

Abstract
Hyperarousal and sleep disturbances are common, debilitating symptoms of post-traumatic stress disorder (PTSD). PTSD patients also exhibit abnormalities in quantitative electroencephalography (qEEG) power spectra during wake as well as rapid eye movement (REM) and non-REM (NREM) sleep. Selective serotonin reuptake inhibitors (SSRIs), the first-line pharmacological treatment for PTSD, provide modest remediation of the hyperarousal symptoms in PTSD patients, but have little to no effect on the sleep–wake architecture deficits. Development of novel therapeutics for these sleep–wake architecture deficits is limited by a lack of relevant animal models. Thus, the present study investigated whether single prolonged stress (SPS), a rodent model of traumatic stress, induces PTSD-like sleep–wake and qEEG spectral power abnormalities that correlate with changes in central serotonin (5-HT) and neuropeptide Y (NPY) signaling in rats. Rats were implanted with telemetric recording devices to continuously measure EEG before and after SPS treatment. A second cohort of rats was used to measure SPS-induced changes in plasma corticosterone, 5-HT utilization, and NPY expression in brain regions that comprise the neural fear circuitry. SPS caused sustained dysregulation of NREM and REM sleep, accompanied by state-dependent alterations in qEEG power spectra indicative of cortical hyperarousal. These changes corresponded with acute induction of the corticosterone receptor co-chaperone FK506-binding protein 51 and delayed reductions in 5-HT utilization and NPY expression in the amygdala. SPS represents a preclinical model of PTSD-related sleep–wake and qEEG disturbances with underlying alterations in neurotransmitter systems known to modulate both sleep–wake architecture and the neural fear circuitry.
Keywords: Single prolonged stress (SPS), post-traumatic stress disorder (PTSD), electroencephalography (EEG), FKBP5, serotonin (5-HT), neuropeptide Y (NPY)
Post-traumatic stress disorder (PTSD) is a debilitating psychiatric condition precipitated by experiencing a traumatic event and characterized by symptoms of reexperiencing, avoidance, and hyperarousal, which include disturbances in sleep–wake architecture.1,2 Polysomnographic studies in chronic PTSD patients as well as recently traumatized individuals have revealed deficits in both rapid eye movement (REM) and non-REM (NREM) sleep, including reduced and fragmented NREM and REM sleep, shortened latency to REM sleep, and increased REM density.2–6 Abnormalities in state-dependent quantitative electroencephalographic (qEEG) power spectra indicative of heightened arousal during wakefulness, such as increased high-frequency beta power, and inappropriate cortical activation during NREM sleep, including reduced low-frequency delta power (aka slow wave activity; SWA), have also been observed in individuals with PTSD.7–11 These abnormalities are correlated with structural and functional alterations in several brain regions that comprise the neural fear circuitry, including the hypothalamic–pituitary–adrenal (HPA) axis, amygdala, prefrontal cortex (PFC), and hippocampus.12 PTSD-related hyperarousal symptoms are specifically linked to impairments in prefrontal cortical and hippocampal functioning and a corresponding disinhibition of amygdala activity.12
Selective serotonin reuptake inhibitors (SSRIs) are the only currently approved pharmacological treatment for PTSD patients.13 Moreover, PTSD susceptibility and severity has been associated with a serotonin (5-HT) transporter gene polymorphism,14–17 further implicating disrupted serotonergic neurotransmission in the pathophysiology of the disorder. Unfortunately, while SSRIs ameliorate some symptoms of hyperarousal and comorbid depression,13 these drugs leave many PTSD patients with treatment-resistant insomnia and nightmares.13,18 Recent studies suggest that SSRIs may partially exert their therapeutic effects through modulation of neuropeptide Y (NPY) and its Y1 and Y2 receptor subtypes.19 NPY has anxiolytic20,21 and sleep-promoting22,23 properties, and is significantly decreased in the plasma and cerebrospinal fluid (CSF) of PTSD patients.24,25 Previous anatomical studies have shown that serotonergic terminals synapse onto NPY-expressing inhibitory interneurons in the amygdala,26 suggesting the possibility that combined disruption of these neurotransmitter systems may contribute to hyperarousal symptoms and sleep–wake disruptions in PTSD patients. Testing of this hypothesis, however, requires a valid animal model of traumatic stress and resulting sleep–wake and qEEG disturbances.
Single prolonged stress (SPS) is a rodent model of traumatic stress that has been shown to induce multiple PTSD-like physiological and behavioral abnormalities.27–29 However, it is not known whether SPS induces accompanying alterations in sleep–wake architecture and state-dependent qEEG power spectra. In the present study, we telemetrically recorded EEG from rats in their home cage to determine whether SPS causes reduced and fragmented NREM and REM sleep that persists beyond the day of traumatic stress, similar to PTSD patients. In addition, we performed qEEG spectral power analysis to evaluate whether SPS induces markers of chronically increased cortical activation during wake and NREM sleep consistent with PTSD-like hyperarousal. To determine whether alterations in sleep–wake architecture coincided with activation of the HPA axis, we assessed changes in several validated physiologic measures of the rodent stress response including hyperthermia, increases in plasma corticosterone,30 and induction of FK506-binding protein 51 (FKBP5), an early stress-responsive gene that acts as a co-chaperone of the glucocorticoid receptor complex.31 Finally, we evaluated the effects of SPS on regional 5-HT utilization and expression of NPY and its receptors to assess whether disruption of these neurotransmitter systems may be involved in mediating SPS-induced sleep–wake and qEEG spectral power alterations.
RESULTS
SPS Induced Acute and Persistent PTSD-Like Alterations in Sleep–Wake Architecture
SPS induced robust acute increases in percent time awake (Figure 1A) (time [F18,162 = 7.99, P < 0.0001], interaction [F18,162 = 12.65, P < 0.0001]) with concurrent reductions in time spent in NREM (Figure 1B) (time [F18,162 = 8.55, P < 0.0001], treatment [F1,9 = 5.35, P = 0.04], interaction [F18,162 = 13.84, P < 0.0001]), and REM sleep (Figure 1C) (time [F18,162 = 5.08, P < 0.0001], interaction [F18,162 = 12.18, P < 0.0001]) during the light (rodent quiescent) phase. The reductions in NREM and REM sleep during the light phase were followed by a rebound in these states during the dark (rodent active) phase. In contrast, SHAM treatment produced minor reductions in percent time awake relative to BL (Figure 1D) (time [F18,162 = 33.98, P < 0.0001], treatment [F1,9 = 51.75, P < 0.0001]), and increased time spent in NREM (Figure 1E) (time [F18,162 = 33.62, P < 0.0001], treatment [F1,9 = 40.15, P = 0.000l]), and REM sleep (Figure 1F) (time [F18,162 = 11.8, P < 0.0001], treatment [F1,9 = 42.3, P = 0.0001]).
Figure 1.
