

Abstract
Hemoglobin (Hb) is a highly conserved molecule present in all life forms and functionally tied to the complexity of aerobic organisms on earth in utilizing oxygen from the atmosphere and delivering to cells and tissues. This primary function sustains the energy requirements of cells and maintains cellular homeostasis. Decades of intensive research has presented a paradigm shift that shows how the molecule also functions to facilitate smooth oxygen delivery through the cardiovascular system for cellular bioenergetic homeostasis and signaling for cell function and defense. These roles are particularly highlighted in the binding of Hb to gaseous molecules carbon dioxide (CO2), nitric oxide (NO) and carbon monoxide (CO), while also serving indirectly or directly as sources of these signaling molecules. The functional activities impacted by Hb outside of bioenergetics homeostasis, include fertilization, signaling functions, modulation of inflammatory responses for defense and cell viability. These activities are efficiently executed while Hb is sequestered safely within the confines of the red blood cell (rbc). Outside of rbc confines, Hb disaggregates and becomes a danger molecule to cell survival. In these perpectives, Hb function is broadly dichotomous, either a friend in its natural environment providing and facilitating the means for cell function or foe when dislocated from its habitat under stress or pathological condition disrupting cell function. The review presents insights into how this dichotomy in function manifests.
Keywords: hemoglobin, gaseous molecules, intravascular and extravascular hemolysis, inflammation, ROS
Introduction
Hemoglobin (Hb), a protein that is found in all life forms was discovered by F L Hunefeld in 1840 at Liepzig University (Sheftel et al., 2012). It consists of a prosthetic heme molecule [Fe II coordinated to a tetrapyrole ring (protoporphyrin IX)] joined to globin chains (Schechter, 2008, 2012). Vincenzo Menghini first established that iron in blood is concentrated in the rbc (Busacchi, 1958). Depending on the species or organism Hb functions to transport or store oxygen, offer protection against reactive oxygen species and detoxify nitric oxide (NO), nitrite and peroxides (Nishi et al., 2008; Schechter, 2008; Tiso et al., 2011). In humans and vertebrates, it lies at the cross road of energy transduction from the external environment to the internal milieu of living systems. Its concentration in blood is 150 g/l so that an adult human body contains approximately 1 kg of Hb (Olsson et al., 2012)The functional activities of hemoglobin are essentially dichotomous: (a) to provide a means for binding to oxygen and facilitate its delivery for cellular bioenergetics homeostasis, (b) to be a direct or indirect source of other gaseous molecules, bind to them and in the process moderate red blood cell (rbc) and cellular health, function, defense or damage to the cells. These activities are friendly, salutary and sustaining to cell health and tissue homeostasis when considered vis-à-vis the high concentration of hemoglobin in rbc or outright inimical to cell survival. This review will first present an overview of the general function and reactivity of Hb highlighting aspects that are friendly to cell health and those that are foe like and damaging. The later part will then have a detailed presentation on the synthesis of hemoglobin in humans, its importance for cell function in relation to binding to gaseous molecules and the harm it causes when released outside of its confinement in rbc.
General Hb function and reactivity
The history of life on earth is closely linked to the evolution of oxygen supply in the atmosphere. This link is central to energy needs of cells and cellular homeostasis in energy transformation (Taylor and Cummins, 2011). The evolution of cyanobacteria with the potential of harnessing energy from the sun with transduction into stored macromolecules in photosynthesis and the release of oxygen, started the process of oxygenation of the atmosphere on the earth (Semenza, 2007). Life forms with increasing complexity including humans have adapted to oxygen usage through Hb in respiration where oxygen serves as the final electron acceptor in the electron transport chain, localized within the mitochondrion (Rich, 2003). While the structure of Hb is optimal to capture oxygen and deliver it to the tissues, it also presents a challenge of auto-oxidation that can modify its structure and lead to generation of reactive oxygen species (ROS) and cellular damage (Reeder et al., 2008). This dichotomy in function makes hemoglobin a double edged sword with one face as a friend and the other a foe under stress.
The primary function of Hb in the capture and release of oxygen is facilitated by heme (with Fe II) which also enables the reversible binding of carbon monoxide (CO) and NO through heme based sensor proteins (Chay and Brillhart, 1974; Girvan and Munro, 2013) while binding of carbon dioxide (CO2) is allosterically regulated in a heterotropic interaction with the terminal α-amino groups of Hb through rbc band 3 protein complex and soluble adenylcyclase sensing (sAC) (Bauer and Kurtz, 1977; De Rosa et al., 2008; Buck and Levin, 2011). The binding of hemoglobin to oxygen molecule does not lead to a change in the oxidative state of iron (Rifkind and Nagababu, 2013). However, because at physiological pH oxygen has a higher redox potential than Fe, sometimes hemoglobin undergoes auto-oxidation with oxygen being reduced to the superoxide anion (O·−2) and generation of metHb/hemin (Fe3+) (Bonaventura et al., 2013). This reaction is particularly enhanced under hypoxia (Abugo and Rifkind, 1994). In hemin/metHb, the binding affinity of globin to heme is weak and can lead to loss of heme. This activity is known to affect about 1–3% of Hb under normal physiological conditions (Alayash, 2004). The generation of superoxide and hemin may be beneficial or detrimental, but the release of heme from Hb binding can be damaging when in excess (Aft and Mueller, 1983; Schmitt et al., 1993; Girvan and Munro, 2013). Superoxide being a reactive oxygen species (ROS) is important for signaling function under normal physiology when present in moderate amounts (Reth, 2002; Taniyama and Griendling, 2003). Similarly hemin has a net positive charge, so it can react with nitrites (anions) whether in oxyhemoglobin or deoxyhemoglobin to regenerate hemin and NO (Cosby et al., 2003; Huang et al., 2005). Because hemin is unable to bind oxygen, it does not take part in oxygen transport. NO on the other hand is important for signaling functions, regulation of vascular tone, modulation of the immune response and moderation of endothelial adhesion molecule expression among others (Guzik et al., 2003; Forstermann and Sessa, 2012). So a limited release of heme under normal circumstances is useful. In addition anti-oxidants in the rbc or in circulation are able to offset any potentially harmful degradative products. These include glutathione, ascorbic acid, glutathione peroxidase and peroxiredoxin in the rbc (Ascenzi et al., 2005; Lutz and Bogdanova, 2013). In circulation the major scavengers that limit hemoglobin and heme damage are haptoglobin and hemopexin respectively (Nielsen and Moestrup, 2009; Reeder, 2010; Tolosano et al., 2010; Belcher et al., 2013; Schaer et al., 2014). However, under stress or pathological conditions, the moderating effect of the scavengers is overwhelmed so that a cascade of ROS generated from Hb auto-oxidation is not dampened. In that circumstance NO can react with superoxide to generate peroxynitrite (ONOO−) which sets up a cascade of ROS generation that are damaging to cells and tissues (Griendling and FitzGerald, 2003; Pacher et al., 2007). This is augmented by the release of heme intravascularly or extravascularly (Schaer et al., 2013).
Hemoglobin binding to CO and CO2 also serves important signaling functions (Maines, 1997; Otterbein et al., 2000; Schechter, 2008). CO has been shown as a signaling molecule in the brain, a messenger molecule in the gastrointestinal tract, and an inducer of hyperpolarization in circular smooth muscle cells (Maines, 1997; Amano and Camara, 2013). CO2 which is produced largely in the TCA cycle in cellular metabolism affects tissue homeostasis (e.g., disrupted pH changes) that can lead to death or play an important role in inhibiting inflammation (Buck and Levin, 2011; Cummins et al., 2014). So Hb not only ensures cellular energy supply through oxygen delivery, but also facilitates cellular homeostasis through signaling functions of the gases to which it binds. These functional activities of hemoglobin are friendly, sustaining and salubrious to cell health and tissue homeostasis when considered vis-à-vis the high concentration of hemoglobin in rbc. The review will presently give details on the synthesis of hemoglobin in humans, its importance for cell function and the harm it causes when released outside of its confinement in rbc.
Human hemoglobin synthesis
Human hemoglobin is produced in erythroid cells and in adults is made up of two major tetramers, HbA (97%) consisting of two alpha and two beta globin polypeptides (α2β2) and HbA2 (2%) consisting of two alpha and two delta globin polypeptides (α2δ2) (Perutz, 1979). In the fetus, the Hb molecule referred to as HbF consists of two alpha and two gamma chains (α2γ2). This phenotype can be present in adults up to 1% (Schechter, 2008). After birth, the gamma chains are replaced with beta chains gradually (Perutz, 1979).
The genes for the alpha globin chains are located on chromosome 16 whereas chromosome 11 harbors the genes for the beta globin chains (Huehns et al., 1964). During ontogenesis, the embryonic alpha chains (ζ) are replaced by the adult type (α) which is maintained throughout adulthood (Huehns et al., 1964). However, the expression of the genes for the globin chains in erythroid cells differ based on the developmental stage. The β-globin genes are arranged sequentially from the 5′ region closest to the locus control region (LCR), in the order in which they are expressed during ontogenesis toward the 3′ region (Huehns et al., 1964). This order is ε (HBE), Gγ(HBG2), Aγ(HBG1), δ(HBD), and β(HBB)-3′) expressed depending on switches in gene expression (Figure 1) (Huehns et al., 1964). These switches are from embryonic (HBE) to fetal (HBG2 and HBG1) stage and fetal to adult stage (HBD and HBB), regulated by the LCR of the globin gene cluster (Schechter, 2008; Razin et al., 2012; Moleirinho et al., 2013). The HBB gene is more highly expressed in adults than the HBD gene which is why adult hemoglobin is made up largely (97%) of HbA and about 2% of HbA2. The prosthetic group heme is synthesized through a series of eight step enzymatic reactions half of which occurs in the cytosol and a half in mitochondria (Heinemann et al., 2008; Chiabrando et al., 2014). After the final step which occurs in the mitochondria matrix, heme is targeted into hemoproteins either membrane bound or in the cytosol (Schultz et al., 2010; Larsen et al., 2012; Korolnek and Hamza, 2014). Hemoglobin and myoglobin acquire heme in the cytosol (Bruns and London, 1965). In these compounds heme (type b) is non-covalently coordinated to each globin chain (alpha and beta) through Fe II. It is postulated that heme acquired into cytosolic globin molecules may be derived from mitochondria or imported through plasma membrane importers or the endocytic pathway (Wu et al., 2006; Korolnek and Hamza, 2014).
Figure 1.
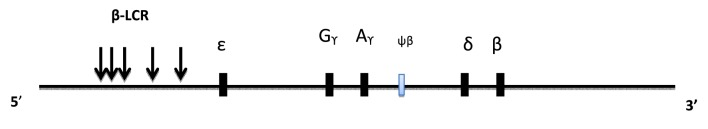
Organization of the β globin genes on chromosome 11. Gene expression occurs sequentially from the 5′ region in ontogenesis. The lightly shaded rectangle represent a pseudogene.
