

Maturation of the large ribosomal subunit (LSU) in eukaryotes is a complex and highly coordinated process that requires the concerted action of a large, dynamic, ribonucleoprotein complex, the LSU processome. To interrogate its organization and architecture, McCann et al. assayed 4800 protein–protein interactions and identified 232 high-confidence, binary-interacting protein pairs, representing a fourfold increase from current knowledge. The resulting LSU processome interactome map enhances our understanding of the organization and function of the biogenesis factors within the LSU processome.
Keywords: LSU processome, RNA, ribosome, yeast two-hybrid
Abstract
Maturation of the large ribosomal subunit (LSU) in eukaryotes is a complex and highly coordinated process that requires the concerted action of a large, dynamic, ribonucleoprotein complex, the LSU processome. While we know that >80 ribosome biogenesis factors are required throughout the course of LSU assembly, little is known about how these factors interact with each other within the LSU processome. To interrogate its organization and architecture, we took a systems biology approach and performed a semi-high-throughput, array-based, directed yeast two-hybrid assay. Assaying 4800 protein–protein interactions, we identified 232 high-confidence, binary-interacting protein pairs, representing a fourfold increase from current knowledge. The resulting LSU processome interactome map has enhanced our understanding of the organization and function of the biogenesis factors within the LSU processome, revealing both novel and previously identified subcomplexes and hub proteins, including Nop4.
Eukaryotic ribosomes are complex cellular machines that are comprised of four different ribosomal RNAs (rRNAs) and >70 ribosomal proteins (r-proteins) (Woolford and Baserga 2013). Ribosome assembly begins in the nucleolus with the transcription of the polycistronic pre-rRNA, termed the 35S in Saccharomyces cerevisiae, by RNA polymerase I. The 35S pre-rRNA undergoes numerous cleavage and modification steps to generate the mature 18S, 5.8S, and 25S rRNAs that are assembled with the r-proteins to form the small and large ribosomal subunits. Accurate and efficient production of ribosomes requires the coordinated activity of >150 biogenesis factors, including a number of proteins with enzymatic activity (Fatica and Tollervey 2002; Fromont-Racine et al. 2003; Tschochner and Hurt 2003; Henras et al. 2008; Strunk and Karbstein 2009; Kressler et al. 2010, 2012; Rodríguez-Galán et al. 2013; Thomson et al. 2013; Woolford and Baserga 2013; Fernandez-Pevida et al. 2015). A subset of these biogenesis factors assembles cotranscriptionally (Osheim et al. 2004; Lebaron et al. 2013) on the 35S pre-rRNA, forming the small subunit (SSU) processome (Dragon et al. 2002) or 90S preribosome (Grandi et al. 2002). After the separating cleavage in internal transcribed spacer 1 (ITS1), SSU and large subunit (LSU) assembly proceed independently (Liang and Fournier 1997).
LSU assembly is driven by a large, dynamic, ribonucleoprotein complex, recently termed the LSU processome (Woolford and Baserga 2013; Grob et al. 2014). Assembly occurs in a number of successive steps as the LSU processome travels from the nucleolus to the nucleoplasm and through the nuclear pore to the cytoplasm (Harnpicharnchai et al. 2001; Milkereit et al. 2001; Kressler et al. 2010). Purification and comparison of the biogenesis factor composition of the LSU processome from different stages of LSU maturation has revealed the highly dynamic nature of this process (Nissan et al. 2002; Saveanu et al. 2003; Lebreton et al. 2008; Merl et al. 2010). The earliest steps in assembly occur in the nucleolus and require the greatest number of biogenesis factors. As the LSU processome exits the nucleolus and enters the nucleoplasm, it undergoes significant compositional changes. A large number of nucleolar factors are released, and a new complement of nuclear biogenesis factors are acquired (Harnpicharnchai et al. 2001; Saveanu et al. 2003; Kressler et al. 2008; Ulbrich et al. 2009; Bassler et al. 2010). The last steps of LSU assembly are completed in the cytoplasm and require comparatively few biogenesis factors (Nissan et al. 2002; Bradatsch et al. 2012; Kressler et al. 2012).
In the past decades, much work has been done to dissect the steps of LSU assembly. The cleavage sites of the pre-rRNA have been mapped, a large number of biogenesis factors have been identified, and, in many cases, the biogenesis factors have been assigned to specific steps in assembly. For example, seven biogenesis factors are involved in processing the 27SA3 pre-rRNA (A3 factors) (Sahasranaman et al. 2011), while processing of the 27SB pre-rRNA requires the action of 14 biogenesis factors (B factors) (Talkish et al. 2012). Furthermore, the utilization of cross-linking and analysis of cDNA (CRAC) technology to identify the sequences of the pre-rRNA that are bound by select assembly factors has provided additional insight into the spatial organization of the LSU processome (Granneman et al. 2011).
Even so, the precise molecular details of LSU assembly remain largely unknown. How the biogenesis proteins interact with each other and how these interactions influence and coordinate LSU assembly has yet to be systematically determined for all LSU processome proteins. For example, while we know that the SSU processome is comprised of several subcomplexes (Gallagher et al. 2004; Krogan et al. 2004; Pérez-Fernández et al. 2007), few complexes have been identified for the LSU processome (Milkereit et al. 2001; Galani et al. 2004; Krogan et al. 2004; Miles et al. 2005). Furthermore, even though they copurify in a large ribonucleoprotein complex, it is not clear whether the A3 factors, B factors, and other LSU biogenesis proteins interact among themselves as LSU processome building blocks or with one another within the LSU processome. Even a recent proteome-scale human interactome failed to yield information on the interactions among the orthologous LSU biogenesis factors (Rolland et al. 2014).
To define the LSU processome interactome and probe its organization, we took a systems biology approach and carried out a semi-high-throughput, array-based yeast two-hybrid (Y2H) assay. We assayed 4800 individual bait–prey pairs one by one and identified 232 binary, high-confidence interactions. Coimmunoprecipitation experiments independently validated 98% of the tested interactions. Subsequent analysis of the resulting LSU processome interactome by Markov clustering led to the prediction of both novel and previously identified subcomplexes. Additionally, the LSU processome interactome revealed the presence of several important hubs, including Nop4. Thus, the LSU processome interactome map has provided novel insight into the organization and function of the biogenesis factors within the LSU processome.
Results
Generation of an LSU processome interactome map by Y2H analysis
Our goal was to systematically map the protein–protein interactions (PPIs) among the nucleolar LSU processome components via an array-based Y2H screen. We chose to test 70 LSU biogenesis factors (Table 1) that were selected based on their presence in copurifications of nucleolar LSU processome complexes as determined by affinity purification and mass spectrometry (AP/MS) (Bassler et al. 2001; Harnpicharnchai et al. 2001; Fatica et al. 2002; Nissan et al. 2002; Lebreton et al. 2008; Li et al. 2009). The 46 LSU r-proteins, while important for ribosome assembly and function, were not included due to technical limitations. We focused on the nucleolar LSU processome because it contains the largest complement of biogenesis factors and because its assembly and organization are not well understood. Of the 70 selected proteins, 78% are essential in yeast, and 90% are conserved to humans (Woolford and Baserga 2013). Prior to this work, only 56 Y2H interactions had been reported previously among the 70 proteins (Supplemental Table S1), representing only 22%–26% of the predicted LSU processome interactome (Blow 2009; Lim et al. 2011).
Table 1.
List of LSU processome proteins included in Y2H screen

To generate libraries for a directed, array-based Y2H screen, all 70 full-length ORFs were cloned into Y2H bait and prey vectors using Gateway cloning from either the MORF collection (Gelperin et al. 2005) or through PCR amplification and cloning into a Gateway Entry vector. We used a semi-high-throughput, array-based Y2H assay to individually screen each bait against each prey (Fig. 1A; de Folter and Immink 2011). An empty prey vector was mated with each bait as a negative control. Pair-wise interactions were assayed on two different selective plates with increasing numbers of reporter genes (Fig. 1A,B). Growth on selective medium greater than that observed for the negative control after 3 wk was scored as a positive interacting pair. The screen was carried out three times. One iteration was performed with the yeast strains PJ69-4a/α (James et al. 1996), and two iterations were performed with the yeast strains Y2H Gold/Y187 (Clontech). Of the 4800 assayed PPIs, 641 PPIs were detected in at least one screen and 257 (40%) PPIs were detected in multiple screens (Fig. 1C; Supplemental Table S2). Interestingly, the number of PPIs observed when the PJ69-4a/α strains were used was approximately twice that observed when the Y2H Gold/Y187 strains were used. Of the 641 observed PPIs, 171 were observed in all three iterations of the screen (Fig. 1C).
Figure 1.

