

Abstract
A growing list of insulin gene mutations causing a new form of monogenic diabetes has drawn increasing attention over the past seven years. The mutations have been identified in the untranslated regions of the insulin gene as well as the coding sequence of preproinsulin including within the signal peptide, insulin B-chain, C-peptide, insulin A-chain, and the proteolytic cleavage sites both for signal peptidase and the prohormone convertases. These mutations affect a variety of different steps of insulin biosynthesis in pancreatic beta cells. Importantly, although many of these mutations cause proinsulin misfolding with early onset autosomal dominant diabetes, some of the mutant alleles appear to engage different cellular and molecular mechanisms that underlie beta cell failure and diabetes. In this article, we review the most recent advances in the field and discuss challenges as well as potential strategies to prevent/delay the development and progression of autosomal dominant diabetes caused by INS-gene mutations. It is worth noting that although diabetes caused by INS gene mutations is rare, increasing evidence suggests that defects in the pathway of insulin biosynthesis may also be involved in the progression of more common types of diabetes. Collectively, the (pre)proinsulin mutants provide insightful molecular models to better understand the pathogenesis of all forms of diabetes in which preproinsulin processing defects, proinsulin misfolding, and ER stress are involved.
Keywords: Diabetes, Pancreatic beta cell, Insulin biosynthesis, Insulin gene mutation, Endoplasmic reticulum stress, Proinsulin misfolding
1. Introduction
Secreted insulin in the circulation regulates body metabolism to homeostatically maintain blood glucose. In pancreatic beta cells, three major steps lasting 30–150 minutes are needed to synthesize mature bioactive insulin: 1) The insulin precursor, preproinsulin, is translated in the cytoplasm, targeted to and translocated across the endoplasmic reticulum (ER) membrane, and proteolytically processed by signal peptidase on the luminal side of the ER membrane, forming proinsulin. 2) In the oxidizing ER environment, proinsulin undergoes oxidative folding, forming three evolutionarily conserved disulfide bonds (B7-A7, B19-A20, and A6-A11), which allow proinsulin to achieve transport-competence for exit from the ER. 3) Intracellular trafficking of proinsulin through the Golgi apparatus to secretory granules allows proinsulin to be proteolytically processed by prohormone convertases (PC1/3 and PC2) and carboxypeptidase E (CPE) to form C-peptide and mature insulin that are stored in insulin secretory granules and released upon stimulation (Fig. 1) (Alarcon et al., 1995; Dodson and Steiner, 1998; Liu et al., 2014; Steiner, 2011).
Fig. 1.
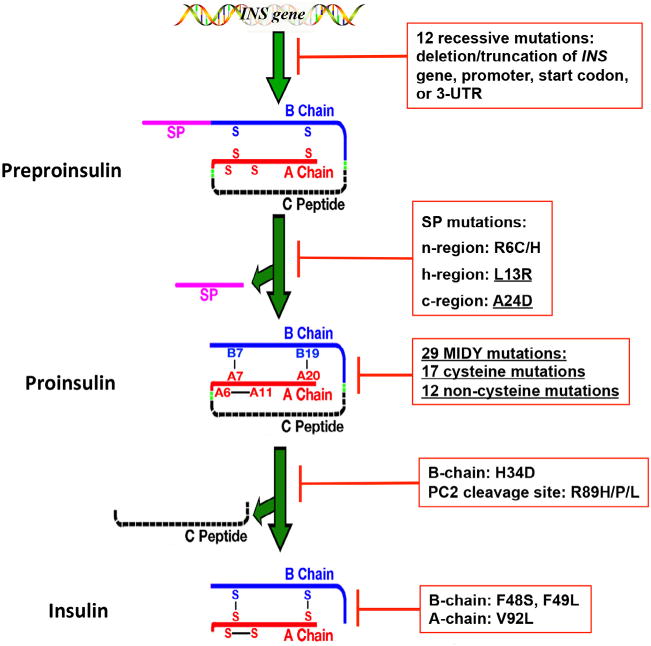
The effects of INS-gene mutations on the major steps of insulin biosynthesis. INS-gene mutations have been identified in the untranslated regions of INS gene and the coding sequence encoding all functional domains of preproinsulin molecule, including signal peptide (SP, pink), insulin B-chain (blue), C-peptide (black), insulin A-chain (red), and the proteolytic cleavage sites of signal peptidase (SPase) as well as prohormone convertases (PC1/3 and PC2, green). The mutations affect all major steps of insulin biosynthesis. Twelve recessive mutations in the untranslated regions result in more than 80% decrease of insulin production due to either INS gene deletion or truncation, or failure of insulin translation initiation, or instability of insulin mRNA. SP mutations in the n-region (R6C/H) or h-region (L13R) cause defective translocation of preproinsulin into the ER. The mutation at SP cleavage site (A24D) impairs SP cleavage. The largest group of INS-gene mutations are the mutations that affect proinsulin folding in the endoplasmic reticulum (ER), impairing formation of three evolutionarily conserved native disulfide bonds, B7-A7, B19-A20, and A6-A11. H34D affects sorting efficiency of proinsulin into regulated secretory pathway. The non-cysteine mutations at the PC2 cleavage site (R89H/P/L) impairs processing of proinsulin. The mutations in the B-chain (F48S and F49L) and A-chain (V92L) affect insulin binding to the insulin receptor. All mutations that cause Mutant INS-gene-induced Diabetes of Youth (MIDY) are underlined.
Following the discovery of proinsulin in 1967 (Steiner and Oyer, 1967; Steiner et al., 1967), many studies have focused on proinsulin intracellular trafficking and processing as well as the formation of granules that lead to insulin storage and secretion (Guest et al., 1991; Huang and Arvan, 1994; Kuliawat and Arvan, 1992; Orci et al., 1985, 1986; Quinn et al., 1991; Rhodes and Halban, 1987; Steiner et al., 1980). As the ER has been increasingly recognized as a central regulator of secretory pathway dynamics (Barlowe and Miller, 2013), the early events of insulin biosynthesis occurring in the ER have drawn increasing attention. These studies have gained greater urgency since 2007 (see below), when several groups have reported new insulin gene mutations that cause permanent neonatal diabetes in humans (Colombo et al., 2008; Edghill et al., 2008; Polak et al., 2008; Stoy et al., 2007).
The concept of insulinopathies was originally introduced to describe rare monogenic causes of adult onset diabetes associated with insulin gene mutations (Gabbay, 1980; Steiner et al., 1990). Those mutations are located in the insulin moiety or at the dibasic cleavage sites of proinsulin (Gabbay et al., 1976; Oohashi et al., 1993; Shoelson et al., 1983b; Tager, 1984). The mutations underlying these classical insulinopathies impair insulin binding to its receptor (Sakura et al., 1986; Vinik and Bell, 1988) or result in alteration of proinsulin trafficking for storage in insulin secretory granules (Carroll et al., 1988) or failure of endoproteolytic processing to insulin (Barbetti et al., 1990; Tager, 1984; Yano et al., 1992).
In 2007, the first study reporting 10 new insulin gene mutants causing neonatal diabetes (Stoy et al., 2007) set off a wave of new discovery about insulin biosynthesis and mutations that perturb insulin production. Over the past seven years, the list of insulin gene mutations associated with human diabetes has significantly expanded and is likely to grow further as genetic analysis becomes increasingly available. At the time of this writing, 51 INS-gene mutations have been identified to cause monogenic diabetes (Table 1). These mutations have been identified in the untranslated regions of the INS gene, the sequence encoding the preproinsulin signal peptide, the proteolytic cleavage sites of signal peptidase (SPase), the insulin B-chain, C-peptide, insulin A-chain, and the cleavage sites for prohormone convertases PC1/3 and PC2. Diabetogenic INS-gene mutations have a broad spectrum of clinical presentations ranging from severe neonatal onset to mild adult onset, suggesting that the product of different mutant INS alleles behave differently and utilize distinct mechanisms to cause diabetes. Accumulating evidence suggests that clinical severity of diabetogenic INS-gene mutations is linked to the nature of the particular mutation and the steps in the pathway of insulin biosynthesis affected by these mutants (Fig. 1 and Table 1) (Liu et al., 2010b, 2014; Støy et al., 2010). Herein, we review the most recent advances in this field, and discuss challenges and potential strategies to prevent/delay the development and progression of this form of monogenic diabetes.
Table 1.
Insulin gene mutations and their affects on insulin biosynthesis.
| Affects on insulin biosynthesis | Mutations | Re/Dn | Age of onset | Ref | |
|---|---|---|---|---|---|
| Transcription and/or Translationa | Deletion/truncation | c.−366_−343del, c.−370-_186 + del, c.−65_581del | Re | <6 moc | 1–2 |
| Promoter | c.−331 C > A/G, c.−332C>G + c.−331 C > G, c.−218 A > C | Re | <6 mob | 1 | |
| 3′UTR | c.*59 A > G | Re | <6 mob | 1 | |
| Translational initiation | c. 3G>T, c.3G>A | Re | <6 mob | 1 | |
| Premature stop | c. 184C>T (Q62X) | Re | <6 mob | 1 | |
| Translocation and/or SP cleavage | Targeting/translocation | L13R | Dn | <6 mo | 3 |
| Translocation | R6C/H | Dn | 15–65 yr | 4–6 | |
| SP cleavage | A24D | Dn | <6 moc | 6–8 | |
| Proinsulin folding in the ER | Non-cysteine mutations | H29D, L30M/P/V/Q, G32R/S, L35P, R46Q, L39Y40delinsH, G47V | Dn | <6 moc | 6–12 |
| Cysteine mutations | C31Y, C43G, F48C, R55C, G84R, R89C, G90C, C95Y, S101C, C96Y/S/R, Y103C, Y108C/X, C109F/Y/R | Dn | <6 moc | 6–14 | |
| Proinsulin trafficking/processing | Sorting/trafficking | H34D | Dn | Hyperpro-insulinimia | 15–16 |
| Processing | R89H/P/L | Dn | Variabled | 17–20 | |
| Insulin bioactivity | Binding to IR | V92L, F48S, F49L | Dn | Adult-onset | 21–23 |
Re: recessive; Dn: Dominant; 3-UTR: 3′-untranslated region; IR: insulin receptor; ER: endoplasmic reticulum; SP: signal peptide; Ref: References.
The nomenclature of the nucleotide changes is based on the INS gene coding sequence where nucleotide 1 represents translational start site.
The patients that are homozygote or compound heterozygote for those recessive INS gene mutations develop diabetes in 6 mo after birth.
Few patients carrying those mutations develop diabetes after 6 mo.
Hyperproinsulinemia, borderline glucose intolerance, or late-onset mild diabetes.
References: 1 (Garin et al., 2010); 2 (Raile et al., 2011); 3 (Hussain et al., 2013); 4 (Boesgaard et al., 2010); 5 (Meur et al., 2010); 6 (Edghill et al., 2008); 7 (Stoy et al., 2007); 8 (Polak et al., 2008); 9 (Colombo et al., 2008); 10 (Molven et al., 2008); 11 (Rubio-Cabezas et al., 2009);12 (Bonfanti et al., 2009); 13 (Moritani et al., 2013); 14 (Ozturk Mehmet et al., 2014); 15 (Chan et al., 1987); 16 (Gruppuso et al., 1984); 17 (Oohashi et al., 1993); 18 (Yano et al., 1992); 19 (Ozturk Mehmet et al., 2014); 20 (Warren-Perry et al., 1997) 21 (Sakura et al., 1986); 22 (Shoelson et al., 1983a); 23 (Tager et al., 1979).
