

Abstract
Conventional MRI is frequently used during the diagnosis of multiple sclerosis but provides only little additional pathological information. Proton MRS (1H-MRS), however, provides biochemical information on the lesion pathology by visualization of a spectrum of metabolites. In this study we aimed to better understand the changes in metabolite concentrations following demyelination of the white matter. Therefore, we used the cuprizone model, a well-established mouse model to mimic type III human multiple sclerosis demyelinating lesions. First, we identified CX3CL1/CX3CR1 signaling as a major regulator of microglial activity in the cuprizone mouse model. Compared with control groups (heterozygous CX3CR1+/− C57BL/6 mice and wild type CX3CR1+/+ C57BL/6 mice), microgliosis, astrogliosis, oligodendrocyte cell death and demyelination were shown to be highly reduced or absent in CX3CR1−/− C57BL/6 mice. Second, we show that 1H-MRS metabolite spectra are different when comparing cuprizone-treated CX3CR1−/− mice showing mild demyelination with cuprizone-treated CX3CR1+/+ mice showing severe demyelination and demyelination-associated inflammation. Following cuprizone treatment, CX3CR1+/+ mice show a decrease in the Glu, tCho and tNAA concentrations as well as an increased Tau concentration. In contrast, following cuprizone treatment CX3CR1−/− mice only showed a decrease in tCho and tNAA concentrations. Therefore, 1H-MRS might possibly allow us to discriminate demyelination from demyelination-associated inflammation via changes in Tau and Glu concentration. In addition, the observed decrease in tCho concentration in cuprizone-induced demyelinating lesions should be further explored as a possible diagnostic tool for the early identification of human MS type III lesions. Copyright © 2015 John Wiley & Sons, Ltd.
Keywords: CX3CR1, cuprizone, demyelination, spectroscopy, MRI
Introduction
Multiple sclerosis (MS) is generally defined as a chronic inflammatory disease of the central nervous system (CNS) that is characterized by focal demyelinating lesions (1). As four different lesion subtypes can be discriminated, MS lesions are often histologically classified by the presence of an autoimmune reaction, reactive gliosis, oxidative damage and deterioration of axonal integrity (2). Due to this lesion heterogeneity, no single test currently exists for the diagnosis of MS, and therefore MS is mainly diagnosed following a battery of tests. MRI is one of the frequently used diagnostic tools for MS, as it allows elimination of other diseases such as spinal stenosis or brain tumors (3). However, most of the frequently applied conventional MRI sequences focus on the detection of white matter abnormalities, while providing only little additional pathological information. As such, these techniques do not allow discrimination between demyelination, inflammation, gliosis, edema and compromised axonal integrity.
Proton MRS (1H-MRS), however, has the advantage over conventional MRI techniques that it provides biochemical information on the lesion pathology by visualization of a spectrum of metabolites (4). For example, N-acetylaspartate (NAA) is found in high concentrations in the CNS and is almost exclusively localized to neurons (5). Therefore, NAA concentrations are often linked to neuronal and axonal integrity (6), and a decrease over time of NAA is observed in MS patients (7). Several rodent studies have shown that decreased NAA levels often occur together with an increase in taurine (Tau). Tau has been linked to astrogliosis, as it was seen upregulated in activated astrocytes (8,9). However, Tau is known to fulfill an osmoregulatory function and, as such, following release of Tau by astrocytes it might function as a neuroprotective agent against inflammation-associated edema (8–10). NAA and Tau are just two of the many metabolites that 1H-MRS can visualize, offering valuable information related to the lesion pathology.
To further unravel the pathological meaning of the different 1H-MRS metabolites during demyelination, we used the cuprizone (CPZ) mouse model for human MS. CPZ induces a perturbation of oligodendrocyte metabolism characterized by a disease pathology resembling type III MS lesions, which are highly suggestive of a primary oligodendrocyte dystrophy rather than autoimmunity (2,11–14). Microglia are known to play a major role in the occurrence of demyelinating lesions in the CNS, and are considered a driving factor behind oligodendrocyte apoptosis and efficient phagocytosis of myelin debris following CPZ treatment (14–18). CX3CL1/CX3CR1 signaling has previously been identified as a major regulator of microglial activity (19,20). With regard to MS, the role of CX3CL1/CX3CR1 signaling was previously investigated in the EAE mouse model. Unexpectedly, CX3CR1−/− mice displayed an exacerbated disease course due to dysfunctional natural killer (20) cell recruitment towards CNS lesions. The latter resulted in a defective immune reaction, and subsequently a failure to initiate disease remission (21). In the CPZ mouse model of MS, however, a toxin-induced mouse model, the role of CX3CL1/CX3CR1 signaling has thus far not been investigated.
In this context, we here investigated whether CX3CL1/CX3CR1 signaling contributes to inflammation and demyelination in the CPZ mouse model. To this end, we performed an extensive quantitative histological analysis to determine total cell infiltration, microgliosis, astrogliosis, oligodendrocyte survival and myelination in the splenium of 12-week-old CX3CR1+/+, CX3CR1+/− and CX3CR1−/− mice on a normal rodent diet or a four-week CPZ-supplemented rodent diet. In the second part of this study, we investigated the possibility of 1H-MRS to discriminate in vivo between the CPZ-induced severe demyelination and gliosis as observed in CX3CR1+/+ mice, compared with the mild demyelination without gliosis as observed in CX3CR1−/− mice.
Materials and Methods
Animal experiments
Female wild type C57BL/6 J mice (denoted CX3CR1+/+ mice, n = 34), 8 weeks of age, were obtained via Charles River Laboratories (L'Arbresle Cedex, France) (strain code 027). Female B6.129P-Cx3cr1tm1Litt/J mice (denoted CX3CR1−/− mice, n = 33) were obtained via Jackson Laboratories (Bar Harbor, Maine, USA) (strain code 5582) and further bred in the specific pathogen free facility of the University of Antwerp. According to the information available upon purchase, the donating investigator has reported to Jackson Laboratories that original CX3CR1+/− chimeric animals were backcrossed to C57BL/6 background for 10 generations before being made homozygous. Female B6.129P-Cx3cr1tm1Litt/J mice were mated with male wild type C57BL/6 J mice and female offspring were used (denoted CX3CR1+/− mice, n = 15). For all experiments, mice were kept in a normal day–night cycle (12/12) with free access to food and water. For induction of CNS inflammation and demyelination, CX3CR1+/+, CX3CR1+/− and CX3CR1−/− mice received standard rodent chow mixed with 0.2% w/w CPZ (Sigma-Aldrich, Diegem, Belgium) for 4 weeks between the ages of 8 and 12 weeks. All experimental procedures were performed in accordance with European guidelines (2010/63/EU) and were approved by the Ethics Committee for Animal Experiments of the University of Antwerp (approval nos 2011/13 and 2013/57).
