonies (70 ng/ml rifampin) divided by the total number of colonies. The median values from 10 experiments are shown, with short black bars designating the first and third quartiles. Statistics were performed using the Wilcoxon rank sum test, where ΔmutS, mutS-F32A, and mutS-S661A strains were significantly different from the parent (*, P < 0.025) but not statistically different from each other. (B) Colony morphologies of the phase-variable parent and phase-variable mutS-F32A mutant after 22 h of growth on medium with ITPG (recA+) or without IPTG (recA−). Results identical to those from the phase-variable parent were obtained with the phase-variable Kanr parent. The arrows indicate emerging nonpiliated (P−) blebs.
N. gonorrhoeae mutS-F32A shows increased pilus phase variation.
Piliated gonococcus cells form small domed colonies due to interactions between adjacent pili, while nonpiliated cells, which do not have the intercell connections mediated by pili, grow faster and spread out to form large flat colonies (Fig. 1C) (44). As piliated (P+) colonies grow over time, a subset of bacteria convert to a nonpiliated (P−) phenotype, leading to irregular borders or blebs (Fig. 3B) (15). P− variants arise from three major mechanisms: (i) the nonreversible deletion of the pilE gene (45), (ii) introduction of variant pilE sequences by pilin AV that encode a nonfunctional or poorly expressed pilus (Fig. 1C), and (iii) loss of expression of the minor pilin protein PilC by phase variation (46). The P− blebs that form due to pilS incorporation or pilE deletion are RecA dependent, while the P− blebs that occur due to the loss of PilC are RecA independent. All strains used in this study contain an IPTG-inducible recA allele (37) to control pilin AV. In the absence of IPTG, P− blebs due to pilin AV are eliminated; however, P− blebs due to pilC phase variation are unaffected (12). Like the previously described mutS loss-of-function mutant (32), the mutS-F32A mutant formed numerous P− blebs in the absence of pilin AV (Fig. 3B, recA−), confirming a deficiency in the MMC of slipped-strand mispairs.
N. gonorrhoeae mutS-F32A and mutS-S661A mutants show increased pilin AV by PMCDC.
The colony variation assay, which follows pilus-dependent colony morphology changes (PDCMC), measures the rate of formation of the faster-growing P− blebs that emerge on the edge of a P+ colony over time (Fig. 3B). An increase in the number of emerging P− cells usually corresponds to an increase in the pilin AV frequency. In the absence of IPTG, P− blebs rarely form in the parent strain over the course of an 8-h experiment, but due to the absence of slipped-strand correction at pilC in MMC mutants, most (∼70%) of the mutS mutant colonies contain at least one P− bleb after 22 h of growth (data not shown). This makes it difficult to perform the PDCMC assay, which requires completely P+ colonies at the start, and also obscures the results, since both pilin AV and slipped-strand mispairing at pilC contribute to nonpiliated blebs.
To reduce the background level of nonpiliated cells, the mutS alleles were placed into a strain where every third G of the pilC1 gene has been changed to a synonymous substitution to make PilC production constitutive (the phase-locked pilC1PL allele) (47). In the phase-locked pilC strain, very few blebs emerge without IPTG, even in the MMC mutants. In the presence of IPTG, the colony variation score of the ΔmutS was significantly increased over that of the mutS+ parent strain, and both the mutS-F32A and mutS-S661A alleles had a similarly elevated rate of pilin AV compared to the mutS+ allele (Fig. 4A). The same level of colony variation between the ΔmutS mutant and the two site-directed mutS-F32A and mutS-S661A mutants supports the hypothesis that G4 binding by MutS is not required for pilin AV.
FIG 4.