2. Diabetogenic INS-gene mutations: recessive vs. dominant and early-onset vs. late-onset
Based on their pathological consequences, the insulin gene mutations can be divided into two major groups: recessive and dominant. Recessive INS gene mutations are “loss-of-function” mutations, which affect insulin biosynthesis at transcriptional and/or translational levels through different mechanisms including insulin gene deletion or truncation with failure of insulin transcription; instability of insulin mRNA; or defective translational initiation due to loss of the natural start codon (Garin et al., 2010; Raile et al., 2011). Such mutations may result in a >80% decrease of insulin production from the mutant allele. The fact that these mutations are inherited in a recessive mode indicates that the remaining normal INS gene allele is sufficient to maintain normoglycemia. This conclusion is consistent with earlier studies of insulin gene knockout mice (Leroux et al., 2001). Rodents have two functional insulin genes: Ins1 and Ins2 (Deltour et al., 1993). It has been shown that homozygous deletion of Ins1 or Ins2 plus heterozygous deletion of the second Ins gene does not lead to diabetes, supporting the notion that the product of even one functional insulin gene allele is ordinarily sufficient to maintain blood glucose in the normal range (Leroux et al., 2001) [although loss of a functional insulin allele may predispose individuals to a higher risk of developing diabetes in adulthood (Raile et al., 2011)].
About 80% (39 of 51) of insulin gene mutations are inherited in an autosomal dominant fashion. Most of them (30 of 39) cause the syndrome called Mutant INS-gene-induced Diabetes of Youth (MIDY) (Liu et al., 2010b). Since insulin haploinsufficiency cannot itself account for early-onset insulin-deficient diabetes, the development and progression of MIDY are attributed to a gain-of-toxic function from the mutant gene product. Indeed, studies using Akita mice, which carry the C96Y mutation in one of two Ins2 alleles, demonstrate that expressing mutant proinsulin leads to ER stress and ultimately, beta cell death (Oyadomari et al., 2002; Ron, 2002; Wang et al., 1999; Yoshioka et al., 1997). Beta cell death culminating from the expression of other MIDY mutants was also evident (Colombo et al., 2008). In addition to cytotoxicity, increasing evidence has suggested that the gain-of-toxic function of MIDY mutants (leading to insulin deficiency in MIDY patients) may be initiated by abnormal interactions between co-expressed mutant and wild-type proinsulin molecules in the ER of pancreatic beta cells. Indeed, the expression of MIDY mutants blocks co-expressed wild-type proinsulin exit from the ER, and therefore decreases insulin production from the wild-type INS allele (Liu et al., 2010a, 2012; Park et al., 2010; Wright et al., 2013b) prior to any decrease in beta cell mass, suggesting that in the MIDY syndrome, blockade of wild-type proinsulin by mutant proinsulin is a triggering event in the onset of insulin deficiency (Gupta et al., 2010; Hodish et al., 2010; Renner et al., 2013).
In addition to mutations causing MIDY, a few autosomal dominant insulin gene mutations (9 out of 39) are associated with late-onset diabetes. These include the original and now classic insulinopathies plus two new preproinsulin signal peptide mutations (Boesgaard et al., 2010; Edghill et al., 2008; Meur et al., 2010; Molven et al., 2008). Unlike the MIDY mutants, the mutations associated with late-onset diabetes do not appear to be linked to proinsulin folding events that occur in the ER. Herein, we describe the distinct groups of insulin gene mutations that are clustered based largely on experimentally confirmed defects in the cell biology of insulin biosynthesis. The mutations that have not been experimentally tested are integrated into these groups based on predicted defects associated with these mutations (Fig. 1 and Table 1).
3. Lessons from INS-gene mutations: the links between biological defects and diabetes phenotypes
Diabetogenic INS gene mutations present a broad spectrum of diabetes severity ranging from neonatal onset severe insulin deficient diabetes to late-onset mild diabetes. One example of this comes from a family carrying proinsulin-C43G mutation. The mutation disrupts one of critical interchain disulfide bonds, leading to proinsulin misfolding in the ER, and the proband carrying this mutation developed very severe diabetes at 43 weeks of age. However, his father who carries the identical mutation was diagnosed with mild type 2 diabetes at 30 years of age (Stoy et al., 2007). Thus, heterogeneity of clinical presentation depends both on the biological behavior of the INS mutants themselves and other genetic and environmental factors.
3.1. Insulin gene mutations affecting insulin gene transcription and translation
In pancreatic beta cells, insulin biosynthesis is tightly regulated at both transcriptional and translational levels (Hay and Docherty, 2006; Itoh and Okamoto, 1980; Leibiger et al., 2000; Sharma and Stein, 1994). Upon glucose stimulation, while general protein synthesis increases 2-fold, preproinsulin biosynthesis can increase as much as 30-fold in one hour, indicating specific control mechanisms (Alarcón et al., 1993; German et al., 1995; Guest et al., 1991; Wicksteed et al., 2001). The physiological and pathological importance of regulatory elements within untranslated region of the INS gene is genetically demonstrated by the newly discovered insulin gene mutations that affect insulin gene transcription/translation (Garin et al., 2010). Twelve such mutations are inherited in a recessive manner (Garin et al., 2010; Raile et al., 2011). Among these mutations, 5 are located in the insulin promoter region, resulting in either a deletion of the promoter region regulated by MAFA and NEUROD1, or disruption of binding sites for additional DNA binding proteins. Those mutations that result in 90% reduction of promoter activity provide the first human genetic evidence that discrete INS cis regulatory elements are essential for regulating insulin biosynthesis (Garin et al., 2010). Another interesting mutation, c.*59 A>G, is found in the polyadenylation signal of 3′ untranslated region (3′-UTR) of insulin mRNA. This mutation results in severe instability of insulin mRNA, supporting previous findings that the 3′-UTR of insulin mRNA plays an essential role in regulating insulin biosynthesis through modulating insulin mRNA stability (Fred and Welsh, 2009; Tillmar et al., 2002). Two mutations (c. 3G>T and c.3G>A) found at the 3rd nucleotide of preproinsulin start codon abolish the native translational initiation site for the preproinsulin. Although there is an ATG encoding another methionine at the 5th residue of preproinsulin that may potentially function as an alternative start codon, the nucleotide sequence upstream of this ATG does not favor translational initiation resulting in an ~80% decrease of insulin production (Garin et al., 2010).
3.2. Insulin gene mutations affecting preproinsulin ER targeting and translocation
The first step of insulin biosynthesis involves the targeting and translocation of newly synthesized preproinsulin from the cytosol into the ER. This process is led by the signal peptide of preproinsulin at its N-terminus. Preproinsulin has a 24 residue signal peptide, which comprises three regions: a positive charged n-region; a central core hydrophobic h-region; and a polar c-region containing a cleavage site of the SPase (for reviews, see Liu et al., 2014; Rapoport, 2007). To date, four new mutations located in the preproinsulin signal peptide have been reported to cause diabetes (Boesgaard et al., 2010; Hussain et al., 2013; Meur et al., 2010; Stoy et al., 2007). Mutations are found in all three regions of the signal peptide (Fig. 2). Interestingly, the clinical diabetes phenotypes associated with these mutants range from severe neonatal-onset insulin-deficient diabetes caused by L13R or A24D, to mild adult onset diabetes associated with R6C or R6H, suggesting the possibility that different cellular defects or molecular mechanisms may underlie the onset and development of diabetes in these patients. Indeed, we have recently shown that the mutation at the site of signal peptide cleavage, A24D, leads not only to inefficient signal peptide cleavage, but the cleavage occurs at an alternative site producing an abnormal proinsulin. Thus, although the proinsulin moiety of preproinsulin-A24D has the same predicted sequence as that of wild-type proinsulin, the combination of uncleaved and improperly cleaved preproinsulin-A24D causes misfolding of both species within the ER. This highlights the physiological and pathological significance of the coordination of signal peptide cleavage and downstream proinsulin folding (for detail, see Liu et al., 2012; Liu et al., 2014). In the next paragraphs, we focus on the most recent findings revealing preproinsulin targeting and translocation defects caused by R6C and R6H.
Fig. 2.
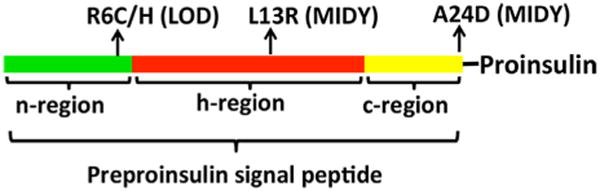
Three functional regions of preproinsulin signal peptide and the mutations associated with diabetes. Preproinsulin signal peptide has three functional regions: n-region (green), h-region (red), and c-region (yellow). Diabetogenic INS-gene mutations have been found in all three regions. The mutations in the n-region (R6C/H) cause inefficient translcocation of preproinsulin across the ER membrane and lead to late-onset diabetes (LOD). The mutation in the h-region (L13R) causes mutant INS-gene-induced diabetes of young (MIDY). The cellular defect caused by L13R remains to be determined, but it is predicted to affect the targeting and translocation of the mutant preproinsulin. The mutation at the signal peptide cleavage site (A24D) impairs normal signal peptide cleavage and causes proinsulin misfolding in the ER, leading to MIDY.
In mammalian cells, most newly synthesized secretory proteins undergo co-translational targeting and translocation into the ER. In this process, when the signal peptide of the nascent polypeptide chain emerges from the ribosome, it is first recognized and bound by the signal recognition particle (SRP), forming the SRP-ribosome-nascent polypeptide complex. The complex is then targeted to the ER membrane via the docking of SRP to the SRP receptor on the ER membrane. Upon targeting to the ER membrane, the signal peptide interacts with the Sec61 translocon, through which the signal sequences of secretory proteins (or signal anchors of membrane proteins) are positioned and oriented for subsequent translocation of the remaining polypeptide (Pilon et al., 1998; Plath et al., 1998; Zimmermann et al., 2011). At least three factors play roles in determining the orientation of the signal sequence within the Sec61 translocon: the length of the h-region; the charge gradient of signal sequence flanking the h-region; and the charge gradient of the translocon spanning the ER membrane (Goder et al., 2004; Hartmann et al., 1989; Parks and Lamb, 1991; von Heijne, 1986; Wahlberg and Spiess, 1997). Since the Sec61 translocon possesses a conserved charge gradient with a positive charge toward the ER lumen (Goder et al., 2004), a signal sequence with a positive charge in the n-region tends to make a “loop”, orienting the N-terminus of the signal sequence towards the cytosol and the C-terminus oriented towards the ER lumen, referred as the “positive-inside” rule (Iino et al., 1987; Peterson et al., 2003; Sasaki et al., 1990; von Heijne, 1989). However, although this “positive-inside” rule is considered as a general principle for orienting the signal sequence during preprotein translocation across the ER membrane, the potential physiological or pathological significance of the n-region positive charge in the signal peptide of secretory and membrane preproteins has only recently been examined.
Preproinsulin-R6C or R6H mutates the highly conserved positively charged residue in the n-region of the signal peptide that is thought to be important in targeting/translocation of secretory and membrane proteins (Fujita et al., 2010; Goder and Spiess, 2001; Parks and Lamb, 1991; Sakaguchi et al., 1992; Sasaki et al., 1990). Surprisingly, using a GFP-tagged preproinsulin construct, one study reported no defect of preproinsulin-R6C/R6H in targeting to insulin granules (Meur et al., 2010). We also examined the translocation efficiency of preproinsulin-R6C/H with a large GFP-tag (248 amino acids), and compared that to preproinsulin-R6C/H with a small Myc-tag (10 amino acids) or without any tag. We confirmed that GFP-tagged preproinsulin-R6C/H has no detectable defect in ER targeting and translocation (Guo et al., 2014). By contrast, at least 50% of newly synthesized Myc-tagged or untagged preproinsulin-R6C/H failed to be efficiently translocated after initial targeting of the mutant preproinsulin to the ER membrane (Guo et al., 2014). The results suggest that larger secretory proteins may be less sensitive than short secretory polypeptides to the effect of loss of positive charge in the n-region of the signal peptide. The underlying mechanism of this length dependence – and whether this behavior is specific to preproinsulin – remains unknown. A recent study shows that polypeptide length is an important factor for determining whether secretory polypeptides undergo co-translational translocation or post-translational translocation (Lakkaraju et al., 2012). Importantly, because of the short time window for signal peptide recognition by SRP, the signal peptide of small secretory proteins may not be accessible for co-translational recognition and targeting by SRP before the completion of synthesis of the full-length precursor. Thus, post-translational modes of targeting and translocation may provide an important backup mechanism to enhance overall translocation efficiency for small secretory proteins including preproinsulin (Johnson et al., 2012, 2013; Shao and Hegde, 2011).
