

Summary
Synapsin III (SynIII) is a phosphoprotein that is highly expressed at early stages of neuronal development. Whereas in vitro evidence suggests a role for SynIII in neuronal differentiation, in vivo evidence is lacking. Here, we demonstrate that in vivo downregulation of SynIII expression affects neuronal migration and orientation. By contrast, SynIII overexpression affects neuronal migration, but not orientation. We identify a cyclin-dependent kinase-5 (CDK5) phosphorylation site on SynIII and use phosphomutant rescue experiments to demonstrate its role in SynIII function. Finally, we show that SynIII phosphorylation at the CDK5 site is induced by activation of the semaphorin-3A (Sema3A) pathway, which is implicated in migration and orientation of cortical pyramidal neurons (PNs) and is known to activate CDK5. Thus, fine-tuning of SynIII expression and phosphorylation by CDK5 activation through Sema3A activity is essential for proper neuronal migration and orientation.
Graphical Abstract
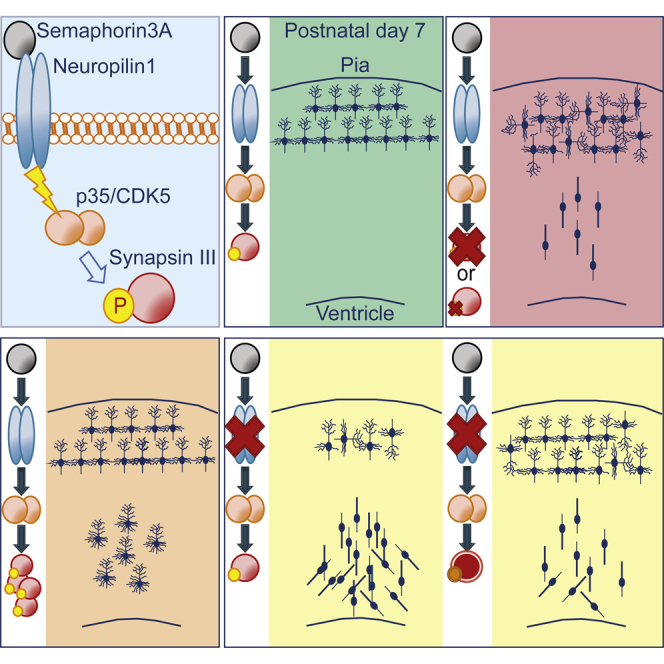
Highlights
-
•
Precise regulation of SynIII expression is essential during brain development
-
•
SynIII regulates neuronal migration, orientation, and morphological maturation
-
•
SynIII acts downstream of the Sema3A pathway, which involves NP1 and kinase CDK5
-
•
Phosphorylation of SynIII by CDK5 on Ser404 is essential for SynIII function
During brain development, newborn pyramidal neurons radially migrate to shape the neocortex. Perlini et al. show that the neuronal phosphoprotein Synapsin III (SynIII) is part of the Sema3A/NP1/CDK5 pathway and contributes to the regulation of proper neuronal migration and orientation/morphological maturation of cortical neurons.
Introduction
The synapsin (Syn) family consists of three neuronal phosphoproteins encoded in mammals by distinct genes (i.e., SynI, II, and III). Although the biology of SynI and II has been extensively studied, the functions of SynIII are still largely uncharacterized. In vitro studies show that SynIII is involved in axonal elongation and growth-cone formation during early neurodevelopment (Feng et al., 2002; Ferreira et al., 2000). Accordingly, SynIII is the earliest expressed Syn isoform during development (Porton et al., 1999, 2004). Moreover, single-nucleotide polymorphisms in SynIII have been linked to neurodevelopmental disorders (i.e., schizophrenia; Chen et al., 2009).
Similar to the other family members, SynIII is a substrate for protein kinases (PKs) (Cesca et al., 2010). Remarkably, many pathways that are essential for the migration and lamination of cortical neurons during brain development involve PKs and lead to the phosphorylation of specific substrates (Ayala et al., 2007). Thus, the developmental expression of SynIII, its role in neuronal developmental processes in vitro, and its phosphorylation profile suggest that it may be a downstream effector in neuronal migration.