An array based Y2H screen identifies novel interactions among LSU processome proteins. (A) Work flow of the array-based Y2H screen. (B) Results from Y2H screen 2 performed in Y2H Gold/Y187 for Rsa3, Mak21/Noc1, and Brx1 bait proteins. Each bait was assayed against all 70 arrayed preys on two different selective media: SD−Leu−Trp−His + 6 mM 3-amino-1,2,4-triazole (3-AT) + 40 μg/mL 5-bromo-4-chloro-3-indolyl α-D-galactopyranoside (X-α-Gal) or SD−Leu−Trp−His−Ade + 6 mM 3-AT + 40 μg/mL X-α-Gal. Growth of a blue colony on either selective medium is indicative of a positive Y2H interaction. (C) A Venn diagram summarizing the PPIs that were identified in each of the three iterations of the array-based Y2H screen. Screen 1 was performed in the PJ69-4a/α yeast strains, while screens 2 and 3 were performed in the Y2H Gold/Y187 yeast strains from Clontech. The number of interactions that were unique to each screen and the number of interactions that were identified in more than one screen are indicated. (D) Histogram of the confidence score distribution for the 641 PPIs identified in this study. The confidence score reflects the reproducibility of an observed interaction across all screens and selective media. Interactions with a confidence score >50% were included in the high-confidence LSU processome interactome data set and are listed in Supplemental Table S3.
Since we performed the array-based Y2H screen to detect the physical interactions among yeast proteins found in the same macromolecular complex, it is plausible that a subset of the observed PPIs are mediated by bridging proteins of the LSU processome. However, only 641 PPIs were collectively observed among all three screens, representing just ∼4% of the total interactions tested. If the observed PPIs were bridged by other proteins in the LSU processome, we would expect there to be fewer negatives and a greater proportion of interactions. Therefore, while it is possible that bridging proteins mediate some of the observed PPIs, the results of the screen suggest that this phenomenon is not general.
To identify the PPIs that are most likely to be biologically relevant, a confidence score was calculated for each observed interaction (Giot et al. 2003; Li et al. 2004; Stelzl et al. 2005). The confidence score is a weighted average that takes the following parameters into account: the number of reporters activated by the PPI (HIS3, ADE2, and MEL1, which encodes α-galactosidase), the reproducibility of the PPI, the observation of a reciprocal PPI, the observation of the PPI in both Y2H strains, and the identification of the PPI in previous studies. Each PPI was evaluated based on these criteria, ranked based on their confidence score, and graphed on a scale of 10%–100% (Fig. 1D). Of the 641 observed PPIs, 409 had a confidence score between 0% and 49% and were classified as low-confidence interactions, and 232 PPIs had a confidence score >50% and were classified as high-confidence interactions (Supplemental Tables S2, S3). The 232 high-confidence PPIs represent a fourfold increase from the 56 previously identified PPIs among these LSU biogenesis factors (Supplemental Table S1).
Validation of the high-confidence interactome
To assess the validity of the LSU processome interactome, we used a coimmunoprecipitation method akin to the cross and capture system to assay a subset of these PPIs (Fig. 2A; Suter et al. 2007; Suter 2012). As each type of PPI assay, including Y2H, has its own inherent false negative and false positive rates, the assays are complementary (Venkatesan et al. 2009; Hegele et al. 2012). Thus, interactions that can be recapitulated with an orthogonal method such as coimmunoprecipitations are less likely to be false positives (Li et al. 2004). The cross and capture system was specifically developed to identify and confirm yeast PPIs detected by other methods, including Y2H (Suter et al. 2007; Suter 2012), and has successfully recapitulated interactions previously detected by Y2H (Suter et al. 2007). Furthermore, similar coimmunoprecipitation assays have been used to validate Y2H data sets (Wang et al. 2011; Hegele et al. 2012).
Figure 2.

Validation of the high-confidence LSU processome interactome by coimmunoprecipitation. (A) Work flow depicting the coimmunoprecipitation approach used to validate novel PPIs. (B) Validation of known interacting proteins as proof of principle. Yeast extract was incubated with α-Flag resin. Coimmunoprecipitations were assessed by α-HA Western blot. (C) Validation of a subset of the novel high-confidence PPIs by coimmunoprecipitation. Yeast extract was incubated with α-Flag resin. Coimmunoprecipitations were assessed by α-HA Western blot. (D) Table summarizing the results of the validation coimmunoprecipitations. Approximately 25% of the high-confidence interactions were assayed by coimmunoprecipitations. Several interactions that were not observed by Y2H were also assayed as negative controls.
To benchmark the coimmunoprecipitation assay, we examined four known PPIs: Brx1–Ebp2, Erb1–Ytm1, Grc3–Las1, and Ssf2–Rrp15 (Miles et al. 2005; Yu et al. 2008; Shimoji et al. 2012; Castle et al. 2013). These ORFs were cloned into either Gateway-converted p415GPD-3xHA or Gateway-converted p414GPD-3xFlag (Mumberg et al. 1995) and cotransformed into yeast. Immunoprecipitations were performed with anti-Flag resin, and the copurifying proteins were visualized by Western blotting with an antibody to the 3xHA tag (Fig. 2A). As expected, all four of the known PPIs were positive by coimmunoprecipitation (Fig. 2B). In contrast, no signal was observed in the absence of a Flag-tagged protein.
Using the coimmunoprecipitation method, we tested ∼25% (58 of 232) of the high-confidence PPIs (Fig. 2C,D; Supplemental Table S4). Of the 58 assayed PPIs, 57 PPIs were recapitulated, giving a validation rate of ∼98%. We also tested nine PPIs that had not been identified in our Y2H screen and were not previously reported. Only one of the nine negative control PPIs was positive by coimmunoprecipitation. We would have expected more of the negative controls to have been positive if the interactions detected by coimmunoprecipitation were mediated by bridging proteins of the LSU processome. Our results support the idea that the validation method is largely detecting direct interactions between two proteins rather than interactions mediated by bridging proteins. Thus, this method successfully validates the observed LSU processome Y2H interactions.
Comparison of the high-confidence interactions with published data sets
The quality of the high-confidence interactome is also supported by the degree of overlap with previously published Y2H interactions. Published Y2H interactions were identified through two databases: the Saccharomyces Genome Database (http://www.yeastgenome.org) and Uniprot (http://www.uniprot.org). Prior to this work, only 56 Y2H interactions had been reported among the 70 proteins included in this screen (Supplemental Table S1). Of the 56 previously reported interactions, 21 (37.5%) were recapitulated in our high-confidence data set, including those among the Ytm1–Erb1–Nop7 complex members and between Brx1–Ebp2 and Rpf2–Rrs1 (Fig. 3A; Supplemental Table S5; Morita et al. 2002; Miles et al. 2005; Shimoji et al. 2012). While the high-confidence data set does not contain all of the previously reported interactions, we did recapitulate 75% of the interactions that were identified using similar or identical screening conditions (Supplemental Table S5), indicating a low false negative rate for our Y2H screen (Brückner et al. 2009; Venkatesan et al. 2009; Hegele et al. 2012). In total, we identified 21 PPIs that were previously identified and 211 novel PPIs, which represent an approximately fourfold increase in our knowledge of the PPIs among the LSU processome proteins.
Figure 3.

Analysis of the high-confidence LSU processome interactome. (A) Network of high-confidence interactions identified in this study that have been previously identified by Y2H. The previously identified interactions are listed in Supplemental Table S1. (B) The interactions among the Rrp5–Mak21/Noc1–Noc2 and Noc2–Noc3 modules were recapitulated in the high-confidence data set (Milkereit et al. 2001; Hierlmeier et al. 2013). (C) The direct, physical interactions of the Urb1/Npa1 subcomplex were identified with high confidence in the LSU processome interactome screen. The Urb1/Npa1 subcomplex was originally identified using genetic screening (Rosado and de la Cruz 2004; Rosado et al. 2007). The dotted lines indicate high-confidence Y2H interactions that were also observed by genetic screening. The solid line indicates an interaction that was only observed in the Y2H screen. (D) Network of high-confidence, reciprocal interactions identified in this study. Dotted lines indicate that the interaction was observed previously by Y2H. (E) Markov cluster algorithm (MCL) clustering of the high-confidence LSU processome interactome predicted subcomplexes that have been described previously. (F) MCL clustering of the high-confidence LSU processome interactome predicted three novel subcomplexes. Circles represent proteins with no known enzymatic activity, diamonds represent helicases, squares represent methyltransferases, parallelograms represent PPIases, hexagons represent GTPases, and octagons represent kinases. The interaction networks were created using Cytoscape. MCL clustering was performed using the clusterMaker plug-in for Cytoscape.