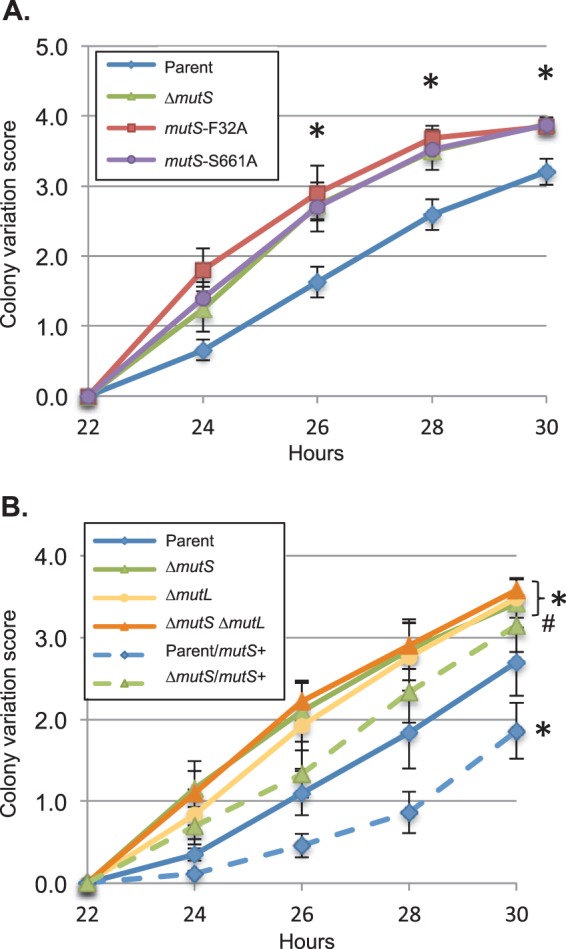
Pilin-dependent colony morphology changes (PDCMC) of MMC mutants. (A) PDCMC of the Kanr parent and the Kanr ΔmutS, mutS-F32A, and mutS-S661A derivatives grown in the presence of IPTG. Shown is the average of results from 5 to 7 assays (of 10 colonies each) with error bars representing the SEM. Statistical significance was determined using Student's two-tailed t test. Asterisks indicate P <0.001 relative to the parent strain for the indicated time point. There was no statistically significant difference between the ΔmutS, mutS-F32A, and mutS-S661A strains. (B) PDCMC assay of the parent and ΔmutS, ΔmutL, and ΔmutS ΔmutL derivatives grown in the presence of IPTG. The parent and mutants are depicted by solid lines, with a copy of the wild-type mutS+ gene at the NICS locus represented by a dashed line. Shown is the average of results from 5 to 6 assays (of 10 colonies each) with error bars representing the SEM. Statistical significance was determined using Student's two-tailed t test. Asterisks indicate P < 0.01, and # denotes P = 0.05 relative to the parent strain.
When complemented with a copy of the wild-type mutS allele at an ectopic locus, the AV frequency of the ΔmutS mutant was reduced close to parental levels, and when two copies of mutS were present (one at its normal position and one at an external locus), the AV frequency was significantly lower than that in the strain with one copy of mutS (Fig. 4B). The increased frequency of PDCMC observed in MMC mutants without PilC phase variation and the lowered frequency of PDCMC with additional MutS suggest that the antirecombination properties of MutS act to reduce the recombination at the pilE locus.
MutL and MutS are in the same pathway for pilin AV.
Although MutS and MutL both act in MMC, they have different functions; MutS directly binds to the mismatch or perturbation, while MutL binds to MutS to initiate the repair of the improper DNA strand (31). Individual disruption of mutS or mutL in gonococci resulted in a similar increase in the colony variation assay compared to the parent strain in the locked pilC background (Fig. 4B), consistent with previous results using the phase-variable pilC background (32). By PDCMC, the ΔmutS ΔmutL double mutant exhibited the same level of pilin AV as either of the single mutants, suggesting that both MutS and MutL are required for the MMC-dependent increase in the level of pilin AV.
mutS-F32A and mutS-S661A mutants show the same level of pilin AV as the ΔmutS mutant by pilE sequencing assays.
The most direct way to measure pilin antigenic variation is to sequence the pilE genes of multiple independently isolated progeny to determine the percentage that have changed and the donor silent copies used. In traditional Sanger sequencing, over 200 individual pilE PCR products are sequenced from cells grown under conditions that allow pilin AV. After 19 generations, ∼12% of progeny have a pilin gene that differs from the starting sequence (12). A previous study showed that the ΔmutS strain had a 2- to 3-fold increase in pilin AV compared to its parental mutS+ strain by a sequencing assay (32). Using a similar assay, we found that the mutS-F32A mutant showed a similar 3-fold increase in pilin AV over the mutS+ strain (Table 3). Although both strains were in the background with phase-variable pilC, only P+ colonies were sequenced, and the results were not affected by P− pilC variation (12). Analysis of the profile of silent copies incorporated into pilE showed there were no significant differences between the mutS-F32A mutant and the ΔmutS mutant (data not shown).