Histological analysis – immunofluorescence staining
For histological analysis, we used a total of 14 CX3CR1+/+ mice, 15 CX3CR1+/− mice and 14 CX3CR1−/− mice. At the age of 12 weeks, after 4 weeks on standard rodent chow or a 0.2% CPZ-supplemented rodent chow diet, mice were deeply anaesthetized via an intraperitoneal injection of 60 mg/kg BW pentobarbital (Nembutal, Ceva Santé Animale, Brussel, Belgium), transcardially perfused with ice cold 0.9% NaCl and perfused–fixed with 4% paraformaldehyde. Whole brains were surgically removed and post-fixed in 4% paraformaldehyde for 2 h. Fixed brains were freeze-protected via a sucrose gradient (2 h at 5%, 2 h at 10% and overnight at 20%), snap frozen in liquid nitrogen and stored at −80 °C until further processing. Consecutive 10 µm thick cryosections were prepared using a microm HM500 cryostat (Prosan, Merelbeke, Belgium) at the level of the splenium. Immunofluorescent analysis was performed according to previously optimized procedures (22) using the following antibodies: a rabbit anti-IBA1 antibody (Wako, 019-19741; 1/400 dilution, Neuss, Germany) in combination with an FITC-labeled goat anti-rabbit secondary antibody (Jackson ImmunoResearch, 111-096-114; 1/600 dilution, Suffolk, UK), a mouse anti-APC/CC1 antibody (Calbiochem, OP80; 1/200 dilution, Darmstadt, Germany) in combination with an AF555-labeled goat anti-mouse secondary antibody (Invitrogen, AF21425; 1/300 dilution, Ghent, Belgium), a mouse anti-GFAP antibody (Millipore Bioscience, MAB360; 1/400 dilution, Overijse, Belgium) in combination with a Pacific Blue-labeled goat anti-mouse secondary antibody (Invitrogen, P31581; 1/200 dilution), a rabbit anti-S100β antibody (Chemicon, MAB377; 1/200 dilution, Temecula, California, US) in combination with an AF555-labeled donkey anti-rabbit secondary antibody (Invitrogen, A31572; 1/1000 dilution) and a chicken anti-MBP (myelin basic protein) antibody (Millipore, AB9348; 1/200 dilution) in combination with a DyLight549 donkey anti-chicken secondary antibody (Jackson ImmunoResearch, 703-506-155; 1/1000 dilution). Slides were counterstained with TOPRO-3 (Invitrogen, T3605, 1/200 dilution) and were then mounted using Prolong Gold Antifade (Invitrogen, P36930). Immunofluorescence images were acquired using a standard research fluorescence microscope (Olympus BX51 fluorescence microscope, Aartselaar, Belgium) equipped with an Olympus DP71 digital camera. Olympus cellSens software was used for image acquisition.
Histological analysis – quantitative analysis
Quantitative analysis of obtained immunofluorescence images was performed using NIH ImageJ analysis software (ImageJ) and TissueQuest immunofluorescence analysis software (TissueGnostics, Vienna, Austria), according to previously established procedures (22). The following parameters were determined: (a) total cell density in the splenium provided as no of cells/mm2 (3–12 slides with 6–24 data counts per mouse brain analyzed), (b) IBA1+ microglial density in the splenium provided as no of cells/mm2 (2–9 slides with 4–17 data counts per mouse brain analyzed), (c) CC1+ oligodendrocyte density in the splenium provided as no of cells/mm2 (1–4 slides with 2–8 data counts per mouse brain analyzed), (d) the degree of MBP+ myelination in the splenium provided as % optical density (O.D.) MBP staining (1–2 slides with 2–4 data counts per mouse brain analyzed), (e) S100β + astrocyte density in the splenium provided as no of cells/mm2 (1 slide with 2 data counts per mouse brain analyzed) and (f) the degree of GFAP+ astrogliosis in the splenium provided as % O.D. GFAP staining (1–4 slides with 2–8 data counts per mouse brain analyzed).
MRI and MRS acquisition
For 1H-MRS analysis, we used a total of 20 CX3CR1+/+ mice and 19 CX3CR1−/− mice. At the age of 12 weeks, after 4 weeks on standard rodent chow or a 0.2% CPZ-supplemented rodent chow diet, mice were subjected to 1H-MRS analysis. All MRI measurements were made using a 9.4 T BioSpec MR scanner with a 20 cm diameter horizontal bore (BioSpec 94/20 USR, Bruker BioSpin, Ettlingen, Germany). This system is equipped with a standard Bruker cross-coil setup, using a quadrature volume coil for excitation and quadrature mouse surface coil for signal detection. The system was interfaced to a Linux PC running Topspin 2.0 and ParaVision 5.1 software (Bruker BioSpin). Mouse anesthesia is induced using 3% isoflurane (Forane, Abbott, West Berkshire, UK) in a gas mixture of 30% O2 and 70% N2 at a flow rate of 600 ml/min. During MR acquisition, isoflurane concentration was maintained at 2% and mouse respiration rate was constantly monitored using a pressure sensitive pad. In addition, mouse body temperature was monitored via a rectal probe and was held constant at 37.0 ± 0.5 °C using warm air coupled to a feedback unit (SA Instruments, New York, USA). Both respiration and body temperature control systems were controlled by pcSam monitoring software (SA Instruments). To localize the volume of interest (VOI), we acquired axial, coronal and sagittal multislice rapid acquisition and relaxation enhancement (RARE) images using the following parameters: repetition time (TR) = 2500 ms, echo time (TE) = 33 ms, matrix size 256 × 256, field of view = 20 × 20 mm, 16 slices, slice thickness = 0.4 mm, RARE factor = 8. The 1H-MRS VOI (0.9 mm × 2.5 mm × 1 mm = 2.25 mm3) was placed in the splenium of mice brains, corresponding to the region chosen for histological analysis. The VOI is indicated on the T2 images shown later in Fig.3(A) (by a thin white dashed box) or in Supplementary File 2, which shows the correspondence between the histological region of interest and the 1H-MRS VOI. Based on the RARE images we were able to estimate the volume fractions of cerebrospinal fluid (fcsf) as well as gray and white matter (fgm and fwm) within the VOI, allowing us to perform a partial volume correction using LCModel. The VOI first and second order shim terms were automatically adjusted using FASTMAP (23,24). Water line widths were obtained between 18 and 20 Hz. 1H-MR spectra were acquired using a point-resolved spectroscopy sequence in combination with outer volume suppression (OVS) and VAPOR water suppression (25,26). The following parameters were used: TR = 4000 ms, TE = 15 ms, number of averages = 720, total acquisition time = 48 min. For each animal, an unsuppressed water signal (TE = 15 ms, TR = 4000 ms, 64 averages, scanning time = 5 min, VAPOR and OVS turned off) was acquired immediately after acquiring the water-suppressed spectrum. During post-processing, this unsuppressed water signal was used for eddy-current correction by LCModel, where the water-suppressed MRS signal (in the time domain) is pointwise divided by the phase part of the unsuppressed water signal (27). In addition, the unsuppressed water signal served as a concentration reference to determine absolute metabolite concentrations by means of water scaling in LCModel.