Also identified with high confidence by the LSU processome interactome screen were interactions among components of known subcomplexes or modules. Previously, coaffinity purification had been used to interrogate the underlying organization of the LSU processome (Bassler et al. 2001; Harnpicharnchai et al. 2001; Fatica et al. 2002; Nissan et al. 2002; Saveanu et al. 2003; Miles et al. 2005; Lebreton et al. 2006, 2008; Rosado et al. 2007; Merl et al. 2010; Hierlmeier et al. 2013). While coaffinity purification does not solely detect direct, physical PPIs, it can sometimes allow for the detection of protein modules or subcomplexes. For example, the protein modules Rrp5–Mak21/Noc1–Noc2 and Noc2–Noc3 were previously detected by coaffinity purification (Milkereit et al. 2001; Hierlmeier et al. 2013). The direct PPIs underlying the Rrp5–Mak21/Noc1–Noc2 module were then mapped through affinity purifications of recombinant proteins (Hierlmeier et al. 2013). Importantly, we identified all of the direct interactions within this module for the first time by Y2H in the high-confidence LSU processome interactome data set (Fig. 3B).
Similarly, in the LSU processome interactome map, we identified the physical interactions of a subcomplex that so far has been only described by genetic interaction mapping. Genetic interactions among DBP6, DBP7, DBP9, NOP8, RSA3, URB1 (NPA1), and URB2 (NPA2) were identified in several genetic screens, suggesting that they function together (Rosado and de la Cruz 2004; Rosado et al. 2007). These genes encode proteins that copurify with Urb2 in a discrete subcomplex, as seen by gel filtration chromatography, yet the direct, physical interactions among the proteins in this subcomplex had not yet been determined (Rosado et al. 2007). Significantly, we identified interactions among the subcomplex members in the high-confidence LSU processome interactome data set, allowing the direct, physical interactions to be mapped for the first time (Fig. 3C).
Analysis of the high-confidence LSU processome interactome map
We focused on the 232 high-confidence interactions for further investigation. We constructed an interactome map of the observed high-confidence PPIs using Cytoscape (Supplemental Fig. S1; Shannon et al. 2003). Of the 70 bait proteins assayed in the Y2H, 55 interacted with at least one prey with high confidence. The number of interacting partners of each bait ranged from one to 24, with a mean of 6.2. This mean is slightly higher than the conservative estimate of three to 3.5 interactions per protein (Blow 2009; Lim et al. 2011), which could be due to the presence of several hub proteins, each of which has a large number of interacting partners. Interestingly, 29 baits interacted reciprocally with high confidence with at least one prey (Fig. 3D). These high-confidence, reciprocal interactions are highly likely to be biologically relevant and important for proper assembly of the LSU.
To identify novel subcomplexes among the LSU processome interactome, we used the Markov cluster algorithm (MCL) option in the clusterMaker plug-in for Cytoscape (Shannon et al. 2003; Morris et al. 2011). The MCL is an unsupervised, agglomerative algorithm designed to reveal natural groups within a highly connected graph (Enright et al. 2002). The MCL predicts more subcomplexes and has a higher accuracy rate for prediction than many of the other algorithms (Brohee and van Helden 2006; Moschopoulos et al. 2011). MCL clustering has been used previously to successfully predict subcomplexes from both AP/MS and Y2H data sets (Pereira-Leal et al. 2004; Krogan et al. 2006; Hart et al. 2007; Pu et al. 2007; Nastou et al. 2014). We applied the MCL to our high-confidence LSU processome interactome data set and found that it clustered 56 of the 59 LSU biogenesis factors in the high-confidence LSU processome interactome into seven clusters or subcomplexes (Fig. 3E,F). Of the seven clusters, four represent subcomplexes or interactions that have been described previously (Fig. 3E; Krogan et al. 2004; Miles et al. 2005; Rosado et al. 2007; Tang et al. 2008; Castle et al. 2013; Hierlmeier et al. 2013).
MCL clustering predicted the existence of three novel subcomplexes within the LSU processome interactome. The smallest predicted novel subcomplex contains the biogenesis factors Mak11 and Rrp15 and the functionally redundant proteins Ssf1 and Ssf2 (Fig. 3F). Previously, it had been shown that genetic depletion of each of Rrp15, Ssf1, and Ssf2 inhibits processing of ITS1 (Fatica et al. 2002; De Marchis et al. 2005). In contrast, depletion of Mak11inhibits processing of ITS2 (Saveanu et al. 2007; Talkish et al. 2012). Therefore, not all components of this predicted subcomplex function at the same pre-rRNA processing step. However, Ssf1 and Rrp15 both coaffinity-purify with Mak11 (Saveanu et al. 2007). Thus, the predicted Mak11–Rrp15–Ssf1–Ssf2 subcomplex may exist in vivo.
The other two novel, predicted subcomplexes are much larger. The largest putative subcomplex is comprised of 23 biogenesis factors and contains the hub proteins Loc1, Ebp2, and Nop4 (Fig. 3F). Interestingly, the other components of this complex have been shown to function at various diverse stages of assembly (Nissan et al. 2002; Sahasranaman et al. 2011; Talkish et al. 2012). As Y2H analysis does not provide temporal information, it is plausible that, as hubs, Loc1, Ebp2, and Nop4 act as central coordinators and that the interactions depicted in this cluster are not all occurring at once but are an integration of the activity of the hubs over time. The second large, putative subcomplex contains 17 biogenesis factors, seven of which are B factors (Talkish et al. 2012). Interestingly, this complex contains six DExD/H-box RNA helicases, two GTPases, and a putative methyltransferase, suggesting that these biogenesis factors may function together to drive or regulate LSU assembly.
Biochemical analysis of the subcomplexes of the LSU processome
Several subcomplexes of the LSU processome that had been proposed previously were also predicted by MCL clustering of the high-confidence LSU processome interactome (Fig. 3E; Milkereit et al. 2001; Miles et al. 2005; Rosado et al. 2007; Tang et al. 2008; Merl et al. 2010; Castle et al. 2013; Hierlmeier et al. 2013). The majority of the published evidence supporting the existence of these subcomplexes in vivo is the presence of the predicted components in a complex that can be affinity-purified after disruption of the LSU processome (Milkereit et al. 2001; Krogan et al. 2004; Miles et al. 2005; Castle et al. 2013; Hierlmeier et al. 2013). These subcomplexes have not yet been examined by glycerol gradient sedimentation, a technique that would separate the subcomplexes from the larger LSU processome without genetic or biochemical disruption of ribosome assembly.
To determine whether the putative LSU processome subcomplexes predicted by MCL clustering exist under normal cellular conditions, we used glycerol gradient sedimentation analysis, which separates complexes on the basis of buoyant density. To this end, we created a series of endogenously tagged yeast strains and subjected extract from these strains to glycerol gradient sedimentation. The resulting fractions were analyzed for the presence of the tagged protein by Western blot. Subcomplexes would be less dense than the LSU processome and would differentially sediment on the gradient, thereby enabling their detection under native conditions.
We analyzed the subcomplexes that have been shown previously to copurify after disruption of the LSU processome and were predicted by MCL clustering of the high-confidence LSU processome interactome: Grc3–Las1, Rrp5–Mak21/Noc1, and Erb1–Nop7–Ytm1 (Fig. 3E; Milkereit et al. 2001; Miles et al. 2005; Castle et al. 2013; Hierlmeier et al. 2013). All three complexes were found to sediment in the less dense fractions, which supports their existence as discrete complexes outside of the LSU processome (Fig. 4). For example, endogenously tagged Grc3 and Las1 comigrate in peak fractions 5 and 6 (Fig. 4A). Comparison of the sedimentation profile of Grc3 and Las1 with molecular weight (MW) markers of indicated S-values shows that the putative Grc3–Las1 complex migrates at ∼11S (predicted MW, 210 kDa).
Figure 4.