TABLE 3.
Pilin AV by traditional sequencing of mutS+ and mutS-F32A mutants
| Progenitor no.a | No. of colonies sequenced | No. of variants | Frequency of AVb |
|---|---|---|---|
| Parent strain | |||
| 1 | 30 | 3 | 0.100 |
| 2 | 32 | 3 | 0.094 |
| 3 | 32 | 2 | 0.063 |
| 4 | 28 | 0 | 0.000 |
| 5 | 28 | 0 | 0.000 |
| 6 | 28 | 1 | 0.036 |
| 7 | 28 | 3 | 0.107 |
| mutS-F32A mutant | |||
| 1 | 31 | 6 | 0.194 |
| 2 | 32 | 2 | 0.063 |
| 3 | 32 | 8 | 0.250 |
| 4 | 30 | 7 | 0.233 |
| 5 | 30 | 9 | 0.300 |
| 6 | 30 | 8 | 0.267 |
| 7 | 29 | 5 | 0.172 |
The strain backgrounds are phase-variable Kanr FA1090 recA6.
The median frequency for the parent strain is 0.063, and that for the mutS-F32A mutant is 0.233.
We used a new version of the pilin AV sequencing assay that uses next-generation sequencing to analyze pilin AV of the mutS mutants. After 22 h of growth under IPTG induction of RecA expression, the pilE gene was amplified from the total DNA of a pool of ∼300 colonies. The sequence of the variable portion of the pilE gene of each population was determined by multiplex 454 sequencing, and the reads were analyzed by an in-house-generated program that scored whether a read was a variant and also determined which pilS copy was the donor. The parental locked pilC strain showed a 12.8% frequency of pilE variants, similar to the results using the phase-variable pilC strain in a traditional sequencing assay (12). All of the MMC mutants showed about a 3-fold increase in pilin AV frequency, with 34.67% for the ΔmutL mutant, 38.11% for the ΔmutS mutant, 39.87% for the mutS-F32A mutant, and 38.95% for the mutS-S661A mutant, consistent with the frequencies reported by the traditional sequencing assay (Table 3) (12). The mutS-F32A and mutS-S661A mutant frequencies were not statistically different from the ΔmutS mutant frequency. Additionally, analysis of the silent copies used to produce the variant pilE genes of all the MMC mutants showed similar profiles to the parent, primarily containing pilS2c1/6c1 (orange), pilS3c1 (blue), and pilS1c1 (dark gray) donor silent copies (where “c1” represents “copy 1”) (Fig. 5). One difference between the MMC mutants and the parental MMC-proficient strain is the number of variants designated as double crossovers (green). These variant pilE sequences, containing two or more stretches of DNA donated from different silent copies, were increased in the MMC mutants, as would be expected from an increased number of recombination events. The similarities between the donor copies used, whether MMC is disabled or not, suggest that while the silent copies are exchanged more frequently, recombination is likely to be by the same mechanism as in the MMC-proficient strains.
FIG 5.

Donor silent copy profile of MMC mutants. Shown is the analysis of variant pilE sequences of MMC mutants. The silent copy profile was derived from the 454 sequencing of the parent and its ΔmutS, mutS-F32A, mutS-S661A, and ΔmutL derivatives grown for 22 h in the presence of IPTG. The charts do not include the sequences retaining the parental pilE 1-81-S2 sequence. The legend depicts the silent copies by position (e.g., “2c1” represents “pilS2 copy 1”). The “var” category includes changes that could have originated from more than one silent copy, and “unassigned” indicates reads that were unable to be reliably assigned to a donor copy.
Loss of MutS exacerbates the ruvB recG synthetic lethality.