Preproinsulin-L13R mutation was identified in a baby girl with in utero growth retardation who developed severe diabetes on the second day after birth (Hussain et al., 2013). The mutation is located in the h-region of preproinsulin signal peptide. An arginine (charged residue) substitution of the hydrophobic leucine disrupts the characteristic hydrophobic core of the h-region that normally plays a critical role in SRP recognition, ER targeting and translocation of secretory proteins (Bird et al., 1990; Haeuptle et al., 1989; Janda et al., 2010). Therefore, the L13R mutation is expected to affect insulin biosynthesis at the earliest step of entry of newly synthesized preproinsulin into the ER. However, the actual cellular defect underlying beta cell failure and diabetes caused by preproinsulin-L13R remains to be experimentally determined.
3.3. Insulin gene mutations affecting proinsulin folding in the ER
Among all autosomal dominant insulin gene mutations, more than 70% (30 out of 39) are predicted to affect the normal folding pathway of proinsulin in the ER (Table 1). To date, about half of these mutants have been experimentally confirmed to cause proinsulin misfolding in the ER (Colombo et al., 2008; Liu et al., 2010a, 2012; Park et al., 2010). The most studied insulin gene mutation of this type is preproinsulin-C96Y mutation. It was found first in the Akita mouse (thus, C96Y mutation is often referred as “the Akita mutation”), which carries a spontaneous C96Y mutation in one of two Ins2 alleles. The mouse develops diabetes shortly after weaning (Wang et al., 1999; Yoshioka et al., 1997). The C96Y mutates one of the six conserved Cys residues of proinsulin, disrupting the formation of the B7-A7 disulfide bond, and creating an unpaired B7 cysteine. Both in vivo and in vitro studies demonstrate that the C96Y mutation causes proinsulin misfolding in the ER, induces ER stress and leads ultimately to beta cell death (Hodish et al., 2010; Izumi et al., 2003; Liu et al., 2007, 2010a; Park et al., 2010; Wang et al., 1999). Another diabetic mouse line, the Munich mouse expressing preproinsulin-C95S, was generated by chemical (N-ethyl-N-nitrosourea) mutagenesis (Herbach et al., 2007). Importantly, both C95S and C96Y mutations are found in humans and cause the MIDY syndrome (Colombo et al., 2008; Stoy et al., 2007). Therefore, the two mouse lines are appropriate animal models in which to study cellular defects and stress caused by misfolded proinsulin in vivo. Besides C95S and C96Y mutations, 16 additional mutations also generate novel unpaired Cys residues – either because of mutations at the site of native cysteines, or because the mutations create new Cys residues (Table 1). Since native disulfide bond formation among the cysteine residues is the most well recognized event in the proinsulin folding pathway in the ER, all of the mutations with unpaired cysteines are likely to seriously interfere with disulfide maturation, leading to proinsulin misfolding (Liu et al., 2005, 2010a, 2012; Rajpal et al., 2012).
In addition to mutations that create unpaired cysteines, there are 12 mutations that are also expected to cause proinsulin misfolding in the ER. Eleven of these have been found within the insulin B-chain while one has been located at the preproinsulin signal peptide cleavage site (none has yet been reported within the A-chain; see Table 1). More work is still needed to understand how these point mutations affect proinsulin folding in the ER. However, it is worth noting that nearly all of these mutations are located at highly conserved residues in the preproinsulin molecule, some of which have already been shown to be important for local folding of the insulin moiety, which may help to align the B- and A-chains in order to facilitate native proinsulin disulfide pairing (Hua et al., 2006b; Liu et al., 2009, 2010c; Sohma et al., 2010; Weiss, 2009). Thus, even these mutations that do not directly alter proinsulin Cys residues are likely to perturb normal disulfide pairing of proinsulin. Indeed, all experimentally tested mutants, including A24D, H29D, L30P, G32R, L35P, R46Q, and G47V, have been shown to exhibit defective disulfide maturation of newly synthesized proinsulin, leading to an increase of mispaired disulfide isomers (Liu et al., 2010a, 2012, 2014). Studies focusing on the pathway of native proinsulin folding and disulfide maturation in the ER are still needed in order to understand how these processes are affected by this subset of insulin gene mutations.
3.4. Insulin gene mutations affecting trafficking and processing of proinsulin
Following oxidative folding and dimerization within the ER, properly folded proinsulin exits from the ER for delivery to the Golgi apparatus. As the Zn2+ concentration begins to rise in the lumen of the trans-Golgi network (TGN) (Dunn, 2005), proinsulin can initiate zinc-dependent hexamerization (Dodson and Steiner, 1998; Huang and Arvan, 1995; Orci et al., 1986). Upon entry into immature secretory granules, proinsulin hexamers undergo endoproteolytic cleavage via the actions of PC1/3 and PC2 in concert with carboxypeptidase E (CPE) to process proinsulin into mature insulin and C-peptide (Alarcón et al., 1993; Bailyes et al., 1992; Davidson and Hutton, 1987; Fricker et al., 1986; Steiner, 1998; for detailed reviews, please see Dodson and Steiner, 1998; Liu et al., 2014. Five insulin gene mutations are located at the junctions between the proinsulin B-chain and C-peptide, or between the C-peptide and A-chain, corresponding to PC1/3-preferred and PC2-preferred cleavage sites, respectively. The clinical phenotypes caused by mutations at the cleavage sites vary greatly depending upon the different substitutions by particular residues. Specifically, substitutions of arginine with either leucine, histidine, or proline at C-A junction results in proinsulin processing defects that are accompanied by relatively asymptomatic hyperproinsulinemia (Gabbay et al., 1979; Steiner et al., 1990), borderline glucose intolerance (Gruppuso et al., 1984), or late-onset mild diabetes (Oohashi et al., 1993; Yano et al., 1992). In contrast, substitution of arginine with cysteine at either B-C junction or C-A junction causes severe early onset insulin-deficient diabetes (Colombo et al., 2008; Edghill et al., 2008; Rubio-Cabezas et al., 2009; Stoy et al., 2007). Direct side-by-side comparison indicates that while the leucine substitution R89L is efficiently secreted from the cells, the cysteine substitution R89C causes proinsulin misfolding and retention in the ER (Colombo et al., 2008). Thus, although R89C mutates the PC2 cleavage site, the underling mechanism of diabetes caused by this mutation is not due to a defect of PC2-mediated proinsulin processing, but rather due to proinsulin misfolding and ER stress. This further highlights severe pathological consequences caused by proinsulin misfolding in the ER.
Surprisingly, another mutation that causes hyperproinsulinemia is located at neither the PC1/3 or PC2 cleavage site, but rather stems from a substitution of the conserved histidine with aspartic acid at position 34, H34D, corresponding to the 10th residue of the proinsulin B chain (Chan et al., 1987; Gruppuso et al., 1984). Studies using a transgenic mouse line expressing this mutation revealed that more than 50% of proinsulin-H34D is normally transferred to the secretory granules and processed to insulin. However, a significant proportion of the newly synthesized mutant proinsulin (~15%) is secreted from the cells through an unregulated or constitutive pathway, and ~20% is rapidly degraded within beta cells (Burgess and Kelly, 1987; Carroll et al., 1988). A similar result is also obtained in At-T20 cells transiently expressing proinsulin-H34D (Gross et al., 1989). In beta cells, almost all of the newly synthesized proinsulin that exits the ER is ultimately packaged into the regulated secretory granules where proinsulin processing occurs (Liu et al., 2007; Rhodes and Halban, 1987; Steiner et al., 1990). The sorting defects of proinsulin-H34D mutant suggest that proinsulin has structural information that plays a role in its intracellular sorting, trafficking and storage in pancreatic beta cells.
3.5. Mutations affecting insulin binding to the insulin receptor
Three of the classical insulinopathies, F48S, F49L, and V92L [corresponding to F(B24)S, F(B25)L, and V(A3)L], identified in the early 1980s (Given et al., 1980; Sakura et al., 1986; Shoelson et al., 1983a, 1983b; Tager et al., 1979), are mutations located in regions thought to be important for insulin binding to its receptor (Hua et al., 1993; Menting et al., 2013; Xu et al., 2004). Indeed, the binding efficiency of those mutant insulins to the insulin receptor ranges from about 14% to 0.2% of that of wild-type insulin (Assoian et al., 1982; Sakura et al., 1986; Shoelson et al., 1983a, 1983b; Steiner et al., 1990). Defective binding of these secreted mutant insulins to insulin receptors leads to a significant impairment of insulin clearance – resulting in an increased half-life of the mutant insulins in the circulation, leading to an elevated ratio of circulating insulin to C-peptide (Shoelson et al., 1984). These findings provide direct evidence that receptor-mediated uptake leading to physiological degradation of insulin is a major pathway of insulin clearance in vivo (Steiner et al., 1990).
In addition to defective binding to the insulin receptor, a recent study reveals surprising intracellular defects of proinsulin-F48S, corresponding to F(B24)S. Compared to proinsulin-V92L or the mutants causing MIDY, F48S presents an intermediate secretory phenotype, including a moderate defect in oxidative folding in the ER, a limited decrease in secretion, a modest activation of unfolded protein response (UPR), and a partial dominant-negative effect on co-expressed proinsulin wild-type (Liu et al., 2010a). Studies have shown that phenylalanine at the 24th residue of the insulin B-chain plays an important role in stabilizing the native-like cluster of hydrophobic side chains, increasing the efficiency of B19-A20 disulfide pairing (Hua et al., 2006a). Computer modeling from NMR spectroscopic data indicates F48S mutation results in surprising variation (ie, instability) in the structure of residues B20-B30, consistent with perturbation of B19-A20 disulfide pairing. By contrast, the structure of another classical mutation insulin-V92L is essentially identical to wild-type insulin and causes only trivial decrements in thermodynamic stability (Fig. 3, Liu et al., 2010a).
Fig. 3.
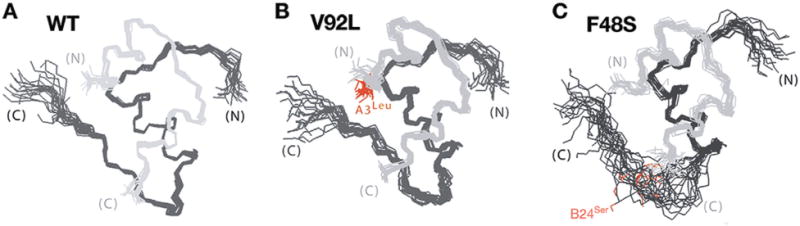
Solution structures of insulin analogs. A. Ensemble of NMR-derived structures DKP-insulin wild-type (WT). The A- and B-chains are shown in light and dark gray, respectively. B. Solution structures of V92L-DKP-insulin. The side chain of the mutant residue leucine (Leu) at the 3rd residue of insulin A-chain is shown in red. C. Solution structures of F48S-DKP-insulin. The side chain of the mutant residue serine (Ser) at the 24th residue of insulin B-chain is shown in red. Whereas V92L is compatible with native-like structure in accord with results of X-ray crystallography, F48S destabilizes the C-terminal strand of the B-chain. This figure is modified from Liu et al. (2010a).