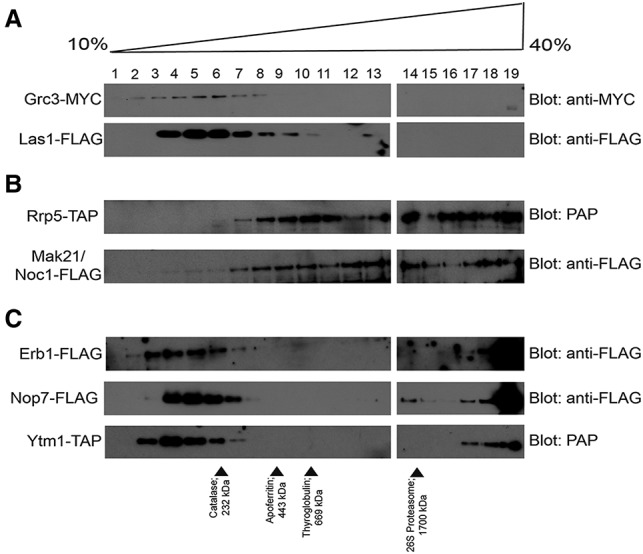
Glycerol gradient sedimentation analysis supports the existence of LSU processome subcomplexes. (A) Grc3 and Las1 comigrate independently of the LSU processome. Yeast extract made from either YPH500 Grc3-MYC or NOY504 Las1-Flag was layered onto a 10%–40% glycerol gradient. The gradients were spun at 150,000g for 18 h, and fractions were harvested from the top of the gradient. All fractions were analyzed by either α-MYC or α-Flag Western blot. (B) Rrp5 and Mak21/Noc1 comigrate in multiple complexes. Yeast extract made from either YPH499 Rrp5-TAP or YPH499 Mak21/Noc1-Flag was layered onto a 10%–40% glycerol gradient. The gradients were spun at 150,000g for 18 h, and fractions were harvested from the top of the gradient. All fractions were analyzed by either PAP or α-Flag Western blot. (C) Erb1, Nop7, and Ytm1 comigrate independently of the LSU processome. Yeast extract made from either NOY504 Erb1-Flag, NOY504 Nop7-Flag, or NOY504 Ytm1-TAP was layered onto a 10%–40% glycerol gradient. The gradients were spun at 150,000g for 18 h, and fractions were harvested from the top of the gradient. All fractions were analyzed by either α-Flag or PAP Western blot.
Similarly, Rrp5 and Mak21/Noc1 comigrate in two smaller complexes. Both Rrp5 and Mak21/Noc1 comigrate in peak fractions 8–10 at ∼18S (predicted MW, 500 kDa) and in peak fractions 13–14 at ∼24S (predicted MW, 1400 kDa) (Fig. 4B), which suggests that Rrp5 and Mak21/Noc1 are found in two distinct complexes. As these complexes are predicted to be much larger than a heterodimeric complex of Rrp5 and Mak21/Noc1 (MW, 209 kDa), it is likely that these complexes contain other LSU biogenesis factors or more than one copy of either protein. Unlike the Grc3–Las1 complex, Rrp5 and Mak21/Noc1 also comigrate in peak fractions 17–19 as part of a putative complex that is larger than the 26S proteasome (Fig. 4A,B). Given the size, this larger complex is likely to be at least in part constituted by the LSU processome.
Likewise, Erb1, Nop7, and Ytm1 comigrate both in a discrete, smaller complex and as part of the LSU processome (Fig. 4C). All three proteins comigrate in peak fractions 4–6. Comparison of the sedimentation profiles of Erb1, Nop7, and Ytm1 with MW markers of the indicated S-values shows that the putative Erb1–Nop7–Ytm1 complex migrates at ∼10S with a predicted MW of 185 kDa. The predicted MW suggests that no more than one copy of each protein is present in this subcomplex. Thus, glycerol gradient sedimentation analysis reveals that the complexes previously identified by copurification and subsequently predicted by MCL clustering of the LSU processome interactome form complexes separate from the LSU processome, suggesting that they may be building blocks for LSU processome assembly.
The LSU processome interactome map uncovers several hub proteins that coordinate LSU assembly
The presence of one or more highly connected proteins, termed hub proteins, is a common, conserved property of interactome maps (Jeong et al. 2001; Yu et al. 2008; Ivanic et al. 2009). In addition to having many more interacting partners than the average protein, hub proteins are also more likely to be essential and more abundant and cause more pleiotropic phenotypes when deleted or mutated. As hub proteins are often found in the center of interactome networks and hold the network together, it has been speculated that hub proteins are more likely to be involved in human disease (Vidal et al. 2011).
The LSU processome interactome map uncovered a number of highly connected hub proteins, including Loc1, Nop4, Ebp2, Mak21/Noc1, and Noc2. These proteins have three to four times as many interacting partners as the average protein (6.2) in the high-confidence data set. For example, Loc1 has 25 interacting partners, and Nop4 has 23 interacting partners (Fig. 5A,C; Supplemental Fig. S1). Using the coimmunoprecipitation validation method (Fig. 2A), we assayed 16 of the 25 Loc1 PPIs and found that all 16 were positive by coimmunoprecipitation (Fig. 5B; Supplemental Table S4). Additionally, as controls, we performed three coimmunoprecipitations with proteins that did not interact with Loc1 in the Y2H screen, none of which was positive. Thus, Loc1 is a hub within the LSU processome.
Figure 5.
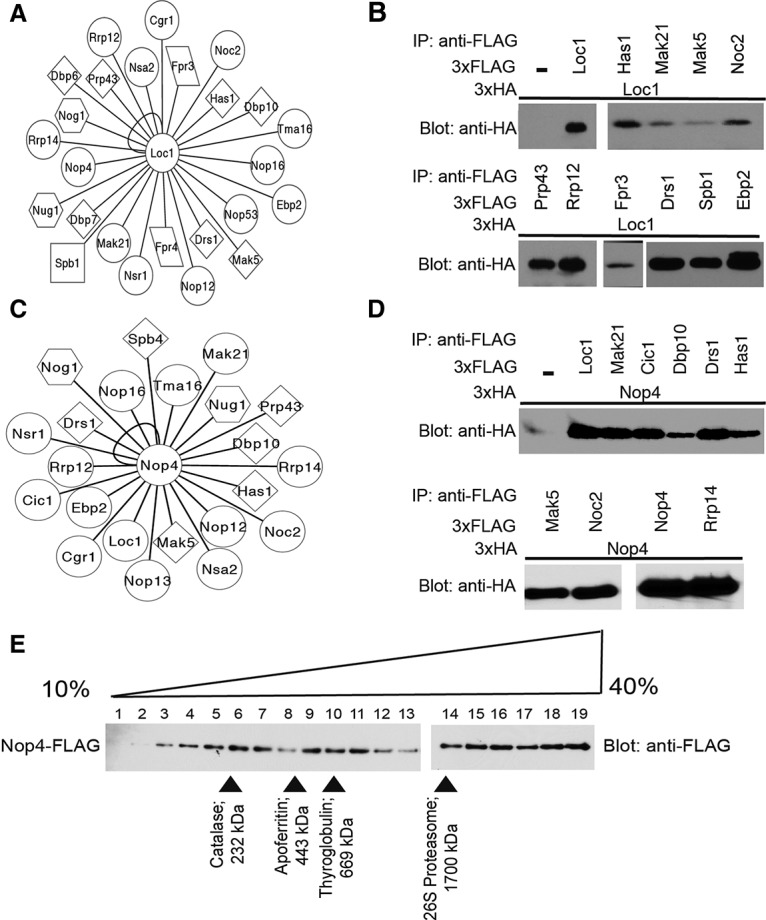
Loc1 and Nop4 are hub proteins within the LSU processome interactome. (A) Network of novel, high-confidence Y2H interactions identified for Loc1. Circles represent proteins with no known enzymatic activity, diamonds represent helicases, squares represent methyltransferases, hexagons represent GTPases, and parallelograms represent PPIases. The interaction network was created using Cytoscape. (B) Validation of the novel Loc1 interactions by coimmunoprecipitation. Yeast extract was incubated with α-Flag resin. Coimmunoprecipitations were assessed by α-HA Western blot. (C) Network of novel, high-confidence Y2H interactions identified for Nop4. Circles represent proteins with no known enzymatic activity, diamonds represent helicases, and hexagons represent GTPases. The interaction network was created using Cytoscape. (D) Validation of the novel Nop4 interactions by coimmunoprecipitation. Yeast extract was incubated with α-Flag resin. Coimmunoprecipitations were assessed by α-HA Western blot. (E) Glycerol gradient sedimentation analysis of Nop4. Yeast extract made from YPH499 Nop4-Flag was layered onto a 10%–40% glycerol gradient. The gradients were spun at 150,000g for 18 h, and fractions were harvested from the top of the gradient. All fractions were analyzed by α-Flag Western blot.
Similarly, we found that Nop4 is also a hub protein within the LSU processome. Nop4 is the yeast ortholog of RBM28, which is mutated in alopecia, neurological defects, and endocrinopathy (ANE) syndrome (Nousbeck et al. 2008). In the LSU interactome map, Nop4 interacts with 23 LSU processome proteins with high confidence. We assayed 14 of the 23 Nop4 PPIs using the coimmunoprecipitation validation method and found that all 14 were positive (Fig. 5D). Like Loc1, Nop4 functions as a hub protein within the LSU processome.
As hubs, Loc1 and Nop4 may interact with their partners as part of multiple subcomplexes or in the context of the LSU processome. To ascertain whether Loc1 or Nop4 form subcomplexes, we created endogenously tagged Loc1 and Nop4 yeast strains and subjected extract from these strains to glycerol gradient sedimentation. While we were unable to extract endogenous Loc1 under native conditions, we were able to analyze endogenously tagged Nop4 on a glycerol gradient. Since the tag on Nop4 does not confer a growth defect (Supplemental Fig. S2), it is not likely to interfere with Nop4 function or sedimentation on a glycerol gradient. We found that Nop4 sediments broadly over all of the gradient fractions and therefore is likely present in multiple protein complexes, consistent with a hub function (Fig. 5E).