The friendly role of hemoglobin in cell physiology
The friendly role of Hb is defined in part by its ability to bind gases in particular O2, NO, CO and CO2 for transport. The binding affinity of the gases to Hb through heme are in the order NO>>CO>> O2 (Chapman and Cokelet, 1997; Reeder, 2010; Olsson et al., 2012). This activity is fundamental to the survival of multicellular organisms. Indeed the principal function of the cardiovascular system is essentially to deliver oxygen and nutrients, while facilitating the removal of carbon dioxide and metabolic waste in the process (Mairbaurl and Weber, 2012).
Hemoglobin and O2
The transport of oxygen from the lungs to tissues is mediated by the Hb tetramer with heme iron in the ferrous state (Fe2+) binding four oxygen molecules (Reedy and Gibney, 2004; Ascenzi et al., 2005; Buehler and Alayash, 2005). In the lungs oxygen diffuses across the alveolar barrier into the blood where it is bound by Hb (Eaton, 1974). Through the activity of the cardiovascular system, oxygen is delivered into tissues by way of the microcirculation comprising the arterioles, venules and capillaries to maintain cellular bioenergetics homeostasis (Haldar and Stamler, 2013). The blood oxygen content which invariably depends on hemoglobin oxygen saturation determines the amount of oxygen delivered to tissues (Mairbaurl and Weber, 2012; Haldar and Stamler, 2013). A decrease in pH or an increase in the partial pressure of CO2 (P CO2) in the systemic capillaries decreases the affinity of oxygen for Hb (Bohr Effect) leading to tissue delivery of oxygen (Dash and Bassingthwaighte, 2006). Tissue oxygen gradient in turn regulates blood flow in the microcirculation, levels of erythropoietin synthesized for erythropoiesis and oxygen delivery (Siri et al., 1966; Hebbel et al., 1978). Hypoxia defined as decreased oxygen levels in the microenvironment of cells or tissues relative to the normal physiological state (demand does not match supply), increases tissue perfusion, erythropoiesis and oxygen delivery (Mairbaurl, 1994; Semenza, 2009, 2011). Hyperoxia has the reverse effect (Piantadosi, 1999b). When oxygen is delivered in the tissues, cytochrome oxidase, the final enzyme in the mitochondria electron transport chain uses the oxygen as the final electron acceptor to generate ATP.
In addition nitric oxide synthase isoforms utilize molecular oxygen together with NADPH and arginine as substrates to generate NO, an important signaling molecule in the central nervous system (CNS) and peripheral nervous system (Robinson et al., 2011). NO can also be generated from a reaction between deoxyhemoglobin and nitrite following oxygen delivery or in hypoxic conditions (Huang et al., 2005).
The microcirculation is facilitated by cellular adaptations to hypoxia which includes improved ventilation and lung perfusion, augmented cardiac contractility, increased vasodilation and regulated ATP release (Jagger et al., 2001; Sprague et al., 2007; Gladwin and Kim-Shapiro, 2012). At the molecular level, a transcription factor, hypoxia inducible factor (HIF) coordinates the responses to hypoxia (Semenza, 2009; Scholz and Taylor, 2013) through the activity of hydroxylases. When oxygen is available, hydroxylases (which are absolutely dependent on molecular oxygen) are activated to hydroxylate HIF for degradation, whereas limited oxygen supply leads to HIF stabilization and expression of genes affecting vascular tone, erythropoiesis, angiogenesis and metabolism (Schofield and Ratcliffe, 2005; Taylor, 2008; Eltzschig and Carmeliet, 2011). These coordinated activities ensure a smooth link for delivery of oxygen and nutrients to tissues with removal of waste and carbon dioxide, so that adequate blood flow through the microcirculation and attendant oxygen supply is maintained for cell health and survival. Decreases in rbc hemoglobin as occurs in hemolysis, or pathological states such as sickle cell disease, malaria, paroxysmal nocturnal hemoglobinuria (PNH), directly leads to decrease in tissue oxygen delivery with attendant pathological consequences (Sikora et al., 2014). The process of oxygen delivery is also regulated by allosteric factors including protons (H+), CO2, organic phosphates, chloride (Cl-) and 2,3-bisphosphoglycerate (Mairbaurl and Weber, 2012). Structural changes at the molecular level in hemoglobin that alters the affinity of the molecule for oxygen or its response to the allosteric modulators, also contribute to the reversible binding to oxygen. The primary function of Hb in oxygen delivery is clearly also coupled to maintaining energy homeostasis, through processes that augment oxygen availability to cells and tissues.
Hemoglobin and CO2
In contrast to oxygen, carbon dioxide is generated in multicellular organisms through the TCA cycle and during aerobic metabolism (Buck and Levin, 2011; Cummins et al., 2014). Carbon dioxide not only contributes to the buffering system in blood but also engages in signaling activity in critical life processes including sperm activation, blood flow and dampening inflammation (Boatman and Robbins, 1991; Buck and Levin, 2011; Taylor and Cummins, 2011; Cummins et al., 2014). As part of the buffering systems in the blood, the enzyme carbonic anhydrase hydrates CO2 to H2CO3 which is transported to the lungs where it readily dissociates through the activity of carbonic anhydrase to HCO−3 and H+, with CO2 release and exhalation. Any remaining carbon dioxide in tissues is bound by hemoglobin and sent through the circulation to the lungs for exhalation. A decrease in pH or an increase in the partial pressure of oxygen (PO2) in the pulmonary capillaries decreases the affinity of CO2 for Hb leading to the release of CO2 from the blood and exhalation (Haldane effect), (Dash and Bassingthwaighte, 2006).
Elevation in carbon dioxide modulates immune response through inhibition of NF-κB, IL-6, TNF-alpha and mast cell degranulation (Oliver et al., 2012; Cummins et al., 2014). These activities lead to reduced inflammatory response. Carbon dioxide sensing is also known to inhibit innate immune response by altering NF-kB signaling (Helenius et al., 2009; Cummins et al., 2014). One of the major roles of CO2 sensing is the regulation of the signaling activity of adenylyl cyclase to generate cAMP (Kamenetsky et al., 2006). The latter function is particularly important as cAMP is generated in microdomains throughout the cell to fine tune intracellular signals through the activity of soluble adenylyl cyclase (sAC) (Oliver et al., 2012). sAC is not dependent on G-protein signaling but rather intracellular signals such as HCO−3, calcium and ATP (Cummins et al., 2014). In this regard sAC also participates in CO2 dependent regulation of ATP generation depending on the amount of CO2 released in the metabolic activity of the cell (Buck and Levin, 2011). Two signaling activities of sAC that requires mentioning in this review are the bicarbonate dependent activation of sperm to swim in the female reproductive tract in a process referred to as capacitation and bicarbonate dependent beating of cilia in the alveolar epithelial cells that help in defense against inhaled pathogens or particles (Huckstepp and Dale, 2011). So capacitation for life's beginnings and salutary cellular activities are important functions of carbon dioxide sensing modulated by Hb. If one considers that the partial pressure of CO2 regulates ventilation rate and variations in pH and HCO−3 in the cell can elicit responses such as changes in breathing rate, metabolism, fertility, immune response and gene expression, Hb binding to CO2 indirectly modulates these processes that contribute to the foundation of life, its sustenance and maintenance of optimal health through signaling function that protects against pathogens and subdue inflammation.
Hemoglobin and NO
Nitric oxide (NO) is an important signaling molecule, recognized in the Nobel Prize award in 1998 for Physiology and Medicine (Butler, 2006). It is produced in mammalian cells mainly by enzymatic and non-enzymatic mechanisms. The enzymatic mechanism is through nitric oxide synthase which is found in three different isoforms (nNOS:NOS I, iNOS:NOS II, eNOS) depending on the cell source; n:neurons, i: inducible (macrophages mainly and sometimes hepatocytes), and e: endothelium (Alderton et al., 2001). The main substrate for the enzymes is L-arginine, which is transported into cells by the cationic amino acid transporters (CAT) (Alderton et al., 2001; Hill et al., 2010). The co-substrates are reduced nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen while tetrahydrobiopterin (BH4), flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) function as cofactors modulated by calmodulin in the redox reaction (Stuehr, 2004; Stuehr et al., 2004). The non-enzymatic source of NO is largely from nitrites (Lundberg and Weitzberg, 2010). Deoxyhemoglobin and deoxymyoglobin by virtue of their nitrite reductase activity (Curtis et al., 2012; Gladwin and Kim-Shapiro, 2012; Kapil et al., 2014; Gao et al., 2015) react with nitrites to generate NO under tissue hypoxia or ischemia. NO acts as a toxin and signaling molecule. The toxic effect is in the generation of ROS (peroxynitrite) from interaction with heme (ferrous iron) or hemin (ferric iron) following Hb auto-oxidation to generate superoxide (Pacher et al., 2007; Beckman, 2009; Hill et al., 2010). This activity functions in the killing of microbes but can also lead to damage of proteins, DNA and lipids derived from peroxynitrite release (Koppenol, 2001). For example NO is toxic to aconitase in the TCA cycle and heme-copper terminal oxidases in the respiratory chain, responsible for peroxynitrites generation, damage to DNA and release of lipid radicals (Nathan and Ding, 2010; Forstermann and Sessa, 2012). This obviously is a problem. Fortunately Hb also functions as nitric oxide dioxygenase (NOD) converting nitric oxide and oxygen to nitrates using NADPH as cofactor so that NO as a ROS source is attenuated (Gardner, 2012). The NOD function of Hb ensures continual energy supply necessary for homeostasis in cellular bioenergetics and reduction in potential damage to cells from NO mediated ROS.
NO signaling is either through soluble guanylate cyclase (sGC) to facilitate vascular homeostasis (blood flow) by modulating vascular tone, inhibiting thrombosis and regulating the expression of endothelial adhesion molecules (Shiva et al., 2011) or through modification of proteins by nitrosylation in metalloproteins, nitrosation or oxidation of protein side chains (Hill et al., 2010; Haldar and Stamler, 2013). It is also important in skeletal muscle contraction, CNS signaling and host resistance to infection. Hb NOD function also means that the role of NO as a signaling molecule can be negatively affected, leading to reduced blood flow. However, NO removal through NOD mediated activity appears to correlate with activation of sGC so that steady state levels are achieved. As a compensatory mechanism, Hb and myoglobin act as a sink for NO releasing it during times of need and generate it from nitrites (Gardner, 2012).