Figure 3.
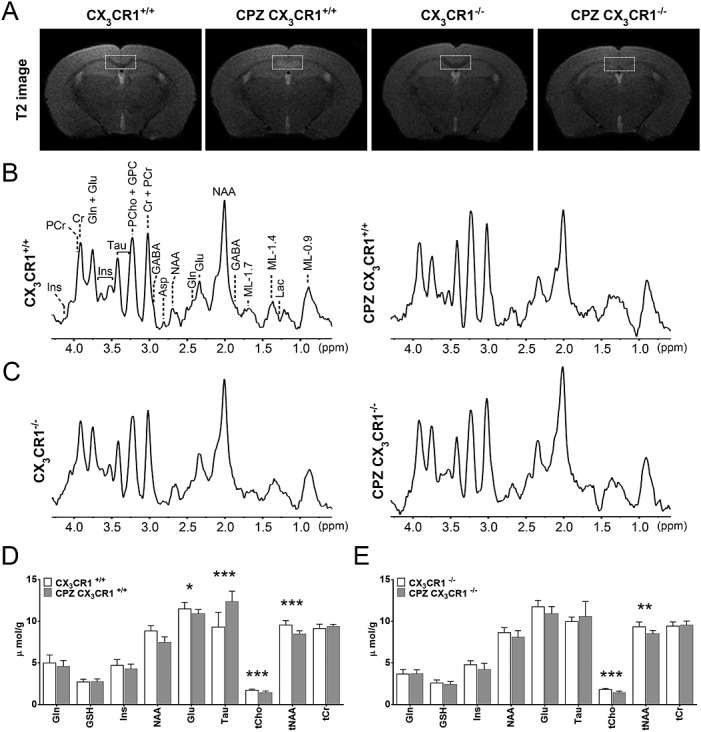
1H-MRS analysis of control and CPZ-treated CX3CR1+/+ and CX3CR1−/− mice. (A) Representative T2 images showing edema, inflammation and demyelination in CPZ-treated CX3CR1+/+ mice but only demyelination in CPZ-treated CX3CR1−/− mice. The thin white dashed boxes indicate the placement of the 1H-MRS VOI. (B), (C) 1H-MRS spectra of control and CPZ-treated CX3CR1+/+ and CX3CR1−/− mice respectively. (D), (E) Bar charts showing the quantification of the different metabolites as seen in the 1H-MRS spectra of control and CPZ-treated CX3CR1+/+ and CX3CR1−/− mice respectively (shown in µmol/g ± SD, *p < 0.05 and **p < 0.01 and *** p < 0.001).
MRI and MRS analysis
1H-MR spectra were analyzed using an automated deconvolution program as described previously (LCModel) (23,28,29). Absolute metabolite concentrations (in µmol/g) were determined using the unsuppressed water signal as internal reference for quantification with water scaling. In addition, absolute metabolite concentrations were corrected for partial volume effects by taking fwm and fgm into account in the quantification (28,30). These volume fractions were used to correct the total water concentration for each VOI using the following equation: water concentration = (43 300fgm + 35 880fwm + 55 556fcsf)/(1 − fcsf). The water concentrations for each of these fractions were taken from Ernst et al. (31). As such, the following metabolites were analyzed: alanine (Ala), aspartate (Asp), choline (Cho), creatine (Cr), phosphocreatine (PCr), γ-aminobutyric acid (GABA), glucose, glutamate (Glu), glutamine (Gln), glutathione, glycerophosphorylcholine (GPC), phosphorylcholine (PCho), myo-inositol, lactate (Lac), NAA, N-acetylaspartylglutamate (NAAG), phosphorylethanolamine, scyllo-inositol and Tau. The LCModel software package also provided Cramér–Rao lower bounds (CRLBs, estimated error of quantification) for each metabolite, which allowed assessment of the precision of the metabolite quantification. Stringent criteria were applied to ensure that metabolites were successfully quantified: metabolites quantified with CRLB > 20% were classified as not detected, and metabolites quantified with CRLB ≤ 20% in at least 50% of the spectra were included in the neurochemical profile. In most spectra, the present analysis was unable to reliably discern PCh from GPC, NAA from NAAG or Cr from PCr. More accurate are the sums expressed as total choline-containing compounds (GPC + PCh, or tCho), total N-acetylaspartate (NAA + NAAG, or tNAA) and total creatine (Cr + PCr, or tCr), as recommended by the LCModel manual (28).
Statistical analysis
For analysis of histology data, generalized estimating equations (32) were used for within and between group comparisons, taking within and between mouse variability into account. A post-hoc Bonferroni correction for multiple comparisons was performed. Both the uncorrected and Bonferroni-corrected p-values for all possible within and between group comparisons are provided in Supplementary File S1. A p-value less than 0.05 was considered to be statistically significant. For analysis of 1H-MRS data, a two-way ANOVA was used to identify significant differences in metabolite ratios in CX3CR1+/+ and CX3CR1−/− mouse groups. A post-hoc Bonferroni correction for multiple comparisons was performed. A p-value less than 0.05 was considered to be statistically significant.