Discussion
Advances in AP/MS in the past decade have enabled the identification of >200 ribosome biogenesis factors. A major challenge has been to determine how these proteins interact with one another to facilitate ribosome assembly, as AP/MS techniques largely report indirect interactions (Vidal et al. 2011). To define the binary PPIs among these proteins, we took a systems biology approach and performed a semi-high-throughput, array-based Y2H screen of 70 nucleolar LSU processome components to define the LSU processome interactome. The resulting interactome is comprised of 232 high-confidence interactions. Of the 232 high-confidence interactions, 211 have not been reported previously. We used an orthogonal, coimmunoprecipitation method to independently validate ∼25% of the high-confidence interactions with a 98% validation rate. Data set analysis via Markov clustering has provided novel insight into the organization, function, and regulation of the LSU processome, as it has led to the prediction of both previously identified and novel subcomplexes. The LSU processome interactome has also revealed the presence of five highly connected hub proteins.
This work has increased the number of PPIs among LSU processome components by fourfold. Despite the completion of three genome-wide, high-throughput Y2H studies and one genome-wide protein fragment complementation assay (PCA) to examine PPIs in S. cerevisiae (Uetz et al. 2000; Ito et al. 2001; Tarassov et al. 2008; Yu et al. 2008), only 56 Y2H interactions had previously been identified among the 70 nucleolar LSU processome components (Supplemental Table S1). It has been conservatively estimated that most proteins interact with approximately three or four other proteins (Blow 2009; Lim et al. 2011). Thus, the 56 previously identified Y2H interactions represent only ∼22%–26% of the predicted interactions, indicating that the LSU processome interactome had been far from complete. The lack of coverage in the published high-throughput, genome-wide screens in yeast is likely due to the use of pooled prey clones, an approach that may be less sensitive and confer a higher number of false negative interactions than directed, array-based screens (Koegl and Uetz 2007; Rajagopala and Uetz 2008; Lim et al. 2011). Directed, array-based Y2H screens such as this one allow for the generation of more complete interactomes because they assay each bait–prey pair individually. Similar directed, array-based Y2H approaches have been used successfully to map the interactomes of yeast cell polarity development, the yeast mitotic spindle, and the human spliceosome (Drees et al. 2001; Wong et al. 2007; Hegele et al. 2012). Additionally, a directed, array-based Y2H screen was conducted on a proteome-wide scale to map the PPIs of the human interactome (Rolland et al. 2014).
The principle goal of mapping the LSU processome interactome was to systematically interrogate the underlying organization of the LSU biogenesis factors within the LSU processome. Thus, to maximize the utility and information content of the LSU processome interactome, we integrated the high-confidence LSU processome interactome with the data from previously published studies (Supplemental Fig. S3). Previously, two functional clusters of LSU biogenesis factors had been defined by their common pre-rRNA processing defect observed upon mutational perturbation or genetic depletion (Sahasranaman et al. 2011; Talkish et al. 2012). Upon depletion or mutation of the seven A3 factors, the 27SA3 pre-rRNA intermediate accumulates, suggesting that these proteins are important for subsequent 27SA3 processing (Sahasranaman et al. 2011). Similarly, depletion or mutational perturbation of the 14 B factors results in the accumulation of the 27SB pre-rRNA intermediates, indicating that these proteins are important for 27SB processing (Talkish et al. 2012). However, with the exception of the subcomplex containing the A3 factors Erb1, Ytm1, and Nop7, these two functional clusters do not appear to form discrete subcomplexes in the LSU processome interactome, as both the A3 factors and the B factors have many more interactions with proteins outside their functional clusters than within their functional clusters (Supplemental Fig. S3). Interestingly, there are a number of interactions between the A3 factors and B factors that would physically link the two functional clusters and could potentially facilitate the advancement of assembly.
Markov clustering analysis of the high-confidence LSU processome interactome revealed novel insight into the organization of the LSU processome, as it predicted the existence of seven putative subcomplexes (Fig. 3E,F). Of the seven predicted subcomplexes, four are subcomplexes that have been described previously (Fig. 3E; Krogan et al. 2004; Miles et al. 2005; Rosado et al. 2007; Tang et al. 2008; Castle et al. 2013; Hierlmeier et al. 2013). Using glycerol gradient sedimentation analysis, we demonstrated that the Erb1–Nop7–Ytm1 subcomplex, the Grc3–Las1 subcomplex, and the Rrp5–Mak21/Noc1 subcomplex exist outside the LSU processome (Fig. 4), thereby validating their identification as subcomplexes. However, MCL analysis also predicted three novel subcomplexes (Fig. 3F) that warrant further investigation. Through the powerful combination of glycerol gradient sedimentation analysis and AP/MS, the existence of these putative subcomplexes outside the LSU processome and the complex members could be tested.
The LSU processome interactome has also revealed the presence of five hub proteins: Loc1, Nop4, Ebp2, Mak21/Noc1, and Noc2. Hub proteins have been classified as either date or party hubs based on how they interact with their partners (Han et al. 2004). Date hubs are thought to have multiple, sequential interactions separated in time or space, while party hubs are thought to have multiple simultaneous interactions (Han et al. 2004). As Y2H does not provide temporal information and assays only one PPI at a time, the assignation of date versus party hub cannot be confidently determined from the interactome alone. However, it is tempting to speculate that Loc1, Nop4, and Ebp2 are date hubs, as some of their interaction partners have been shown to function at different stages of LSU assembly (Figs. 3F, 5A,C; Supplemental Table S3; Sahasranaman et al. 2011; Talkish et al. 2012). As date hubs, Loc1, Nop4, and Ebp2 would coordinate and potentially drive assembly by sequentially interacting with different biogenesis factors.
Interestingly, hub proteins are predicted to preferentially encode disease-related genes (Vidal et al. 2011; De Las Rivas and Fontanillo 2012). Consistent with this, RBM28, the human ortholog of the hub protein Nop4, is mutated in the ribosomopathy ANE syndrome (Nousbeck et al. 2008). We propose that disruption of hub function may underlie the pathogenesis of this disease.
AP/MS studies have provided significant advances in our understanding of the composition and dynamic nature of the LSU processome, as they are designed to identify the members of protein complexes. Our understanding of the organization and function of the biogenesis factors within the LSU processome, though, has been stymied by the lack of knowledge of how these factors interact within the LSU processome. A Y2H approach such as the one described here is different from AP/MS in that it is more likely to map direct, binary interactions. Indeed, our Y2H analysis of the nucleolar LSU processome components has uncovered >200 novel, high-confidence interactions; identified key hub proteins, including one with a connection to human disease; and provided a blueprint for LSU processome organization. The LSU processome interactome will serve as a guide for future experiments designed to further probe the function of the 70 ribosome biogenesis factors dedicated to LSU assembly.
Materials and methods
Y2H analysis
ORFs encoding factors involved in the nucleolar steps in LSU assembly were either obtained in a Gateway Entry vector (pBG1805 or pDONR221) from the MORF library (Gelperin et al. 2005) or amplified using PCR and cloned into a Gateway Entry vector (pDONR221) (Supplemental Table S6). All ORFs were subsequently shuttled into both bait (pAS2-1) and prey (pACT2) Y2H destination vectors adapted for Gateway cloning (Life Technologies), as in Charette and Baserga (2010). All clones were fully sequenced by the DNA Analysis Facility at Yale University or Genewiz, Inc. ORFs cloned into the Gateway Entry vector have been deposited in AddGene.
All bait vectors were individually transformed into either PJ69-4α (MATα trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (James et al. 1996) or Y2H Gold (MATa trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1UAS-Gal1TATA-His3 GAL2UAS-Gal2TATA-Ade2 URA3::MEL1UAS-Mel1TATA AUR1-C MEL1) (Clontech). All prey vectors were individually transformed into either PJ69-4a (MATa trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (James et al. 1996) or Y187 (MATα trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ met-, URA3::GAL1-LacZ, MEL1) (Clontech) in a 96-well array format. All baits were screened for autoactivation on SD−Trp−His plates containing concentrations of 3-amino-1,2,4-triazole (3-AT) ranging from 0 mM to 100 mM. The optimal concentration of 3-AT was 6 mM, which is the concentration used for screening. An empty prey vector was included in the prey array as a negative control, and previously identified Y2H interactions served as positive controls. All baits were mated against all preys in a semi-high-throughput Y2H matrix screen (de Folter and Immink 2011). The mated yeast were transferred to SD−Leu−Trp plates to select for diploids. Diploids were then transferred to the selective medium SD−Leu−Trp−His + 6 mM 3-AT + 40 μg/mL 5-bromo-4-chloro-3-indolyl α-D-galactopyranoside (X-α-Gal) and SD−Leu−Trp−His−Ade + 6 mM 3-AT + 40 μg/mL X-α-Gal. Growth on the selective medium greater than that of the negative control after 3 wk was indicative of an interacting bait–prey pair.