Hemoglobin and CO
CO is produced in cells by heme oxygenase (HO encoded by HMOX genes) mediated breakdown of heme to give CO, biliverdin and iron (Fe2+) (Tenhunen et al., 1969). Two main HO isoforms, HO-1 and HO-2 are known which are induced and constitutive respectively (Tenhunen et al., 1969; Maines et al., 1974; Yoshida and Kikuchi, 1974; Maines, 2000). HO-1 is expressed basally in the liver and spleen while HO-2 is basally and largely expressed in the brain (Immenschuh et al., 2010; Farrugia and Szurszewski, 2014). Other minor sources include lipid peroxidation and progesterone activity (Delivoria-Papadopoulos et al., 1974; Wolff, 1976). Induced HO-1 expression is through a wide variety of stimuli including inflammation, injury, ROS, hypoxia, ischemia and hypothermia (Beckman et al., 2009; Immenschuh et al., 2010; Kacimi et al., 2011). So cells that express HO-2 stably produce CO, while stimuli dependent CO generation from HO-1 expression varies depending on cellular environment (Farrugia and Szurszewski, 2014).
CO is a signaling molecule with antioxidant and anti-inflammatory activities (Farrugia and Szurszewski, 2014). It activates soluble guanylyl cyclase (Ramos et al., 1989; Lutz and Bogdanova, 2013) to generate cyclic GMP (cGMP) for signaling based on the cellular environment (Maines, 2000). In general, the signaling function of CO parallels that of NO (although not as potent) as a neuromessenger and regulator of vascular tone (Furchgott and Jothianandan, 1991). The two gases are involved in a negative feedback regulation with NO activating HO-1 expression, while CO and HO-1 inhibit NO synthesis, (White and Marletta, 1992; Durante et al., 1997; Zuckerbraun et al., 2005). CO is biologically more stable than NO and therefore can exert effects distant from its site of production in addition to local effects (Farrugia and Szurszewski, 2014). The functional activity of CO relates to its high affinity for transition metals. Since iron is the most abundant transition metal in cells, CO competes for binding to various heme proteins, particularly Hb. It binds to hemoglobin with an affinity that is 210–250 times that of molecular oxygen (Haldane, 1895; Nasmith and Graham, 1906; Amano and Camara, 2013). Partial occupation of CO at the oxygen binding site of Hb, inhibits oxygen release from other heme sites (Ryter and Choi, 2013). Hemoglobin can therefore scavenge cellular CO to reduce its physiological impact but at the same time render it incapable of carrying oxygen. The baseline carboxyhemoglobin (CO-Hb) level in man is 0.1–1.0% (Rudra et al., 2010). If this level reaches 10–30%, it may cause headache, dizziness and shortness of breath (Gorman et al., 2003; Sen et al., 2010; La Fauci et al., 2012). Levels above 30% are harmful and can lead to severe headache, vomiting, syncope, cardiac arrhythmias and death (Piantadosi, 1999a; Gorman et al., 2003).
The brain maintains the highest expression of HO, highlighting the importance of CO as a signaling molecule in the brain (Leffler et al., 2011). In the brain, CO dilates cerebral arteries and arterioles by activating smooth muscle large conductance Ca2+ activated K+ channels or “Big potassium” channel (BKca) (Leffler et al., 2011) by a mechanism that is unrelated to sGC activation (Koneru and Leffler, 2004; Telezhkin et al., 2011). The activation of BKca channel activity leads to vasodilation promoting brain vascular system health. CO also provides protection for the cerebral vasculature (Parfenova et al., 2005). In hemolysis, when heme availability is augmented, heme binding to the BKca channel deactivates it, which is reversed through CO binding. Therefore, CO generation in hemolysis makes regulation of the CO concentration particularly important during ischemia-reperfusion not only in the brain but also in transplantation (Sata et al., 2001). CO has emerged as a choice therapeutic agent in cerebral malaria (CM). CM is a life threatening neurological complication of Plasmodium falciparum infection in children and adults, with the majority of cases (>90%) occurring in children <5 years old in sub-Saharan Africa (Pena et al., 2012; Murray et al., 2014a; Christensen and Eslick, 2015). The incidence rate is 1120/100,000 annually in endemic Africa, with mortality of 13-20% (Idro et al., 2010b; Postels and Birbeck, 2013). Surviving children from the disease endure long term neurological and cognitive deficits (Brewster et al., 1990; Idro et al., 2010a; Postels et al., 2012). Although the pathophysiology is not completely known, current understanding outlines sequestration of infected RBC leading to changes in brain microcirculation, cerebral inflammation, vascular dysfunction and breakdown in blood brain barrier (Shikani et al., 2012; Postels and Birbeck, 2013). Using P. berghei ANKA mouse models, HO-1/CO was shown to completely inhibit CM and facilitate the resolution of inflammation in the disease state (Pamplona et al., 2007; Pena et al., 2012). Because of the potential danger of CO binding to heme containing molecules in particular Hb, CO releasing molecules with limited formation of CO-Hb are now being developed (Motterlini et al., 2005; Vera et al., 2005) for CM treatment. CO is also under evaluation for use as a vasodilator and inhibitor of ischemia reperfusion injury, an immunosuppressant in transplantation, (Sato et al., 2001; Otterbein et al., 2003), and as an anticancer agent (Wegiel et al., 2013). Additional roles of CO that are gaining attention include modulation of the innate and adaptive immune response, inflammation (Otterbein et al., 2000), cell proliferation, (Morita et al., 1997) apoptosis (Brouard et al., 2000), mitochondrial biogenesis (Suliman et al., 2007) and autophagy (Lee et al., 2014). Clearly Hb as a source of CO and scavenger of CO confers significant salutary impact on brain health (facilitating nutrient and oxygen delivery, limiting inflammation) and enabling maintenance of healthy cerebral vasculature.
Hemoglobin as an antioxidant/defense molecule
Hemoglobin in other vertebrates contains a number of thiols that exert antioxidant defenses (Moleirinho et al., 2013). In humans only one reactive cysteine is present at the 93rd position of the β-chain (Vitturi et al., 2013). The Hb β93 cysteine has been observed to moderate heme reactivity leading to auto-oxidation (Balagopalakrishna et al., 1998). It scavenges reactive species (hydrogen peroxide and superoxide anion) generated in the heme pocket of the β-globin chain while inhibiting the activity of nitrosating and alkylating agents (Bonaventura et al., 1999; Vitturi et al., 2013). The mechanism of scavenging of hydrogen peroxide and superoxide anion appears to be the provision of safe transfer of electrons to traditional antioxidant systems in the rbc (Winterbourn and Carrell, 1977). This activity is most effective under high oxygen tension (21%), when the risk of auto-oxidation is high, being lost at 4% oxygen saturation (Benesch and Benesch, 1962). When the total concentration of Hb in circulation is taken into consideration, this will have a significant bearing on cellular defense against hydrogen peroxide reactivity, generation of ROS and hemoglobin auto-oxidation.
Hb in cellular immune modulation
Central to cellular defense is the host immune response which adapts to confer protection against infection or tissue damage. The cells at the center of this adaptation are the myelomonocytic cell lineage (de Back et al., 2014; Hamilton et al., 2014; Muraille et al., 2014). In the event of an inflammation or infection, the blood monocytes are recruited into tissues where they mature into macrophages or dendritic cells (Geissmann et al., 2003). Macrophages are polarized into M1 and M2 phenotypes depending on the type of stimuli they receive and cytokine profile they generate. In general M1 macrophages are elicited by pro-inflammatory stimuli, while M2 macrophages by anti-inflammatory stimuli (Lawrence and Natoli, 2011; Muraille et al., 2014). In the scheme of activities of hemoglobin modulation of the generation and function of the gaseous molecules NO, CO, and CO2, hemoglobin will indirectly influence the M1 and M2 macrophage phenotype and therefore the TH1 and TH2 immune cytokine profiles, (Lee and Chau, 2002; Alcaraz et al., 2003; Murray et al., 2014b). This will have significant implication for cellular defense against damage and pathogens through modulation of TH1 and TH2 responses (Figure 2). This function requires further interrogation.
Figure 2.

Hemoglobin functional activities in promoting rbc and cellular health. (see text for details). *Binding of Hb to CO2 is through terminal α-amino groups of Hb in contrast to the other gases.
Summary: hemoglobin: life's pivotal heart beat
In the natural environment hemoglobin is the support base for life, ensuring that vital solar energy captured by plants for photosynthesis with release of oxygen is channeled to humans and vertebrates to sustain the energy requirements of cells and life. It supports life processes by binding to gaseous molecules with major functions of efficient blood flow in the vasculature, oxygen delivery and bioenergetics homeostasis. The cumulative processes for oxygen supply and delivery to tissues is associated with modulation of activities that not only constitute a threat to vascular health but also cellular health in general. These activities which include mitochondria biogenesis, signaling functions, regulation of inflammation, immune responses and cell viability, palpably projects Hb as life's pivotal heart beat (Figure 3) without which evolution of life on earth would have been impossible.
Figure 3.
Hemoglobin: life's core friend in sustaining cardiovascular health. Hb binding to gaseous molecules through heme (O2, CO, NO) or terminal α-amino groups of Hb (CO2) facilitates cardiovascular function and health.
Hemoglobin mediated tissue injury: the foe
The damaging role of Hb under stress or pathological conditions fall under four categories:
Hemoglobin auto-oxidation within the rbc
Hemoglobin release outside of its confines in rbc to act as ROS source in the vasculature and tissues
Hemoglobin scavenging of NO
Heme release from Hb to augment free radical generation
For reviews covering these topics the reader is referred to Olsson et al. (2012), Schaer et al. (2014), Rother et al. (2005), Larsen et al. (2012), Gladwin and Sachdev (2012) and Tolosano et al. (2010). So, how does Hb that provides such a strong support base for life turn into an enemy?
Hb auto-oxidation
In general oxygen reversibly binds to Hb without any change in the redox state of iron (Bonaventura et al., 2013). However, at physiological pH, O2 has a higher redox potential than aqueous iron (Fe3+/Fe2+) so oxygen coordinated to heme in hemoglobin accepts an electron from Fe2+ heme to form superoxide (O−2) with oxidation of Fe2+ to Fe3+ generating met-hemoglobin (hemin) in a stable complex (HbFe3+)O−2, (Weiss, 1964; Faivre et al., 1998; Gardner, 2012). This process is Hb auto-oxidation which is enhanced at acidic pH (low oxygen tension) but reduced at high oxygen tension (Faivre et al., 1998; Aranda et al., 2009; Bonaventura et al., 2013). Within the confines of the rbc, antioxidants enable reduction of Ferric ion to the Fe2+ state (Antonini, 1974; Edsall, 1986) so that Fe3+ is depleted to cause no damage. Outside of the rbc auto-oxidation is high posing significant danger.