Results
CX3CL1/CX3CR1 signaling contributes to CPZ-induced demyelination
In order to investigate the role of CX3CL1/CX3CR1 signaling during the inflammatory phase of CPZ-induced CNS demyelination, six experimental mouse groups were included in two independent experiments, and shown here are the pooled data from (a) wild type CX3CR1+/+ C57BL/6 mice at the age of 12 weeks (n = 7), (b) wild type CX3CR1+/+ C57BL/6 mice at the age of 12 weeks that received a 0.2% CPZ-supplemented diet between the age of 8 and 12 weeks (n = 7), (c) transgenic CX3CR1+/− C57BL/6 mice, in which one copy of the CX3CR1 gene is replaced by the eGFP reporter gene, at the age of 12 weeks (n = 7), (d) transgenic CX3CR1+/− C57BL/6 mice at the age of 12 weeks that received a 0.2% CPZ-supplemented diet between the age of 8 and 12 weeks (n = 8), (e) transgenic CX3CR1−/− C57BL/6 mice, in which both copies of the CX3CR1 gene are replaced by the eGFP reporter gene, at the age of 12 weeks (n = 7) and (f) transgenic CX3CR1+/− C57BL/6 mice at the age of 12 weeks that received a 0.2% CPZ-supplemented diet between the age of 8 and 12 weeks (n = 7). Note that mice receiving the 0.2% CPZ-supplemented diet do not lose weight, but fail to gain weight when compared with mice on standard rodent chow. Fig.1 provides an overview of representative immunofluorescence images taken from the splenium at the age of 12 weeks for each of the different groups. For clarity, the exact anatomical localization of the images is provided in Supplementary File S2 and larger images of those provided in Fig.1 are provided in Supplementary File S3. On each of the images, the splenium is delineated by a white dotted line. Note that thickness of the splenium varies between experimental groups, as the presence of an inflammatory reaction causes swelling of the splenium. Within the splenium, total cell density (no of nuclei/mm2), oligodendrocyte density (no of CC1+ cells/mm2), microglial density (no of IBA1+ or eGFP+ cells/mm2), astrocyte density (no of S100β + cells/mm2), degree of astrogliosis (% O.D. GFAP) and degree of myelination (% O.D. MBP) were quantified and plotted in Fig.2 for each of the groups.
Figure 1.
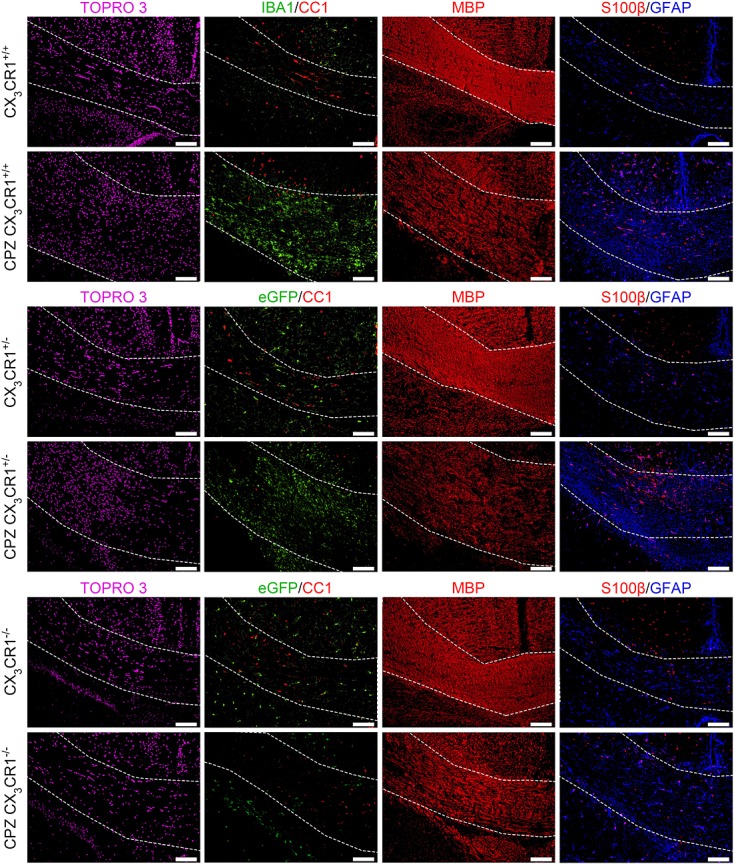
Representative immunofluorescence images taken from the splenium of control and CPZ-treated CX3CR1+/+, CX3CR1+/− and CX3CR1−/− mice. All images were taken from the same reference point and the splenium is delineated using thin white lines. TOPRO-3 staining (first column) visualizes total cell density (in purple, false color image). IBA1 staining or direct eGFP fluorescence (second column) visualizes microglial density (in green). CC1 staining (second column) visualizes oligodendrocyte density (in red). MBP staining (third column) visualizes myelination (in red). S100β staining (fourth column) visualizes oligodendrocyte density (in red). GFAP staining (fourth column) visualizes astrogliosis (in blue). Scale bars indicate 100 µm.
Figure 2.
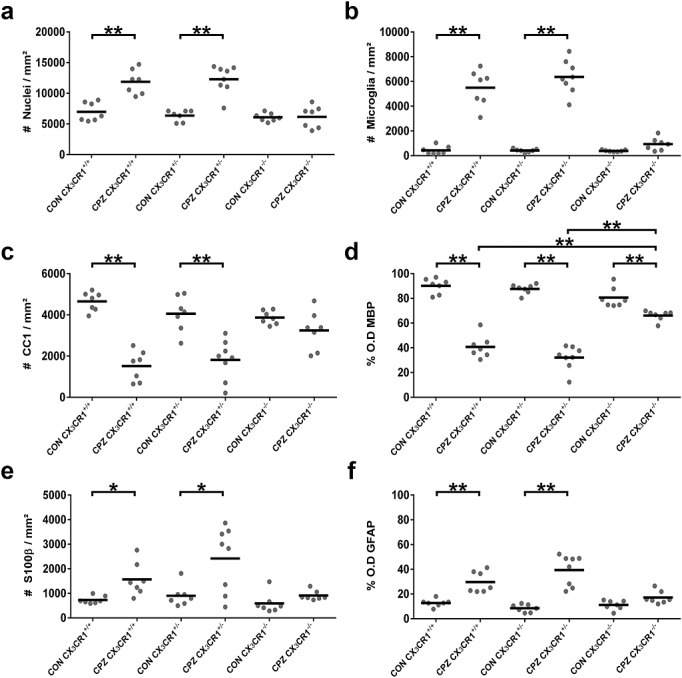
Quantitative analysis of the immunofluorescence images. Dot-plot charts indicate within the delineated splenium and per mouse analyzed (a) total nuclei/mm2, (b) IBA1+ microglia/mm2, (c) CC1+ oligodendrocytes/mm2, (d) % MBP+ myelination status as determined by O.D., (e) S100β+ astrocytes/mm2 and (f) % GFAP+ astrogliosis as determined by O.D. (*p < 0.05; **p < 0.0001).