PPIs were assayed three times. All observed interactions were assigned a confidence score. The confidence score represents the likelihood that the interaction is biologically relevant and was calculated using the following formula:
where x1 is growth in screen 1 (PJ69-4a/α), x2 is growth in screen 2 (Y2H Gold/Y187), x3 is growth in screen 3 (Y2H Gold/Y187), x4 is growth on SD−Leu−Trp−His + X-α-Gal + 6 mM 3-AT in screen 1, x5 is growth on SD−Leu−Trp−His−Ade + X-α-Gal + 6 mM 3-AT in screen 1, x6 is growth on SD−Leu−Trp−His + X-α-Gal + 6 mM 3-AT in screen 2, x7 is growth on SD−Leu−Trp−His−Ade + X-α-Gal + 6 mM 3-AT in screen 2, x8 is growth on SD−Leu−Trp−His + X-α-Gal + 6 mM 3-AT in screen 3, x9 is growth on SD−Leu− Trp−His−Ade + X-α-Gal + 6 mM 3-AT in screen 3, x10 represents observed in multiple strains, x11 is reciprocal interaction observed, x12 is known interaction, w1 is 0.12, w2 is 0.12, w3 is 0.12, w4 is 0.05, w5 is 0.07, w6 is 0.05, w7 is 0.07, w8 is 0.05, w9 is 0.07, w10 is 0.12, w11 is 0.15, and w12 is 0.01. When x is true, it has a value of 1. When x is not true, it has a value of 0. The complete set of observed interactions and the corresponding confidence scores are in Supplemental Table S2. The high-confidence interactions from this study have been submitted to the International Molecular Exchange (IMEx) Consortium (http://www.imexconsortium.org) through IntAct (Orchard et al. 2014) and assigned the identifier IM-23972.
Y2H result validation
Select ORFs were shuttled into the Gateway-modified yeast expression vectors p415GPD-3xHA-GW (LEU2 marker) and p414GPD-3xFlag-GW (TRP1 marker) (Mumberg et al. 1995). The Gateway-modified yeast expression vectors have been deposited in AddGene. The yeast expression vectors were cotransformed into YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1) (Sikorski and Hieter 1989). The resulting transformed strains were grown in medium containing 2% dextrose and lacking leucine and tryptophan (SD−Leu−Trp) at 30°C. Negative control strains were only transformed with p415GPD-3xHA-GW clones and were grown in medium containing 2% dextrose and lacking leucine (SD−Leu) at 30°C. For each coimmunoprecipitation, 20 mL of cells at an OD600 of ∼0.5 was collected, washed with water, and resuspended in NET2 (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 0.01% Nonidet P-40) with protease inhibitors (Roche mix). Cells were lysed with 0.5-mm glass beads. The lysate was cleared by centrifugation at 15,000g for 10 min at 4°C. Aliquots of 500 μL of lysate or ultracentrifuged lysate were incubated with α-Flag beads (Sigma) for 1 h at 4°C. The beads were washed a total of five times with NET2 to remove unbound protein and then resuspended in 20 μL of SDS loading dye. Immunoprecipitates were separated by 10% SDS-PAGE and transferred to a PVDF membrane. Western blot analysis was carried out with a 1:20,000 dilution of α-HA HRP (Roche).
Markov clustering analysis
The high-confidence LSU processome interactome was imported into Cytoscape (Shannon et al. 2003) and clustered using the MCL option in the Cytoscape plug-in clusterMaker (Morris et al. 2011) with the following settings: granularity parameter = 2.5 and array sources = confidence score. The MCL advanced settings were weak edge weight pruning threshold = 1 × 10−15, maximum residual value = 0.001, and iterations = 16.
Yeast strains
Strains carrying chromosomally integrated C-terminal TAP tag fusions were constructed in the parental strains YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3-Δ200 leu2-Δ1) or NOY504 (MATα rpa12::LEU2 leu2-3, 112 ura3-1 trp-1 his3-11 CAN1-100) (Nogi et al. 1992) using primers complementary to Ytm1 and Rrp5 and the plasmid pBS1479 (Puig et al. 2001). A strain carrying a chromosomally integrated C-terminal 13xMYC tag fusion was constructed in the parental strain YPH500 (MATα ura3-52 lys2-801 ade2-101 trp1Δ63 his3-Δ200 leu2-Δ1) (Sikorski and Hieter 1989) using primers complementary to Grc3 and the plasmid pFA6a-13Myc-His3MX6 (Longtine et al. 1998). Strains carrying chromosomally integrated C-terminal 3xFlag tag fusions were constructed in the parental strains YPH499 or NOY504 using primers complementary to Nop7, Erb1, Las1, Mak21/Noc1, and Nop4 and the plasmid p3Flag-KanMX (Gelbart et al. 2001). All strains carrying chromosomal integrations were verified by Western blot.
Glycerol gradient sedimentation analysis
For sedimentation analysis of YPH499 Nop4-Flag, Mak21/Noc1-Flag, and Rrp5-TAP, 60 mL of cells grown at 30°C to an OD600 of ∼0.5 was collected, washed with water, and resuspended in 1 mL of NET2. Cells were lysed with 0.5-mm glass beads. Aliquots of 800 μL of cleared lysate were run on 10%–40% glycerol gradients in NET2. Glycerol gradients were centrifuged in an SW41 rotor at 35,000 rpm for 18 h at 4°C and harvested in 600-μL fractions starting from the top of the gradient. Fractions were evaluated for the presence of the tagged protein by Western blot analysis with α-Flag HRP (1:20,000 dilution; Sigma) or PAP (1:6000 dilution; Sigma).
For glycerol gradient sedimentation analysis of Las1-Flag, Erb1-Flag, Nop7-Flag, and Ytm1-TAP, 60 mL of cells grown at 23°C to an OD600 of ∼0.5 was collected, washed with water, and resuspended in 10% glycerol RNPB (20 mM HEPES at pH 7.5 110 mM KOAc, 0.5% Triton, 0.1% Tween, 4 μg/mL pepstatin A, 180 μg/mL PMSF, 1:5000 anti-foam B [Sigma], 1:5000 protector RNase inhibitor [Roche]) (Oeffinger et al. 2007). For glycerol gradient sedimentation analysis of Grc3-MYC, 60 mL of cells grown at 30°C to an OD600 of ∼0.5 was collected, washed with water, and resuspended in 10% glycerol RNPB. Cells were lysed with 0.5-mm glass beads. Aliquots of 800 μL of cleared lysate were run on 10%–40% glycerol gradients in 20 mM HEPES (pH 7.5), 110 mM KOAc, 0.5% Triton, and 0.1% Tween. Glycerol gradients were centrifuged in an SW41 rotor at 35,000 rpm for 18 h at 4°C and harvested in 600-μL fractions starting from the top of the gradient. Fractions were evaluated for the presence of the tagged protein by Western blot analysis with α-MYC (1:5000 dilution; 9E10), α-Flag HRP (1:20,000 dilution; Sigma), or PAP (1:6000 dilution; Sigma).
The sedimentation of the endogenously tagged proteins in 10%–40% glycerol gradients was compared with sedimentation of protein markers of known S coefficient and MW, as in Bleichert and Baserga (2007). The 26S proteasome was the fourth protein marker. The yeast strain MHY6952 (Sa-Moura et al. 2013) was grown to mid-log phase (OD600 = 0.5) in YPD (1% yeast extract, 2% peptone, 2% dextrose) at 30°C and harvested. The cell pellet was washed once with water, frozen in liquid nitrogen, and ground in a Retsch mill according to Wälde and King (2014). Approximately 100 μL of cryolysate of the yeast strain MHY6952 was rapidly thawed in 900 μL of 10% glycerol RNPB. Cell debris was removed by centrifugation at 15,000g for 10 min. An aliquot of 800 μL of cleared lysate was run on a 10%–40% glycerol gradient in 20 mM HEPES (pH 7.5), 110 mM KOAc, 0.5% Triton, and 0.1% Tween. The glycerol gradient was centrifuged in an SW41 rotor at 35,000 rpm for 18 h at 4°C and harvested in 600-μL fractions from the top of the gradient. Fractions were evaluated for the presence of the tagged protein by Western blot analysis with α-Flag HRP (1:20,000 dilution; Sigma).