Injury associated with Hb auto-oxidation and release outside of the rbc
Outside of the rbc, the major antioxidants are haptoglobin for Hb (Schaer et al., 2014) and hemopexin, albumin, and α-1 microglobulin (Fasano et al., 2007; Nordberg et al., 2007; Tolosano et al., 2010) for heme. When hemolysis is severe the capacity of the antioxidants to scavenge is overwhelmed, so metHb and superoxide persist from Hb oxidation to set a cascade of free radical generation to be described presently. First released hemoglobin is closer to the vascular endothelium and can also extravasate into tissues as dimers so that ROS generated in auto-oxidation readily access macromolecules and cause damages (Nathan and Cunningham-Bussel, 2013; Dutra et al., 2014). In that scenario tissue NADPH oxidases and vascular NADPH oxidases facilitate the generation of hydrogen peroxide from superoxide (Klebanoff, 2005). Superoxide also undergoes dismutation by dismutase (superoxide dismutase) to generate hydrogen peroxide which can further oxidize oxyHb/Met-Hb to the ferryl (Fe4+ = O2−) form and in the process produce hydroxyl radical (OH−) through Fenton chemistry (O•−2 + H2O2 → O2 + OH− + OH−), (Brawn and Fridovich, 1980; Klebanoff et al., 1989; Pastor et al., 2000; Liochev and Fridovich, 2002). These are highly reactive species that react with globin chains and porphyrin rings to form globin and porphyrin ring radicals (Alayash, 2011), DNA to form adducts and lipids to form peroxides (Cadet et al., 1999; Marnett et al., 2003). The oxidized hemoglobin species may resist scavenging and sustain tissue injury, particularly the vascular endothelium (Goldman et al., 1998; Rother et al., 2005). Moreover when superoxide is formed in large quantities, because NO has a free electron in its outer shell it can react with the superoxide to form peroxynitrite radical (ONOO−) which has a high reactivity with heme or hemin and cellular macromolecules (Brawn and Fridovich, 1980; Halliwell, 1996; Burney et al., 1999; Marnett et al., 2003). Hydrogen peroxide can also cause intermolecular cross linking of tyrosine residues in the globin chains which are reported to contribute to atherosclerotic plaques in cardiovascular disease (Doyle et al., 1985; Halliwell et al., 1999). So severe intravascular and extravascular hemolysis lead to the damage of the vasculature as are exposed cells and tissues at the cell membrane, mitochondria and cytoskeleton, a very unfriendly, foe type activity arising from Hb.
Hb and NO scavenging
Hemoglobin reacts rapidly and irreversibly with nitric oxide (107 m−1 s−1) to generate metHb and nitrate. The rate of this reaction is such that a very small quantity of Hb can cause significant NO depletion (Olson et al., 2004). The significance of this activity was demonstrated in the reversal of acetylcholine mediated aortic arc relaxation in rabbits by cell free Hb at concentrations >1 mg/ml (Nakai et al., 1996). NO is an important signaling molecule in maintaining vascular tone and blood pressure, angiogenesis, platelet function, vascular smooth muscle cell proliferation and inflammation (Shaul, 2002; Sessa, 2004; Atochin and Huang, 2011) as alluded to previously. By scavenging NO, cell free Hb significantly impair vascular function, bioenergetics homeostasis and portend significant morbidity.
Heme release from cell free Hb
Oxidized and denatured hemoglobins lose their attached heme readily leading to heme mediated ROS generation (Balla et al., 1991a,b). Heme mediated ROS generation is largely attributed to the redox function of iron, which mediates reductive or oxidative reactions involving the transfer or acceptance of electrons (Dutra et al., 2014). This redox function makes Hb also function as a major inducer of ROS because of its large concentration in the rbc (Jeney et al., 2013). Heme is lipophilic and can intercalate into cell membranes to disrupt membrane integrity (Schmitt et al., 1993). Other downstream processes follow similar path to that outlined for Hb auto oxidation which involves generation of free radicals (Huertas and Palange, 2011; Dutra and Bozza, 2014) pro-inflammatory activities and cytotoxicity (Larsen et al., 2012). The scavenger proteins haptoglobin (Hp) and hemopexin (Hpx) will normally scavenge cell free Hb and heme respectively obviating any ROS related activity if not in excess (Hvidberg et al., 2005; Quaye, 2008; Tolosano et al., 2010; Schaer et al., 2014). Hb bound haptoglobin is targeted for elimination through the monocyte/macrophage scavenger receptor CD163 (Schaer et al., 2014) while is heme first bound by albumin, transferred to Hpx and the heme-Hpx complex targeted for clearance by the LDL receptor related protein 1 (LRP 1/CD91 in hepatocytes in particular (Ascenzi and Fasano, 2007; Schaer et al., 2014). In addition heme can also be scavenged by α-1 microglobulin (Allhorn et al., 2002) and high density lipoproteins (Miller and Gladwin, 2012). When levels of these proteins fall due to excessive hemolysis then cellular/tissue damage is cascaded. For further reading the reader is referred to Schaer et al. (2014), Tolosano et al. (2010), Alayash (2011), Kristiansen et al. (2001), Nielsen and Moestrup (2009), Hrkal and Klementova (1984), Balla et al. (1991b), Hvidberg et al. (2005), Dutra and Bozza (2014) and Dutra et al. (2014).
Mechanism of Hb release
Red blood cells lack a nucleus or mitochondria so injury mainly affects the cell membrane leading to cell rupture and release of hemoglobin either extra-vascularly or intra-vascularly (Balla et al., 1991b). In severe insults (infection, complement fixation and activation, trauma) where the rbc membrane is compromised, hemoglobin is released outside of its confines in the rbc posing danger not only to the vasculature but also to exposed tissues following extravasation (Jeney et al., 2013; Dutra et al., 2014; Schaer et al., 2014).
Extravascular and intravascular hemolysis
Human red blood cells have a normal life span of 120 days (Dhaliwal et al., 2004; Sugawara et al., 2010). If this life span is curtailed either through breakdown of the cell within the blood vessel or outside of the vessels leading to the release of hemoglobin, the process is referred to as intravascular or extravascular hemolysis respectively (Dhaliwal et al., 2004; Sugawara et al., 2010; Mairbaurl, 2013). Normally when hemoglobin reaches a life span of 120 days, it is removed by the spleen, bone marrow macrophage phagocytosis and liver Kupffer cells. Part of the trigger for removal is the presence of Heinz bodies which are aggregates of oxidized or denatured hemoglobin in the rbc, Howell-jolly bodies (inclusions of nuclear chromatin), siderocytes (rbcs with granules of iron that is not part of Hb) and Pappenheimer bodies (inclusion bodies formed by phagosomes ingesting large amounts of iron) (Winslow et al., 1978; Weatherall, 1995, 2011; Sugawara et al., 2010). The process of rbc removal by the mononuclear phagocyte system in the spleen in particular, does not lead to lysis so endogenous antioxidants including glutathione, ascorbic acid, catalase, glutathione peroxidase and peroxiredoxin-2 in the rbc limit the release of hemoglobin extravascularly (Crespo et al., 1987; Mohandas and Gallagher, 2008). Indeed some hemoglobin may leak and trigger the release of reactive oxygen species but this is limited with respect to severity as the hemoglobin scavenger receptor CD163, and heme scavenger receptor CD91 effectively scavenge the released hemoglobin through haptoglobin and hemopexin respectively (Kristiansen et al., 2001; Hvidberg et al., 2005; Tolosano et al., 2010; Schaer et al., 2013, 2014). Hb can also be released extravascularly from internal bleedings (trauma) due to damaged micro- and macro blood vessels.
In intravascular hemolysis, rbcs break up during circulation from oxidative stress, aging or infection releasing Hb directly into the vasculature and subsequently heme (Rother et al., 2005; Jeney et al., 2013; Schaer et al., 2014). Intravascular and extravascular hemolysis serve as potent sources of free radicals that define the hazardous nature of Hb. The amount of Hb released depends on the degree of severity of the oxidative stress or pathological condition which overwhelms natural antioxidants and scavengers in the rbc and plasma.
Summary: hemoglobin as a danger molecule outside of its confines
In severe extravascular and intravascular hemolysis Hb assumes a foe like nature serving to propagate ROS generation, disrupt cell membranes and cell function, the very activities that it supports when confined. Fortunately, this foe like activities present under abnormal conditions requiring external intervention. The nature and type of the interventions present a challenge but also a major focus of research the world over.
Conclusion
Hemoglobin is a support base for all life processes physiologically. In the normal, physiological conditions it is salutary, under stress (infection or trauma) the nature that promotes cell health is disrupted leading to real threat and danger to cell function and survival.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author acknowledges the unflinching support of my family (Dr Larysa Aleksenko and Noah Quaye,) during the write up of this review.