Holliday junctions are central intermediates that arise during homologous recombination, and RuvAB and RecG are helicases that catalyze the branch migration of the Holliday junction intermediates to extend or remove heteroduplexes (48). Initiation of pilin AV in a ruvB recG double mutant results in a synthetic lethality due to unresolved recombination intermediates at the pilE locus (49). Cells that cannot undergo pilin AV (e.g., due to a mutation of recA, deletion of the pilE gene, or disruption to the upstream G4) are rescued from the synthetic lethality (13, 49). Since MutS acts to prevent recombination at the pilE locus, we reasoned that the absence of mutS would exacerbate the ruvB recG synthetic lethality. In the locked pilC background, the viability of a double ruvB recG mutant was reduced an order of magnitude when grown under AV-permissible conditions (IPTG induction of RecA), while a single ruvB mutant was only mildly affected (Fig. 6). This result is consistent with previous reports using a phase-variable pilC strain (49). Both the ΔmutS and the mutS-F32A mutations decreased the viability of the IPTG-induced ruvB recG mutant an additional order of magnitude relative to the MMC-proficient parent (Fig. 6). These results confirm that MMC limits the formation of Holliday junctions and heteroduplexes during pilin AV and lend support to the idea that there is no additional role of MutS in pilin AV.
FIG 6.
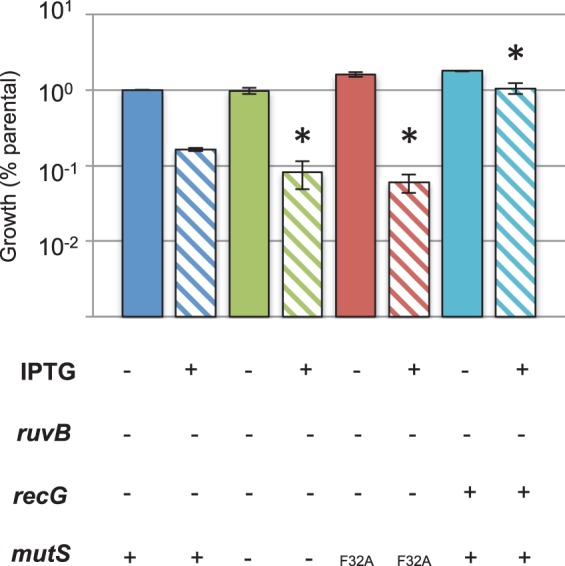
Effect of mutS on ruvB recG synthetic lethality. Viability of strains grown in the presence of IPTG. Shown are the ruvB recG (blue), ruvB recG ΔmutS (green), and ruvB recG mutS-F32A (red) strains and the single ruvB mutant (turquoise). The cells were grown on plates for 22 h in the absence (solid bars) and presence (striped bars) of IPTG, with the number or CFU per colony measured for 3 to 4 colonies each. The assay was performed 6 times, except for the assay for the single ruvB mutant, which was done 4 times. Error bars represent the SEM, and the asterisk denotes a Student's two-tailed t test value of P < 0.05 compared to the ruvB recG strain grown with IPTG.
One means to escape the ruvB recG synthetic lethality is the chance deletion of the pilE locus or the partial duplication of the pilE gene (49). In the parental locked pilC strain, only 2.7% of analyzed P− variants were nonpiliated due to a deletion of the pilE locus. In contrast, the majority (88%) of the P− colonies that arose in the ruvB recG mutant under AV-permissible conditions were deleted for pilE (Table 4), consistent with previous results (49). Both the ΔmutS and mutS-F32A mutants in the ruvB recG strain showed 96% deleted escape variants, suggesting that the failure to disrupt heteroduplexes formed at pilE during pilin AV accumulates a greater percentage of deletions to escape the increased lethality in an MMC mutant.