With regard to total cell density in the splenium (Fig.1 TOPRO-3 staining and Fig.2(a)), a significant increase was observed in CPZ-treated CX3CR1+/+ and CX3CR1+/− mice as compared with control CX3CR1+/+ and CX3CR1+/− mice (both p < 0.0001). In contrast, total cellular density in the splenium of CX3CR1−/− mice did not significantly increase following CPZ treatment as compared with control CX3CR1−/− mice, despite slight edema in the splenium due to the inherent toxicity of CPZ administration (12). The increase in cellular density in the splenium of CPZ-treated CX3CR1+/+ and CX3CR1+/− mice can mainly be ascribed to the transmigration of microglia (Fig.1 IBA1 staining or direct eGFP fluorescence and Fig.2(b), p < 0.0001). Although a small increase in the number of microglia was observed in CPZ-treated CX3CR1−/− mice as compared with control CX3CR1−/− mice, the increase in microglial density was not statistically significant after correction for multiple testing (p = 0.1974; p = 0.0132 without multiple testing correction).
In the CPZ model, it has previously been shown that microglia effectively contribute to oligodendrocyte cell death (16). Therefore, the severe transmigration of microglia to the splenium of CPZ-treated CX3CR1+/+ mice and CX3CR1+/− mice logically resulted in a notable decrease of CC1+ oligodendrocytes (Fig.1 CC1 staining and Fig.2(c), p < 0.0001). In the absence of microglial accumulation in CPZ-treated CX3CR1−/− mice, the number of CC1+ oligodendrocytes did not significantly differ from that in control CX3CR1−/− mice (p = 0.7939). The degree of oligodendrocyte survival is obviously linked to myelination, and as such CPZ-treated CX3CR1+/+ and CX3CR1+/− mice display severe demyelination of the splenium in comparison to control CX3CR1+/+ and CX3CR1+/− mice (Fig.1 MBP staining and Fig.2(d), p < 0.0001). In contrast, CPZ-treated CX3CR1−/− mice displayed only minimal demyelination of the splenium as compared with control CX3CR1−/− mice (p < 0.0001), which was markedly reduced compared with the demyelination observed in CX3CR1+/+ and CX3CR1+/− mice (p < 0.0001). This small degree of demyelination, despite survival of the CC1+ oligodendrocytes in CPZ-treated CX3CR1−/− mice, can however be attributed to the metabolic stress CPZ treatment induces on oligodendrocytes (12,33).
Finally, in addition to the absence of microglial cell responses and demyelination in CPZ-treated CX3CR1−/− mice, astroglial cell responses were highly reduced or absent. While the number of S100β + astrocytes (Fig.1 S100β staining and Fig.2(e)) is significantly increased in the splenium of CX3CR1+/+ and CX3CR1+/− mice following CPZ treatment as compared with control CX3CR1+/+ and CX3CR1+/− mice (p = 0.0270 and p = 0.0159 respectively), the small increase in astrocyte density within the splenium of CPZ-treated CX3CR1−/− mice was not statistically significant after correction for multiple testing (p = 0.6375; p = 0.0425 without multiple testing correction). In addition, similar results were observed at the level of astroglial cell activity, as determined by the degree of astrogliosis observed within the splenium (Fig.1 GFAP staining and Fig.2(f)). While CPZ-treated CX3CR1+/+ and CX3CR1+/− mice display severe astrogliosis in the splenium in comparison with control CX3CR1+/+ and CX3CR1+/− mice (p < 0.0001), astrogliosis detected in the splenium of CPZ-treated CX3CR1−/− mice was not statistically significant after correction for multiple testing (p = 0.3112; p = 0.0208 without multiple testing correction).
1H-MRS analysis allows discrimination of demyelination from demyelination-associated inflammation
As we observed that CPZ treatment of CX3CR1−/− mice induces mild demyelination without microglial accumulation or astrogliosis, CX3CR1−/− mice offer a valuable tool to identify the contribution of specific metabolites to changes in the 1H-MRS spectrum. As such, we performed 1H-MRS on two groups of mice: (i) wild type CX3CR1+/+ C57BL/6 mice (n = 20) and (ii) transgenic CX3CR1−/− C57BL/6 mice (n = 19). 1H-MRS analysis was performed at 12 weeks of age in control mice (n = 10 for CX3CR1+/+ C57BL/6 mice and n = 11 for CX3CR1−/− C57BL/6 mice) or in mice that had been on a 0.2% CPZ-supplemented diet for 4 weeks between the age of 8 and 12 weeks (CPZ, n = 10 for CX3CR1+/+ C57BL/6 mice and n = 8 for CX3CR1−/− C57BL/6 mice). Fig.3(A) shows representative T2 MR images for the respective groups, which were acquired alongside the 1H-MRS spectra. The thin white dashed box each time indicates the placement of the 1H-MRS VOI. On these T2 images, both control CX3CR1+/+ and CX3CR1−/− mice show a densely myelinated splenium (hypointense contrast). Following 4 weeks of CPZ treatment, CX3CR1+/+ mice show hyperintense contrast, indicative of demyelination, inflammation and edema. In contrast, CPZ-treated CX3CR1−/− mice do not show hyperintense contrast following 4 weeks of CPZ treatment, and instead only lose the hypointense contrast. Fig.3(B) and (C) shows representative 1H-MRS spectra of the different groups, acquired from the VOI within the splenium. The quantification of the absolute metabolite concentration (µmol/g) is then shown in Fig.3(D) for CX3CR1+/+ mice and in Fig.3(E) for CX3CR1−/− mice. For CX3CR1+/+ mice, we observed that following CPZ treatment a significant decrease occurs in the absolute concentration of Glu (p = 0.0437), tCho (p = 0.0005) and tNAA (p < 0.0001), while the absolute concentration of Tau was significantly increased (p < 0.0001). In contrast, following CPZ treatment of CX3CR1−/− mice, we only observed a significant decrease in the absolute concentration of tCho (p < 0.0001) and tNAA (p = 0.0022), while other metabolites were unaffected.