4. Molecular mechanisms underlying pancreatic beta cell failure and diabetes caused by insulin gene mutations
4.1. Cellular response to misfolded proinsulin in the ER
Upon delivery into the ER lumen, preproinsulin is immediately processed into proinsulin, which undergoes rapid oxidative folding. As discussed above, the subset of INS-gene mutations that lead to the autosomal dominant syndrome of MIDY cause proinsulin misfolding in the ER. These MIDY mutants can be recognized by the ER quality control system, with the misfolded molecules bound by ER chaperones. The immunoglobulin heavy chain-binding protein (BiP) is one of the major chaperones that has been experimentally confirmed to bind to misfolded proinsulin and proinsulin folding intermediates (Liu et al., 2005; Scheuner et al., 2005; Schmitz et al., 1995). As a member of the heat shock protein 70 (HSP70) superfamily, BiP is a master chaperone that recognizes unfolded and misfolded proteins, sensing changes in the ER environment, and maintaining ER homeostasis by contributing indirectly to the activation of three unfolded protein response (UPR) signaling pathways (Hendershot, 2004; Liu and Kaufman, 2003; Volchuk and Ron, 2010; Wang et al., 2009). Those pathways are initiated by three ER transmembrane proteins, including protein kinase (PKR)-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (for review, please see Cavener et al., 2010; Walter and Ron, 2011; Zhang and Kaufman, 2008). In normal beta cells, the lumenal domain of all three transmembrane sensor proteins have the potential to be bound by BiP. However, in beta cells expressing MIDY mutants, BiP preferentially relocates its binding to misfolded proinsulin concomitant with the activation of IRE1 (Gardner and Walter, 2011), PERK (Bertolotti et al., 2000) and ATF6 (Shen et al., 2005), triggering the tripartite UPR. Recent studies suggest that, at least for IRE1 pathway, unfolded proteins can also directly bind and activate IRE1 (Bernales et al., 2006; Gardner and Walter, 2011; Kimata et al., 2007). Acute activation of UPR is considered as a defense mechanism that could potentially alleviate ER stress by attenuating protein translation, selectively upregulating ER chaperones, and accelerating retrotranslocation of misfolded protein for proteasomal degradation, a process called ER-associated degradation (ERAD) (Smith et al., 2011). However, in Akita mice (and likely in MIDY patients), continuous expression of misfolded mutant proinsulin in the ER results in an enlarged ER and prolonged activation of the UPR, which could disrupt ER protein homeostasis, leading to chronic ER stress and activation of apoptosis signaling, and promoting beta cell death (Hartley et al., 2010; Izumi et al., 2003). ER stress-related apoptosis appears to be mediated by transcriptional activation of the C/EBP homologus protein (CHOP: gene name GADD153). Targeted disruption of this gene decreases the rate of beta cell death and delays the onset of diabetes in heterozygous Akita mice by 8–10 weeks (Oyadomari et al., 2002).
4.2. Bystander effects of misfolded proinsulin on co-expressed wild-type proinsulin
It has been a commonly held view that diabetes in Akita mice is caused solely by ER stress-mediated beta cell death. However, given the fact that decreased beta cell death (by disrupting GADD153) in Akita mice delays but does not prevent diabetes onset, factors other than decreased beta cell mass are likely to be involved in the development of diabetes caused by misfolded proinsulin. Using a compound heterozygous mouse line co-expressing Akita mutant proinsulin and proinsulin fused with GFP in beta cells, several recent studies find that there is no decrease in beta cell mass in newborn Akita mice (Hodish et al., 2010) or even at the time of initial presentation of hyperglycemia – indeed, Gupta et al. argued that the pancreatic islets of Akita mice may even exhibit beta cell hyperplasia at the time of initial onset of diabetes (Gupta et al., 2010). More recently, a transgenic pig model expressing Akita mutant proinsulin in beta cells has been established. Hyperglycemia is evident within 24 hours after birth in the transgenic pigs, a direct sign of insulin deficiency. Yet the transgenic pigs exhibit virtually no difference of beta cell mass with that of littermate controls more than one week after birth (Renner et al., 2013). Altogether, these studies indicate that insulin deficiency precedes the decrease in beta cell mass during diabetes onset as a consequence of the expression of misfolded MIDY mutant proinsulin – and this is likely to be true in humans as well.
As noted in section 2 above, complete homozygous knockout of either Ins1 or Ins2 in conjunction with heterozygous deletion of the other INS gene does not cause diabetes in mice (Leroux et al., 2001), just as several human INS gene mutations silencing one INS allele are recessive (Garin et al., 2010; Raile et al., 2011). Thus, neither loss of beta cell mass nor loss of one normal INS allele is likely to be a sufficient reason for onset of diabetes in MIDY. As discussed above, the beta cells of Akita mice do experience ER stress due to expression of misfolded mutant proinsulin. However, neither overall protein synthesis nor specific proinsulin synthesis in Akita beta cells is decreased compared with that of wild-type islets, indicating that decreased insulin production is not caused by attenuation of proinsulin synthesis (Izumi et al., 2003; Liu et al., 2007). However, pulse-chase experiments have shown that, two hours after synthesis, at a time when the majority of proinsulin from wild-type islets is processed to mature insulin – a significant amount of newly synthesized wild-type proinsulin in Akita islets has been degraded without being processed, leading to dramatically decreased insulin production (Liu et al., 2007). Altogether, the data in in Akita mice (and probably in humans with MIDY) indicate that insulin deficiency is initiated by a defect in the biosynthesis pathway from proinsulin to insulin.
Based on the foregoing reasoning, cellular defects in normal proinsulin processing, caused by MIDY mutant proinsulin at a time before there is significant loss of beta cell mass, is likely to be responsible for the initial failure of insulin production in MIDY. In Akita mice, a significant amount of wild-type proinsulin itself becomes misfolded and becomes recognized by the ER chaperone, BiP (Liu et al., 2010a), suggesting that misfolded mutant proinsulin can affect the normal folding of co-expressed wild-type proinsulin in the ER. There may be at least two reasons for this. First, ER stress caused by the expression of misfolded mutant proinsulin may change the ER environment, altering the availability of free chaperones, the redox environment, and perhaps even the kinetics with which preproinsulin is translocated and its signal peptide cleaved at the ER membrane – all of which may directly or indirectly affect the normal folding pathway of wild-type proinsulin in the ER. However, in cells pretreated with a low dose of tunicamycin for 16 hours to induce ER stress, wild-type proinsulin continues to exit the ER and proceeds on to secretion (Liu et al., 2010a). Thus, at least mild acute ER stress does not seem to be sufficient to block proinsulin trafficking (although the effect of severe prolonged ER stress on proinsulin folding and trafficking remains to be determined).
Second, misfolded mutant proinsulin directly acts on co-expressed wild-type proinsulin, affecting its folding pathway in the ER. Indeed, both co-immunoprecipitation and an-isotropy FRET microscopy have shown that misfolded mutant proinsulin can intimately interact with co-expressed wild-type proinsulin in the ER (Haataja et al., 2013; Hodish et al., 2010; Liu et al., 2010a, 2012). Importantly, these abnormal interactions appear to include, among others, an intermolecular thiol attack in which cysteine residues from the mutant proinsulin form intermolecular disulfide bonds with wild-type proinsulin. As proinsulin normally uses all of its 6 Cys residues for intramolecular disulfide pairing, any intermolecular disulfide bonds are by definition non-native, and will leave the wild-type partner with at least one unpaired Cys residue that itself may produce further inappropriate interactions, thereby affecting wild-type proinsulin folding and resulting in formation of disulfide-linked complexes that contain both mutant and wild-type proinsulin molecules (Liu et al., 2010a, 2012). The consequence of the formation of aberrant protein complexes between mutant and wild-type proinsulin is that the wild-type partner becomes retained in the ER. Thus, even though Akita beta cells continuously synthesize normal amounts of proinsulin (Izumi et al., 2003; Liu et al., 2007), the failure of ER export of wild-type proinsulin is a direct cause of diminished insulin production, which is an initiating factor in the onset of insulin-deficient diabetes (Liu et al., 2007, 2012).
The dominant-negative blockade by misfolded mutant proinsulin imposed upon co-expressed wild-type proinsulin appears to be dose-dependent. While the male Akita Ins2 heterozygous mice begin to have hyperglycemia shortly after weaning, the male Akita Ins2 homozygous mice develop more severe diabetes shortly after birth (Yoshioka et al., 1997). In contrast, when Akita mutant proinsulin is expressed at a lower level from a transgene (not more than 4% of endogenous Ins2), diabetes prevalence is low (less than 10% of males) but the animals display pre-diabetes by glucose tolerance testing (Hodish et al., 2011). Similarly, a diabetic phenotype is not observed in transgenic pig lines that express Akita mutant proinsulin at levels < 15 % of the endogenous INS gene. In contrast, when the expression of Akita proinsulin reaches a relative level equivalent to 75% of the expression of the endogenous INS gene (i.e., 43% of total), then the transgenic pigs exhibit hyperglycemia within 24 hours after birth (Renner et al., 2013). This dose-dependence is also evident in cell culture studies in which dominant-negative blockade of wild-type proinsulin gradually decreases as the expression ratio of mutant : wild-type proinsulin drops (Liu et al., 2012). Collectively, these studies indicate that diabetogenic MIDY mutants act in a dose-dependent manner.
4.3. Bidirectional effects of mutant and wild-type proinsulin partners in the ER
A recent study not only confirmed the dose-dependent dominant blockade of wild-type proinsulin by MIDY mutants, but also revealed a surprising bidirectional effect of wild-type proinsulin on mutant proinsulin (Wright et al., 2013b). Indeed, by changing the stoichiometry of wild-type and mutant proinsulins to further favor the wild-type, secretion of some of the MIDY mutant gene product was found to be partially rescued by co-expression with wild-type proinsulin. This may explain different results that have been observed in transfected beta cells compared to results in non-beta cells. In transfected beta cells, due to a large amount of endogenous proinsulin, some MIDY mutants (e.g. R89C) have been found to egress from the ER and be transported to insulin secretory granules (Rajan et al., 2010). In contrast, in cells lacking wild-type proinsulin, the same MIDY mutants are entrapped in the ER without detectable secretion (Liu et al., 2010a, 2012).
Intriguingly, this rescue effect appears to be protein specific. Whereas wild-type proinsulin rescued some MIDY mutants, wild-type proinsulin could not rescue secretion of co-expressed mutant thyroglobulin (Tg). Reciprocally, wild-type thyroglobulin could not rescue secretion of co-expressed MIDY mutants, but wild-type thyroglobulin could promote the secretion of two recessive thyroglobulin mutants known as cog-Tg and rdw-Tg, which are misfolded and retained in the ER when expressed alone (Wang et al., 2010; Wright et al., 2013b). Evidently, specific interaction between dimerization partner proteins is needed to attain this rescue effect. The physiological and pathological significance of the bidirectional effect in the development and progression of MIDY remains to be further evaluated in vivo. However, studies seem to suggest that this bidirectional effect may play a role in the pathogenesis of some genetic diseases. In the heterozygotes of autosomal dominant diseases such as MIDY, the predominant direction is that the mutants block the co-expressed wild-type proteins. Indeed, there are some MIDY mutants that cannot be rescued at all by wild-type proinsulin (Wright et al., 2013b), so in these heterozygous patients, there will be no bidirectionality. In contrast, in autosomal recessive diseases, the predominant direction is that wild-type proteins are not only efficiently exported from ER; they may also promote ER export of their mutant dimerization partners, alleviating potential ER stress caused by these misfolded mutant proteins.
4.4. Cellular responses to untranslocated/mislocated preproinsulin
As discussed in section 3.2, based on the general function and properties of the signal peptide, three preproinsulin signal peptide mutations, R6C, R6H, and L13R are expected to affect SRP recognition, ER targeting, and/or translocation of preproinsulin across the ER membrane. Specifically, our recent study shows that after targeting to the ER, preproinsulin-R6C/H fails to be efficiently translocated across the ER membrane. Untranslocated secretory proteins usually are unstable and rapidly degraded in cells (Ulmer and Palade, 1989a, 1989b). Indeed, most of the newly synthesized untranslocated preproinsulin-R6C/H molecules are degraded (Guo et al., 2014). However, a certain fraction of untranslocated preproinsulin is relocated intracellularly and accumulated within an undefined juxtanuclear compartment with the proinsulin moiety exposed to the cytosol (Fig. 4).