Here, we showed that SynIII is involved in neocortical development in vivo; specifically, both the knockdown (KD) of SynIII and its genetic deletion lead to defective radial migration and orientation of layer II/III pyramidal neurons (PNs). Proper development requires SynIII phosphorylation by cyclin-dependent kinase-5 (CDK5), putting SynIII downstream of the semaphorin-3A (Sema3A)-signaling cascade.
Results
SynIII Expression Is Required for Radial Migration of PNs In Vivo
To investigate the role of SynIII in cortical development, we first confirmed its expression in the rat brain cortex at developmental stages (Supplemental Results; Figures S1A–S1F). Subsequently, we designed two short-hairpin (sh)RNAs against SynIII—but not SynI/II (Figures S2A and S2B)—to examine the effects of SynIII KD on the radial migration of newly generated cortical PNs in vivo. Using in utero electroporation (IUE) at embryonic day 17 (E17), we expressed active shRNAs (shRNA no. 1 and shRNA no. 2) or a control scrambled shRNA vector (shRNAscr; Figures S2A and S2B) in a subpopulation of neural progenitors that would normally migrate to layer II/III of the somatosensory cortex (dal Maschio et al., 2012). We analyzed the radial migration of layer II/III PNs derived from shRNA+ progenitors at E21 and postnatal day 7 (P7) (the first and last time points during the peak of SynIII expression, respectively) as well as at P14 (the first time point at which SynIII expression begins to be endogenously downregulated; see Figures S1A and S1B).
At E21, control shRNAscr+ cells were primarily found in the cortical plate (CP) and intermediate zone (IZ), whereas only few cells remained in the ventricular zone/subventricular zone (VZ/SVZ) (Figures 1A and 1B). The migrating PNs located at the IZ expressed SynIII at E21 (Figures S1G and S1H). Interestingly, we observed a significant delay in radial migration when either shRNA no. 1 or no. 2 was electroporated (Figures 1A and 1B). As the effect on migration was larger in shRNA no. 1 experiments, we performed all subsequent experiments with this construct (shRNA onward).
Figure 1.

SynIII KD Affects Radial Migration and Neuronal Orientation In Vivo
(A) Confocal images of rat somatosensory cortices at E21 after IUE at E17 with control scrambled shRNA (shRNAscr, control) or SynIII shRNA no. 1. The scale bar represents 100 μm.
(B) Quantification of the average number of PNs transfected with shRNAscr (control), shRNA no. 1, or shRNA no. 2 and residing at the VZ/SVZ, IZ, or CP (marked in A). The data are mean percentages (±SEM) of the total number of fluorescent cells (one-way ANOVA; post hoc Holm-Sidak: ∗∗∗p < 0.001).
(C) Confocal images of rat somatosensory cortices at P7 after IUE at E17 with control scrambled shRNA (shRNAscr, control) or SynIII shRNA no. 1. Arrowheads: sample neurons that did not complete radial migration at P7 (black, UP neurons; gray, IN neurons). The scale bar represents 50 μm.
(D) Schematics of apical dendrite orientation used for classification in (E)–(G). HOR, horizontal orientation; IN, inverted orientation; UR, upright orientation.
(E) High-magnification image of the boxed region in (C). The scale bar represents 50 μm.
(F) Reconstruction of representative control and shRNA+ cells in the CP. The scale bar represents 50 μm.
(G) Quantification of the distribution of CP cells in the three distinct classes, as in (D) and (F) (Student's t test; ∗∗∗p < 0.001).
(H) Reconstruction of representative control and shRNA+ cells in ectopic positions. The scale bar represents 50 μm.
(I) Quantification of the number of control or shRNA+ cells that did not complete their migration, as in (H) (Student's t test; ∗∗∗p < 0.001).
(J) Quantification of the number of ectopic control or shRNA+ cells that displayed inverted orientation, as in (H). The data in (G)–(J) are mean percentages (±SEM) of the total number of ectopic cells (Student's t test; ∗∗∗p < 0.001).
(K) Quantification of the percentage of shRNA+ cells that did not complete migration and stopped in the upper or lower half of deep cortical layers. The pie histogram indicates the mean percentage of IN or UP cells observed in the two groups (Student’s t test; ∗∗∗p < 0.001).