TABLE 4.
pilE PCR products from nonpiliated colonies
| Straina | Total no. of colonies | % of colonies |
||
|---|---|---|---|---|
| Normal | With l-pilin | With deletion | ||
| Parent | 224 | 96 | 1.3 | 2.7 |
| ΔmutL mutant | 221 | 94.6 | 2.7 | 2.7 |
| ΔmutS mutant | 232 | 87.9 | 5.6 | 6.5 |
| mutS-F32A mutant | 228 | 94.3 | 4.8 | 0.9 |
| mutS-S661A mutant | 216 | 80.6 | 14.8 | 4.6 |
| ΔruvB mutant | 25 | 80 | 0 | 20 |
| ΔruvB ΔrecG mutant | 25 | 12 | 0 | 88 |
| ΔruvB ΔrecG ΔmutS mutant | 25 | 4 | 0 | 96 |
| ΔruvB ΔrecG mutS-F32A mutant | 25 | 4 | 0 | 96 |
The background for all strains is FA1090 recA6 pilC1PL, with the first five derived from FA7458-1A.
DISCUSSION
Pilin AV is a major factor in the pathogenicity of N. gonorrhoeae, as it mediates changes to the amino acid sequence and expression levels of the main type IV pilin protein throughout the course of infection. It was previously reported that loss of MMC increased the level of pilin AV, and we have confirmed that result here. MutS is known to have two main functions in the cell as part of MMC: one is to recognize mismatches and small perturbations (like the loops of slipped strands and G4 structures) that occur during recombination, and the other is antirecombination, by preventing recombination with homeologous DNA. As pilS and pilE sequences are homeologous to each other, MMC is expected to limit pilin AV. However, because MutS proteins have been shown to preferentially bind to G4 sequences over a canonical mismatch, it was a plausible hypothesis that MutS could have an alternative role in pilin AV by interacting with the pilE G4.
This study focused on two mutS mutants (F32A and S661A) that are predicted to encode proteins that are unable to perform MMC but should still bind the pilE G4. Although the F36A mutation in E. coli can still bind to G4s, albeit at a lower level than the wild type, we have been unable to test whether the GcMutS-F32A mutant binds to G4 structures like the corresponding EcMutS-F36A mutant. We thus examined a second mutation, mutS-S661A, that was reported to disrupt the ATPase domain in the E. coli ortholog. The mutator phenotypes of both the mutS-F32A and mutS-S661A mutants confirm that they are each deficient in MMC, similarly to a mutS deletion mutant, and as expected, both the mutS-F32A and mutS-S661A mutants exhibited an increase in pilC phase variation. We used pilus-dependent colony morphology changes in a locked pilC background to measure the pilin AV levels of the mutants. Both site-directed mutS mutants and the ΔmutS showed an elevated level of nonpiliated blebs compared to the mutS+ parent, confirming the defect in MMC, and both site-directed mutants showed similar levels of pilin AV to the ΔmutS strain. We also confirmed that MutL acts in the same pathway as MutS, since the phenotype of a double mutant was the same as that of each of the single mutants. Moreover, overexpression of MutS (by supplying two chromosomal copies) further reduced pilin AV, strongly suggesting that MutS is limiting for pilin AV. Combined with the data that showed that loss of MutS exacerbates the synthetic lethality of a ruvB recG mutant, these data show that MutS working through MMC disrupts heteroduplex formed during pilin AV.
The second assay we used to measure pilin AV was direct pilE sequencing. Previous data used traditional sequencing to show that the ΔmutS mutant had a 2- to 3-fold increase in pilin variants compared to the parental strain. We show, using the same method, that the mutS-F32A mutant also has a 2- to 3-fold increase in variants compared to its parental strain. Since the traditional pilin AV sequencing assay is limited by the stochastic nature of variant production and the small number of progeny that can be effectively examined, we analyzed the mutS mutants by a pilin AV deep-sequencing assay. This assay confirmed that there is no difference in variation frequency or type of variants in the mutS mutants and supports the conclusion that it is only the loss of MMC that influences pilin AV, while the ability of MutS to bind to G4s has no discernible role.