Discussion
In the first part of this study we observed that, in contrast to CX3CR1+/+ mice, CPZ treatment of CX3CR1−/− mice does not induce any reactive microgliosis. These results are in line with the currently accepted hypothesis that membrane-bound CX3CL1 can be cleaved from neuronal membranes and that the resulting soluble CX3CL1 drives microglial migration and activation (34). In the course of our experiments we attempted to demonstrate the presence of soluble CX3CL1 in the splenium of CPZ-treated wild type, CX3CR1+/− and CX3CR1−/− mice. Given the technical difficulties associated with this type of analysis, to date we did not yet succeed in doing so. However, as the presence of soluble CX3CL1 has previously been shown in the demyelinating spinal cord of EAE-induced mice (21), we postulate that a similar shedding of soluble CX3CL1 might occur in the demyelinating splenium of CPZ-treated mice. In addition, due to the absence of reactive microgliosis in CPZ-treated CX3CR1−/− mice, oligodendrocyte cell death, and thus also demyelination, were markedly reduced. These results are in line with previous observations that microglia actively contribute to the induction of apoptosis of metabolically stressed oligodendrocytes (16). Of note, the metabolic stress induced by CPZ treatment inhibits myelin lipid and protein synthesis while normal catabolic processes continue (14,35). This process therefore slowly induces myelin sheath disintegration, explaining why we observed a small but significant amount of demyelination in CX3CR1−/− mice following 4 weeks of CPZ treatment. It was recently shown that oligodendrocyte apoptosis can occur within days following CPZ treatment due to a selective reduction in amino acid levels, and thus without extensive microgliosis (36). This finding stresses the fact that, while microglia might actively contribute to oligodendrocyte apoptosis, they cannot be solely held responsible for the apoptosis of all oligodendrocytes following CPZ treatment. Additionally, while it might be argued that the microglial contribution to oligodendrocyte apoptosis is a specific feature of the CPZ mouse model, to date it is still unclear exactly how microglia contribute to the (early) pathophysiology of different types of human MS lesion. Nonetheless, the study presented here is the first to show the important role of CX3CL1/CX3CR1 signaling for efficient microglial accumulation, and thus oligodendrocyte apoptosis and demyelination, following 4 weeks of 0.2% CPZ treatment.
In the second part of this study, we investigated if 1H-MRS was able to discriminate between the different pathological phenotypes observed in CX3CR1+/+ and CX3CR1−/− mice following a 4-week 0.2% CPZ-supplemented diet. 1H-MRS studies in human MS patients have shown that a decrease in NAA is often linked to neuronal and axonal damage (37,38). In addition, it is known that the axon diameter correlates well with myelin sheath thickness (39), and the axon diameter was seen to decrease upon demyelination (40). As such, the observed decrease in tNAA concentration in both CX3CR1+/+ and CX3CR1−/− mice following 4 weeks of 0.2% CPZ treatment can, albeit indirectly, be linked to demyelination, as a loss of myelin at 4 weeks of CPZ treatment was previously shown to result in a compromised axonal integrity (41). The observed decrease in absolute tNAA concentration following 4 weeks of 0.2% CPZ treatment was more pronounced in CX3CR1+/+ mice (from 9.6 ± 0.5 µmol/g to 8.5 ± 0.4 µmol/g, p < 0.0001) as compared with CX3CR1−/− mice (from 9.3 ± 0.6 µmol/g to 8.5 ± 0.4 µmol/g, p = 0.0022). As axonal diameter and integrity is linked to myelination, this larger decrease in absolute tNAA concentration in CX3CR1+/+ mice can be explained by the more pronounced demyelination in these mice (as was observed histologically).
In line with tNAA, the absolute concentration of tCho was also decreased for both CX3CR1+/+ and CX3CR1−/− mice following 4 weeks of 0.2% CPZ treatment. The tCho spectrum peak is complex and consists of several metabolites, including PCho and GPC, which are often difficult to separate. As such, studies often only report on the total Cho concentration. Nonetheless, GPC has previously been suggested as a marker for membrane turnover and, as such, alterations in GPC levels represent altered membrane synthesis and catabolism and are closely linked to myelination (42). Cho, on the other hand, was shown to be increased in human MS patients who suffered inflammatory, demyelinating lesions (43). The decrease in tCho concentration that we observed following 0.2% CPZ treatment is therefore in line with the current consensus concerning GPC, but contradictory to the previous findings on Cho in human MS patients. This contradiction can however be explained by the severe disruption of normal mitochondrial functioning by CPZ treatment (11,14), as Cho concentrations have frequently been observed to decrease in various mitochondrial diseases (44,45). In particular, the copper-chelating properties of CPZ disrupt the cupro-enzymes of the electron transport chain, which results in mitochondrial dysfunction and thus ATP shortage. This specifically results in oligodendrocyte cell death, as these cells have a very high metabolic demand while synthesizing myelin sheaths (11,12,14). Interestingly, type III MS lesions (which are mimicked by the CPZ mouse model) also show signs of mitochondrial damage and are seen very early during MS pathology (2,46). While it is tempting to speculate that these lesions are a precursor for the development of type I and II MS lesions, no causal link has been found despite profound testing of this hypothesis (47). While type II MS lesions are the predominant type of lesion, type III MS lesions are the second most abundant lesion type and occur in nearly half of the patients within months of receiving the diagnosis of MS. As such, the early occurrence of mitochondrial damage in type III MS lesions, coupled to the ability of 1H-MRS to detect a decreased tCho concentration in the CPZ model, makes the latter an interesting finding, which, if developed further, could possibly allow an earlier diagnosis of MS.
While tNAA and tCho were seen to change concentration in both CX3CR1+/+ and CX3CR1−/− mice following 4 weeks of 0.2% CPZ treatment, the concentration of Tau and Glu only changed in CX3CR1+/+ mice following 4 weeks of 0.2% CPZ treatment. In mice, Tau has been identified as a marker for gliosis since it was found to be upregulated in reactive astrocytes. Here Tau, an important osmoregulator, is thought to fulfil a neuroprotective role in response to for example oxidative stress and ischemia (8,9). Therefore, the observed upregulation of Tau in CX3CR1+/+ mice following 4 weeks of 0.2% CPZ treatment fits well with the histologically observed increase in S100β+ astrocytes and the presence of pronounced astrogliosis in CX3CR1+/+ mice. We also observed a decreased concentration of Glu in CX3CR1+/+ mice, but not CX3CR1−/− mice, following 4 weeks of 0.2% CPZ. This decrease however contrasts findings in human MS patients, which observe an increased Glu concentration in acute lesions (48), which was linked to the acute inflammatory reaction (e.g. gliosis). As Glu and Gln are closely linked, an increased Gln concentration could be expected as well, but was however not observed. Our observations on Glu and Gln could possibly be explained by the way CPZ works. First, as mentioned above, CPZ inhibits efficient mitochondrial ATP production. While CPZ affects the mitochondria of all cells, not all cells undergo apoptosis. Neurons are metabolically very active but are protected from CPZ-induced ATP shortage via the metabolic coupling to astrocytes (Glu–Gln shuttle) (14). Second, a recent study has also shown that CPZ treatment induces liver dysfunction, which results in reduced plasma levels of Ala, glycine and proline (36). A shortage of these three amino acids might induce their synthesis within the brain, with Glu being either a direct precursor (proline) or a mediator in their synthesis (Ala and glycine). This might eventually result in a reduced Glu concentration within the brain. While unproven, both these hypotheses could offer a possible explanation for the observed decrease of Glu and absence of increased Gln.