See Figures S1–S4.
At P7, almost all shRNAscr+ cells reached cortical layer II/III, whereas many shRNA+ cells were misplaced in deep layers (Figures 1C and 1I). We defined these cells as “ectopic” PNs (Figure 1H). The severity of misplacement was proportional to the severity of SynIII downregulation (Supplemental Results; Figure S2E). To investigate whether the effect of SynIII KD was long lasting, we examined neuronal migration at P14, when endogenous expression of SynIII is low (see Figures S1A and S1B). We found that SynIII shRNA+ cortices exhibited ectopic cells at P7 (Figures 1C, 1H, and 1I) and at P14 (Figures S3D and S3E). SynIII downregulation did not affect neuronal identity, proliferation, or radial glia (RG) scaffold organization (Supplemental Results; Figures S2C, S2D, and S2F).
SynIII Regulates the Orientation of PNs In Vivo
Cell migration, orientation, and polarization share molecular mechanisms, and often the same proteins are involved in all three processes (Chen et al., 2008; Polleux et al., 2000; Shelly et al., 2011). To further explore the role of SynIII in cortical development in vivo, we investigated whether its KD affected the orientation of PNs in the layer II/III. Control cells were typically characterized by an upright (UR) orientation with a pyramid-shaped cell body, one thick apical dendrite directed toward the pial surface, and one axon pointing to the ventricle. Interestingly, ∼20% of SynIII shRNA+ PNs were misoriented in layer II/III (Figure 1D; Polleux et al., 2000), ∼10% of PNs were inverted (IN), and ∼10% of PNs were horizontal (HOR) (Figures 1F and 1G). Similar results were obtained at P14 (Figures S3A–S3C).
Given the common mechanisms involved in migration and orientation (Chen et al., 2008), we expected a larger effect on the orientation of ectopic cells. Indeed, ∼1/3 of ectopic cells depleted of SynIII were IN (Figures 1E and 1H–1J). When we divided the cortical region where the ectopic cells were located (from layer IV to layer VI) in two equal parts, we found that, whereas ∼65% of the ectopic cells were located in the upper half, the IN ectopic cells were evenly distributed in the two halves, suggesting that the cortical location and neuronal orientation were not causally linked (Figure 1K). Moreover, when we immunostained against the γ-tubulin centrosomal marker, we found that the centrosome colocalized with the apical dendrite regardless of cell orientation in both shRNAscr+ and SynIII shRNA+ cells (Figure S4A). Whereas SynIII regulated the morphological maturation of PNs both in the axonal and dendritic compartments, it did not affect the basic electrophysiological proprieties of PNs (Supplemental Results; Figures S3F–S3L and S4C–S4E).
Finally, we investigated whether SynIII KD also affected the orientation and migration of PNs in deep cortical layers (i.e., IV/V) by performing IUE at earlier embryonic stages (E15.5). In P7 SynIII shRNA-transfected brains, we found migration defects and significant cell misorientation similar to that observed in superficial layers (Figure S4B).
The Co-expression of shRNA-Resistant SynIII Rescues the Defects Associated with Endogenous SynIII KD
To exclude off-target effects (De Paula et al., 2007), we generated a shRNA-resistant SynIII cDNA with four silent mutations in the shRNA target sequence (rSynIII; Figure 2A). After confirming rSynIII insensitivity to shRNA (Figure 2B), we performed rescue experiments with rSynIII/shRNA co-expression in E17 embryos. The co-expression of rSynIII and shRNA at a ratio of 0.5:1 rescued the delay in radial migration at E21 (Figures 2C and 2D) as well as the number and orientation of ectopic cells (Figures 2E–2G); however, the phenotype of HOR and IN cells in the CP at P7 was not rescued (Figures 2H and 2I). It is possible that the lack of rescue in the orientation phenotype in the CP was due to the concentration gradient of SynIII expression across the cortex, which shows the highest levels in the CP (Figures S1C and S1F). Indeed, by increasing the SynIII/shRNA ratio to 1:1, we achieved complete rescue of the orientation defect of layer II/III PNs (Figures 2H and 2I).