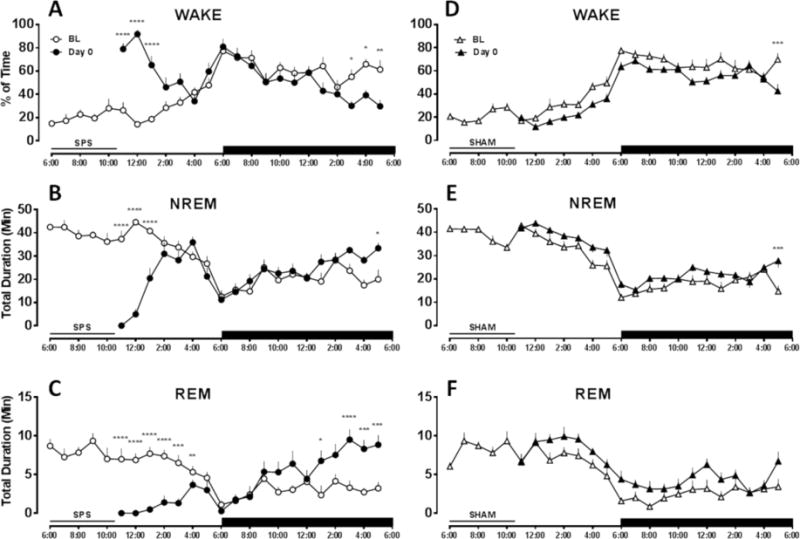
SPS induced acute alterations in sleep–wake architecture the day of treatment. SPS (left panels, n = 10) increased (A) % time spent in wake and suppressed (B) time in NREM and (C) time in REM sleep during the light phase. Both NREM and REM sleep rebounded during the dark phase. SHAM treatment (right panels, n = 10) caused the opposite effect, moderately decreasing (D) % time spent in wake and increasing (E) time in NREM and (F) time in REM sleep during the light phase. Black bar indicates dark phase. Missing values occur while the rats were removed from the recording room for treatment. No significant differences were detected between SPS BL and SHAM BL. Data are depicted as the mean + SEM. Comparison between treatment and BL performed by repeated measures two-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 in Bonferroni posthoc test compared to BL.
We then determined the time spent in wake as well as NREM and REM sleep on days 1, 2, and 7 post-SPS or SHAM treatment to determine whether SPS-induced sleep–wake alterations persisted beyond the day of traumatic stress. Increased wake and decreased NREM and REM sleep during the light phase persisted for at least 2 days post-SPS but normalized by day 7 (see Table 1 for statistical analysis). On day 2, SPS caused reductions in NREM bout length and increases in NREM bout number, indicative of sleep fragmentation. SHAM treatment produced no sustained effect on sleep–wake architecture (Table S1).
Table 1.
SPS Induced Persistent Disturbances in Sleep–Wake Architecturea
| light phase
| ||||||
|---|---|---|---|---|---|---|
| SPS BL | SPS day 1 | SPS day 2 | SPS day 7 | F | P | |
| WAKE (min/h) | 15.6 ± 0.7 | 18.7 ± 0.6*** | 17.8 ± 0.4* | 16.2 ± 0.8 | 7.80 | <0.001 |
| NREM (min/h) | 37.2 ± 0.8 | 35.7 ± 0.5* | 35.9 ± 0.5 | 36.9 ± 0.6 | 3.11 | 0.043 |
| REM (min/h) | 7.1 ± 0.3 | 5.6 ± 0.4*** | 6.1 ± 0.3* | 6.9 ± 0.4 | 10.0 | <0.001 |
| WAKE bouts/h | 10.8 ± 0.7 | 11.4 ± 0.8 | 12.4 ± 0.7 | 10.9 ± 1.0 | 2.21 | 0.110 |
| NREM bouts/h | 11.1 ± 0.7 | 11.8 ±0.8 | 13.0 ± 0.6* | 11.1 ± 1.0 | 3.63 | 0.026 |
| REM bouts/h | 4.1 ± 0.2 | 3.4 ± 0.2 | 4.1 ± 0.2 | 4.0 ± 0.4 | 2.25 | 0.106 |
| WAKE bout (min) | 1.3 ± 0.1 | 1.6 ±0.1* | 1.4 ± 0.1 | 1.4 ± 0.1 | 2.88 | 0.054 |
| NREM bout (min) | 3.5 ± 0.3 | 3.1 ± 0.2 | 2.8 ± 0.2** | 3.1 ± 0.2 | 5.43 | 0.005 |
| REM bout (min) | 1.8 ± 0.1 | 1.6 ± 0.1 | 1.5 ± 0.1 | 1.7 ± 0.1 | 2.62 | 0.071 |
| dark phase
| ||||||
| SPSBL | SPS day 1 | SPS day 2 | SPS day 7 | F | P | |
|
| ||||||
| WAKE (min/h) | 37.2 ± 1.0 | 30.6 ± 2.2*** | 31.6 ± 1.4* | 33.6 ± 1.7 | 7.09 | 0.001 |
| NREM (min/h) | 19.9 ± 0.8 | 23.8 ± 1.8** | 24.1 ± 1.2* | 22.8 ± 1.5 | 6.27 | 0.002 |
| REM (min/h) | 2.9 ± 0.3 | 5.5 ± 0.7*** | 4.3 ± 0.3 | 3.6 ± 0.3 | 7.34 | <0.001 |
| WAKE bouts/h | 8.4 ± 0.3 | 8.8 ± 0.6 | 8.9 ± 0.9 | 8.6 ± 0.6 | 0.257 | 0.856 |
| NREM bouts/h | 8.4 ± 0.3 | 9.0 ± 0.6 | 9.1 ± 0.9 | 8.8 ± 0.6 | 0.508 | 0.680 |
| REM bouts/h | 2.5 ± 0.2 | 3.0 ± 0.2 | 2.8 ± 0.2 | 2.7 ± 0.3 | 1.14 | 0.350 |
| WAKE bout (min) | 4.3 ± 0.3 | 3.5 ± 0.3 | 4.1 ± 0.5 | 4.7 ± 0.7 | 1.86 | 0.160 |
| NREM bout (min) | 2.4 ± 0.1 | 2.8 ± 0.1 | 2.6 ± 0.2 | 2.5 ± 0.2 | 1.80 | 0.172 |
| REM bout (min) | 1.2 ± 0.1 | 1.5 ±0.1*** | 1.3 ± 0.0 | 1.2 ± 0.1 | 10.7 | <0.001 |
Statistical analysis
P < 0.0S
P < 0.01
P < 0.001 in Bonferroni posthoc test compared to BL.
SPS Induced Acute and Sustained PTSD-Like Alterations in State-Dependent qEEG Power Spectra in the Frontal Cortex
We next tested the hypothesis that SPS would disrupt the normal qEEG power spectra within each sleep–wake state in a manner similar to that exhibited by PTSD patients. On day 0, SPS significantly altered qEEG power spectra in the frontal cortex during light phase wake, causing an increase in relative theta and high gamma power (black line, Figure 2A) (frequency [F100,900 = 5.91, P < 0.0001], interaction [F100,900 = 5.91, P < 0.0001]). In addition, SPS induced qEEG power spectra changes during dark phase wake, resulting in increased alpha, beta, and low gamma power and decreased high gamma power (black line, Figure 2D) (frequency [F100,900 = 33, P < 0.0001], interaction [F100,900 = 33, P < 0.0001]).
Figure 2.