There are a number of cellular proteins involved in the formation, stability, and unwinding of G4s (50). The N. gonorrhoeae RecA and RecQ proteins have already been shown to bind to the pilE G4 in vitro (51, 52). RecA is required for pilin AV through promotion of single-strand annealing and DNA strand exchange in homologous recombination, and the pilE G4 may serve to recruit RecA, facilitating strand exchange. RecQ is a 3′→5′ helicase that is proposed to unwind secondary structures, and mutations to recQ severely decrease the rate of pilin AV (51, 53). Another protein candidate for binding to G4s is MutS, whose eukaryotic MutSα homolog activates the immunoglobulin switch recombination through G4 binding (54). In vitro, the E. coli MutS protein has been shown to bind more strongly to G4 structures than to its canonical G:T mismatch (36). The most definitive way to confirm whether GcMutS binds to G4s (and in particular to the pilE G4) was purification of the protein and in vitro experiments. We have observed that wild-type GcMutS (a generous gift from Erik Larson) binds both a folded pilE G4 and mismatched double-stranded DNA using fluorescence anisotropy but not to a random oligonucleotide or double-stranded DNA (data not shown). These studies were complicated by the fact that the wild-type GcMutS protein had solubility issues at high concentrations, and the mutant proteins were even less soluble (E. Larson, personal communication). Additional binding assays using purified wild-type, F32A, and S661A versions of GcMutS would be required to conclude that these are true separation-of-function mutations. However, since both separation-of-function mutations have identical phenotypes that phenocopy the loss-of-function mutant, we conclude that there is not likely to be a positive role for MutS in pilin AV. It is possible that the limitation of pilin AV by MMC is a way to regulate the frequency of variation, and this hypothesis will be explored in the future.
ACKNOWLEDGMENTS
We thank Erik Larson of Southern Illinois University for suggesting the project and providing purified GcMutS and for helpful conversations. We thank Carl Gunderson, Jing Xu, and Liz Stohl for critical reading of the manuscript.
Funding was provided by grants from the National Institutes of Health: R37 AI033493 and R01 AI044239 to H.S.S. and F32 AI0I94945 to E.R.
REFERENCES
- 1.CDC. 2013. Sexually transmitted disease surveillance 2012. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.Seifert HS, Wright CJ, Jerse AE, Cohen MS, Cannon JG. 1994. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J Clin Invest 93:2744–2749. doi: 10.1172/JCI117290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saunders NJ, Jeffries AC, Peden JF, Hood DW, Tettelin H, Rappuoli R, Moxon ER. 2000. Repeat-associated phase variable genes in the complete genome sequence of Neisseria meningitidis strain MC58. Mol Microbiol 37:207–215. doi: 10.1046/j.1365-2958.2000.02000.x. [DOI] [PubMed] [Google Scholar]
- 4.Snyder LA, Butcher SA, Saunders NJ. 2001. Comparative whole-genome analyses reveal over 100 putative phase-variable genes in the pathogenic Neisseria spp. Microbiology 147:2321–2332. [DOI] [PubMed] [Google Scholar]
- 5.Jordan PW, Snyder LA, Saunders NJ. 2005. Strain-specific differences in Neisseria gonorrhoeae associated with the phase variable gene repertoire. BMC Microbiol 5:21. doi: 10.1186/1471-2180-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moxon R, Bayliss C, Hood D. 2006. Bacterial contingency loci: the role of simple sequence DNA repeats in bacterial adaptation. Annu Rev Genet 40:307–333. doi: 10.1146/annurev.genet.40.110405.090442. [DOI] [PubMed] [Google Scholar]
- 7.Rotman E, Seifert HS. 2014. The genetics of Neisseria species. Annu Rev Genet 48:405–431. doi: 10.1146/annurev-genet-120213-092007. [DOI] [PubMed] [Google Scholar]
- 8.Sparling PF. 1966. Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J Bacteriol 92:1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swanson J. 1973. Studies on gonococcus infection. IV. Pili: their role in attachment of gonococci to tissue culture cells. J Exp Med 137:571–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. 1998. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol 29:321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- 11.Stohl EA, Dale EM, Criss AK, Seifert HS. 2013. Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. mBio 4(4):e00399–13. doi: 10.1128/mBio.00399-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Criss AK, Kline KA, Seifert HS. 2005. The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol 58:510–519. doi: 10.1111/j.1365-2958.2005.04838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cahoon LA, Seifert HS. 2009. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science 325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kline KA, Criss AK, Wallace A, Seifert HS. 2007. Transposon mutagenesis identifies sites upstream of the Neisseria gonorrhoeae pilE gene that modulate pilin antigenic variation. J Bacteriol 189:3462–3470. doi: 10.1128/JB.01911-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sechman EV, Rohrer MS, Seifert HS. 2005. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol 57:468–483. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- 16.Hamrick TS, Dempsey JA, Cohen MS, Cannon JG. 2001. Antigenic variation of gonococcal pilin expression in vivo: analysis of the strain FA1090 pilin repertoire and identification of the pilS gene copies recombining with pilE during experimental human infection. Microbiology 147:839–849. [DOI] [PubMed] [Google Scholar]
- 17.Maizels N, Gray LT. 2013. The G4 genome. PLoS Genet 9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bochman ML, Paeschke K, Zakian VA. 2012. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 13:770–780. doi: 10.1038/ni.2363,10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todd AK, Neidle S. 2011. Mapping the sequences of potential guanine quadruplex motifs. Nucleic Acids Res 39:4917–4927. doi: 10.1093/nar/gkr104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaume N, Pathak R, Yadav VK, Kota S, Misra HS, Gautam HK, Chowdhury S. 2013. Genome-wide study predicts promoter-G4 DNA motifs regulate selective functions in bacteria: radioresistance of D. radiodurans involves G4 DNA-mediated regulation. Nucleic Acids Res 41:76–89. doi: 10.1093/nar/gks1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Leeuwen HC, Bakker D, Steindel P, Kuijper EJ, Corver J. 2013. Clostridium difficile TcdC protein binds four-stranded G-quadruplex structures. Nucleic Acids Res 41:2382–2393. doi: 10.1093/nar/gks1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rawal P, Kummarasetti VB, Ravindran J, Kumar N, Halder K, Sharma R, Mukerji M, Das SK, Chowdhury S. 2006. Genome-wide prediction of G4 DNA as regulatory motifs: role in Escherichia coli global regulation. Genome Res 16:644–655. doi: 10.1101/gr.4508806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahoon LA, Seifert HS. 2013. Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog 9:e1003074. doi: 10.1371/journal.ppat.1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez V, Guo K, Hurley L, Sun D. 2009. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J Biol Chem 284:23622–23635. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marinus MG. 2012. DNA mismatch repair. EcoSal Plus doi: 10.1128/ecosalplus.7.2.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukui K. 2010. DNA mismatch repair in eukaryotes and bacteria. J Nucleic Acids 2010:260512. doi: 10.4061/2010/260512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su SS, Modrich P. 1986. Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc Natl Acad Sci U S A 83:5057–5061. doi: 10.1073/pnas.83.14.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polosina YY, Cupples CG. 2010. Wot the 'L—does MutL do?. Mutat Res 705:228–238. doi: 10.1016/j.mrrev.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Duppatla V, Bodda C, Urbanke C, Friedhoff P, Rao DN. 2009. The C-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochem J 423:265–277. doi: 10.1042/BJ20090626. [DOI] [PubMed] [Google Scholar]
- 30.Modrich P. 2006. Mechanisms in eukaryotic mismatch repair. J Biol Chem 281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schofield MJ, Hsieh P. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 32.Criss AK, Bonney KM, Chang RA, Duffin PM, LeCuyer BE, Seifert HS. 2010. Mismatch correction modulates mutation frequency and pilus phase and antigenic variation in Neisseria gonorrhoeae. J Bacteriol 192:316–325. doi: 10.1128/JB.01228-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worth L Jr, Clark S, Radman M, Modrich P. 1994. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc Natl Acad Sci U S A 91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fabisiewicz A, Worth L Jr. 2001. Escherichia coli MutS, L modulate RuvAB-dependent branch migration between diverged DNA. J Biol Chem 276:9413–9420. doi: 10.1074/jbc.M005176200. [DOI] [PubMed] [Google Scholar]