Figure 2.

The Expression of shRNA-Resistant SynIII Rescues the shRNA-Dependent Defects in Neuronal Migration and Orientation
(A) Illustration showing the silent mutations in the shRNA-resistant variant of SynIII (rSynIII).
(B) Western blot showing insensitivity of rSynIII versus sensitivity of WT SynIII to shRNA. The proteins were exogenously expressed in COS7 cells.
(C) Confocal images of rat cortical coronal sections at E21 from animals transfected with shRNAscr (control), shRNA, or rSynIII/shRNA 0.5:1. The scale bar represents 100 μm.
(D) Quantification of the number of PNs transfected with shRNAscr (control), rSynIII/shRNA, or shRNA and residing at the VZ/SVZ, IZ, and CP. The data are mean percentages (±SEM) of the total number of fluorescent cells. ANOVA on ranks; post hoc Dunn’s test; ∗∗∗p < 0.001.
(E) Confocal images of rat cortical coronal sections at P7 from animals transfected with shRNAscr (control) or rSynIII/shRNA 0.5:1. The scale bar represents 100 μm.
(F) Quantification of the number of shRNAscr, rSynIII/shRNA (0.5:1 ratio), or shRNA+ PNs that did not complete migration (one-way ANOVA; post hoc Tukey’s test; ∗∗p < 0.01; ∗∗∗p < 0.001).
(G) Quantification of the number of ectopic PNs that displayed inverted orientation in shRNAscr, rSynIII/shRNA (0.5:1 ratio), and shRNA-treated cortices. The data are mean percentages (±SEM) of the total number of ectopic cells (one-way ANOVA; post hoc Tukey’s test; ∗∗∗p < 0.001).
(H) Confocal images of rat cortical coronal sections at P7 from animals transfected with rSynIII/shRNA 0.5:1 or rSynIII/shRNA 1:1. The scale bar represents 50 μm.
(I) Quantification of the distribution of CP cells in the three orientation classes shown in Figure 1D in animals transfected with shRNAscr, rSynIII/shRNA (0.5:1 ratio), rSynIII/shRNA (1:1 ratio), or shRNA. The data are mean percentages (±SEM) of the total number of fluorescent CP cells (one-way ANOVA; post hoc Tukey’s test; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
See Figure S6.
SynIII KO Mice Display a Cortical Phenotype Similar to SynIII shRNA-Electroporated Animals
Although SynIII knockout (KO) mice display behavioral defects and changes in synaptic transmission, no gross brain abnormalities have been described (Feng et al., 2002; Porton et al., 2010). Nevertheless, no study has specifically investigated whether subtler developmental defects occur in the SynIII KO somatosensory cortex.
To address this issue, we first performed immunostaining with layer-specific markers (i.e., CUX1, layer II/III/IV; CTIP2, layer V; FOXP2, layer VI) in somatosensory cortex slices of SynIII KO and control littermates. We did not observe any overt abnormality in cortical layering at P7 in SynIII KO mice (Figure S5). Thus, we considered two possible scenarios: (1) given the high homology among Syn isoforms, other members of the family, such as SynI, which is also expressed at embryonic ages, may compensate for SynIII’s absence during development and (2) immunostaining with layer-specific markers may not be sufficiently sensitive to detect mild migration/orientation defects.
To investigate possibility (1), we performed co-electroporation of SynI with SynIII shRNA and assessed the migration/orientation defect in P7 mouse slices. We found that SynI could not rescue SynIII shRNA effects, indicating that SynI cannot compensate for loss of SynIII-specific functions during development (Figures 3A–3D). To investigate possibility (2), we performed IUE with a pCAGGs-IRES-tdTomato (Tom) vector to visualize migrating PNs in E15.5 SynIII KO and wild-type (WT) mice. In agreement with the cortical phenotype of shRNA-electroporated animals, we observed a misorientation of an ∼20% of PNs at the level of the CP in all KO brains (Figures 3E–3G). Interestingly, in contrast to controls, in two out of seven KO animals analyzed, we observed PNs in ectopic positions, with these cells occasionally exhibiting IN orientation (Figures 3E and 3H).
Figure 3.