References
- Abugo O. O., Rifkind J. M. (1994). Oxidation of hemoglobin and the enhancement produced by nitroblue tetrazolium. J. Biol. Chem. 269, 24845–24853. [PubMed] [Google Scholar]
- Aft R. L., Mueller G. C. (1983). Hemin-mediated DNA strand scission. J. Biol. Chem. 258, 12069–12072. 10.1203/PDR.0b013e3181eee738 [DOI] [PubMed] [Google Scholar]
- Alayash A. I. (2004). Redox biology of blood. Antioxid. Redox Signal. 6, 941–943. 10.1089/ars.2004.6.941 [DOI] [PubMed] [Google Scholar]
- Alayash A. I. (2011). Haptoglobin: old protein with new functions. Clin. Chim. Acta 412, 493–498. 10.1016/j.cca.2010.12.011 [DOI] [PubMed] [Google Scholar]
- Alcaraz M. J., Fernandez P., Guillen M. I. (2003). Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr. Pharm. Des. 9, 2541–2551. 10.2174/1381612033453749 [DOI] [PubMed] [Google Scholar]
- Alderton W. K., Cooper C. E., Knowles R. G. (2001). Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357(Pt 3), 593–615 10.1042/0264-6021:3570593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allhorn M., Berggard T., Nordberg J., Olsson M. L., Akerstrom B. (2002). Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood 99, 1894–1901. 10.1182/blood.V99.6.1894 [DOI] [PubMed] [Google Scholar]
- Amano M. T., Camara N. O. (2013). The immunomodulatory role of carbon monoxide during transplantation. Med. Gas Res. 3:1. 10.1186/2045-9912-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini E. (1974). Heme proteins: hemoglobin. FEBS Lett. 40 Suppl., S98–S104 10.1016/0014-5793(74)80691-5 [DOI] [PubMed] [Google Scholar]
- Aranda R. IV., Cai H., Worley C. E., Levin E. J., Li R., Olson J. S., et al. (2009). Structural analysis of fish versus mammalian hemoglobins: effect of the heme pocket environment on autooxidation and hemin loss. Proteins 75, 217–230. 10.1002/prot.22236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascenzi P., Bocedi A., Visca P., Altruda F., Tolosano E., Beringhelli T., et al. (2005). Hemoglobin and heme scavenging. IUBMB Life 57, 749–759. 10.1080/15216540500380871 [DOI] [PubMed] [Google Scholar]
- Ascenzi P., Fasano M. (2007). Heme-hemopexin: a ‘chronosteric’ heme-protein. IUBMB Life 59, 700–708. 10.1080/15216540701689666 [DOI] [PubMed] [Google Scholar]
- Atochin D. N., Huang P. L. (2011). Role of endothelial nitric oxide in cerebrovascular regulation. Curr. Pharm. Biotechnol. 12, 1334–1342. 10.2174/138920111798280974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balagopalakrishna C., Abugo O. O., Horsky J., Manoharan P. T., Nagababu E., Rifkind J. M. (1998). Superoxide produced in the heme pocket of the beta-chain of hemoglobin reacts with the beta-93 cysteine to produce a thiyl radical. Biochemistry 37, 13194–13202. 10.1021/bi980941c [DOI] [PubMed] [Google Scholar]
- Balla G., Jacob H. S., Eaton J. W., Belcher J. D., Vercellotti G. M. (1991a). Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 11, 1700–1711. 10.1203/PDR.0b013e3181eee738 [DOI] [PubMed] [Google Scholar]
- Balla G., Vercellotti G. M., Muller-Eberhard U., Eaton J., Jacob H. S. (1991b). Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Invest. 64, 648–655. 10.1203/PDR.0b013e3181eee738 [DOI] [PubMed] [Google Scholar]
- Bauer C., Kurtz A. (1977). Oxygen-linked carbon dioxide binding to isolated β subunits of human hemoglobin. J. Biol. Chem. 252, 2952–2955. [PubMed] [Google Scholar]
- Beckman J. D., Belcher J. D., Vineyard J. V., Chen C., Nguyen J., Nwaneri M. O., et al. (2009). Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease. Am. J. Physiol. Heart Circ. Physiol. 297, H1243–H1253. 10.1152/ajpheart.00327.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J. S. (2009). Understanding peroxynitrite biochemistry and its potential for treating human diseases. Arch. Biochem. Biophys. 484, 114–116. 10.1016/j.abb.2009.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher J. D., Nath K. A., Vercellotti G. M. (2013). Vasculotoxic and proinflammatory effects of plasma heme: cell signaling and cytoprotective responses. ISRN Oxidative Med. 2013, 831596. 10.1155/2013/831596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch R. E., Benesch R. (1962). The influence of oxygenation on the reactivity of the—SH groups of hemoglobin. Biochemistry 1, 735–738. 10.1021/bi00911a002 [DOI] [PubMed] [Google Scholar]
- Boatman D. E., Robbins R. S. (1991). Bicarbonate: carbon-dioxide regulation of sperm capacitation, hyperactivated motility, and acrosome reactions. Biol. Reprod. 44, 806–813. 10.1095/biolreprod44.5.806 [DOI] [PubMed] [Google Scholar]
- Bonaventura C., Godette G., Tesh S., Holm D. E., Bonaventura J., Crumbliss A. L., et al. (1999). Internal electron transfer between hemes and Cu(II) bound at cysteine beta93 promotes methemoglobin reduction by carbon monoxide. J. Biol. Chem. 274, 5499–5507. 10.1074/jbc.274.9.5499 [DOI] [PubMed] [Google Scholar]
- Bonaventura C., Henkens R., Alayash A. I., Banerjee S., Crumbliss A. L. (2013). Molecular controls of the oxygenation and redox reactions of hemoglobin. Antioxid. Redox Signal. 18, 2298–2313. 10.1089/ars.2012.4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawn K., Fridovich I. (1980). Superoxide radical and superoxide dismutases: threat and defense. Acta Physiol. Scand. Suppl. 492, 9–18. [PubMed] [Google Scholar]
- Brewster D. R., Kwiatkowski D., White N. J. (1990). Neurological sequelae of cerebral malaria in children. Lancet 33622, 1039–1043 10.1016/0140-6736(90)92498-7 [DOI] [PubMed] [Google Scholar]
- Brouard S., Otterbein L. E., Anrather J., Tobiasch E., Bach F. H., Choi A. M., et al. (2000). Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 192, 1015–1026. 10.1084/jem.192.7.1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns G. P., London I. M. (1965). The effect of hemin on the synthesis of globin. Biochem. Biophys. Res. Commun. 18, 236–242. 10.1016/0006-291X(65)90746-1 [DOI] [PubMed] [Google Scholar]
- Buck J., Levin L. R. (2011). Physiological sensing of carbon dioxide/bicarbonate/pH via cyclic nucleotide signaling. Sensors 11, 2112–2128. 10.3390/s110202112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler P. W., Alayash A. I. (2005). Redox biology of blood revisited: the role of red blood cells in maintaining circulatory reductive capacity. Antioxid. Redox Signal. 7, 1755–1760. 10.1089/ars.2005.7.1755 [DOI] [PubMed] [Google Scholar]
- Burney S., Caulfield J. L., Niles J. C., Wishnok J. S., Tannenbaum S. R. (1999). The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424, 37–49. 10.1016/S0027-5107(99)00006-8 [DOI] [PubMed] [Google Scholar]
- Busacchi V. (1958). Vincenzo Menghini and the discovery of iron in the blood. Bull. Sci. Med. 130, 202–205. [PubMed] [Google Scholar]
- Butler A. R. (2006). ‘The heart less bounding’: treating angina pectoris. J. R. Coll. Phys. Edinb. 36, 185–189. [PubMed] [Google Scholar]
- Cadet J., Carvalho V. M., Onuki J., Douki T., Medeiros M. H., Di Mascio P. D. (1999). Purine DNA adducts of 4,5-dioxovaleric acid and 2,4-decadienal. IARC Sci. Publ. 150, 103–113. [PubMed] [Google Scholar]
- Chapman G. B., Cokelet G. R. (1997). Model studies of leukocyte-endothelium-blood interactions. II. Hemodynamic impact of leukocytes adherent to the wall of post-capillary vessels. Biorheology 34, 37–56. 10.1016/S0006-355X(97)00003-6 [DOI] [PubMed] [Google Scholar]
- Chay T. R., Brillhart D. (1974). Mechanism of cooperative oxygen binding to hemoglobin: kinetic aspects. Biochemistry 13, 5311–5317. 10.1021/bi00723a009 [DOI] [PubMed] [Google Scholar]
- Chiabrando D., Mercurio S., Tolosano E. (2014). Heme and erythropoieis: more than a structural role. Haematologica 99, 973–983. 10.3324/haematol.2013.091991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen S. S., Eslick G. D. (2015). Cerebral malaria as a risk factor for the development of epilepsy and other long-term neurological conditions: a meta-analysis. Trans. R. Soc. Trop. Med. Hyg. 109, 233–238. 10.1093/trstmh/trv005 [DOI] [PubMed] [Google Scholar]
- Cosby K., Partovi K. S., Crawford J. H., Patel R. P., Reiter C. D., Martyr S., et al. (2003). Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9, 1498–1505. 10.1038/nm954 [DOI] [PubMed] [Google Scholar]
- Crespo L. M., Novak T. S., Freedman J. C. (1987). Calcium, cell shrinkage, and prolytic state of human red blood cells. Am. J. Physiol. 252(Pt 1), C138–C1352. [DOI] [PubMed] [Google Scholar]
- Cummins E. P., Selfridge A. C., Sporn P. H., Sznajder J. I., Taylor C. T. (2014). Carbon dioxide-sensing in organisms and its implications for human disease. Cell. Mol. Life Sci. 71, 831–845. 10.1007/s00018-013-1470-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis E., Hsu L. L., Noguchi A. C., Geary L., Shiva S. (2012). Oxygen regulates tissue nitrite metabolism. Antioxid. Redox Signal. 17, 951–961. 10.1089/ars.2011.4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R. K., Bassingthwaighte J. B. (2006). Simultaneous blood-tissue exchange of oxygen, carbon dioxide, bicarbonate, hydrogen ion. Ann. Biomed. Eng. 34, 1129–1148. 10.1007/s10439-005-9066-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Back D. Z., Kostova E. B., van Kraaij M., van den Berg T. K., van Bruggen R. (2014). Of macrophages and red blood cells; a complex love story. Front. Physiol. 5:9. 10.3389/fphys.2014.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delivoria-Papadopoulos M., Coburn R. F., Forster R. E. (1974). Cyclic variation of rate of carbon monoxide production in normal women. J. Appl. Physiol. 36, 49–51. [DOI] [PubMed] [Google Scholar]
- De Rosa M. C., Alinovi C. C., Galtieri A., Russo A., Giardina B. (2008). Allosteric properties of hemoglobin and the plasma membrane of the erythrocyte: new insights in gas transport and metabolic modulation. IUBMB Life 60, 87–93. 10.1002/iub.15 [DOI] [PubMed] [Google Scholar]
- Dhaliwal G., Cornett P. A., Tierney L. M., Jr. (2004). Hemolytic anemia. Am. Fam. Physician 69, 2599–2606. [PubMed] [Google Scholar]
- Doyle M. P., Herman J. G., Dykstra R. L. (1985). Autocatalytic oxidation of hemoglobin induced by nitrite: activation and chemical inhibition. J. Free Rad. Biol. Med. 1, 145–153. 10.1016/0748-5514(85)90019-4 [DOI] [PubMed] [Google Scholar]
- Durante W., Kroll M. H., Christodoulides N., Peyton K. J., Schafer A. I. (1997). Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ. Res. 80, 557–564. 10.1161/01.RES.80.4.557 [DOI] [PubMed] [Google Scholar]