Supplementary Material
Acknowledgments
We thank Megan King for the helpful suggestions, and Baserga laboratory members for critical discussion and reading the manuscript. K.L.M. was supported by the National Institute of Health (NIH) predoctoral training grant in genetics (T32 GM 007499). J.M.C was supported by a postdoctoral NIH-Ruth L. Kirschstein National Research Service Award and an Institutional Research Training grant (Radiation Therapy, Biology, Physics T32 CA 009259). N.G.V. was supported by the NIH predoctoral training grant in cellular and molecular biology (T32 GM 007223). This work was supported by National Institute of General Medical Sciences grant 52581 to S.J.B.. S.J.B. is a Bohmfalk Scholar in Medical Research.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.256370.114.
References
- Bassler J, Grandi P, Gadal O, Lessmann T, Petfalski E, Tollervey D, Lechner J, Hurt E. 2001. Identification of a 60S preribosomal particle that is closely linked to nuclear export. Mol Cell 8: 517–529. [DOI] [PubMed] [Google Scholar]
- Bassler J, Kallas M, Pertschy B, Ulbrich C, Thoms M, Hurt E. 2010. The AAA–ATPase Rea1 drives removal of biogenesis factors during multiple stages of 60S ribosome assembly. Mol Cell 38: 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Baserga SJ. 2007. The long unwinding road of RNA helicases. Mol Cell 27: 339–352. [DOI] [PubMed] [Google Scholar]
- Blow N. 2009. Systems biology: untangling the protein web. Nature 460: 415–418. [DOI] [PubMed] [Google Scholar]
- Bradatsch B, Leidig C, Granneman S, Gnädig M, Tollervey D, Böttcher B, Beckmann R, Hurt E. 2012. Structure of the pre-60S ribosomal subunit with nuclear export factor Arx1 bound at the exit tunnel. Nat Struct Mol Biol 19: 1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohee S, van Helden J. 2006. Evaluation of clustering algorithms for protein–protein interaction networks. BMC Bioinformatics 7: 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner A, Polge C, Lentze N, Auerbach D, Schlattner U. 2009. Yeast two-hybrid, a powerful tool for systems biology. Int J Mol Sci 10: 2763–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle CD, Sardana R, Dandekar V, Borgianini V, Johnson AW, Denicourt C. 2013. Las1 interacts with Grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae. Nucleic Acids Res 41: 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette JM, Baserga SJ. 2010. The DEAD-box RNA helicase-like Utp25 is an SSU processome component. RNA 16: 2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Folter S, Immink RG. 2011. Yeast protein–protein interaction assays and screens. Methods Mol Biol 754: 145–165. [DOI] [PubMed] [Google Scholar]
- De Las Rivas J, Fontanillo C. 2012. Protein–protein interaction networks: unraveling the wiring of molecular machines within the cell. Brief Funct Genomics 11: 489–496. [DOI] [PubMed] [Google Scholar]
- De Marchis ML, Giorgi A, Schinina ME, Bozzoni I, Fatica A. 2005. Rrp15p, a novel component of pre-ribosomal particles required for 60S ribosome subunit maturation. RNA 11: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, et al. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417: 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees BL, Sundin B, Brazeau E, Caviston JP, Chen G-C, Guo W, Kozminski KG, Lau MW, Moskow JJ, Tong A, et al. 2001. A protein interaction map for cell polarity development. J Cell Biol 154: 549–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Tollervey D. 2002. Making ribosomes. Curr Opin Cell Biol 14: 313–318. [DOI] [PubMed] [Google Scholar]
- Fatica A, Cronshaw AD, Dlakic M, Tollervey D. 2002. Ssf1p prevents premature processing of an early pre-60S ribosomal particle. Mol Cell 9: 341–351. [DOI] [PubMed] [Google Scholar]
- Fernandez-Pevida A, Kressler D, de la Cruz J. 2015. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip Rev RNA 6: 191–209. [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M, Senger B, Saveanu C, Fasiolo F. 2003. Ribosome assembly in eukaryotes. Gene 313: 17–42. [DOI] [PubMed] [Google Scholar]
- Galani K, Nissan TA, Petfalski E, Tollervey D, Hurt E. 2004. Rea1, a dynein-related nuclear AAA–ATPase, is involved in late rRNA processing and nuclear export of 60 S subunits. J Biol Chem 279: 55411–55418. [DOI] [PubMed] [Google Scholar]
- Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ. 2004. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev 18: 2506–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Rechsteiner T, Richmond TJ, Tsukiyama T. 2001. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol Cell Biol 21: 2098–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19: 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. 2003. A protein interaction map of Drosophila melanogaster. Science 302: 1727–1736. [DOI] [PubMed] [Google Scholar]
- Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schäfer T, Kuster B, Tschochner H, Tollervey D, et al. 2002. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell 10: 105–115. [DOI] [PubMed] [Google Scholar]
- Granneman S, Petfalski E, Tollervey D. 2011. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. EMBO J 30: 4006–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob A, Colleran C, McStay B. 2014. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev 28: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, et al. 2004. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 430: 88–93. [DOI] [PubMed] [Google Scholar]
- Harnpicharnchai P, Jakovljevic J, Horsey E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, et al. 2001. Composition and functional characterization of yeast 66S ribosome assembly intermediates. Mol Cell 8: 505–515. [DOI] [PubMed] [Google Scholar]
- Hart GT, Lee I, Marcotte ER. 2007. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. 2012. Dynamic protein–protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. [DOI] [PubMed] [Google Scholar]
- Henras AK, Soudet J, Gérus M, Lebaron S, Caizergues-Ferrer MM, Mougin A, Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65: 2334–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierlmeier T, Merl J, Sauert M, Perez-Fernandez J, Schultz P, Bruckmann A, Hamperl S, Ohmayer U, Rachel R, Jacob A, et al. 2013. Rrp5p, Noc1p and Noc2p form a protein module which is part of early large ribosomal subunit precursors in S. cerevisiae. Nucleic Acids Res 41: 1191–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci 98: 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanic J, Yu X, Wallqvist A, Reifman J. 2009. Influence of protein abundance on high-throughput protein–protein interaction detection. PLoS One 4: e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabási AL, Oltvai ZN. 2001. Lethality and centrality in protein networks. Nature 411: 41–42. [DOI] [PubMed] [Google Scholar]
- Koegl M, Uetz P. 2007. Improving yeast two-hybrid screening systems. Brief Funct Genomic Proteomic 6: 302–312. [DOI] [PubMed] [Google Scholar]
- Kressler D, Roser D, Pertschy B, Hurt E. 2008. The AAA ATPase Rix7 powers progression of ribosome biogenesis by stripping Nsa1 from pre-60S particles. J Cell Biol 181: 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressler D, Hurt E, Bassler J. 2010. Driving ribosome assembly. Biochim Biophys Acta 1803: 673–683. [DOI] [PubMed] [Google Scholar]
- Kressler D, Hurt E, Bergler H, Bassler J. 2012. The power of AAA–ATPases on the road of pre-60S ribosome maturation--molecular machines that strip pre-ribosomal particles. Biochim Biophys Acta 1823: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, et al. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell 13: 225–239. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- Lebaron S, Segerstolpe A, French SL, Dudnakova T, de Lima Alves F, Granneman S, Rappsilber J, Beyer AL, Wieslander L, Tollervey D. 2013. Rrp5 binding at multiple sites coordinates pre-rRNA processing and assembly. Mol Cell 52: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton A, Saveanu C, Decourty L, Jacquier A, Fromont-Racine M. 2006. Nsa2 is an unstable, conserved factor required for the maturation of 27 SB pre-rRNAs. J Biol Chem 281: 27099–27108. [DOI] [PubMed] [Google Scholar]
- Lebreton A, Rousselle J-C, Lenormand P, Namane A, Jacquier A, Fromont-Racine M, Saveanu C. 2008. 60S ribosomal subunit assembly dynamics defined by semi-quantitative mass spectrometry of purified complexes. Nucleic Acids Res 36: 4988–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al. 2004. A map of the interactome network of the metazoan C. elegans. Science 303: 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lee I, Moradi E, Hung NJ, Johnson AW, Marcotte EM. 2009. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol 7: e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WQ, Fournier MJ. 1997. Synthesis of functional eukaryotic ribosomal RNAs in trans: development of a novel in vivo rDNA system for dissecting ribosome biogenesis. Proc Natl Acad Sci 94: 2864–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YH, Charette JM, Baserga SJ. 2011. Assembling a protein–protein interaction map of the SSU processome from existing datasets. PLoS One 6: e17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Merl J, Jakob S, Ridinger K, Hierlmeier T, Deutzmann R, Milkereit P, Tschochner H. 2010. Analysis of ribosome biogenesis factor-modules in yeast cells depleted from pre-ribosomes. Nucleic Acids Res 38: 3068–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles TD, Jakovljevic J, Horsey EW, Harnpicharnchai P, Tang L, Woolford JL Jr. 2005. Ytm1, Nop7, and Erb1 form a complex necessary for maturation of yeast 66S preribosomes. Mol Cell Biol 25: 10419–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkereit P, Gadal O, Podtelejnikov A, Trumtel S, Gas N, Petfalski E, Tollervey D, Mann M, Hurt E, Tschochner H. 2001. Maturation and intranuclear transport of pre-ribosomes requires Noc proteins. Cell 105: 499–509. [DOI] [PubMed] [Google Scholar]