Fig. 4.
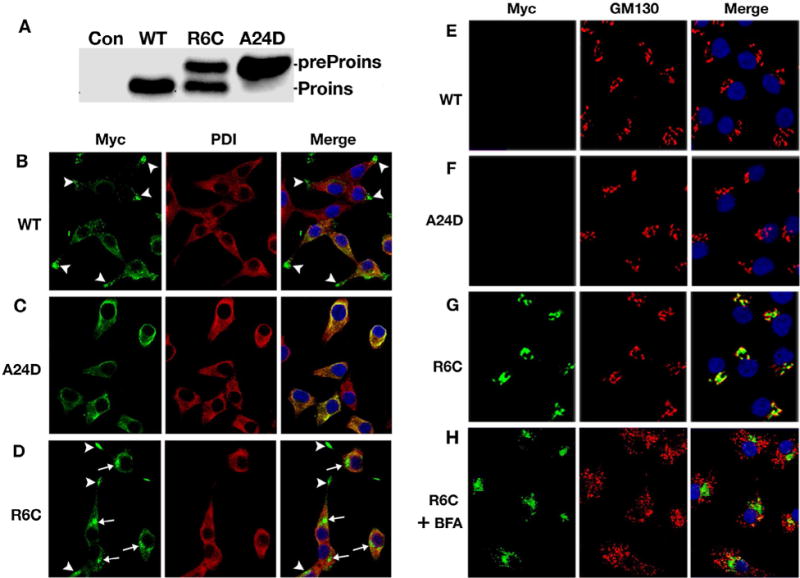
Two preproinsulin signal peptide mutations cause distinct cellular defects in beta cells. A. Processing of Myc-tagged preproinsulin R6C and A24D in INS1 cells were examined by western blotting using anti-Myc antibody While most of preproinsulin-A24D presents as uncleaved preproinsulin, preproinsulin-R6C produces two populations: proinsulin and unprocessed preproinsulin. C–D. INS1 cells expressing Myc-tagged preproinsulin-WT, R6C or A24D were fully permeabilized and immunoblotted with anti-Myc (green) and anti-PDI (red) antibodies. Nuclei were counterstained with DAPI. In most cells expressing Myc-tagged preproinsulin-WT (A), anti-myc immunoreactable molecules presented as punctate insulin granule-like pattern (arrowheads) that were distinct from PDI. Preproinsulin-A24D lost the granule pattern and largely overlapped with PDI (B). Preproinsulin-R6C produces two major intracellular pools: one did indeed concentrate in distal tips (arrowheads) while another accumulated in a juxtanuclear location (arrows), and neither pool overlapped with PDI (C). E–G. The parallel sets of INS1 cells were selectively permeabilized by 0.01% digitonin and immunoblotted with anti-Myc (green) and anti-GM130 (red) antibodies. Nuclei were counterstained with DAPI. Unlike B–D, anti-myc immunoreactable molecules were detected only in the cells expressing Myc-tagged R6C (G). Such molecules appeared to accumulate in a juxtanuclear region close to Golgi marker GM130. H. A parallel well of the cells of G were pre-treated with 5uM brefeldin A (BFA) before being partially permeabilized and immunoblotted as in G. This figure is modified from Guo et al. (2014).
Accumulation of juxtanuclear puncta has been observed upon expression of cytosolic misfolded and aggregation-prone proteins. Such a disturbance in cytoplasmic proteostasis with juxtanuclear accumulation is thought to induce cytoplasmic stress (Kaganovich et al., 2008; Kocik et al., 2012). In contradistinction to the ER stress response, preproinsulin-R6C leads to induced expression of HSP70 (Guo et al., 2014), a major cytosolic stress-inducible chaperone, which can bind to misfolded proteins to prevent their aggregation and promote their degradation (Kakkar et al., 2012; Saibil, 2013). Importantly, cytosolic/juxtanuclear accumulation of misfolded proteins has been linked to cell death that contributes to the development and progression of various neurodegenerative diseases including Huntington’s, Parkinson’s, Alzheimer’s and prion diseases (Debure et al., 2003; Kaganovich et al., 2008; Miesbauer et al., 2010; Rane et al., 2010; Reiner et al., 2011; Rubinsztein, 2006; Schaffar et al., 2004; Strom et al., 2006; Viswanathan et al., 2011). Consistent with this, the expression of preproinsulin-R6C leads to increased beta cell death, which contributes to late-onset diabetes caused by inefficient preproinsulin translocation (Guo et al., 2014). Thus, unlike defects in the secretory pathway caused by other autosomal dominant insulin gene mutations, the preproinsulin cytosolic accumulation caused by R6C/H appears to lead to beta cell failure and diabetes through a completely novel mechanism (Guo et al., 2014).
The cellular defect caused by preproinsulin-L13R has not been experimentally examined. Unlike preproinsulin-R6C/H associated with late-onset diabetes, preproinsulin-L13R causes in utero growth retardation and severe insulin-deficient diabetes on the second day after birth (Hussain et al., 2013). Although the L13R mutation is predicted to affect preproinsulin translocation, the much more severe clinical presentation than that seen with two other signal peptide mutations suggests that additional mechanisms may be involved in beta cell failure and diabetes. Similar to the findings noted above for preproinsulin-R6C, studies of other proteins have shown that blocking SRP-dependent protein translocation induces the expression of heat shock responses, attenuates cell growth, and promotes cell death (Hall et al., 2014; Mutka and Walter, 2001). Since the L13R mutation may affect SRP-dependent translocation, it will be important to determine whether preproinsulin-L13R results in beta cell growth arrest. Similar mutations that disrupt the hydrophobic core of the signal peptide have been reported to cause several human diseases (Jarjanazi et al., 2008). Examples of this include a heterozygous L25R mutation of type V collagen proα1-chain, which results in Ehlers-Danlos syndrome (Chan et al., 2001); a homozygous L15R mutation of bilirubin UDP-glucoronyltransferase (B-UGT), which causes type II Crigler-Najjar disease (Seppen et al., 1996); and a heterozygous C18R mutation of preproparathyroid hormone (preProPTH), which leads to hypoparathyroidism (Arnold et al., 1990). Among these mutants, preProPTH-C18R has been experimentally examined. The preProPTH-C18R exhibits a translocation defect due to an impaired recognition of the mutant signal peptide by SRP (Karaplis et al., 1995). Interestingly, however, the intracellular accumulation of untranslocated preProPTH-C18R is largely colocalized with the ER, inducing ER stress, and promoting cell death (Datta et al., 2007). Reasoning by analogy, it remains possible that preproinsulin-L13R might cause beta cell failure and diabetes through ER stress, with beta cell death – this possibility has yet to be investigated.
5. Perspectives and strategies for preventing diabetes caused by defects in the early steps of insulin biosynthesis
More than 50% of all insulin gene mutations (30 out of 51) cause proinsulin misfolding in the ER and MIDY. Over the past decade, considerable advance has been made to better understand the molecular mechanisms of beta cell failure caused by misfolded proinsulin. This establishes a foundation for developing strategies to delay/prevent the onset of diabetes. As discussed above, although ER stress and beta cell death are a hallmark of MIDY, an earlier event (i.e., before a significant loss of beta cell mass) that contributes to insulin deficiency includes the failure of insulin production from wild-type proinsulin due to its blockade by misfolded mutant proinsulin (Gupta et al., 2010; Hodish et al., 2010; Liu et al., 2007; Renner et al., 2013). Given the fact that one normal INS gene allele is sufficient to produce enough insulin to maintain normoglycemia (Garin et al., 2010; Leroux et al., 2001), strategies that rescue wild-type proinsulin from blockade may therefore potentially restore sufficient insulin production to delay/prevent the onset of insulin-deficient diabetes.
Two distinct approaches might be employed to rescue wild-type proinsulin from blockade by mutant proinsulin. First, because the dominant-negative effect of mutant proinsulin is dose-dependent (Liu et al., 2012; Renner et al., 2013; Wright et al., 2013b), one potential therapeutic strategy for the MIDY syndrome is to target the mutant proinsulin for degradation, so that the stoichiometry of mutant and wild-type proinsulin becomes more favorable to the wild-type – thereby allowing wild-type proinsulin exit from the ER. Compared to the extensive studies of glycoprotein degradation in the secretory pathway (Goldberg, 2003; Hebert and Molinari, 2007; Oda et al., 2003; Smith et al., 2011; Ushioda et al., 2008; Vembar and Brodsky, 2008), the degradation pathway of unglycosylated secretory proteins including proinsulin remains largely unexplored. Although both the ubiquitin-proteasome system and autophagy have been suggested to play roles in degradation of proinsulin in Akita mice (Allen et al., 2004; Bachar-Wikstrom et al., 2013a, 2013b), the key intracellular molecules that regulate degradation of misfolded proinsulin are unknown. Identifying those molecules may provide potential targets to modulate the degradation of misfolded mutant proinsulin and thereby rescue wild-type proinsulin.
The second approach is to target wild-type proinsulin for improved oxidative folding in the ER. Because abnormal inter-molecular disulfide bonds between mutant and wild-type proinsulins contribute to the dominant negative effect of mutant proinsulin (Liu et al., 2010a, 2012), approaches that accelerate the oxidative folding of wild-type proinsulin may promote its folding, limiting its interactions with co-expressed mutant proinsulin, and allowing it to escape from the ER prior to its retention induced by aberrant interaction with the mutant gene products. Protein disulfide isomerase (PDI) and ER oxidoreducin-1 (ERO1) play central roles in maintaining the oxidizing environment to promote disulfide pairing (Bulleid, 2012; Sevier, 2010). Recently, studies show that overexpressing (ERO1) can improve oxidative folding of wild-type proinsulin even when it is co-expressed with MIDY mutants (Liu et al., 2012; Wright et al., 2013a). This results in a significant increase of wild-type proinsulin export from the ER, accompanied by increased insulin production. Mutation of the cysteines in ERO1 that negatively regulate this enzyme (to create a constitutively active oxidase) enhances this effect, while mutation of the catalytic cysteines of ERO1 abolishes most of the rescue, suggesting that enhanced oxidation of proinsulin downstream of ERO1 contributes to proinsulin escape from the ER.
One concern about strategies to enhance ERO1 activity in order to rescue proinsulin export from the ER is that ER hyperoxidation by ERO1 may potentially cause ER stress. However, in cells expressing mutant proinsulin, although overexpressing wild-type or activated ERO1 increases oxidation of the ER lumen, it does not augment ER stress induced by mutant proinsulin – indeed, this maneuver appears to lead to a significant diminution of mutant proinsulin-induced ER stress (Wright et al., 2013a). Decreased ER stress may result from improved oxidative folding of proinsulin with its increased export from the ER. Despite these promising initial studies, the long-term effect of increasing oxidation of the ER lumen on beta cell function still needs to be evaluated in vivo.
Surprisingly, overexpression of PDI does not improve oxidative folding of proinsulin or improve insulin production. As PDI does not mediate rescue of proinsulin, PDI per se might not play a role in native proinsulin disulfide bond formation in vivo (Liu et al., 2012; Rajpal et al., 2012). Of course, there are many other PDI-like ER oxidoreductase family members that might serve this function (Bulleid, 2012; Ellgaard and Ruddock, 2005).