- Dutra F. F., Alves L. S., Rodrigues D., Fernandez P. L., de Oliveira R. B., Golenbock D. T., et al. (2014). Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. U.S.A. 111, E4110–E4118. 10.1073/pnas.1405023111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra F. F., Bozza M. T. (2014). Heme on innate immunity and inflammation. Front. Pharmacol. 5:115. 10.3389/fphar.2014.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton J. W. (1974). Oxygen affinity and environmental adaptation. Ann. N.Y. Acad. Sci. 241, 491–497. 10.1111/j.1749-6632.1974.tb21905.x [DOI] [PubMed] [Google Scholar]
- Edsall J. T. (1986). Understanding blood and hemoglobin: an example of international relations in science. Perspect. Biol. Med. 29(Pt 2), S107–S1023. 10.1353/pbm.1986.0052 [DOI] [PubMed] [Google Scholar]
- Eltzschig H. K., Carmeliet P. (2011). Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665. 10.1056/NEJMra0910283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre B., Menu P., Labrude P., Vigneron C. (1998). Hemoglobin autooxidation/oxidation mechanisms and methemoglobin prevention or reduction processes in the bloodstream. Literature review and outline of autooxidation reaction. Artif. Cells Blood Substit. Immobil. Biotechnol. 26, 17–26. 10.3109/10731199809118943 [DOI] [PubMed] [Google Scholar]
- Farrugia G., Szurszewski J. H. (2014). Carbon monoxide, hydrogen sulfide, and nitric oxide as signaling molecules in the gastrointestinal tract. Gastroenterology 147, 303–313. 10.1053/j.gastro.2014.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano M., Fanali G., Leboffe L., Ascenzi P. (2007). Heme binding to albuminoid proteins is the result of recent evolution. IUBMB Life 59, 436–440. 10.1080/15216540701474523 [DOI] [PubMed] [Google Scholar]
- Forstermann U., Sessa W. C. (2012). Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837, 837a–837d. 10.1093/eurheartj/ehr304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott R. F., Jothianandan D. (1991). Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels 28, 52–61. [DOI] [PubMed] [Google Scholar]
- Gao X., Yang T., Liu M., Peleli M., Zollbrecht C., Weitzberg E., et al. (2015). NADPH oxidase in the renal microvasculature is a primary target for blood pressure-lowering effects by inorganic nitrate and nitrite. Hypertension 65, 161–170. 10.1161/HYPERTENSIONAHA.114.04222 [DOI] [PubMed] [Google Scholar]
- Gardner P. R. (2012). Hemoglobin: a nitric-oxide dioxygenase. Scientifica 2012:683729. 10.6064/2012/683729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F., Jung S., Littman D. R. (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. 10.1016/S1074-7613(03)00174-2 [DOI] [PubMed] [Google Scholar]
- Girvan H. M., Munro A. W. (2013). Heme sensor proteins. J. Biol. Chem. 288, 13194–13203. 10.1074/jbc.R112.422642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin M. T., Kim-Shapiro D. B. (2012). Vascular biology: nitric oxide caught in traffic. Nature 491, 344–345. 10.1038/nature11640 [DOI] [PubMed] [Google Scholar]
- Gladwin M. T., Sachdev V. (2012). Cardiovascular abnormalities in sickle cell disease. J. Am. Coll. Cardiol. 59, 1123–1133. 10.1016/j.jacc.2011.10.900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D. W., Breyer R. J., III., Yeh D., Brockner-Ryan B. A., Alayash A. I. (1998). Acellular hemoglobin-mediated oxidative stress toward endothelium: a role for ferryl iron. Am. J. Physiol. 275(Pt 2), H1046–H1053. [DOI] [PubMed] [Google Scholar]
- Gorman D., Drewry A., Huang Y. L., Sames C. (2003). The clinical toxicology of carbon monoxide. Toxicology 187, 25–38. 10.1016/S0300-483X(03)00005-2 [DOI] [PubMed] [Google Scholar]
- Griendling K. K., FitzGerald G. A. (2003). Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108, 1912–1916. 10.1161/01.CIR.0000093660.86242.BB [DOI] [PubMed] [Google Scholar]
- Guzik T. J., Korbut R., Adamek-Guzik T. (2003). Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 54, 469–487. [PubMed] [Google Scholar]
- Haldane J. (1895). The action of carbonic oxide on man. J. Physiol. 18, 430–462. 10.1113/jphysiol.1895.sp000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar S. M., Stamler J. S. (2013). S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J. Clin. Invest. 123, 101–110. 10.1172/JCI62854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. (1996). Free radicals, proteins and DNA: oxidative damage versus redox regulation. Biochem. Soc. Trans. 24, 1023–1027. [DOI] [PubMed] [Google Scholar]
- Halliwell B., Zhao K., Whiteman M. (1999). Nitric oxide and peroxynitrite. The ugly, the uglier and the not so good: a personal view of recent controversies. Free Radic. Res. 31, 651–669. 10.1080/10715769900301221 [DOI] [PubMed] [Google Scholar]
- Hamilton T. A., Zhao C., Pavicic P. G., Jr., Datta S. (2014). Myeloid colony-stimulating factors as regulators of macrophage polarization. Front. Immunol. 5:554. 10.3389/fimmu.2014.00554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbel R. P., Eaton J. W., Berger E. M., Kronenberg R. S., Zanjani E. D., Moore L. G. (1978). Hemoglobin oxygen affinity and adaptation to altitude: evidence for pre-adaptation to altitude in humans with left-shifted oxyhemoglobin dissociation curves. Trans. Assoc. Am. Physicians 91, 212–228. [PubMed] [Google Scholar]
- Heinemann I. U., Jahn M., Jahn D. (2008). The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 474, 238–251. 10.1016/j.abb.2008.02.015 [DOI] [PubMed] [Google Scholar]
- Helenius I. T., Krupinski T., Turnbull D. W., Gruenbaum Y., Silverman N., Johnson E. A., et al. (2009). Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 106, 18710–18715. 10.1073/pnas.0905925106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill B. G., Dranka B. P., Bailey S. M., Lancaster J. R., Jr., Darley-Usmar V. M. (2010). What part of NO don't you understand? Some answers to the cardinal questions in nitric oxide biology. J. Biol. Chem. 285, 19699–19704. 10.1074/jbc.R110.101618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrkal Z., Klementova S. (1984). Bilirubin and haeme binding to human serum albumin studied by spectroscopy methods. Int. J. Biochem. 16, 799–804. 10.1016/0020-711X(84)90192-7 [DOI] [PubMed] [Google Scholar]
- Huang Z., Shiva S., Kim-Shapiro D. B., Patel R. P., Ringwood L. A., Irby C. E., et al. (2005). Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J. Clin. Invest. 115, 2099–2107. 10.1172/JCI24650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp R. T., Dale N. (2011). Redefining the components of central CO2 chemosensitivity—towards a better understanding of mechanism. J. Physiol. 589(Pt 23), 5561–5579 10.1113/jphysiol.2011.214759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huehns E. R., Dance N., Beaven G. H., Hecht F., Motulsky A. G. (1964). Human Embryonic Hemoglobins. Cold Spring Harb. Symp. Quant. Biol. 29, 327–331. 10.1101/SQB.1964.029.01.035 [DOI] [PubMed] [Google Scholar]
- Huertas A., Palange P. (2011). Circulating endothelial progenitor cells and chronic pulmonary diseases. Eur. Respir. J. 37, 426–431. 10.1183/09031936.00034810 [DOI] [PubMed] [Google Scholar]
- Hvidberg V., Maniecki M. B., Jacobsen C., Hojrup P., Moller H. J., Moestrup S. K. (2005). Identification of the receptor scavenging hemopexin-heme complexes. Blood 106, 2572–2579. 10.1182/blood-2005-03-1185 [DOI] [PubMed] [Google Scholar]
- Idro R., Kakooza-Mwesige A., Balyejjussa S., Mirembe G., Mugasha C., Tugumisirize J., et al. (2010a). Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res. Notes 3:104. 10.1186/1756-0500-3-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idro R., Marsh K., John C. C., Newton C. R. (2010b). Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 68, 267–274. 10.1203/PDR.0b013e3181eee738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immenschuh S., Baumgart-Vogt E., Mueller S. (2010). Heme oxygenase-1 and iron in liver inflammation: a complex alliance. Curr. Drug Targets 11, 1541–1550. 10.2174/1389450111009011541 [DOI] [PubMed] [Google Scholar]
- Jagger J. E., Bateman R. M., Ellsworth M. L., Ellis C. G. (2001). Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am. J. Physiol. Heart Circ. Physiol. 280, H2833–H2839. [DOI] [PubMed] [Google Scholar]
- Jeney V., Eaton J. W., Balla G., Balla J. (2013). Natural history of the bruise: formation, elimination, and biological effects of oxidized hemoglobin. Oxid. Med. Cell. Longev. 2013, 703571. 10.1155/2013/703571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacimi R., Giffard R. G., Yenari M. A. (2011). Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. 8:7. 10.1186/1476-9255-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetsky M., Middelhaufe S., Bank E. M., Levin L. R., Buck J., Steegborn C. (2006). Molecular details of cAMP generation in mammalian cells: a tale of two systems. J. Mol. Biol. 362, 623–639. 10.1016/j.jmb.2006.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil V., Weitzberg E., Lundberg J. O., Ahluwalia A. (2014). Clinical evidence demonstrating the utility of inorganic nitrate in cardiovascular health. Nitric Oxide 38, 45–57. 10.1016/j.niox.2014.03.162 [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J. (2005). Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77, 598–625. 10.1189/jlb.1204697 [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J., Waltersdorph A. M., Michel B. R., Rosen H. (1989). Oxygen-based free radical generation by ferrous ions and deferoxamine. J. Biol. Chem. 264, 19765–19771. [PubMed] [Google Scholar]
- Koneru P., Leffler C. W. (2004). Role of cGMP in carbon monoxide-induced cerebral vasodilation in piglets. Am. J. Physiol. Heart Circ. Physiol. 286, H304–H309. 10.1152/ajpheart.00810.2003 [DOI] [PubMed] [Google Scholar]
- Koppenol W. H. (2001). 100 years of peroxynitrite chemistry and 11 years of peroxynitrite biochemistry. Redox Rep. 6, 339–341. 10.1179/135100001101536517 [DOI] [PubMed] [Google Scholar]
- Korolnek T., Hamza I. (2014). Like iron in the blood of the people: the requirement for heme trafficking in iron metabolism. Front. Pharmacol. 5:126. 10.3389/fphar.2014.00126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M., Graversen J. H., Jacobsen C., Sonne O., Hoffman H. J., Law S. K., et al. (2001). Identification of the haemoglobin scavenger receptor. Nature 409, 198–201. 10.1038/35051594 [DOI] [PubMed] [Google Scholar]
- La Fauci G., Weiser G., Steiner I. P., Shavit I. (2012). Carbon monoxide poisoning in narghile (water pipe) tobacco smokers. CJEM 14, 57–59. [DOI] [PubMed] [Google Scholar]
- Larsen R., Gouveia Z., Soares M. P., Gozzelino R. (2012). Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol. 3:77. 10.3389/fphar.2012.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T., Natoli G. (2011). Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761. 10.1038/nri3088 [DOI] [PubMed] [Google Scholar]