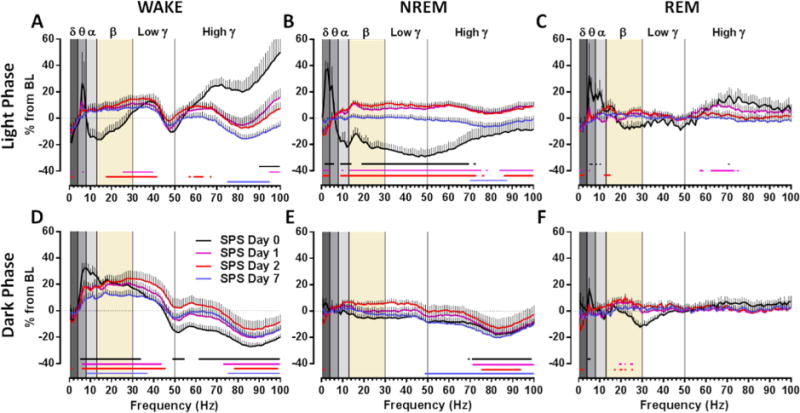
SPS induced acute and sustained alterations in qEEG power spectra in the frontal cortex. In the light phase (top panels), SPS caused (A) a transient increase in high gamma and a prolonged increase in low gamma during wake, (B) an acute rebound, but a persistent subsequent reduction in delta power during NREM sleep, and (C) a prolonged decrease in delta power during REM sleep. In the dark phase (bottom panels), SPS caused (D) an increase in theta, alpha, and low gamma with a sustained increase in beta and a sustained decrease in high gamma during wake, (E) a prolonged reduction in high gamma during NREM sleep, and (F) an acute increase in theta during REM sleep. Day 0 only includes values from remaining hours of the light phase immediately after SPS treatment. Data are depicted as the mean + SEM (n = 9–10). Background shades delineate power bands delta (δ), theta (θ), alpha (α), beta (β), and low and high gamma (γ). Comparison between treatment and BL was performed by repeated measures two-way ANOVA. Colored lines below data points correspond to each day and indicate P < 0.01 in Bonferroni posthoc test.
During light phase NREM sleep on day 0, SPS increased delta and theta power and decreased relative power in the higher frequencies (black line, Figure 2B) (frequency [F100,900 = 9.31, P < 0.0001], treatment [F1,9 = 11.82, P = 0.0074], interaction [F100,900 = 9.31, P < 0.0001]), whereas during dark phase NREM sleep, SPS caused a selective reduction in high gamma (black line, Figure 2E) (frequency [F100,900 = 4.90, P < 0.0001], treatment [F1,9 = 7.61, P = 0.0222], interaction [F100,900 = 4.90, P < 0.0001]). Finally, during light phase REM sleep, SPS increased theta and alpha power on day 0 (black line, Figure 2C) (frequency [F100,800 = 3.11, P < 0.0001], interaction [F100,800 = 3.11, P < 0.0001]) but decreased beta power during the dark phase (black line, Figure 2F) (frequency [F100,900 = 2.52, P < 0.0001], interaction [F100,900 = 2.52, P < 0.0001]).
These alterations in qEEG power spectra were sustained for multiple days after SPS. Thus, SPS increased beta and low gamma power and decreased high gamma power during light phase wake (colored lines, Figure 2A) (frequency [F100,900 = 4.56, P < 0.0001], interaction [F300,2700 = 3.81, P < 0.0001]) and dark phase wake (colored lines, Figure 2D) (frequency [F100,900 = 17.96, P < 0.0001], interaction [F300,2700 = 10.32, P < 0.0001]) over the entire 7 day time course. Delta power was significantly reduced for at least 2 days post-SPS during light phase NREM sleep (colored lines, Figure 2B) (frequency [F100,900 = 5.02, P < 0.0001], treatment [F3,27 = 9.07, P = 0.0003], interaction [F300,2700 = 5.46, P < 0.0001]) and was more moderately reduced during dark phase NREM sleep (colored lines, Figure 2E) (frequency [F100,900 = 6.05, P < 0.0001], treatment [F3,27 = 3.34, P = 0.0341], interaction [F300,2700 = 4.44, P < 0.0001]). Most alterations in qEEG power spectra during REM sleep normalized by day 2 with the exception of a sustained decrease in delta during the light phase (colored lines, Figure 2C) (interaction [F300,2700 = 2.16, P < 0.0001]) and dark phase (colored lines, Figure 2F) (interaction [F300,2700 = 1.15, P = 0.0465]). SPS treatment had similar effects in the parietal cortex (Figure S1), but it did not decrease waking high gamma in this brain region. SHAM treatment produced only minor effects on qEEG power spectra in the frontal (Figure S2) and parietal cortex (Figure S3).
SPS Induced Prolonged Reductions in SWA
SWA was highest during the early hours of the light phase and gradually reduced across the quiescent period (Figure 3), consistent with dissipation of sleep drive.32 Relative to BL, SPS increased SWA on day 0 (treatment [F1,50 = 42.74, P < 0.0001]), consistent with the rebound effects of sleep deprivation,32 but produced significantly decreased SWA on days 1 and 2 post-SPS treatment (Figure 3) (time [F15,215 = 24.33, P < 0.0001], treatment [F15,215 = 14.19, P < 0.0001]), especially during the early hours of the light phase. SHAM treatment had no prolonged effect on SWA (Figure S4). No SWA differences were detected between SPS BL and SHAM BL.
Figure 3.
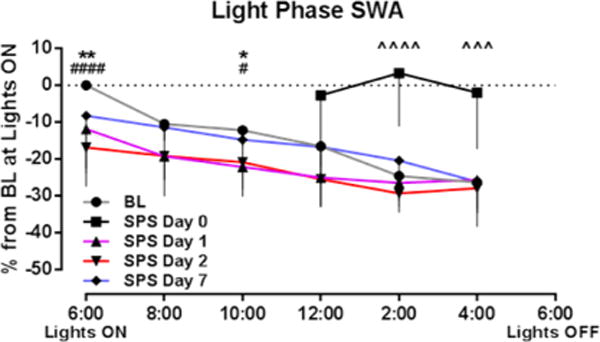
SPS induced prolonged reductions in slow wave activity (SWA). During the light (rodent quiescent) phase, SPS caused an initial rebound in SWA immediately after SPS but subsequently reduced SWA for up to 2 days post-SPS. Data are depicted as the mean – SEM (n = 9–10). ˆˆˆ, P < 0.001 and ˆˆˆˆ, P < 0.0001 (day 0 vs BL); *,P < 0.05 and **, P < 0.01 (day 1 vs BL); #, P < 0.05 and ####, P < 0.0001 (day 2 vs BL) in Bonferroni posthoc test.
SPS Induced an Acute and Persistent Physiological Stress Response
Given the magnitude and duration of SPS-induced sleep–wake and qEEG disruptions, we measured concomitant changes in several validated measures of the rodent stress response, including hyperthermia, corticosterone release, and brain regional FKBP5 induction.30,31 SPS induced acute and persistent hyperthermia for several days post-SPS (Figure 4A) during wake (day 0: hour [F8,158 = 3.2, P = 0.0022], hour [F8,158 = 3.2, P = 0.0022]; days 1, 2, and 7: interaction [F33,430 = 1.69, P = 0.0116], treatment [F3,430 = 4.67, P < 0.0032], hour [F11,430 = 5.29, P < 0.0001]), NREM (day 0: hour [F8,155 = 2.13, P = 0.0357], treatment [F1,155 = 4.11, P < 0.0443], interaction [F8,155 = 2.13, P = 0.0357]; days 1, 2, and 7: interaction [F33,424 = 1.85, P = 0.0035], treatment [F3,424 = 6.5, P < 0.0003], hour [F11,424 = 5.78, P < 0.0001]), and REM sleep (day 0: hour [F8,118 = 2.84, P = 0.0064], treatment [F1,118 = 16.82, P < 0.0001], interaction [F8,118 = 2.84, P = 0.0064]; days 1, 2, and 7: interaction [F33,398 = 1.56, P = 0.0279], treatment [F3,398 = 5.91, P < 0.0006], hour [F11,398 = 6.63, P < 0.0001]), specifically during the light phase. SHAM treatment had only minor effects on body temperature (Figure S5). In parallel with body temperature increases, SPS rats exhibited robust acute HPA axis activation, as evidenced by elevated plasma corticosterone (Figure 4B) [F3,29 = 14.67, P < 0.0001]. In addition, there was a concurrent acute induction of FKBP5 mRNA levels in the brain regions that comprise the neural fear circuitry (Figure 4C), including the PFC [F3,28 = 25.43, P < 0.0001], hippocampus [F3,28 = 40.84, P < 0.0001], and amygdala [F3,27 = 36.46, P < 0.0001].