6. Conclusion
The discovery of new insulin gene mutations has stimulated renewed interest in insulin biosynthesis in pancreatic beta cells. These mutants provide unique tools with which to explore the molecular and cellular processes that go awry during the onset and progression of diabetes. Although many questions remain unanswered, we are beginning to get a deeper understanding of the molecular mechanisms of beta cell failure and diabetes caused by defects in the early events of insulin biosynthesis, including preproinsulin targeting and translocation across the ER membrane, signal-peptide cleavage to proinsulin, and the folding of newly made proinsulin molecules in the ER (Fig. 5). Studies are still needed to understand the proinsulin folding pathway in the ER of pancreatic beta cells, because accruing evidence suggests that misfolded proinsulin may be able to “propagate” its misfolding onto “bystander” proinsulin molecules. Factors including the stoichiometry of misfolded and bystander proinsulin forms, the redox environment of the ER lumen, and the activity of ERAD machinery in pancreatic beta cells, are all essential components to the development of novel therapeutic approaches for MIDY. At the same time, a new realization that proinsulin misfolding may occur under pathological conditions in the absence of any mutations, opens the door to the possibility that MIDY may be an extreme example of the kind of ER stress generated in more common forms of type 2 diabetes. Future work is needed to explore these links.
Fig. 5.
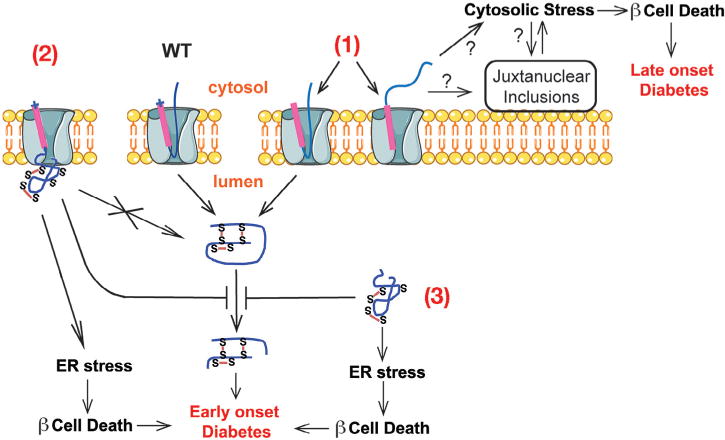
A proposed model of beta cell failure and diabetes caused by the defects in the early events of insulin biosynthesis. The preproinsulin signal peptide mutations in the n-region, i.e. R6C/H, result in a translocation defect of newly synthesized preproinsulin across the ER membrane (1). A certain fraction of untranslocatd preproinsulin relocates and accumulates in the juxtanuclear compartment, activating cytosolic response, promoting beta cell death, and leading to beta cell failure. The preproinsulin mutation at the signal peptide cleavage site, i.e. A24D, impairs normal cleavage of the signal peptide (2). This results in misfolding of downstream proinsulin domain. Twenty-eight reported gene mutations, e.g. C(A7)Y, lead to proinsulin misfolding in the ER (3). Misfolded mutant proinsulins not only induce ER stress and beta cell death, but also block the ER export of co-expressed proinsulin-WT, resulting in decreased insulin production from proinsulin-WT, leading to MIDY. This figure is modified from Guo et al. (2014).
Acknowledgments
This work was supported by NIH RO1-DK088856 (to M.L), NIH RO1-DK-48280 (to P.A.), the research grants from the National Natural Science Foundation of China 81070629 and 81370895 (to M.L), and start-up fund from the University of Michigan Medical School (to M.L). Jinqiu Cui was supported partly by research grants from the Key Project of applied basis research program of Tianjin Scientific and Technical Committee (11JCZDJC18500) and Novo Nordisk β-cell Academy. We acknowledge the support of the Protein Folding Disease Initiative of the University of Michigan. The authors thank Bill and Dee Brehm for helping to establish the Brehm Center for Diabetes Research at the University of Michigan.
References
- Alarcón C, Lincoln B, Rhodes CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J Biol Chem. 1993;268(6):4276–4280. [PubMed] [Google Scholar]
- Alarcon C, Leahy JL, Schuppin GT, Rhodes CJ. Increased secretory demand rather than a defect in the proinsulin conversion mechanism causes hyperproinsulinemia in a glucose-infusion rat model of non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;95(3):1032–1039. doi: 10.1172/JCI117748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JR, Nguyen LX, Sargent KEG, Lipson KL, Hackett A, Urano F. High ER stress in β-cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun. 2004;324(1):166–170. doi: 10.1016/j.bbrc.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. J Clin Invest. 1990;86(4):1084–1087. doi: 10.1172/JCI114811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian RK, Thomas NE, Kaiser ET, Tager HS. [LeuB24]insulin and [AlaB24]insulin: altered structures and cellular processing of B24-substituted insulin analogs. Proc Natl Acad Sci USA. 1982;79(17):5147–5151. doi: 10.1073/pnas.79.17.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachar-Wikstrom E, Wikstrom J, Kaiser N, Cerasi E, Leibowitz G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy. 2013a;9(4):626–628. doi: 10.4161/auto.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, et al. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 2013b;62(4):1227–1237. doi: 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailyes EM, Shennan KI, Seal AJ, Smeekens SP, Steiner DF, Hutton JC, et al. A member of the eukaryotic subtilisin family (PC3) has the enzymic properties of the type 1 proinsulin-converting endopeptidase. Biochem J. 1992;285(2):391–394. doi: 10.1042/bj2850391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbetti F, Raben N, Kadowaki T, Cama A, Accili D, Gabbay KH, et al. Two unrelated patients with familial hyperproinsulinemia due to a mutation substituting histidine for arginine at position 65 in the proinsulin molecule: identification of the mutation by direct sequencing of genomic deoxyribonucleic acid amplified by polymerase chain reaction. J Clin Endocrinol Metab. 1990;71(1):164–169. doi: 10.1210/jcem-71-1-164. [DOI] [PubMed] [Google Scholar]
- Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193(2):383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22(1):487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Bird P, Gething MJ, Sambrook J. The functional efficiency of a mammalian signal peptide is directly related to its hydrophobicity. J Biol Chem. 1990;265(15):8420–8425. [PubMed] [Google Scholar]
- Boesgaard T, Pruhova S, Andersson E, Cinek O, Obermannova B, Lauenborg J, et al. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY) BMC Med Genet. 2010;11(1):42. doi: 10.1186/1471-2350-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfanti R, Colombo C, Nocerino V, Massa O, Lampasona V, Iafusco D, et al. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care. 2009;32(1):123–125. doi: 10.2337/dc08-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulleid NJ. Disulfide bond formation in the mammalian endoplasmic reticulum. Cold Spring Harbor Perspect Biol. 2012;4(11) doi: 10.1101/cshperspect.a013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess TL, Kelly RB. Constitutive and regulated secretion of proteins. Annu Rev Cell Biol. 1987;3(1):243–293. doi: 10.1146/annurev.cb.03.110187.001331. [DOI] [PubMed] [Google Scholar]
- Carroll RJ, Hammer RE, Chan SJ, Swift HH, Rubenstein AH, Steiner DF. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc Natl Acad of Sci USA. 1988;85(23):8943–8947. doi: 10.1073/pnas.85.23.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavener DR, Gupta S, McGrath BC. PERK in beta cell biology and insulin biogenesis. Trends Endocrinol Metab. 2010;21(12):714–721. doi: 10.1016/j.tem.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Ho MSP, Cheah KSE. Aberrant signal peptide cleavage of collagen X in schmid metaphyseal chondrodysplasia. Implications for the molecular basis of the disease. J Biol Chem. 2001;276(11):7992–7997. doi: 10.1074/jbc.M003361200. [DOI] [PubMed] [Google Scholar]
- Chan SJ, Seino S, Gruppuso PA, Schwartz R, Steiner DF. A mutation in the B chain coding region is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc Natl Acad Sci USA. 1987;84(8):2194–2197. doi: 10.1073/pnas.84.8.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J Clin Invest. 2008;118(6):2148–2156. doi: 10.1172/JCI33777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Waheed A, Shah GN, Sly WS. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci USA. 2007;104(50):19989–19994. doi: 10.1073/pnas.0708725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson HW, Hutton JC. The insulin-secretory-granule carboxypeptidase H. Purification and demonstration of involvement in proinsulin processing. Biochem J. 1987;245(2):575–582. doi: 10.1042/bj2450575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debure L, Vayssière JL, Rincheval V, Loison F, Le Dréan Y, Michel D. Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J Cell Sci. 2003;116(15):3109–3121. doi: 10.1242/jcs.00619. [DOI] [PubMed] [Google Scholar]
- Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, et al. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci USA. 1993;90(2):527–531. doi: 10.1073/pnas.90.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson G, Steiner DF. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol. 1998;8(2):189–194. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- Dunn M. Zinc, ligand interactions modulate assembly and stability of the insulin hexamer. Biometals. 2005;18(4):295–303. doi: 10.1007/s10534-005-3685-y. [DOI] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, et al. Insulin mutation screening in 1,044 patients with diabetes. Diabetes. 2008;57(4):1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6(1):28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fred RG, Welsh N. The importance of RNA binding proteins in preproinsulin mRNA stability. Mol Cell Endocrinol. 2009;297:1–2. 28–33. doi: 10.1016/j.mce.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Fricker LD, Evans CJ, Esch FS, Herbert E. Cloning and sequence analysis of cDNA for bovine carboxypeptidase E. Nature. 1986;323(6087):461–464. doi: 10.1038/323461a0. [DOI] [PubMed] [Google Scholar]
- Fujita H, Kida Y, Hagiwara M, Morimoto F, Sakaguchi M. Positive charges of translocating polypeptide chain retrieve an upstream marginal hydrophobic segment from the endoplasmic reticulum lumen to the translocon. Mol Biol Cell. 2010;21(12):2045–2056. doi: 10.1091/mbc.E09-12-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay KH. The insulinopathies. NEJM. 1980;302(3):165–167. doi: 10.1056/NEJM198001173020308. [DOI] [PubMed] [Google Scholar]
- Gabbay KH, DeLuca K, Fisher JN, Mako ME, Rubenstein AH. Familial hyperproinsulinemia. NEJM. 1976;294(17):911–915. doi: 10.1056/NEJM197604222941701. [DOI] [PubMed] [Google Scholar]
- Gabbay KH, Bergenstal RM, Wolff J, Mako ME, Rubenstein AH. Familial hyperproinsulinemia: partial characterization of circulating proinsulin-like material. Proc Natl Acad Sci USA. 1979;76(6):2881–2885. doi: 10.1073/pnas.76.6.2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333(6051):1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin I, Edghill E, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA. 2010;107(7):3105–3110. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German M, Ashcroft S, Docherty K, Edlund H, Edlund T, Goodison S, et al. The insulin gene promoter: a simplified nomenclature. Diabetes. 1995;44(8):1002–1004. doi: 10.2337/diab.44.8.1002. [DOI] [PubMed] [Google Scholar]
- Given BD, Mako ME, Tager HS, Baldwin D, Markese J, Rubenstein AH, et al. Diabetes due to secretion of an abnormal insulin. NEJM. 1980;302(3):129–135. doi: 10.1056/NEJM198001173020301. [DOI] [PubMed] [Google Scholar]
- Goder V, Spiess M. Topogenesis of membrane proteins: determinants and dynamics. FEBS Lett. 2001;504(3):87–93. doi: 10.1016/s0014-5793(01)02712-0. [DOI] [PubMed] [Google Scholar]
- Goder V, Junne T, Spiess M. Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol Biol Cell. 2004;15(3):1470–1478. doi: 10.1091/mbc.E03-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gross DJ, Halban PA, Kahn CR, Weir GC, Villa-Komaroff L. Partial diversion of a mutant proinsulin (B10 aspartic acid) from the regulated to the constitutive secretory pathway in transfected AtT-20 cells. Proc Natl Acad Sci USA. 1989;86(11):4107–4111. doi: 10.1073/pnas.86.11.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruppuso PA, Gorden P, Kahn CR, Cornblath M, Zeller WP, Schwartz R. Familial hyperproinsulinemia due to a proposed defect in conversion of proinsulin to insulin. N Engl J Med. 1984;311(10):629–634. doi: 10.1056/NEJM198409063111003. [DOI] [PubMed] [Google Scholar]