- Lee S., Lee S. J., Coronata A. A., Fredenburgh L. E., Chung S. W., Perrella M. A., et al. (2014). Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 20, 432–442. 10.1089/ars.2013.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. S., Chau L. Y. (2002). Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 8, 240–246. 10.1038/nm0302-240 [DOI] [PubMed] [Google Scholar]
- Leffler C. W., Parfenova H., Jaggar J. H. (2011). Carbon monoxide as an endogenous vascular modulator. Am. J. Physiol. Heart Circ. Physiol. 301, H1–H11. 10.1152/ajpheart.00230.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev S. I., Fridovich I. (2002). The Haber-Weiss cycle—70 years later: an alternative view. Redox Rep. 7, 55–57. author reply 59–60. 10.1179/135100002125000190 [DOI] [PubMed] [Google Scholar]
- Lundberg J. O., Weitzberg E. (2010). The biological role of nitrate and nitrite: the times they are a-changin'. Nitric Oxide 22, 61–63. 10.1016/j.niox.2009.11.004 [DOI] [PubMed] [Google Scholar]
- Lutz H. U., Bogdanova A. (2013). Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 4:387. 10.3389/fphys.2013.00387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines M. D. (1997). The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 37, 517–554. 10.1146/annurev.pharmtox.37.1.517 [DOI] [PubMed] [Google Scholar]
- Maines M. D. (2000). The heme oxygenase system and its functions in the brain. Cell. Mol. Biol. 46, 573–585. [PubMed] [Google Scholar]
- Maines M. D., Anders M. W., Muller-Eberhard U. (1974). Studies on heme transfer from microsomal hemoproteins to heme-binding plasma proteins. Mol. Pharmacol. 10, 204–213. [PubMed] [Google Scholar]
- Mairbaurl H. (1994). Red blood cell function in hypoxia at altitude and exercise. Int. J. Sports Med. 15, 51–63. 10.1055/s-2007-1021020 [DOI] [PubMed] [Google Scholar]
- Mairbaurl H. (2013). Red blood cells in sports: effects of exercise and training on oxygen supply by red blood cells. Front. Physiol. 4:332. 10.3389/fphys.2013.00332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairbaurl H., Weber R. E. (2012). Oxygen transport by hemoglobin. Compr. Physiol. 2, 1463–1489. 10.1002/cphy.c080113 [DOI] [PubMed] [Google Scholar]
- Marnett L. J., Riggins J. N., West J. D. (2003). Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J. Clin. Invest. 111, 583–593. 10.1172/JCI200318022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. C., Gladwin M. T. (2012). Pulmonary complications of sickle cell disease. Am. J. Respir. Crit. Care Med. 185, 1154–1165. 10.1164/rccm.201111-2082CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N., Gallagher P. G. (2008). Red cell membrane: past, present, and future. Blood 112, 3939–3948. 10.1182/blood-2008-07-161166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moleirinho A., Seixas S., Lopes A. M., Bento C., Prata M. J., Amorim A. (2013). Evolutionary constraints in the beta-globin cluster: the signature of purifying selection at the delta-globin (HBD) locus and its role in developmental gene regulation. Genome Biol. Evol. 5, 559–571. 10.1093/gbe/evt029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T., Mitsialis S. A., Koike H., Liu Y., Kourembanas S. (1997). Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 272, 32804–32809. 10.1074/jbc.272.52.32804 [DOI] [PubMed] [Google Scholar]
- Motterlini R., Mann B. E., Foresti R. (2005). Therapeutic applications of carbon monoxide-releasing molecules. Expert Opin. Investig. Drugs 14, 1305–1318. 10.1517/13543784.14.11.1305 [DOI] [PubMed] [Google Scholar]
- Muraille E., Leo O., Moser M. (2014). TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 5:603. 10.3389/fimmu.2014.00603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. J., Ortblad K. F., Guinovart C., Lim S. S., Wolock T. M., Roberts D. A., et al. (2014a). Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 1005–1070. 10.1016/S0140-6736(14)60844-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., et al. (2014b). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K., Ohta T., Sakuma I., Akama K., Kobayashi Y., Tokuyama S., et al. (1996). Inhibition of endothelium-dependent relaxation by hemoglobin in rabbit aortic strips: comparison between acellular hemoglobin derivatives and cellular hemoglobins. J. Cardiovasc. Pharmacol. 28, 115–123. 10.1097/00005344-199607000-00018 [DOI] [PubMed] [Google Scholar]
- Nasmith G. G., Graham D. A. (1906). The haematology of carbon-monoxide poisoning. J. Physiol. 35, 32–52. 10.1113/jphysiol.1906.sp001179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C., Cunningham-Bussel A. (2013). Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat. Rev. Immunol. 13, 349–361. 10.1038/nri3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C., Ding A. (2010). SnapShot: reactive oxygen intermediates (ROI). Cell 140, 951-951.e2. 10.1016/j.cell.2010.03.008 [DOI] [PubMed] [Google Scholar]
- Nielsen M. J., Moestrup S. K. (2009). Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood 114, 764–771. 10.1182/blood-2009-01-198309 [DOI] [PubMed] [Google Scholar]
- Nishi H., Inagi R., Kato H., Tanemoto M., Kojima I., Son D., et al. (2008). Hemoglobin is expressed by mesangial cells and reduces oxidant stress. J. Am. Soc. Nephrol. 19, 1500–1508. 10.1681/ASN.2007101085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg J., Allhorn M., Winqvist I., Akerstrom B., Olsson M. L. (2007). Quantitative and qualitative evaluation of plasma and urine alpha1-microglobulin in healthy donors and patients with different haemolytic disorders and haemochromatosis. Clin. Chim. Acta 386, 31–37. 10.1016/j.cca.2007.07.017 [DOI] [PubMed] [Google Scholar]
- Oliver K. M., Lenihan C. R., Bruning U., Cheong A., Laffey J. G., McLoughlin P., et al. (2012). Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J. Biol. Chem. 287, 14004–14011. 10.1074/jbc.M112.347971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson J. S., Foley E. W., Rogge C., Tsai A. L., Doyle M. P., Lemon D. D. (2004). No scavenging and the hypertensive effect of hemoglobin-based blood substitutes. Free Rad. Biol. Med. 36, 685–697. 10.1016/j.freeradbiomed.2003.11.030 [DOI] [PubMed] [Google Scholar]
- Olsson M. G., Allhorn M., Bulow L., Hansson S. R., Ley D., Olsson M. L., et al. (2012). Pathological conditions involving extracellular hemoglobin: molecular mechanisms, clinical significance, and novel therapeutic opportunities for alpha(1)-microglobulin. Antioxid. Redox Signal. 17, 813–846. 10.1089/ars.2011.4282 [DOI] [PubMed] [Google Scholar]
- Otterbein L. E., Bach F. H., Alam J., Soares M., Tao Lu H., Wysk M., et al. (2000). Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 6, 422–428. 10.1038/74680 [DOI] [PubMed] [Google Scholar]
- Otterbein L. E., Zuckerbraun B. S., Haga M., Liu F., Song R., Usheva A., et al. (2003). Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med. 9, 183–190. 10.1038/nm817 [DOI] [PubMed] [Google Scholar]
- Pacher P., Beckman J. S., Liaudet L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. 10.1152/physrev.00029.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona A., Ferreira A., Balla J., Jeney V., Balla G., Epiphanio S., et al. (2007). Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 13, 703–710. 10.1038/nm1586 [DOI] [PubMed] [Google Scholar]
- Parfenova H., Carratu P., Tcheranova D., Fedinec A., Pourcyrous M., Leffler C. W. (2005). Epileptic seizures cause extended postictal cerebral vascular dysfunction that is prevented by HO-1 overexpression. Am. J. Physiol. Heart Circ. Physiol. 288, H2843–H2850. 10.1152/ajpheart.01274.2004 [DOI] [PubMed] [Google Scholar]
- Pastor N., Weinstein H., Jamison E., Brenowitz M. (2000). A detailed interpretation of OH radical footprints in a TBP-DNA complex reveals the role of dynamics in the mechanism of sequence-specific binding. J. Mol. Biol. 304, 55–68. 10.1006/jmbi.2000.4173 [DOI] [PubMed] [Google Scholar]
- Pena A. C., Penacho N., Mancio-Silva L., Neres R., Seixas J. D., Fernandes A. C., et al. (2012). A novel carbon monoxide-releasing molecule fully protects mice from severe malaria. Antimicrob. Agents Chemother. 56, 1281–1290. 10.1128/AAC.05571-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz M. F. (1979). Regulation of oxygen affinity of hemoglobin: influence of structure of the globin on the heme iron. Annu. Rev. Biochem. 48, 327–386. 10.1146/annurev.bi.48.070179.001551 [DOI] [PubMed] [Google Scholar]
- Piantadosi C. A. (1999a). Diagnosis and treatment of carbon monoxide poisoning. Respir. Care Clin. N. Am. 5, 183–202. [PubMed] [Google Scholar]
- Piantadosi C. A. (1999b). Physiology of hyperbaric hyperoxia. Respir. Care Clin. North Am. 5, 7–19, v. [PubMed] [Google Scholar]
- Postels D. G., Birbeck G. L. (2013). Cerebral malaria. Handb. Clin. Neurol. 114, 91–102. 10.1016/B978-0-444-53490-3.00006-6 [DOI] [PubMed] [Google Scholar]
- Postels D. G., Taylor T. E., Molyneux M., Mannor K., Kaplan P. W., Seydel K. B., et al. (2012). Neurologic outcomes in retinopathy-negative cerebral malaria survivors. Neurology 79, 1268–1272. 10.1212/WNL.0b013e31826aacd4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaye I. K. (2008). Haptoglobin, inflammation and disease. Trans. R. Soc. Trop. Med. Hyg. 102, 735–742. 10.1016/j.trstmh.2008.04.010 [DOI] [PubMed] [Google Scholar]
- Ramos K. S., Lin H., McGrath J. J. (1989). Modulation of cyclic guanosine monophosphate levels in cultured aortic smooth muscle cells by carbon monoxide. Biochem. Pharmacol. 38, 1368–1370. 10.1016/0006-2952(89)90347-X [DOI] [PubMed] [Google Scholar]
- Razin S. V., Ulianov S. V., Ioudinkova E. S., Gushchanskaya E. S., Gavrilov A. A., Iarovaia O. V. (2012). Domains of alpha- and beta-globin genes in the context of the structural-functional organization of the eukaryotic genome. Biochemistry 77, 1409–1423. 10.1134/S0006297912130019 [DOI] [PubMed] [Google Scholar]
- Reeder B. J. (2010). The redox activity of hemoglobins: from physiologic functions to pathologic mechanisms. Antioxid. Redox Signal. 13, 1087–1123. 10.1089/ars.2009.2974 [DOI] [PubMed] [Google Scholar]
- Reeder B. J., Cutruzzola F., Bigotti M. G., Hider R. C., Wilson M. T. (2008). Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radic. Biol. Med. 44, 274–283. 10.1016/j.freeradbiomed.2007.06.030 [DOI] [PubMed] [Google Scholar]
- Reedy C. J., Gibney B. R. (2004). Heme protein assemblies. Chem. Rev. 104, 617–649. 10.1021/cr0206115 [DOI] [PubMed] [Google Scholar]
- Reth M. (2002). Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 3, 1129–1134. 10.1038/ni1202-1129 [DOI] [PubMed] [Google Scholar]
- Rich P. R. (2003). The molecular machinery of Keilin's respiratory chain. Biochem. Soci. Trans. 31(Pt 6), 1095–1105. 10.1042/BST0311095 [DOI] [PubMed] [Google Scholar]