Figure 4.
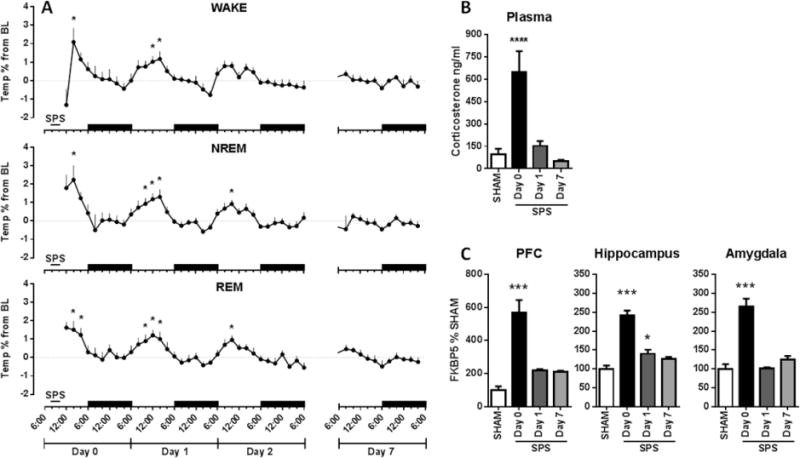
SPS induced an acute and persistent physiological stress response. SPS caused (A) increases in body temperature during the light phase of all sleep–wake states that lasted until day 2 (n = 9–10). Black bars indicate dark phases. SPS also acutely increased (B) plasma corticosterone concentrations and (C) FKBP5 mRNA levels in the PFC, hippocampus, and amygdala (n = 8–9). *, P < 0.05; **, P < 0.01; ****, P < 0.0001 in Bonferroni posthoc test compared to BL (A) or Dunnett’s posthoc test compared to SHAM (B, C).
SPS Caused Acute and Sustained Alterations in Brain Regional 5-HT Utilization
Due to the well-established role of 5-HT in modulating anxiety and sleep–wake architecture,33 we evaluated whether the observed SPS-induced sleep–wake and qEEG power spectra changes were associated with altered 5-HT signaling. SPS produced acute increases in the levels of the 5-HT metabolite, 5-HIAA, in the PFC (Figure 5A) [F3,32 = 29.31, P < 0.0001] and hippocampus (Figure 5B) [F3,32 = 8.70, P = 0.0002] and prolonged reductions in the amygdala (Figure 5C) [F3,32 = 5.98, P = 0.0023], with no effect on 5-HT levels across the three brain regions (Figure 5D–F).
Figure 5.
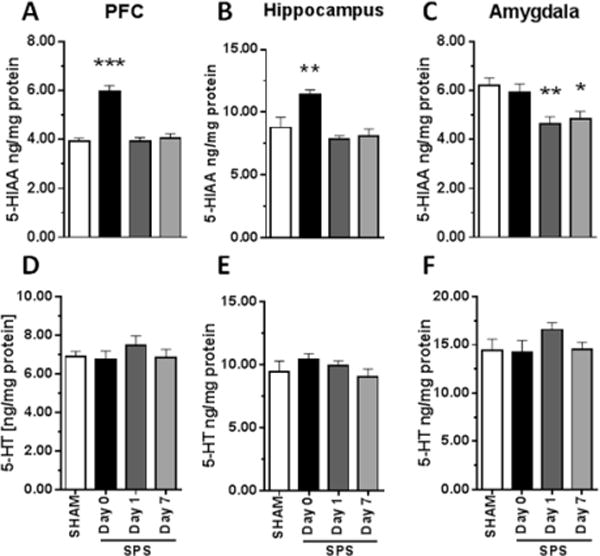
SPS caused acute and sustained alterations in brain regional 5-HT utilization. Concentration of the 5-HT metabolite 5-HIAA was increased by SPS on day 0 in (A) the PFC and (B) the hippocampus but decreased on days 1 and 7 in (C) the amygdala. (D–F) 5-HT levels in these regions were unchanged (n = 8–9) *, P < 0.05; **, P < 0.01 in posthoc Dunnett’s test versus SHAM.
SPS Caused Delayed Reductions in Amygdala Expression of NPY
As previously discussed, NPY signaling in the amygdala plays a critical role in modulating the stress response;20 thus, we hypothesized that SPS would alter expression of NPY and its Y1 and Y2 receptor subtypes specifically in the amygdala. While SPS had no effect on Y1 or Y2 mRNA levels, NPY mRNA levels were significantly reduced in the amygdala by day 7 post-SPS (Figure 6) [F3,26 = 4.94, P = 0.0076].
Figure 6.
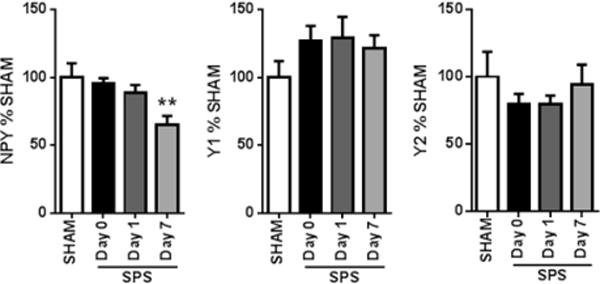
SPS caused acute and sustained alterations in amygdala expression of neuropeptide Y (NPY). mRNA levels of NPY were reduced on day 7 after SPS, while NPY receptor mRNA levels were unchanged in the amygdala (n = 8–9). **, P < 0.01 in posthoc Dunnett’s test versus SHAM.
SPS produced robust alterations in sleep–wake architecture accompanied by state-dependent changes in qEEG power spectra that resemble PTSD symptomatology. These changes corresponded with time-dependent and brain region-specific alterations in physiological markers of HPA axis activation, 5-HT utilization, and NPY expression, suggesting key alterations in the neural fear circuitry that may potentially underlie PTSD-related hyperarousal and sleep–wake disturbances.
DISCUSSION
Our current findings demonstrate SPS-induced dysregulation and fragmentation of NREM and REM sleep that mirror the abnormal sleep–wake patterns of recently traumatized individuals5,34 and patients with chronic PTSD.3 The observed changes in sleep–wake architecture on day 0 post-SPS, however, represent the immediate effects of traumatic stress, which have been not yet been assessed in traumatized clinical populations. In healthy subjects, the amount of NREM and especially REM sleep accumulated immediately following emotional learning imparts strong and lasting benefits to the consolidation and subsequent recall of these new memories,35–37 suggesting that acute SPS-induced reductions in sleep may actually represent a protective response in the hours after traumatic stress on day 0. In contrast, the significant fragmentation of NREM sleep coupled with increased wake time during the quiescent phases observed on days 1 and 2 closely resembles the previously documented sleep disruptions in chronic PTSD patients.2–5,34 Taken together, these alterations in NREM and REM sleep provide attractive windows for testing novel mechanisms of sleep-dependent prophylactic intervention in the aftermath of a traumatic event.