- Guest PC, Bailyes EM, Rutherford NG, Hutton JC. Insulin secretory granule biogenesis. Co-ordinate regulation of the biosynthesis of the majority of constituent proteins. Biochem J. 1991;274(1):73–78. doi: 10.1042/bj2740073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Xiong Y, Witkowski P, Wang L-J, Cui J, Lara-Lemus R, et al. Inefficient translocation of preproinsulin contributes to pancreatic beta cell failure and late onset diabetes. J Biol Chem. 2014;289(23):16290–16302. doi: 10.1074/jbc.M114.562355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, et al. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J Biol Chem. 2013;288(3):1896–1906. doi: 10.1074/jbc.M112.420018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeuptle MT, Flint N, Gough NM, Dobberstein B. A tripartite structure of the signals that determine protein insertion into the endoplasmic reticulum membrane. J Cell Biol. 1989;108(4):1227–1236. doi: 10.1083/jcb.108.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, et al. The pathogenic mechanism of the mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog. 2014;10(4):e1004061. doi: 10.1371/journal.ppat.1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley T, Siva M, Lai E, Teodoro T, Zhang L, Volchuk A. Endoplasmic reticulum stress response in an INS-1 pancreatic beta-cell line with inducible expression of a folding-deficient proinsulin. BMC Cell Biol. 2010;11(1):59. doi: 10.1186/1471-2121-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E, Rapoport TA, Lodish HF. Predicting the orientation of eukaryotic membrane-spanning proteins. Proc Natl Acad Sci USA. 1989;86(15):5786–5790. doi: 10.1073/pnas.86.15.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay CW, Docherty K. Comparative analysis of insulin gene promoters: implications for diabetes research. Diabetes. 2006;55(12):3201–3213. doi: 10.2337/db06-0788. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87(4):1377–1408. doi: 10.1152/physrev.00050. [DOI] [PubMed] [Google Scholar]
- Hendershot L. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71(5):289–297. [PubMed] [Google Scholar]
- Herbach N, Rathkolb B, Kemter E, Pichl L, Klaften M, de Angelis MH, et al. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe {beta}-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56(5):1268–1276. doi: 10.2337/db06-0658. [DOI] [PubMed] [Google Scholar]
- Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, et al. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem. 2010;285(1):685–694. doi: 10.1074/jbc.M109.038042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodish I, Absood A, Liu L, Liu M, Haataja L, Larkin D, et al. In vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes. 2011;60(8):2092–2101. doi: 10.2337/db10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua QX, Mayer JP, Jia W, Zhang J, Weiss MA. The folding nucleus of the insulin superfamily: a flexible peptide model foreshadows the native state. J Biol Chem. 2006a;281(38):28131–28142. doi: 10.1074/jbc.M602616200. [DOI] [PubMed] [Google Scholar]
- Hua QX, Shoelson SE, Inouye K, Weiss MA. Paradoxical structure and function in a mutant human insulin associated with diabetes mellitus. Proc Natl Acad Sci USA. 1993;90(2):582–586. doi: 10.1073/pnas.90.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua QX, Liu M, Hu SQ, Jia W, Arvan P, Weiss MA. A conserved histidine in insulin is required for the foldability of human proinsulin: structure and function of an ALAB5 analog. J Biol Chem. 2006b;281(34):24889–24899. doi: 10.1074/jbc.M602617200. [DOI] [PubMed] [Google Scholar]
- Huang XF, Arvan P. Formation of the insulin-containing secretory granule core occurs within immature beta-granules. J Biol Chem. 1994;269(33):20838–20844. [PubMed] [Google Scholar]
- Huang XF, Arvan P. Intracellular transport of proinsulin in pancreatic β-cells: structural maturation probed by disulfide accessibility. J Biol Chem. 1995;270(35):20417–20423. doi: 10.1074/jbc.270.35.20417. [DOI] [PubMed] [Google Scholar]
- Hussain S, Ali JM, Jalaludin MY, Harun F. Permanent neonatal diabetes due to a novel insulin signal peptide mutation. Pediatr Diabetes. 2013 doi: 10.1111/pedi.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino T, Takahashi M, Sako T. Role of amino-terminal positive charge on signal peptide in staphylokinase export across the cytoplasmic membrane of Escherichia coli. J Biol Chem. 1987;262(15):7412–7417. [PubMed] [Google Scholar]
- Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature. 1980;283:100–102. doi: 10.1038/283100a0. [DOI] [PubMed] [Google Scholar]
- Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the akita diabetic mouse. Diabetes. 2003;52(2):409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465(7297):507–510. doi: 10.1038/nature08870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarjanazi H, Savas S, Pabalan N, Dennis JW, Ozcelik H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins Struct Funct Genet. 2008;70(2):394–403. doi: 10.1002/prot.21548. [DOI] [PubMed] [Google Scholar]
- Johnson N, Vilardi F, Lang S, Leznicki P, Zimmermann R, High S. TRC40 can deliver short secretory proteins to the Sec61 translocon. J Cell Sci. 2012;125(15):3612–3620. doi: 10.1242/jcs.102608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Powis K, High S. Post-translational translocation into the endoplasmic reticulum. Biochim Biophys Acta Mol Cell Res. 2013;1833(11):2403–2409. doi: 10.1016/j.bbamcr.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454(7208):1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkar V, Prins L, LKampinga H. DNAJ proteins and protein aggregation diseases. Curr Top Med Chem. 2012;12(22):2479–2490. doi: 10.2174/1568026611212220004. [DOI] [PubMed] [Google Scholar]
- Karaplis AC, Lim SK, Baba H, Arnold A, Kronenberg HM. Inefficient membrane targeting, translocation, and proteolytic processing by signal peptidase of a mutant preproparathyroid hormone protein. J Biol Chem. 1995;270(4):1629–1635. doi: 10.1074/jbc.270.4.1629. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, et al. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179(1):75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocik L, Junne T, Spiess M. Orientation of internal signal-anchor sequences at the Sec61 translocon. J Mol Biol. 2012;424(5):368–378. doi: 10.1016/j.jmb.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Kuliawat R, Arvan P. Protein targeting via the “constitutive-like” secretory pathway in isolated pancreatic islets: passive sorting in the immature granule compartment. J Cell Biol. 1992;118(3):521–529. doi: 10.1083/jcb.118.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkaraju AKK, Thankappan R, Mary C, Garrison JL, Taunton J, Strub K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol Biol Cell. 2012;23(14):2712–2722. doi: 10.1091/mbc.E12-03-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibiger B, Wåhlander K, Berggren PO, Leibiger IB. Glucose-stimulated insulin biosynthesis depends on insulin-stimulated insulin gene transcription. J Biol Chem. 2000;275(39):30153–30156. doi: 10.1074/jbc.M005216200. [DOI] [PubMed] [Google Scholar]
- Leroux L, Desbois P, Lamotte L, Duvillié B, Cordonnier N, Jackerott M, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes. 2001;50(90001):S150–S153. doi: 10.2337/diabetes.50.2007.s150. [DOI] [PubMed] [Google Scholar]
- Liu CY, Kaufman RJ. The unfolded protein response. J Cell Sci. 2003;116(10):1861–1862. doi: 10.1242/jcs.00408. [DOI] [PubMed] [Google Scholar]
- Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem. 2005;280(14):13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA. 2007;104(40):15841–15846. doi: 10.1073/pnas.0702697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wan ZL, Chu YC, Aladdin H, Klaproth B, Choquette M, et al. Crystal structure of a “nonfoldable” insulin impaired folding efficiency despite native activity. J Biol Chem. 2009;284(50):35259–35272. doi: 10.1074/jbc.M109.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Haataja L, Wright J, Wickramasinghe NP, Hua QX, Phillips NF, et al. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE. 2010a;5(10):e13333. doi: 10.1371/journal.pone.0013333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010b;21(11):652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hua QX, Hu SQ, Jia W, Yang Y, Saith SE, et al. Deciphering the hidden informational content of protein sequences. J Biol Chem. 2010c;285(40):30989–31001. doi: 10.1074/jbc.M110.152645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lara-Lemus R, Shan SO, Wright J, Haataja L, Barbetti F, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes. 2012 doi: 10.2337/db11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wright J, Guo H, Xiong Y, Arvan P. Chapter two – proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. In: Gerald L, editor. Vitamins & Hormones. Academic Press; 2014. pp. 35–62. [DOI] [PubMed] [Google Scholar]
- Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GKW, Smith BJ, et al. How insulin engages its primary binding site on the insulin receptor. Nature. 2013;493(7431):241–245. doi: 10.1038/nature11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meur G, Simon A, Harun N, Virally M, Dechaume AL, Bonnefond AL, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59(3):653–661. doi: 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesbauer M, Rambold AS, Winklhofer KF, Tatzelt J. Targeting of the prion protein to the cytosol: mechanisms and consequences. Curr Issues Mol Biol. 2010;12:109–118. [PubMed] [Google Scholar]
- Molven A, Ringdal M, Nordbã AM, Rã¦der H, Stã¦y J, Lipkind GM, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57(4):1131–1135. doi: 10.2337/db07-1467. [DOI] [PubMed] [Google Scholar]
- Moritani M, Yokota I, Tsubouchi K, Takaya R, Takemoto K, Minamitani K, et al. Identification of INS and KCNJ11 gene mutations in type 1B diabetes in Japanese children with onset of diabetes before 5 yr of age. Pediatr Diabetes. 2013;14(2):112–120. doi: 10.1111/j.1399-5448.2012.00917.x. [DOI] [PubMed] [Google Scholar]
- Mutka SC, Walter P. Multifaceted physiological response allows yeast to adapt to the loss of the signal recognition particle-dependent protein-targeting pathway. Mol Biol Cell. 2001;12(3):577–588. doi: 10.1091/mbc.12.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299(5611):1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- Oohashi H, Ohgawara H, Nanjo K, Tasaka Y, Cao QP, Chan SJ, et al. Familial hyperproinsulinemia associated with NIDDM. A case study. Diabetes Care. 1993;16(10):1340–1346. doi: 10.2337/diacare.16.10.1340. [DOI] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Amherdt MN, Madsen O, Vassalli JD, Perrelet A. Direct identification of prohormone conversion site in insulin-secreting cells. Cell. 1985;42(2):671–681. doi: 10.1016/0092-8674(85)90124-2. [DOI] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Amherdt M, Madsen O, Perrelet A, Vassalli JD, et al. Conversion of proinsulin to insulin occurs coordinately with acidification of maturing secretory vesicles. J Cell Biol. 1986;103(6):2273–2281. doi: 10.1083/jcb.103.6.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk Mehmet A, Kurtoglu S, Bastug O, Korkmaz L, Daar G, Memur S, et al. Neonatal diabetes in an infant of diabetic mother: same novel INS missense mutation in the mother and her offspring. J Pediatr Endocrinol Metab. 2014;745 doi: 10.1515/jpem-2013-0285. [DOI] [PubMed] [Google Scholar]
- Park SY, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391(3):1449–1454. doi: 10.1016/j.bbrc.2009.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks GD, Lamb RA. Topology of eukaryotic type II membrane proteins: importance of N-terminal positively charged residues flanking the hydrophobic domain. Cell. 1991;64(4):777–787. doi: 10.1016/0092-8674(91)90507-u. [DOI] [PubMed] [Google Scholar]
- Peterson JH, Woolhead C, Bernstein HD. Basic amino acids in a distinct subset of signal peptides promote interaction with the signal recognition particle. J Biol Chem. 2003;278(46):46155–46162. doi: 10.1074/jbc.M309082200. [DOI] [PubMed] [Google Scholar]
- Pilon M, Romisch K, Quach D, Schekman R. Sec61p serves multiple roles in secretory precursor binding and translocation into the endoplasmic reticulum membrane. Mol Biol Cell. 1998;9(12):3455–3473. doi: 10.1091/mbc.9.12.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 1998;94(6):795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- Polak M, Dechaume A, Cavé H, Nimri R, Crosnier H, Sulmont V, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy. Diabetes. 2008;57(4):1115–1119. doi: 10.2337/db07-1358. [DOI] [PubMed] [Google Scholar]