- 35.Fukui K, Bessho Y, Shimada A, Yokoyama S, Kuramitsu S. 2013. Thermostable mismatch-recognizing protein MutS suppresses nonspecific amplification during polymerase chain reaction (PCR). Int J Mol Sci 14:6436–6453. doi: 10.3390/ijms14036436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrat EA, Johnson BR, Williams JD, Borchert GM, Larson ED. 2012. G-quadruplex recognition activities of E. coli MutS. BMC Mol Biol 13:23. doi: 10.1186/1471-2199-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seifert HS. 1997. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene 188:215–220. doi: 10.1016/S0378-1119(96)00810-4. [DOI] [PubMed] [Google Scholar]
- 38.Kellogg DS Jr, Peacock WL Jr, Deacon WE, Brown L, Pirkle DI. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J Bacteriol 85:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehr IJ, Long CD, Serkin CD, Seifert HS. 2000. A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics 154:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malkov VA, Biswas I, Camerini-Otero RD, Hsieh P. 1997. Photocross-linking of the NH2-terminal region of Taq MutS protein to the major groove of a heteroduplex DNA. J Biol Chem 272:23811–23817. doi: 10.1074/jbc.272.38.23811. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto A, Schofield MJ, Biswas I, Hsieh P. 2000. Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Res 28:3564–3569. doi: 10.1093/nar/28.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowers J, Sokolsky T, Quach T, Alani E. 1999. A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. J Biol Chem 274:16115–16125. doi: 10.1074/jbc.274.23.16115. [DOI] [PubMed] [Google Scholar]
- 43.Acharya S, Patterson K. 2010. Mutations in the conserved glycine and serine of the MutS ABC signature motif affect nucleotide exchange, kinetics of sliding clamp release of mismatch and mismatch repair. Mutat Res 684:56–65. doi: 10.1016/j.mrfmmm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Swanson J, Kraus SJ, Gotschlich EC. 1971. Studies on gonococcus infection. I. Pili and zones of adhesion: their relation to gonococcal growth patterns. J Exp Med 134:886–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swanson J, Bergstrom S, Barrera O, Robbins K, Corwin D. 1985. Pilus-gonococcal variants. Evidence for multiple forms of piliation control. J Exp Med 162:729–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jonsson AB, Nyberg G, Normark S. 1991. Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J 10:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng Y, Johnson MD, Burillo-Kirch C, Mocny JC, Anderson JE, Garrett CK, Redinbo MR, Thomas CE. 2013. Mutation of the conserved calcium-binding motif in Neisseria gonorrhoeae PilC1 impacts adhesion but not piliation. Infect Immun 81:4280–4289. doi: 10.1128/IAI.00493-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharples GJ, Ingleston SM, Lloyd RG. 1999. Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA J Bacteriol. 181:5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sechman EV, Kline KA, Seifert HS. 2006. Loss of both Holliday junction processing pathways is synthetically lethal in the presence of gonococcal pilin antigenic variation. Mol Microbiol 61:185–193. doi: 10.1111/j.1365-2958.2006.05213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brazda V, Haronikova L, Liao JC, Fojta M. 2014. DNA and RNA quadruplex-binding proteins. Int J Mol Sci 15:17493–17517. doi: 10.3390/ijms151017493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cahoon LA, Manthei KA, Rotman E, Keck JL, Seifert HS. 2013. Neisseria gonorrhoeae RecQ helicase HRDC domains are essential for efficient binding and unwinding of the pilE guanine quartet structure required for pilin antigenic variation. J Bacteriol 195:2255–2261. doi: 10.1128/JB.02217-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuryavyi V, Cahoon LA, Seifert HS, Patel DJ. 2012. RecA-binding pilE G4 sequence essential for pilin antigenic variation forms monomeric and 5′ end-stacked dimeric parallel G-quadruplexes. Structure 20:2090–2102. doi: 10.1016/j.str.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Killoran MP, Kohler PL, Dillard JP, Keck JL. 2009. RecQ DNA helicase HRDC domains are critical determinants in Neisseria gonorrhoeae pilin antigenic variation and DNA repair. Mol Microbiol 71:158–171. doi: 10.1111/j.1365-2958.2008.06513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larson ED, Duquette ML, Cummings WJ, Streiff RJ, Maizels N. 2005. MutSalpha binds to and promotes synapsis of transcriptionally activated immunoglobulin switch regions. Curr Biol 15:470–474. doi: 10.1016/j.cub.2004.12.077. [DOI] [PubMed] [Google Scholar]