The SPS-induced alterations in qEEG power spectra during wake as well as NREM and REM sleep also recapitulate many of the qEEG abnormalities reported in PTSD patients. Augmentation of beta/low gamma power during wake in the active phase was one of the most enduring effects of SPS, lasting for at least 7 days. In chronic PTSD patient populations, increased waking beta/low gamma power has been reported both at rest7,9 and in response to affective stimuli,8 although this finding was absent in one study.38 Reductions in waking high gamma power were also sustained for 7 days post-SPS, a change that was specific to frontal cortical regions, as it was absent in recordings from the parietal cortex. A recent functional magnetic resonance imaging (fMRI) study demonstrated that increases in high gamma power are correlated with activation of corresponding cortical regions.39 The current finding of SPS-induced reductions in this power band specifically in the frontal cortex, therefore, could be indicative of PFC hypoactivity, a commonly reported finding in PTSD patients.12
During NREM sleep, SPS caused a prolonged, but not acute, increase in beta/gamma power and a decrease in SWA that mirrors similar deficits in patients with PTSD10,11 and likely indicates poor sleep quality.40 This finding further supports the interpretation that the previously discussed sleep–wake architecture changes present on day 2 represent sleep disturbances related to established PTSD. Impaired SWA has also been associated with impairments in sleep-dependent fear extinction memory, a robust PTSD-like behavioral effect of SPS.29 Pharmacologically augmenting SWA, therefore, could represent a therapeutic approach for PTSD patients or recently traumatized individuals that can be tested in this model. In the case of REM sleep, the most significant change in frontal cortical qEEG spectral power was an acute increase in frontal theta power, which is thought to promote emotional memory consolidation in rodents41 and humans,35 perhaps contributing to the subsequent development of PTSD-like symptoms in the SPS model. Future experiments correlating SPS-induced alterations in theta power during REM sleep with subsequent anxiety-like behaviors will be critical for understanding the function of these state-specific oscillations in the processing of traumatic events.
In addition to causing sustained qEEG deficits indicative of hyperarousal, we confirmed that SPS concomittantly and robustly induces markers of HPA axis activation. For example, SPS induced hyperthermia during all sleep–wake states and caused substantial increases in plasma corticosterone, consistent with previous findings.42 Corticosterone release was accompanied by induction of FKBP5 expression, a gene that has been associated with PTSD risk, diagnosis, and treatment,43–45 and which may contribute to acute SPS-induced sleep loss.46
SPS-induced reductions in NREM and REM sleep during the quiescent phase could also be explained by acute increases in 5-HT utilization in the PFC and hippocampus, brain structures implicated in the modulation of sleep–wake architecture.47 This hypothesis is supported by reports that other acute stressors cause 5-HT release in multiple brain regions including the cortex, leading to inhibition of sleep.48 In the amygdala, however, SPS did not increase 5-HT utilization but rather induced a delayed decrease on days 1 and 7 that correlated with sustained increases in relative beta/low gamma power during wake. 5-HT exerts a net inhibitory influence on the excitability of lateral amygdala neurons,49,50 which, when directly stimulated, can induce high-frequency EEG oscillations51 highly comparable to the long-lasting effects of SPS. This finding may help to explain the partial efficacy of SSRIs on hyperarousal symptoms in PTSD patients and their behavioral correlates in SPS-treated rats.52,53 Similar to 5-HT, SPS caused a delayed reduction in expression of amygdala NPY, which also acts to inhibit the firing of projection neurons in the lateral amygdala.54 Importantly, this finding is consistent with previous studies demonstrating the therapeutic efficacy of exogenous NPY administration in SPS-treated rats.55,56 Moreover, intracerebroventricular infusion of NPY in rats increases low-frequency and decreases beta-frequency qEEG spectral power,57,58 in direct opposition to the long-term effects of SPS. The changes in NPY expression reported here were at the level of mRNA, however, and may not translate into reductions in amygdala peptide concentration or, more importantly, peptide release.
Collectively, we have demonstrated that SPS, a rodent model of traumatic stress, leads to alterations in sleep–wake architecture and state-dependent qEEG spectral power that correlate with regional changes in 5-HT utilization and NPY expression, providing new insight into the pathophysiology of PTSD-related hyperarousal and sleep–wake disturbances. Ongoing studies are focused on evaluating whether potential novel therapeutic interventions can ameliorate the alterations in sleep–wake architecture and state-dependent qEEG power spectra induced by SPS.
METHODS
Subjects
All male Sprague–Dawley rats (Harlan, Indianapolis, IN) used in the present studies were housed under a 12 h light/12 h dark cycle and given ad libitum access to food and water. All animal experiments were approved by the Vanderbilt University Animal Care and Use Committee, and experimental procedures conformed to guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and the number of animals used.
Surgery
Twenty male rats (250–375 g) were surgically implanted with a telemetry transmitter (4-ET, Data Sciences International, St. Paul, MN) for recording EEG, electromyography (EMG), and body temperature. Under isoflurane anesthesia (3% induction; 1.5–2.5% maintenance), the transmitter was implanted subcutaneously across the back of each rat. Transmitter leads were tunneled subcutaneously to the skull. After holes were drilled in the skull, the exposed wires were placed in contact with the dura and secured in place with dental cement (Butler Schein, Dublin, OH). Three sets of leads were placed bilaterally to record from cortical regions corresponding with the frontal, parietal, and occipital cortices (+2 mm, −2 mm, and −6 mm anterior–posterior from Bregma, respectively, and ±2 mm lateral to the midline). An additional set of leads was placed bilaterally in the nuchal muscles for EMG recording. Rats were individually housed following surgery and allowed to recover and acclimate to the recording room for a minimum of 10 days prior to testing.
Experimental Design
After postoperative recovery, each rat was randomized into either the SPS or SHAM group. Continuous 24 h baseline (BL) recordings were performed for each rat in its home cage to serve as within-subjects comparator for all subsequent sleep–wake, qEEG, and body temperature data. After BL recordings, each rat received either SPS or SHAM treatment. Immediately following treatment, home cage recordings were reinitiated in both groups (day 0) and continued for 2 days (days 1 and 2), after which transmitters were turned off and then reactivated on day 7. Subsequent off-line analysis of sleep–wake and qEEG data was divided into the remaining hours of day 0 or in 24 h intervals comprising days 1, 2, and 7 post-SPS or SHAM treatment. Figure 7 depicts the experimental design for the EEG studies (cohort 1) as well as the time points for tissue collections for the biochemistry and neurochemistry studies (cohort 2). For all experiments, SPS or SHAM treatment occurred within the first 6 h of the light phase.
Figure 7.
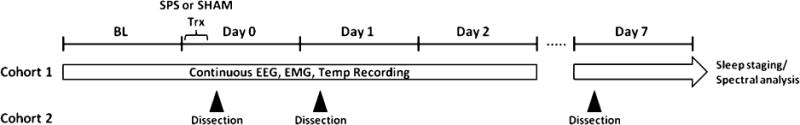
Experimental design for EEG studies and tissue collection. In cohort 1, continuous EEG, EMG, and temperature data were telemetrically recorded from chronically implanted rats throughout successive 24 h light-dark cycles (ON, 6:00 am; OFF, 6:00 pm) before (BL) and several days after (days 0, 1, 2, and 7) either single prolonged stress (SPS) or SHAM treatment. Both treatments were performed within the first 6 h of the light phase on day 0, during which recording was not possible; EEG data from this day was reinitiated when each animal was returned to its home cage. In cohort 2, nonimplanted aged-matched rats underwent either SPS or SHAM treatment. SPS rats were sacrificed either 1 h (day 0), 1 day (day 1), or 7 days (day 7) later; SHAM rats were sacrificed 7 days later.