- Morita D, Miyoshi K, Matsui Y, Toh EA, Shinkawa H, Miyakawa T, Mizuta K. 2002. Rpf2p, an evolutionarily conserved protein, interacts with ribosomal protein L11 and is essential for the processing of 27 SB Pre-rRNA to 25 S rRNA and the 60 S ribosomal subunit assembly in Saccharomyces cerevisiae. J Biol Chem 277: 28780–28786. [DOI] [PubMed] [Google Scholar]
- Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE. 2011. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12: 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschopoulos CN, Pavlopoulos GA, Iacucci E, Aerts J, Likothanassis S, Schneider R, Kossida S. 2011. Which clustering algorithm is better for predicting protein complexes? BMC Res Notes 4: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Müller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122. [DOI] [PubMed] [Google Scholar]
- Nastou KC, Tsaousis GN, Kremizas KE, Litou ZI, Hamodrakas SJ. 2014. The human plasma membrane peripherome: visualization and analysis of interactions. BioMed Res Int 2014: 397145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissan TA, Bassler J, Petfalski E, Tollervey D, Hurt E. 2002. 60S pre-ribosome formation viewed from assembly in the nucleolus until export to the cytoplasm. EMBO J 21: 5539–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi Y, Yano R, Dodd J, Carles C, Nomura M. 1992. Gene RRN4 in Saccharomyces cerevisiae encodes the A12.2 subunit of RNA polymerase I and is essential only at high temperatures. Mol Cell Biol 13: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousbeck J, Spiegel R, Ishida-Yamamoto A, Indelman M, Shani-Adir A, Adir N, Lipkin E, Bercovici S, Geiger D, van Steensel MA, et al. 2008. Alopecia, neurological defects, and endocrinopathy syndrome caused by decreased expression of RBM28, a nucleolar protein associated with ribosome biogenesis. Am J Hum Genet 82: 1114–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP. 2007. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods 4: 951–956. [DOI] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. 2014. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42: D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheim YN, French SL, Keck KM, Champion EA, Spasov K, Dragon F, Baserga SJ, Beyer AL. 2004. Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell 16: 943–954. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal JB, Enright AJ, Ouzounis CA. 2004. Detection of functional modules from protein interaction networks. Proteins 54: 49–57. [DOI] [PubMed] [Google Scholar]
- Pérez-Fernández J, Román A, De Las Rivas J, Bustelo XR, Dosil M. 2007. The 90S preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol Cell Biol 27: 5414–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu S, Vlasblom J, Emili A, Greenblatt J, Wodak SJ. 2007. Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 7: 944–960. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229. [DOI] [PubMed] [Google Scholar]
- Rajagopala SV, Uetz P. 2008. Analysis of protein–protein interactions using array-based yeast two-hybrid screens. Methods Mol Biology 548: 223–245. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Galán O, García-Gómez JJ, de la Cruz J. 2013. Yeast and human RNA helicases involved in ribosome biogenesis: current status and perspectives. Biochim Biophys Acta 1829: 775–790. [DOI] [PubMed] [Google Scholar]
- Rolland T, Taşan M, Charloteaux B, Pevzner Samuel J, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. 2014. A proteome-scale map of the human interactome network. Cell 159: 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado IV, de la Cruz J. 2004. Npa1p, a component of very early pre-60S ribosomal particles, associates with a subset of small nucleolar RNPs required for peptidyl transferase center modification. Mol Cell Biol 24: 6324–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado IV, Dez C, Lebaron S, Caizergues-Ferrer M, Henry Y, de la Cruz J. 2007. Characterization of Saccharomyces cerevisiae Npa2p (Urb2p) reveals a low-molecular-mass complex containing Dbp6p, Npa1p (Urb1p), Nop8p, and Rsa3p involved in early steps of 60S ribosomal subunit biogenesis. Mol Cell Biol 27: 1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasranaman A, Dembowski J, Strahler J, Andrews P, Maddock J, Woolford JL. 2011. Assembly of Saccharomyces cerevisiae 60S ribosomal subunits: role of factors required for 27S pre-rRNA processing. EMBO J 30: 4020–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Moura B, Funakoshi M, Tomko RJ Jr, Dohmen RJ, Wu Z, Peng J, Hochstrasser M. 2013. A conserved protein with AN1 zinc finger and ubiquitin-like domains modulates Cdc48 (p97) function in the ubiquitin-proteasome pathway. J Biol Chem 288: 33682–33696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveanu C, Namane A, Gleizes PE, Lebreton A, Rousselle JC, Noaillac-Depeyre J, Gas N, Jacquier A, Fromont-Racine M. 2003. Sequential protein association with nascent 60S ribosomal particles. Mol Cell Biol 23: 4449–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveanu C, Rousselle JC, Lenormand P, Namane A, Jacquier A, Fromont-Racine M. 2007. The p21-activated protein kinase inhibitor Skb15 and its budding yeast homologue are 60S ribosome assembly factors. Mol Cell Biol 27: 2897–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoji K, Jakovljevic J, Tsuchihashi K, Umeki Y, Wan K, Kawasaki S, Talkish J, Woolford JL Jr, Mizuta K. 2012. Ebp2 and Brx1 function cooperatively in 60S ribosomal subunit assembly in Saccharomyces cerevisiae. Nucleic Acids Res 40: 4574–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. 2005. A human protein–protein interaction network: a resource for annotating the proteome. Cell 122: 957–968. [DOI] [PubMed] [Google Scholar]
- Strunk BS, Karbstein K. 2009. Powering through ribosome assembly. RNA 15: 2083–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B. 2012. The Cross-and-Capture system: a versatile tool in yeast proteomics. Methods 58: 360–366. [DOI] [PubMed] [Google Scholar]
- Suter B, Fetchko MJ, Imhof R, Graham CI, Stoffel-Studer I, Zbinden C, Raghavan M, Lopez L, Beneti L, Hort J, et al. 2007. Examining protein-protein interactions using endogenously tagged yeast arrays: the cross-and-capture system. Genome Res 17: 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkish J, Zhang J, Jakovljevic J, Horsey EW, Woolford JL Jr. 2012. Hierarchical recruitment into nascent ribosomes of assembly factors required for 27SB pre-rRNA processing in Saccharomyces cerevisiae. Nucleic Acids Res 40: 8646–8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Sahasranaman A, Jakovljevic J, Schleifman E, Woolford JL Jr. 2008. Interactions among Ytm1, Erb1, and Nop7 required for assembly of the Nop7-subcomplex in yeast preribosomes. Mol Biol Cell 19: 2844–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. 2008. An in vivo map of the yeast protein interactome. Science 320: 1465–1470. [DOI] [PubMed] [Google Scholar]
- Thomson E, Ferreira-Cerca S, Hurt E. 2013. Eukaryotic ribosome biogenesis at a glance. J Cell Sci 126: 4815–4821. [DOI] [PubMed] [Google Scholar]
- Tschochner H, Hurt E. 2003. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol 13: 255–263. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. 2000. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627. [DOI] [PubMed] [Google Scholar]
- Ulbrich C, Diepholz M, Bassler J, Kressler D, Pertschy B, Galani K, Böttcher B, Hurt E. 2009. Mechanochemical removal of ribosome biogenesis factors from nascent 60S ribosomal subunits. Cell 138: 911–922. [DOI] [PubMed] [Google Scholar]
- Venkatesan K, Rual J-F, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh K-I, et al. 2009. An empirical framework for binary interactome mapping. Nat Methods 6: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Cusick ME, Barabási A-L. 2011. Interactome networks and human disease. Cell 144: 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälde S, King MC. 2014. The KASH protein Kms2 coordinates mitotic remodeling of the spindle pole body. J Cell Sci 127: 3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Huo K, Ma L, Tang L, Li D, Huang X, Yuan Y, Li C, Wang W, Guan W, et al. 2011. Toward an understanding of the protein interaction network of the human liver. Mol Syst Biol 7: 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Nakajima Y, Westermann S, Shang C, Kang J-S, Goodner C, Houshmand P, Fields S, Chan CS, Drubin D, et al. 2007. A protein interaction map of the mitotic spindle. Mol Biol Cell 18: 3800–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. 2008. High-quality binary protein interaction map of the yeast interactome network. Science 322: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.