Conclusions
In this study, we observed that CX3CL1/CX3CR1 signaling is a key contributor to neuro-inflammation in the CPZ mouse model. We have shown that CPZ-induced demyelination and CPZ-induced demyelination-associated inflammation show different 1H-MRS metabolite spectra. More specifically, differences were seen in Tau and Glu concentration. As human MS type III lesions are often seen preceding the occurrence of autoimmune-mediated demyelinating lesions (type II lesions), the use of 1H-MRS tCho analysis as an early diagnostic for MS patients should be further explored.
Conflict of Interest
There is no conflict of interest to declare.
Acknowledgments
JP conceived the study, performed experiments, analyzed the data and wrote the manuscript. JO performed MRI experiments and analyzed MRI data. FK analyzed MRI data. CG contributed to the MRI experiments and study design. JD contributed to histological analyses. ES and NH performed statistical analyses. ZB and MV contributed to study design and acquired funding. PP conceived the study, acquired funding, analyzed the data and wrote the manuscript. AVdL conceived the study, acquired funding and wrote the manuscript.
This work was supported by research grants G.0136.11 and G.0130.11 (granted to ZB, AVdL and PP) of the Fund for Scientific Research – Flanders (FWO – Vlaanderen, Belgium) and in addition by an “FWO – Krediet aan Navorsers” grant of the Fund for Scientific Research – Flanders (FWO – Vlaanderen, Belgium, granted to MV); next, by a Methusalem research grant from the Flemish government (granted to ZB) and in part by funding received from the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement 278850 (INMiND) (granted to AVdL). Caroline Guglielmetti is holder of an IWT PhD grant (Institute for the Promotion of Innovation through Science and Technology in Flanders, IWT – Vlaanderen). Firat Kara is holder of an “FWO – Postdoc” grant of the Fund for Scientific Research – Flanders (FWO – Vlaanderen, Belgium). MRI equipment was funded by the Flemish Impulse funding for heavy scientific equipment (granted to AVdL).
Glossary
- 1H-MRS
proton MRS
- Ala
alanine
- Asp
aspartate
- Cho
choline
- CNS
central nervous system
- CPZ
cuprizone
- Cr
creatine
- CRLB
Cramér–Rao lower bound
- fcsf
cerebrospinal fluid fraction
- fgm
gray matter fraction
- fwm
white matter fraction
- GABA
γ-aminobutyric acid
- Gln
glutamine
- Glu
glutamate
- GPC
glycerophosphorylcholine
- Lac
lactate
- MBP
myelin basic protein
- MS
multiple sclerosis
- NAA
N-acetylaspartate
- NAAG
N-acetylaspartylglutamate
- O.D.
optical density
- OVS
outer volume suppression
- PCho
phosphorylcholine
- PCr
phosphocreatine
- RARE
rapid acquisition and relaxation enhancement
- Tau
taurine
- tCho
total choline
- tCr
total creatine
- TE
echo time
- tNAA
total N-acetylaspartate
- TR
repetition time
- VOI
volume of interest
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web site.
References
- 1.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 2.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 2000;47(6):707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 3.Traboulsee AL, Li DK. The role of MRI in the diagnosis of multiple sclerosis. Adv. Neurol. 2006;98:125–146. [PubMed] [Google Scholar]
- 4.Soares DP, Law M. Magnetic resonance spectroscopy of the brain: review of metabolites and clinical applications. Clin. Radiol. 2009;64(1):12–21. doi: 10.1016/j.crad.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience. 1991;45(1):37–45. doi: 10.1016/0306-4522(91)90101-s. [DOI] [PubMed] [Google Scholar]
- 6.Benarroch EE. N-acetylaspartate and N-acetylaspartylglutamate: neurobiology and clinical significance. Neurology. 2008;70(16):1353–1357. doi: 10.1212/01.wnl.0000311267.63292.6c. [DOI] [PubMed] [Google Scholar]
- 7.De Stefano N, Narayanan S, Matthews PM, Mortilla M, Dotti MT, Federico A, Arnold DL. Proton MR spectroscopy to assess axonal damage in multiple sclerosis and other white matter disorders. J. Neurovirol. 2000;6(2):S121–129. Suppl. [PubMed] [Google Scholar]
- 8.Tkac I, Dubinsky JM, Keene CD, Gruetter R, Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J. Neurochem. 2007;100(5):1397–1406. doi: 10.1111/j.1471-4159.2006.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem. Res. 2002;27(1/2):21–26. doi: 10.1023/a:1014890219513. [DOI] [PubMed] [Google Scholar]
- 10.Pascual JM, Solivera J, Prieto R, Barrios L, Lopez-Larrubia P, Cerdan S, Roda JM. Time course of early metabolic changes following diffuse traumatic brain injury in rats as detected by 1H NMR spectroscopy. J. Neurotrauma. 2007;24(6):944–959. doi: 10.1089/neu.2006.0190. [DOI] [PubMed] [Google Scholar]
- 11.Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118(6):723–736. doi: 10.1007/s00401-009-0591-3. [DOI] [PubMed] [Google Scholar]
- 12.Skripuletz T, Gudi V, Hackstette D, Stangel M. De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol. Histopathol. 2011;26(12):1585–1597. doi: 10.14670/HH-26.1585. [DOI] [PubMed] [Google Scholar]
- 13.Zendedel A, Beyer C, Kipp M. Cuprizone-induced demyelination as a tool to study remyelination and axonal protection. J. Mol. Neurosci. 2013;51(2):567–572. doi: 10.1007/s12031-013-0026-4. [DOI] [PubMed] [Google Scholar]
- 14.Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014;47:485–505. doi: 10.1016/j.neubiorev.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001;4(11):1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 16.Pasquini LA, Calatayud CA, Bertone Una AL, Millet V, Pasquini JM, Soto EF. The neurotoxic effect of cuprizone on oligodendrocytes depends on the presence of pro-inflammatory cytokines secreted by microglia. Neurochem. Res. 2007;32(2):279–292. doi: 10.1007/s11064-006-9165-0. [DOI] [PubMed] [Google Scholar]
- 17.Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60(2):306–321. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- 18.Plemel JR, Manesh SB, Sparling JS, Tetzlaff W. Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia. 2013;61(9):1471–1487. doi: 10.1002/glia.22535. [DOI] [PubMed] [Google Scholar]