Single Prolonged Stress (SPS)
SPS was performed according to Liberzon et al.28 Briefly, rats were restrained for 2 h, followed by forced swim for 15 min in 24°C water. Following a 15 min recovery period, rats were exposed to diethyl ether vapor in a bell jar until anesthesia. The SPS model did not cause mortality. SPS did illicit hallmarks of the rodent stress response such as porphyrin staining of the eyes and urination and defecation. There were no major individual differences observed in these parameters during each experiment, and no inclusion or exclusion criteria were applied prior to the start of EEG recordings or tissue collection. SHAM treatment consisted of placement in a novel procedure room for 2 h followed by brief handling. All animals were placed into fresh cages after treatment.
Tissue Collection
For all biochemical and neurochemical end points, a group of 36 nonimplanted rats was randomly assigned to SHAM treatment or one of three SPS groups (day 0, 1 or 7). Rats were briefly anesthetized with isoflurane and sacrificed by decapitation either immediately (day 0), 1 day (day 1), or 7 days (day 7) after SPS; SHAM rats were sacrificed immediately after SHAM treatment. Hippocampus, amygdala, and PFC were dissected, rapidly frozen on dry ice, and stored at −80°C for tissue mRNA and neurochemistry experiments. Trunk blood was collected into heparin-lined tubes and then centrifuged at 5000 rpm for 9 min at 4°C to obtain plasma.
Plasma Corticosterone
Corticosterone, the rodent analogue of the human glucocorticoid cortisol, was measured using a double antibody radioimmunoassay (RIA) kit (MP Biomedicals, Orangeburg, NY).
Tissue Neurochemistry
Tissue concentrations of 5-HT and its metabolite 5-hydroxyindoleacetic acid (5-HIAA) were determined by HPLC-ECD as described previously.59
qRT-PCR
Alterations in mRNA expression levels of NPY and its Y1 and Y2 receptor subtypes were measured using Aqueous Micro kits (Life Technologies, Grand Island, NY) for RNA extraction, a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) was used for RNA quantification, a QuantiTect reverse transcription kit (QIAGEN, Hilden, Germany) was used for complementary DNA transcription, and a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA) using primers from TaqMan gene expression assays (Life Technologies) was used for qRT-PCR of rat NPY (Rn01410145_m1), Y1 (Rn02769337_s1), and Y2 (Rn00576733_s1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control; data are presented using the comparative cycle threshold (CT) method normalized to SHAM-treated rats.
Data Analysis
Sleep Staging
EEG, EMG, and temperature data were collected with Dataquest A.R.T. 4.3 software (D, Minneapolis, MN) using a continuous sampling method. Telemetric data were sampled at a rate of 500 Hz and transmitted via a receiver (RPC-2, D) placed below the cage of each rat. Each receiver was connected to a data exchange matrix (D) which transferred EEG, EMG, and temperature data to a computer for off-line analysis. Two trained observers used Neuroscore 3.0 software to manually stage each 10 s epoch as wake, NREM, or REM sleep based on accepted characteristic EEG and EMG oscillatory patterns.60 All 10 s epochs were summed into 60 min bins. For the acute effects of SPS or SHAM treatment on day 0, 60 min bins were group-averaged to examine the amount of time spent in wake, NREM sleep, or REM sleep. To assess the prolonged effects of SPS or SHAM treatment (days 1,2, and 7), 12 h bins comprising either the light or dark phase of a given day were group-averaged.
qEEG Spectral Power Analysis
qEEG relative power spectra from frontal and parietal electrodes were computed for each rat and on each day of recording in 10 s epochs in 1 Hz bins from 0.5 to 100 Hz using a fast Fourier transform with a Hamming window and overlap ratio of 0.5. Relative power within each 1 Hz increment was calculated as a percent of total power, binned by stage (wake, NREM, or REM), and averaged across the 12 h light or dark phase to yield the state-dependent relative power spectrum for each rat. To calculate the percent change from BL, the following formula was used
where relative power (post-treatment day) is the relative power value of a frequency bin of a rat on day 0, 1, 2, or 7, and relative power (BL) is the BL value of the same frequency bin for the same rat during the corresponding sleep–wake stage and light–dark phase. The percent change values were then group-averaged. The qEEG changes are discussed in terms of changes in power bands defined based on convention as delta (0.5–4 Hz), theta (5–8 Hz), alpha (9–13 Hz), beta (14–30 Hz), low gamma (31–50 Hz), and high gamma (51–100 Hz).61 Slow wave activity (SWA) was defined as relative delta power in the frontal cortex during NREM sleep; a time course of SWA changes was calculated by normalizing SWA values for each rat, in 2 h bins, to the same rat’s BL SWA value during the first 2 h of the light phase.
Statistical Analysis
For the acute effects of SPS or SHAM on sleep–wake architecture (day 0) and the effect of SPS or SHAM on qEEG spectral power, a repeated measures two-way analysis of variance (ANOVA) was applied; if significant, then a Bonferroni posthoc test was performed with significance defined as P < 0.05 for sleep–wake data and P < 0.01 for qEEG data. For the prolonged effects of SPS or SHAM (days 1, 2, and 7), a repeated measures one-way ANOVA followed by a Dunnett’s posthoc test was used with significance defined as P < 0.05. Two-way ANOVA without repeated measures was used to analyze temperature and SWA changes due to the fact that certain rats did not enter NREM or REM states during various 2 h epochs, resulting in randomly missing values. If significant, then Bonferroni posthoc tests were conducted with significance defined as P < 0.05. Day 0 sleep–wake, qEEG, and temperature data were analyzed separately from days 1, 2, and 7 to distinguish between the acute and prolonged effects of SPS, which could differ due to the short-term rebound effects of sleep deprivation. One rat in the SPS group did not enter REM sleep during the light phase of day 0 and was excluded from spectral and temperature analysis for this period. One rat in the SHAM group was excluded from spectral and temperature analysis on day 7 due to transmitter failure. For qRT-PCR, tissue neurochemistry, and plasma corticosterone data, analysis was performed by one-way ANOVA followed by Dunnett’s posthoc test with significance defined as P < 0.05.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of Mental Health to C.K.J. (MH86601) and P.J.C. (MH97056) as well as the Barrus Foundation to C.K.J, and the Department of Veterans Affairs to C.K.J. at the VA Tennessee Valley Healthcare System (CDA-2-051-07F). We thank Dina McGinnis for performing EEG surgeries. A portion of these studies was presented at the Society for Biological Psychiatry 2014 meeting in New York, NY.
Footnotes
Supporting Information: Effect of SHAM treatment on sleep–wake and quantitative EEG in the frontal and parietal cortices and on slow wave activity and body temperature. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Over the past year, P.J.C. and C.K.J. received research/salary support from Bristol Meyers Squibb, AstraZeneca, and/or Johnson and Johnson.