- Quinn D, Orci L, Ravazzola M, Moore H. Intracellular transport and sorting of mutant human proinsulins that fail to form hexamers. J Cell Biol. 1991;113(5):987–996. doi: 10.1083/jcb.113.5.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raile K, O’Connell M, Galler A, Werther G, Khnen P, Krude H, et al. Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur J Endocrinol. 2011;165(2):255–260. doi: 10.1530/EJE-11-0208. [DOI] [PubMed] [Google Scholar]
- Rajan S, Eames SC, Park SY, Labno C, Bell GI, Prince VE, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab. 2010;298(3):E403–E410. doi: 10.1152/ajpendo.00592.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal G, Schuiki I, Liu M, Volchuk A, Arvan P. Action of protein disulfide isomerase on proinsulin exit from endoplasmic reticulum of pancreatic β cells. J Biol Chem. 2012;287(1):43–47. doi: 10.1074/jbc.C111.279927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol. 2010;188(4):515–526. doi: 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450(7170):663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- Reiner T, Thurber G, Gaglia J, Vinegoni C, Liew CW, Upadhyay R, et al. Accurate measurement of pancreatic islet b-cell mass using a second-generation fluorescent exendin-4 analog. Proce Natl Acad Sci USA. 2011;108(31):12815–12820. doi: 10.1073/pnas.1109859108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner S, Braun-Reichhart C, Blutke A, Herbach N, Emrich D, Streckel E, et al. Permanent neonatal diabetes in INSC94Y transgenic pigs. Diabetes. 2013;62(5):1505–1511. doi: 10.2337/db12-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes CJ, Halban PA. Newly synthesized proinsulin/insulin and stored insulin are released from pancreatic B cells predominantly via a regulated, rather than a constitutive, pathway. J Cell Biol. 1987;105(1):145–153. doi: 10.1083/jcb.105.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109(4):443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–787. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Edghill E, Argente J, Hattersley A. Testing for monogenic diabetes among children and adolescents with antibody-negative clinically defined Type 1 diabetes. Diabetic Med. 2009;26(10):1070–1074. doi: 10.1111/j.1464-5491.2009.02812.x. [DOI] [PubMed] [Google Scholar]
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14(10):630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Tomiyoshi R, Kuroiwa T, Mihara K, Omura T. Functions of signal and signal-anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc Natl Acad Sci USA. 1992;89(1):16–19. doi: 10.1073/pnas.89.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Iwamoto Y, Sakamoto Y, Kuzuya T, Hirata H. Structurally abnormal insulin in a diabetic patient. Characterization of the mutant insulin A3 (Val—Leu) isolated from the pancreas. J Clin Invest. 1986;78(6):1666–1672. doi: 10.1172/JCI112760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Matsuyama S, Mizushima S. In vitro kinetic analysis of the role of the positive charge at the amino-terminal region of signal peptides in translocation of secretory protein across the cytoplasmic membrane in Escherichia coli. J Biol Chem. 1990;265(8):4358–4363. [PubMed] [Google Scholar]
- Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15(1):95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- Schmitz A, Maintz M, Kehle T, Herzog V. In vivo iodination of a misfolded proinsulin reveals co-localized signals for Bip binding and for degradation in the ER. EMBO J. 1995;14(6):1091–1098. doi: 10.1002/j.1460-2075.1995.tb07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppen J, Steenken E, Lindhout D, Bosma PJ, Oude Elferink RPJ. A mutation which disrupts the hydrophobic core of the signal peptide of bilirubin UDP-glucuronosyltransferase, an endoplasmic reticulum membrane protein, causes Crigler-Najjar type IIs. FEBS Lett. 1996;390(3):294–298. doi: 10.1016/0014-5793(96)00677-1. [DOI] [PubMed] [Google Scholar]
- Sevier CS. New insights into oxidative folding. J Cell Biol. 2010;188(6):757–758. doi: 10.1083/jcb.201002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Hegde RS. A calmodulin-dependent translocation pathway for small secretory proteins. Cell. 2011;147(7):1576–1588. doi: 10.1016/j.cell.2011.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Stein R. Glucose-induced transcription of the insulin gene is mediated by factors required for beta-cell-type-specific expression. Mol Cell Biol. 1994;14(2):871–879. doi: 10.1128/mcb.14.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol. 2005;25(3):921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson S, Fickova M, Haneda M, Nahum A, Musso G, Kaiser E, et al. Identification of a mutant human insulin predicted to contain a serine-for-phenylalanine substitution. Proc Natl Acad Sci USA. 1983a;80(24):7390–7394. doi: 10.1073/pnas.80.24.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Inouye K, et al. Three mutant insulins in man. Nature. 1983b;302(5908):540–543. doi: 10.1038/302540a0. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Polonsky K, Zeidler A, Rubenstein AH, Tager HS. Human insulin B24 (Phe—Ser). Secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J Clin Invest. 1984;73(5):1351–1358. doi: 10.1172/JCI111338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohma Y, Hua QX, Liu M, Phillips NB, Hu SQ, Whittaker J, et al. Contribution of residue B5 to the folding and function of insulin and IGF-I: constraints and fine-tuning in the evolution of a protein family. J Biol Chem. 2010;285(7):5040–5055. doi: 10.1074/jbc.M109.062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner D. On the discovery of precursor processing. In: Mbikay M, Seidah NG, editors. Proprotein Convertases. Humana Press; 2011. pp. 3–11. [Google Scholar]
- Steiner D, Oyer P. The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc Natl Acad Sci USA. 1967;57(2):473–480. doi: 10.1073/pnas.57.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2(1):31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence fora precursor. Science. 1967;157(3789):697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- Steiner DF, Quinn PS, Chan SJ, Marsh J, Tager HS. Processing mechanisms in the biosynthesis of proteins. Ann N Y Acad Sci. 1980;343(1):1–16. doi: 10.1111/j.1749-6632.1980.tb47238.x. [DOI] [PubMed] [Google Scholar]
- Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990;13(6):600–609. doi: 10.2337/diacare.13.6.600. [DOI] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA. 2007;104(38):15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Støy J, Steiner D, Park SY, Ye H, Philipson L, Bell G. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11(3):205–215. doi: 10.1007/s11154-010-9151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A, Wang GS, Reimer R, Finegood DT, Scott FW. Pronounced cytosolic aggregation of cellular prion protein in pancreatic [beta]-cells in response to hyperglycemia. Lab Invest. 2006;87(2):139–149. doi: 10.1038/labinvest.3700500. [DOI] [PubMed] [Google Scholar]
- Tager H. Lilly lecture 1983. Abnormal products of the human insulin gene. Diabetes. 1984;33(7):693–699. doi: 10.2337/diab.33.7.693. [DOI] [PubMed] [Google Scholar]
- Tager H, Given B, Baldwin D, Mako M, Markese J, Rubenstein A, et al. A structurally abnormal insulin causing human diabetes. Nature. 1979;13(281(5727)):122–125. doi: 10.1038/281122a0. [DOI] [PubMed] [Google Scholar]
- Tillmar L, Carlsson C, Welsh N. Control of insulin mRNA stability in rat pancreatic islets: regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J Biol Chem. 2002;277(2):1099–1106. doi: 10.1074/jbc.M108340200. [DOI] [PubMed] [Google Scholar]
- Ulmer JB, Palade GE. Anomalies in the translocation and processing of glycophorin precursors in murine erythroleukemia cells. J Biol Chem. 1989a;264(2):1084–1091. [PubMed] [Google Scholar]
- Ulmer JB, Palade GE. Targeting and processing of glycophorins in murine erythroleukemia cells: use of brefeldin A as a perturbant of intracellular traffic. Proc Natl Acad Sci USA. 1989b;86(18):6992–6996. doi: 10.1073/pnas.86.18.6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321(5888):569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Net N-C charge imbalance may be important for signal sequence function in bacteria. J Mol Biol. 1986;192(2):287–290. doi: 10.1016/0022-2836(86)90365-7. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 1989;341(6241):456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinik A, Bell G. Mutant insulin syndromes. Horm Metab Res. 1988;20(1):1–10. doi: 10.1055/s-2007-1010736. [DOI] [PubMed] [Google Scholar]
- Viswanathan J, Haapasalo A, Böttcher C, Miettinen R, Kurkinen KMA, Lu A, et al. Alzheimer’s disease-associated ubiquilin-1 regulates presenilin-1 accumulation and aggresome formation. Traffic. 2011;12(3):330–348. doi: 10.1111/j.1600-0854.2010.01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchuk A, Ron D. The endoplasmic reticulum stress response in the pancreatic β-cell. Diabetes Obes Metab. 2010;12:48–57. doi: 10.1111/j.1463-1326.2010.01271.x. [DOI] [PubMed] [Google Scholar]
- Wahlberg JM, Spiess M. Multiple determinants direct the orientation of signal, Äìanchor proteins: the topogenic role of the hydrophobic signal domain. J Cell Biol. 1997;137(3):555–562. doi: 10.1083/jcb.137.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103(1):27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ye R, Barron E, Baumeister P, Mao C, Luo S, et al. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2009;17(3):488–498. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lee J, Di Jeso B, Treglia AS, Comoletti D, Dubi N, et al. Cis and trans actions of the cholinesterase-like domain within the thyroglobulin dimer. J Biol Chem. 2010;285(23):17564–17573. doi: 10.1074/jbc.M110.111641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren-Perry MG, Manley SE, Ostrega D, Polonsky K, Mussett S, Brown P, et al. A novel point mutation in the insulin gene giving rise to hyperproinsulinemia. J Clin Endocrinol Metab. 1997;82(5):1629–1631. doi: 10.1210/jcem.82.5.3914. [DOI] [PubMed] [Google Scholar]
- Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem. 2009;284(29):19159–19163. doi: 10.1074/jbc.R109.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicksteed B, Herbert TP, Alarcon C, Lingohr MK, Moss LG, Rhodes CJ. Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J Biol Chem. 2001;276(25):22553–22558. doi: 10.1074/jbc.M011214200. [DOI] [PubMed] [Google Scholar]
- Wright J, Birk J, Haataja L, Liu M, Ramming T, Weiss MA, et al. Endoplasmic reticulum oxidoreductin-1α (Ero1α) improves folding and secretion of mutant proinsulin and limits mutant proinsulin-induced endoplasmic reticulum stress. J Biol Chem. 2013a;288(43):31010–31018. doi: 10.1074/jbc.M113.510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J, Wang X, Haataja L, Kellogg AP, Lee J, Liu M, et al. Dominant protein interactions that influence the pathogenesis of conformational diseases. J Clin Invest. 2013b;123(7):3124–3134. doi: 10.1172/JCI67260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Hu SQ, Chu Y-C, Huang K, Nakagawa SH, Whittaker J, et al. Diabetes-associated mutations in insulin: consecutive residues in the B chain contact distinct domains of the insulin receptor. Biochemistry. 2004;43(26):8356–8372. doi: 10.1021/bi0497796. [DOI] [PubMed] [Google Scholar]
- Yano H, Kitano N, Morimoto M, Polonsky K, Imura H, Seino Y. A novel point mutation in the human insulin gene giving rise to hyperproinsulinemia (proinsulin Kyoto) J Clin Invest. 1992;89(6):1902–1907. doi: 10.1172/JCI115795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M, Kayo T, Ikeda T, Koizumi A. A Novel Locus, Mody4, Distal to D7Mit189 on Chromosome 7 Determines Early-Onset NIDDM in Nonobese C57BL/6 (Akita) Mutant Mice. (Non-Insulin-Dependent Diabetes Mellitus), (Non-Insulin-Dependent Diabetes Mellitus) American Diabetes Association. 1997:887–888. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Eyrisch S, Ahmad M, Helms V. Protein translocation across the ER membrane. Biochim Biophys. Acta Biomembranes. 2011;1808(3):912–924. doi: 10.1016/j.bbamem.2010.06.015. [DOI] [PubMed] [Google Scholar]