- 19.Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29(4):305–315. [PubMed] [Google Scholar]
- 20.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 21.Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20(7):896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- 22.De Vocht N, Lin D, Praet J, Hoornaert C, Reekmans K, Le Blon D, Daans J, Pauwels P, Goossens H, Hens N, Berneman Z, Van der Linden A, Ponsaerts P. Quantitative and phenotypic analysis of mesenchymal stromal cell graft survival and recognition by microglia and astrocytes in mouse brain. Immunobiology. 2013;218(5):696–705. doi: 10.1016/j.imbio.2012.08.266. [DOI] [PubMed] [Google Scholar]
- 23.Tkac I, Henry PG, Andersen P, Keene CD, Low WC, Gruetter R. Highly resolved in vivo 1H NMR spectroscopy of the mouse brain at 9.4 T. Magn. Reson. Med. 2004;52(3):478–484. doi: 10.1002/mrm.20184. [DOI] [PubMed] [Google Scholar]
- 24.Gruetter R. Automatic, localized in vivo adjustment of all first- and second-order shim coils. Magn. Reson. Med. 1993;29(6):804–811. doi: 10.1002/mrm.1910290613. [DOI] [PubMed] [Google Scholar]
- 25.Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn. Reson. Med. 1999;41(4):649–656. doi: 10.1002/(sici)1522-2594(199904)41:4<649::aid-mrm2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 26.Bottomley PA. Spatial localization in NMR-spectroscopy in vivo. Ann. N. Y. Acad. Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- 27.Mandal PK. In vivo proton magnetic resonance spectroscopic signal processing for the absolute quantitation of brain metabolites. Eur. J. Radiol. 2012;81(4):e653–664. doi: 10.1016/j.ejrad.2011.03.076. [DOI] [PubMed] [Google Scholar]
- 28.Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001;14(4):260–264. doi: 10.1002/nbm.698. [DOI] [PubMed] [Google Scholar]
- 29.Marjanska M, Curran GL, Wengenack TM, Henry PG, Bliss RL, Poduslo JF, Jack CR, Jr, Ugurbil K, Garwood M. Monitoring disease progression in transgenic mouse models of Alzheimer's disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2005;102(33):11906–11910. doi: 10.1073/pnas.0505513102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med. 1993;30(6):672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- 31.Ernst T, Kreis R, Ross BD. Absolute quantitation of water and metabolites in the human brain. 1. Compartments and water. J. Magn. Reson. B. 1993;102(1):1–8. [Google Scholar]
- 32.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44(4):1049–1060. [PubMed] [Google Scholar]
- 33.Jurevics H, Largent C, Hostettler J, Sammond DW, Matsushima GK, Kleindienst A, Toews AD, Morell P. Alterations in metabolism and gene expression in brain regions during cuprizone-induced demyelination and remyelination. J. Neurochem. 2002;82(1):126–136. doi: 10.1046/j.1471-4159.2002.00954.x. [DOI] [PubMed] [Google Scholar]
- 34.Re DB, Przedborski S. Fractalkine: moving from chemotaxis to neuroprotection. Nat. Neurosci. 2006;9(7):859–861. doi: 10.1038/nn0706-859. [DOI] [PubMed] [Google Scholar]
- 35.Jurevics H, Hostettler J, Muse ED, Sammond DW, Matsushima GK, Toews AD, Morell P. Cerebroside synthesis as a measure of the rate of remyelination following cuprizone-induced demyelination in brain. J. Neurochem. 2001;77(4):1067–1076. doi: 10.1046/j.1471-4159.2001.00310.x. [DOI] [PubMed] [Google Scholar]
- 36.Goldberg J, Daniel M, van Heuvel Y, Victor M, Beyer C, Clarner T, Kipp M. Short-term cuprizone feeding induces selective amino acid deprivation with concomitant activation of an integrated stress response in oligodendrocytes. Cell. Mol. Neurobiol. 2013;33(8):1087–1098. doi: 10.1007/s10571-013-9975-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnold DL, De Stefano N, Narayanan S, Matthews PM. Proton MR spectroscopy in multiple sclerosis. Neuroimaging Clin. N. Am. 2000;10(4):789–798. ix–x. [PubMed] [Google Scholar]
- 38.Sajja BR, Wolinsky JS, Narayana PA. Proton magnetic resonance spectroscopy in multiple sclerosis. Neuroimaging Clin. N. Am. 2009;19(1):45–58. doi: 10.1016/j.nic.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chomiak T, Hu B. What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PLoS One. 2009;4(11):e7754. doi: 10.1371/journal.pone.0007754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mason JL, Langaman C, Morell P, Suzuki K, Matsushima GK. Episodic demyelination and subsequent remyelination within the murine central nervous system: changes in axonal calibre. Neuropathol. Appl. Neurobiol. 2001;27(1):50–58. doi: 10.1046/j.0305-1846.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- 41.VonDran MW, Singh H, Honeywell JZ, Dreyfus CF. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J. Neurosci. 2011;31(40):14182–14190. doi: 10.1523/JNEUROSCI.6595-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podo F. Tumour phospholipid metabolism. NMR Biomed. 1999;12(7):413–439. doi: 10.1002/(sici)1099-1492(199911)12:7<413::aid-nbm587>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 43.Zhu H, Barker PB. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol. Biol. 2011;711:203–226. doi: 10.1007/978-1-61737-992-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bianchi MC, Tosetti M, Battini R, Manca ML, Mancuso M, Cioni G, Canapicchi R, Siciliano G. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance. Am. J. Neuroradiol. 2003;24(10):1958–1966. [PMC free article] [PubMed] [Google Scholar]
- 45.De Stefano N, Matthews PM, Ford B, Genge A, Karpati G, Arnold DL. Short-term dichloroacetate treatment improves indices of cerebral metabolism in patients with mitochondrial disorders. Neurology. 1995;45(6):1193–1198. doi: 10.1212/wnl.45.6.1193. [DOI] [PubMed] [Google Scholar]
- 46.Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131(7):1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, Kruger M, Karram K, Buhlmann C, Bechmann I, Heppner FL, Waisman A, Becher B. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat. Neurosci. 2012;15(4):543–550. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- 48.Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128(5):1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.