References
- 1.Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5. American Psychiatric Association; Arlington, VA: 2013. [Google Scholar]
- 2.Germain A. Sleep disturbances as the hallmark of PTSD: where are we now? Am J Psychiatry. 2013;170:372–382. doi: 10.1176/appi.ajp.2012.12040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi I, Boarts JM, Delahanty DL. Polysomnographically measured sleep abnormalities in PTSD: a meta-analytic review. Psychophysiology. 2007;44:660–669. doi: 10.1111/j.1469-8986.2007.537.x. [DOI] [PubMed] [Google Scholar]
- 4.Yetkin S, Aydin H, Ozgen F. Polysomnography in patients with post-traumatic stress disorder. Psychiatry Clin Neurosci. 2010;64:309–317. doi: 10.1111/j.1440-1819.2010.02084.x. [DOI] [PubMed] [Google Scholar]
- 5.Mellman TA, Bustamante V, Fins AI, Pigeon WR, Nolan B. REM sleep and the early development of posttraumatic stress disorder. Am J Psychiatry. 2002;159:1696–1701. doi: 10.1176/appi.ajp.159.10.1696. [DOI] [PubMed] [Google Scholar]
- 6.Mellman TA, Pigeon WR, Nowell PD, Nolan B. Relationships between REM sleep findings and PTSD symptoms during the early aftermath of trauma. J Trauma Stress. 2007;20:893–901. doi: 10.1002/jts.20246. [DOI] [PubMed] [Google Scholar]
- 7.Begic D, Hotujac L, Jokic-Begic N. Electroencephalographic comparison of veterans with combat-related post-traumatic stress disorder and healthy subjects. Int J Psychophysiol. 2001;40:167–172. doi: 10.1016/s0167-8760(00)00153-7. [DOI] [PubMed] [Google Scholar]
- 8.Ehlers CL, Hurst S, Phillips E, Gilder DA, Dixon M, Gross A, Lau P, Yehuda R. Electrophysiological responses to affective stimuli in American Indians experiencing trauma with and without PTSD. Ann NY Acad Sci. 2006;1071:125–136. doi: 10.1196/annals.1364.011. [DOI] [PubMed] [Google Scholar]
- 9.Jokic-Begic N, Begic D. Quantitative electroencephalogram (qEEG) in combat veterans with post-traumatic stress disorder (PTSD) Nord J Psychiatry. 2003;57:351–355. doi: 10.1080/08039480310002688. [DOI] [PubMed] [Google Scholar]
- 10.Neylan TC, Lenoci M, Maglione ML, Rosenlicht NZ, Metzler TJ, Otte C, Schoenfeld FB, Yehuda R, Marmar CR. Delta sleep response to metyrapone in post-traumatic stress disorder. Neuropsychopharmacology. 2003;28:1666–1676. doi: 10.1038/sj.npp.1300215. [DOI] [PubMed] [Google Scholar]
- 11.Woodward SH, Murburg MM, Bliwise DL. PTSD-related hyperarousal assessed during sleep. Physiol Behav. 2000;70:197–203. doi: 10.1016/s0031-9384(00)00271-7. [DOI] [PubMed] [Google Scholar]
- 12.Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger W, Mendlowicz MV, Marques-Portella C, Kinrys G, Fontenelle LF, Marmar CR, Figueira I. Pharmacologic alternatives to antidepressants in posttraumatic stress disorder: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:169–180. doi: 10.1016/j.pnpbp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HJ, Lee MS, Kang RH, Kim H, Kim SD, Kee BS, Kim YH, Kim YK, Kim JB, Yeon BK, Oh KS, Oh BH, Yoon JS, Lee C, Jung HY, Chee IS, Paik IH. Influence of the serotonin transporter promoter gene polymorphism on susceptibility to posttraumatic stress disorder. Depression Anxiety. 2005;21:135–139. doi: 10.1002/da.20064. [DOI] [PubMed] [Google Scholar]
- 15.Kimbrel NA, Morissette SB, Meyer EC, Chrestman R, Jamroz R, Silvia PJ, Beckham JC, Young KA. Effect of the 5-HTTLPR polymorphism on posttraumatic stress disorder, depression, anxiety, and quality of life among Iraq and Afghanistan veterans. Anxiety, Stress, Coping. 2014:1–24. doi: 10.1080/10615806.2014.973862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wald I, Degnan KA, Gorodetsky E, Charney DS, Fox NA, Fruchter E, Goldman D, Lubin G, Pine DS, Bar-Haim Y. Attention to threats and combat-related posttraumatic stress symptoms: prospective associations and moderation by the serotonin transporter gene. JAMA Psychiatry. 2013;70:401–408. doi: 10.1001/2013.jamapsychiatry.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh K, Uddin M, Soliven R, Wildman DE, Bradley B. Associations between the SS variant of 5-HTTLPR and PTSD among adults with histories of childhood emotional abuse: results from two African American independent samples. J Affective Disord. 2014;161:91–96. doi: 10.1016/j.jad.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nappi CM, Drummond SP, Hall JM. Treating nightmares and insomnia in posttraumatic stress disorder: a review of current evidence. Neuropharmacology. 2012;62:576–585. doi: 10.1016/j.neuropharm.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redrobe JP, Dumont Y, Quirion R. Neuropeptide Y (NPY) and depression: from animal studies to the human condition. Life Sci. 2002;71:2921–2937. doi: 10.1016/s0024-3205(02)02159-8. [DOI] [PubMed] [Google Scholar]
- 20.Gutman AR, Yang Y, Ressler KJ, Davis M. The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci. 2008;28:12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heilig M. The NPY system in stress, anxiety and depression. Neuropeptides. 2004;38:213–224. doi: 10.1016/j.npep.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Antonijevic IA, Murck H, Bohlhalter S, Frieboes RM, Holsboer F, Steiger A. Neuropeptide Y promotes sleep and inhibits ACTH and cortisol release in young men. Neuropharmacology. 2000;39:1474–1481. doi: 10.1016/s0028-3908(00)00057-5. [DOI] [PubMed] [Google Scholar]
- 23.Zini I, Merlo Pich E, Fuxe K, Lenzi PL, Agnati LF, Harfstrand A, Mutt V, Tatemoto K, Moscara M. Actions of centrally administered neuropeptide Y on EEG activity in different rat strains and in different phases of their circadian cycle. Acta Physiol Scand. 1984;122:71–77. doi: 10.1111/j.1748-1716.1984.tb07483.x. [DOI] [PubMed] [Google Scholar]
- 24.Rasmusson AM, Hauger RL, Morgan CA, Bremner JD, Charney DS, Southwick SM. Low baseline and yohimbine-stimulated plasma neuropeptide Y (NPY) levels in combat-related PTSD. Biol Psychiatry. 2000;47:526–539. doi: 10.1016/s0006-3223(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 25.Sah R, Ekhator NN, Jefferson-Wilson L, Horn PS, Geracioti TD., Jr Cerebrospinal fluid neuropeptide Y in combat veterans with and without posttraumatic stress disorder. Psychoneuroendocrinology. 2014;40:277–283. doi: 10.1016/j.psyneuen.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonn M, Schmitt A, Lesch KP, Van Bockstaele EJ, Asan E. Serotonergic innervation and serotonin receptor expression of NPY-producing neurons in the rat lateral and basolateral amygdaloid nuclei. Brain Struct Funct. 2013;218:421–435. doi: 10.1007/s00429-012-0406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberzon I, Krstov M, Young EA. Stress–restress: effects on ACTH and fast feedback. Psychoneuroendocrinology. 1997;22:443–453. doi: 10.1016/s0306-4530(97)00044-9. [DOI] [PubMed] [Google Scholar]
- 28.Liberzon I, Lopez JF, Flagel SB, Vazquez DM, Young EA. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: relevance to post-traumatic stress disorder. J Neuroendocrinol. 1999;11:11–17. doi: 10.1046/j.1365-2826.1999.00288.x. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto S, Morinobu S, Takei S, Fuchikami M, Matsuki A, Yamawaki S, Liberzon I. Single prolonged stress: toward an animal model of posttraumatic stress disorder. Depression Anxiety. 2009;26:1110–1117. doi: 10.1002/da.20629. [DOI] [PubMed] [Google Scholar]