- Rifkind J. M., Nagababu E. (2013). Hemoglobin redox reactions and red blood cell aging. Antioxid. Redox Signal. 18, 2274–2283. 10.1089/ars.2012.4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. A., Baumgardner J. E., Otto C. M. (2011). Oxygen-dependent regulation of nitric oxide production by inducible nitric oxide synthase. Free Rad. Biol. Med. 51, 1952–1965. 10.1016/j.freeradbiomed.2011.08.034 [DOI] [PubMed] [Google Scholar]
- Rother R. P., Bell L., Hillmen P., Gladwin M. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293, 1653–1662. 10.1001/jama.293.13.1653 [DOI] [PubMed] [Google Scholar]
- Rudra C. B., Williams M. A., Sheppard L., Koenig J. Q., Schiff M. A., Frederick I. O., et al. (2010). Relation of whole blood carboxyhemoglobin concentration to ambient carbon monoxide exposure estimated using regression. Am. J. Epidemiol. 171, 942–951. 10.1093/aje/kwq022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter S. W., Choi A. M. (2013). Carbon monoxide: present and future indications for a medical gas. Korean J. Intern. Med. 28, 123–140. 10.3904/kjim.2013.28.2.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata M., Luo Z., Walsh K. (2001). Fas ligand overexpression on allograft endothelium inhibits inflammatory cell infiltration and transplant-associated intimal hyperplasia. J. Immunol. 166, 6964–6971. 10.4049/jimmunol.166.11.6964 [DOI] [PubMed] [Google Scholar]
- Sato K., Balla J., Otterbein L., Smith R. N., Brouard S., Lin Y., et al. (2001). Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J. Immunol. 166, 4185–4194. 10.4049/jimmunol.166.6.4185 [DOI] [PubMed] [Google Scholar]
- Schaer D. J., Buehler P. W., Alayash A. I., Belcher J. D., Vercellotti G. M. (2013). Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. 10.1182/blood-2012-11-451229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer D. J., Vinchi F., Ingoglia G., Tolosano E., Buehler P. W. (2014). Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front. Physiol. 5:415. 10.3389/fphys.2014.00415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter A. N. (2008). Hemoglobin research and the origins of molecular medicine. Blood 112, 3927–3938. 10.1182/blood-2008-04-078188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter A. N. (2012). Introduction to the symposium on synthetic life. Perspect. Biol. Med. 55, 467–469. 10.1353/pbm.2012.0049 [DOI] [PubMed] [Google Scholar]
- Schmitt T. H., Frezzatti W. A., Jr., Schreier S. (1993). Hemin-induced lipid membrane disorder and increased permeability: a molecular model for the mechanism of cell lysis. Arch. Biochem. Biophys. 307, 96–103. 10.1006/abbi.1993.1566 [DOI] [PubMed] [Google Scholar]
- Schofield C. J., Ratcliffe P. J. (2005). Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 338, 617–626. 10.1016/j.bbrc.2005.08.111 [DOI] [PubMed] [Google Scholar]
- Scholz C. C., Taylor C. T. (2013). Targeting the HIF pathway in inflammation and immunity. Curr. Opin. Pharmacol. 13, 646–653. 10.1016/j.coph.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Schultz I. J., Chen C., Paw B. H., Hamza I. (2010). Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 285, 26753–26759. 10.1074/jbc.R110.119503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. (2007). Life with oxygen. Science 318, 62–64. 10.1126/science.1147949 [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2009). Regulation of vascularization by hypoxia-inducible factor 1. Ann. N.Y. Acad. Sci. 1177, 2–8. 10.1111/j.1749-6632.2009.05032.x [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2011). Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547. 10.1056/NEJMra1011165 [DOI] [PubMed] [Google Scholar]
- Sen S., Peltz C., Beard J., Zeno B. (2010). Recurrent carbon monoxide poisoning from cigarette smoking. Am. J. Med. Sci. 340, 427–428. 10.1097/MAJ.0b013e3181ef712d [DOI] [PubMed] [Google Scholar]
- Sessa W. C. (2004). eNOS at a glance. J. Cell Sci. 117(Pt 12), 2427–2429 10.1242/jcs.01165 [DOI] [PubMed] [Google Scholar]
- Shaul P. W. (2002). Regulation of endothelial nitric oxide synthase: location, location, location. Annu. Rev. Physiol. 64, 749–774. 10.1146/annurev.physiol.64.081501.155952 [DOI] [PubMed] [Google Scholar]
- Sheftel A. D., Mason A. B., Ponka P. (2012). The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 1820, 161–187. 10.1016/j.bbagen.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikani H. J., Freeman B. D., Lisanti M. P., Weiss L. M., Tanowitz H. B., Desruisseaux M. S. (2012). Cerebral malaria: we have come a long way. Am. J. Pathol. 181, 1484–1492. 10.1016/j.ajpath.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S., Rassaf T., Patel R. P., Gladwin M. T. (2011). The detection of the nitrite reductase and NO-generating properties of haemoglobin by mitochondrial inhibition. Cardiovasc. Res. 89, 566–573. 10.1093/cvr/cvq327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora J., Orlov S. N., Furuya K., Grygorczyk R. (2014). Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 124, 2150–2157. 10.1182/blood-2014-05-572024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siri W. E., Van Dyke D. C., Winchell H. S., Pollycove M., Parker H. G., Cleveland A. S. (1966). Early erythropoietin, blood, and physiological responses to severe hypoxia in man. J. Appl. Physiol. 21, 73–80. [DOI] [PubMed] [Google Scholar]
- Sprague R. S., Stephenson A. H., Ellsworth M. L. (2007). Red not dead: signaling in and from erythrocytes. Trends Endocrinol. Metab. 18, 350–355. 10.1016/j.tem.2007.08.008 [DOI] [PubMed] [Google Scholar]
- Stuehr D. J. (2004). Enzymes of the L-arginine to nitric oxide pathway. J. Nutrition 134 Suppl., 2748S–2751S. discussion: 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- Stuehr D. J., Wei C. C., Santolini J., Wang Z., Aoyagi M., Getzoff E. D. (2004). Radical reactions of nitric oxide synthases. Biochem. Soc. Symp. 71, 39–49. [DOI] [PubMed] [Google Scholar]
- Sugawara Y., Hayashi Y., Shigemasa Y., Abe Y., Ohgushi I., Ueno E., et al. (2010). Molecular biosensing mechanisms in the spleen for the removal of aged and damaged red cells from the blood circulation. Sensors 10, 7099–7121. 10.3390/s100807099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman H. B., Carraway M. S., Tatro L. G., Piantadosi C. A. (2007). A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 120 (Pt 2), 299–308 10.1242/jcs.03318 [DOI] [PubMed] [Google Scholar]
- Taniyama Y., Griendling K. K. (2003). Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42, 1075–1081. 10.1161/01.HYP.0000100443.09293.4F [DOI] [PubMed] [Google Scholar]
- Taylor C. T. (2008). Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J. Physiol. 586(Pt 17), 4055–4059. 10.1113/jphysiol.2008.157669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. T., Cummins E. P. (2011). Regulation of gene expression by carbon dioxide. J. Physiol. 589(Pt 4), 797–803 10.1113/jphysiol.2010.201467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telezhkin V., Brazier S. P., Mears R., Muller C. T., Riccardi D., Kemp P. J. (2011). Cysteine residue 911 in C-terminal tail of human BK(Ca)alpha channel subunit is crucial for its activation by carbon monoxide. Pflugers Arch. 461, 665–675. 10.1007/s00424-011-0924-7 [DOI] [PubMed] [Google Scholar]
- Tenhunen R., Marver H. S., Schmid R. (1969). The enzymatic conversion of hemoglobin to bilirubin. Trans. Assoc. Am. Physicians 82, 363–371. [PubMed] [Google Scholar]
- Tiso M., Tejero J., Basu S., Azarov I., Wang X., Simplaceanu V., et al. (2011). Human neuroglobin functions as a redox-regulated nitrite reductase. J. Biol. Chem. 286, 18277–18289. 10.1074/jbc.M110.159541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosano E., Fagoonee S., Morello N., Vinchi F., Fiorito V. (2010). Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 12, 305–320. 10.1089/ars.2009.2787 [DOI] [PubMed] [Google Scholar]
- Vera T., Henegar J. R., Drummond H. A., Rimoldi J. M., Stec D. E. (2005). Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J. Am. Soc. Nephrol. 16, 950–958. 10.1681/ASN.2004090736 [DOI] [PubMed] [Google Scholar]
- Vitturi D. A., Sun C. W., Harper V. M., Thrash-Williams B., Cantu-Medellin N., Chacko B. K., et al. (2013). Antioxidant functions for the hemoglobin beta93 cysteine residue in erythrocytes and in the vascular compartment in vivo. Free Radic. Biol. Med. 55, 119–129. 10.1016/j.freeradbiomed.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall D. (2011). The inherited disorders of haemoglobin: an increasingly neglected global health burden. Indian J. Med. Res. 134, 493–497. [PMC free article] [PubMed] [Google Scholar]
- Weatherall D. J. (1995). The molecular basis for phenotypic diversity of genetic disease. Ann. N.Y. Acad. Sci. 758, 245–260. 10.1111/j.1749-6632.1995.tb24832.x [DOI] [PubMed] [Google Scholar]
- Wegiel B., Gallo D., Csizmadia E., Harris C., Belcher J., Vercellotti G. M., et al. (2013). Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 73, 7009–7021. 10.1158/0008-5472.CAN-13-1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. J. (1964). Nature of the iron-oxygen bond in oxyhaemoglobin. Nature 202, 83–84. 10.1038/202083b0 [DOI] [PubMed] [Google Scholar]
- White K. A., Marletta M. A. (1992). Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry 31, 6627–6631. 10.1021/bi00144a001 [DOI] [PubMed] [Google Scholar]
- Winslow R. M., Morrissey J. M., Berger R. L., Smith P. D., Gibson C. C. (1978). Variability of oxygen affinity of normal blood: an automated method of measurement. J. Appl. Physiol. 45, 289–297. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C., Carrell R. W. (1977). Oxidation of human haemoglobin by copper. Mechanism and suggested role of the thiol group of residue beta-93. Biochem. J. 165, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff D. G. (1976). The formation of carbon monoxide during peroxidation of microsomal lipids. Biochem. Biophys. Res. Commun. 73, 850–857. 10.1016/0006-291X(76)90199-6 [DOI] [PubMed] [Google Scholar]
- Wu C. J., Sheu J. R., Chen H. H., Liao H. F., Yang Y. C., Yang S., et al. (2006). Modulation of monocyte-derived dendritic cell differentiation is associated with ischemic acute renal failure. J. Surg. Res. 132, 104–111. 10.1016/j.jss.2005.09.029 [DOI] [PubMed] [Google Scholar]
- Yoshida T., Kikuchi G. (1974). Sequence of the reaction of heme catabolism catalyzed by the microsomal heme oxygenase system. FEBS Lett. 48, 256–261. 10.1016/0014-5793(74)80481-3 [DOI] [PubMed] [Google Scholar]
- Zuckerbraun B. S., McCloskey C. A., Gallo D., Liu F., Ifedigbo E., Otterbein L. E., et al. (2005). Carbon monoxide prevents multiple organ injury in a model of hemorrhagic shock and resuscitation. Shock 23, 527–532. [PubMed] [Google Scholar]