- 30.Groenink L, van der Gugten J, Zethof T, van der Heyden J, Olivier B. Stress-induced hyperthermia in mice: hormonal correlates. Physiol Behav. 1994;56:747–749. doi: 10.1016/0031-9384(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 31.Gillespie CF, Phifer J, Bradley B, Ressler KJ. Risk and resilience: genetic and environmental influences on development of the stress response. Depression Anxiety. 2009;26:984–992. doi: 10.1002/da.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vyazovskiy VV, Cirelli C, Tononi G. Electrophysiological correlates of sleep homeostasis in freely behaving rats. Prog Brain Res. 2011;193:17–38. doi: 10.1016/B978-0-444-53839-0.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ursin R. Serotonin and sleep. Sleep Med Rev. 2002;6:55–69. doi: 10.1053/smrv.2001.0174. [DOI] [PubMed] [Google Scholar]
- 34.Koren D, Arnon I, Lavie P, Klein E. Sleep complaints as early predictors of posttraumatic stress disorder: a 1-year prospective study of injured survivors of motor vehicle accidents. Am J Psychiatry. 2002;159:855–857. doi: 10.1176/appi.ajp.159.5.855. [DOI] [PubMed] [Google Scholar]
- 35.Nishida M, Pearsall J, Buckner RL, Walker MP. REM sleep, prefrontal theta, and the consolidation of human emotional memory. Cereb Cortex. 2009;19:1158–1166. doi: 10.1093/cercor/bhn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner U, Gais S, Born J. Emotional memory formation is enhanced across sleep intervals with high amounts of rapid eye movement sleep. Learn Mem. 2001;8:112–119. doi: 10.1101/lm.36801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner U, Hallschmid M, Rasch B, Born J. Brief sleep after learning keeps emotional memories alive for years. Biol Psychiatry. 2006;60:788–790. doi: 10.1016/j.biopsych.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 38.Cohen DJ, Begley A, Alman JJ, Cashmere DJ, Pietrone RN, Seres RJ, Germain A. Quantitative electroencephalography during rapid eye movement (REM) and non-REM sleep in combat-exposed veterans with and without post-traumatic stress disorder. J Sleep Res. 2013;22:76–82. doi: 10.1111/j.1365-2869.2012.01040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lachaux JP, Fonlupt P, Kahane P, Minotti L, Hoffmann D, Bertrand O, Baciu M. Relationship between task-related gamma oscillations and BOLD signal: new insights from combined fMRI and intracranial EEG. Hum Brain Mapp. 2007;28:1368–1375. doi: 10.1002/hbm.20352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nofzinger EA, Price JC, Meltzer CC, Buysse DJ, Villemagne VL, Miewald JM, Sembrat RC, Steppe DA, Kupfer DJ. Towards a neurobiology of dysfunctional arousal in depression: the relationship between beta EEG power and regional cerebral glucose metabolism during NREM sleep. Psychiatry Res. 2000;98:71–91. doi: 10.1016/s0925-4927(00)00045-7. [DOI] [PubMed] [Google Scholar]
- 41.Popa D, Duvarci S, Popescu AT, Lena C, Pare D. Coherent amygdalocortical theta promotes fear memory consolidation during paradoxical sleep. Proc Natl Acad Sci USA. 2010;107:6516–6519. doi: 10.1073/pnas.0913016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohda K, Harada K, Kato K, Hoshino A, Motohashi J, Yamaji T, Morinobu S, Matsuoka N, Kato N. Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: a putative post-traumatic stress disorder model. Neuroscience. 2007;148:22–33. doi: 10.1016/j.neuroscience.2007.05.041. [DOI] [PubMed] [Google Scholar]
- 43.Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy-Gigi E, Szabo C, Kelemen O, Keri S. Association among clinical response, hippocampal volume, and FKBP5 gene expression in individuals with posttraumatic stress disorder receiving cognitive behavioral therapy. Biol Psychiatry. 2013;74:793–800. doi: 10.1016/j.biopsych.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 45.Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10:500–513. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- 46.Albu S, Romanowski CP, Letizia Curzi M, Jakubcakova V, Flachskamm C, Gassen NC, Hartmann J, Schmidt MV, Schmidt U, Rein T, Holsboer F, Hausch F, Paez-Pereda M, Kimura M. Deficiency of FK506-binding protein (FKBP) 51 alters sleep architecture and recovery sleep responses to stress in mice. J Sleep Res. 2014;23:176–185. doi: 10.1111/jsr.12112. [DOI] [PubMed] [Google Scholar]
- 47.Germain A, Buysse DJ, Nofzinger E. Sleep-specific mechanisms underlying posttraumatic stress disorder: integrative review and neurobiological hypotheses. Sleep Med Rev. 2008;12:185–195. doi: 10.1016/j.smrv.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawlyk AC, Morrison AR, Ross RJ, Brennan FX. Stress-induced changes in sleep in rodents: models and mechanisms. Neurosci Biobehav Rev. 2008;32:99–117. doi: 10.1016/j.neubiorev.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stutzmann GE, McEwen BS, LeDoux JE. Serotonin modulation of sensory inputs to the lateral amygdala: dependency on corticosterone. J Neurosci. 1998;18:9529–9538. doi: 10.1523/JNEUROSCI.18-22-09529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang RY, Aghajanian GK. Inhibiton of neurons in the amygdala by dorsal raphe stimulation: mediation through a direct serotonergic pathway. Brain Res. 1977;120:85–102. doi: 10.1016/0006-8993(77)90499-1. [DOI] [PubMed] [Google Scholar]
- 51.Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi T, Morinobu S, Iwamoto Y, Yamawaki S. Effect of paroxetine on enhanced contextual fear induced by single prolonged stress in rats. Psychopharmacology. 2006;189:165–173. doi: 10.1007/s00213-006-0545-6. [DOI] [PubMed] [Google Scholar]
- 53.Wang W, Liu Y, Zheng H, Wang HN, Jin X, Chen YC, Zheng LN, Luo XX, Tan QR. A modified single-prolonged stress model for post-traumatic stress disorder. Neurosci Lett. 2008;441:237–241. doi: 10.1016/j.neulet.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 54.Sosulina L, Schwesig G, Seifert G, Pape HC. Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci. 2008;39:491–498. doi: 10.1016/j.mcn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Laukova M, Alaluf LG, Serova LI, Arango V, Sabban EL. Early intervention with intranasal NPY prevents single prolonged stress-triggered impairments in hypothalamus and ventral hippocampus in male rats. Endocrinology. 2014 doi: 10.1210/en.2014-1192. en20141192. [DOI] [PubMed] [Google Scholar]
- 56.Serova LI, Laukova M, Alaluf LG, Pucillo L, Sabban EL. Intranasal neuropeptide Y reverses anxiety and depressive-like behavior impaired by single prolonged stress PTSD model. Eur Neuropsychopharmacol. 2014;24:142–147. doi: 10.1016/j.euroneuro.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 57.Ehlers CL, Somes C, Lopez A, Kirby D, Rivier JE. Electrophysiological actions of neuropeptide Y and its analogs: new measures for anxiolytic therapy? Neuropsychopharmacology. 1997;17:34–43. doi: 10.1016/S0893-133X(97)00001-8. [DOI] [PubMed] [Google Scholar]
- 58.Toth A, Zaborszky L, Detari L. EEG effect of basal forebrain neuropeptide Y administration in urethane anaesthetized rats. Brain Res Bull. 2005;66:37–42. doi: 10.1016/j.brainresbull.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 59.Hackler EA, Airey DC, Shannon CC, Sodhi MS, Sanders-Bush E. 5-HT(2C) receptor RNA editing in the amygdala of C57BL/6J, DBA/2J, and BALB/cJ mice. Neurosci Res. 2006;55:96–104. doi: 10.1016/j.neures.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Ivarsson M, Paterson LM, Hutson PH. Antidepressants and REM sleep in Wistar–Kyoto and Sprague–Dawley rats. Eur J Pharmacol. 2005;522:63–71. doi: 10.1016/j.ejphar.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 61.Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.