SynIII KO Mice Exhibit a Cortical Migration Phenotype Similar to SynIII-KD Animals
(A) Confocal images of coronal sections of rat somatosensory cortices at P7 after IUE at E17 with shRNA or SynI/shRNA. Arrowheads, example PNs that did not complete radial migration at P7 (black arrowheads, UP neurons; gray arrowheads, IN neurons). The scale bar represents 100 μm.
(B) Quantification of the distribution of CP cells in the three distinct classes as in Figure 1D.
(C) Quantification of the number of shRNA+ or SynI/shRNA+ cells that did not complete migration in experiments, as in (A).
(D) Quantification of the number of ectopic shRNA+ or SynI/shRNA+ cells that displayed inverted orientation, as in (A). The data in (B)–(D) are mean percentages (±SEM) of the total number of ectopic cells.
(E) Confocal images of coronal sections of somatosensory cortices from WT or SynIII KO C57BL/6 mice at P7, after IUE at E15.5 with tdTomato. The arrowheads point to example PNs that did not complete radial migration. The scale bar represents 100 μm.
(F) Quantification of the distribution of cells in the three classes as in Figure 1D in WT or KO animals. The data are mean percentages (±SEM) of the total number of CP fluorescent cells (Student’s t test: ∗p < 0.05; ∗∗p < 0.01).
(G) Confocal images of coronal sections of somatosensory cortices from WT or SynIII KO C57BL/6 mice at P7, after IUE at E15.5 with tdTomato. The scale bar represents 50 μm.
(H) High-magnification images of the boxed regions in (E). The scale bar represents 50 μm.
See Figure S5.
The Overexpression of SynIII Affects Radial Migration of PNs In Vivo
While rescuing orientation defects in the CP (Figures 2H and 2I), the co-transfection of shRNA rSynIII and shRNA at a ratio 1:1 also led to ectopic cells in deep cortical layers (Figures S6A and S6B); however, these cells did not exhibit orientation defects (Figure S6C). Given the tight temporal regulation of SynIII expression, we hypothesized that not only downregulation/deletion but also overexpression of SynIII could interfere with proper radial migration of PNs. To test this hypothesis, we overexpressed WT SynIII by IUE at E17. At E21, PNs expressing either Tom or SynIII properly migrated through the cortex toward the CP (Figures 4A and 4B). Nonetheless, a significant percentage of SynIII-overexpressing cells were misplaced at P7 (Figures 4C and 4F–4H). The effect of SynIII on neuronal migration was long-lasting, as SynIII overexpression also caused the appearance of ectopic cells at P14 (Figures S6G and S6H).
Figure 4.

Overexpression of SynIII Results in Ectopic Cells without Orientation Defects
(A) Confocal images of coronal sections of the rat somatosensory cortex at E21 transfected with tdTomato (control) or SynIII. The scale bar represents 100 μm.
(B) Quantification of the number of migrating PNs residing in the VZ/SVZ, IZ, or CP for control and SynIII-transfected animals. The data are mean percentages (±SEM) of the total number of fluorescent cells.
(C) Confocal images of coronal sections of the rat somatosensory cortex at P7 transfected with tdTomato (control) or SynIII. The arrowheads point to PNs that did not complete radial migration. The scale bar represents 100 μm.
(D) Representative reconstructions of control or SynIII-overexpressing PNs at CP. The scale bar represents 50 μm.
(E) Quantification of the distribution of cells in the orientation classes shown in Figure 1D for control and SynIII-transfected cortices at the CP. The data are mean percentages (±SEM) of the total number of CP fluorescent cells.
(F) Higher magnification of the boxed region in (C). The scale bar represents 25 μm.
(G) Representative reconstructions of control or SynIII-overexpressing PNs in ectopic positions. The scale bar represents 50 μm.
(H) Quantification of the number of control or SynIII-overexpressing PNs that did not complete migration. The data are mean percentages (±SEM) of the total number of fluorescent cells (Student’s t test: ∗∗p < 0.01).
See Figure S6.
Thus, SynIII overexpression, KD, and KO all affected migration and resulted in the ectopic placement of cells. However, SynIII-overexpressing cells were all properly oriented (Figures 4C–4H and S6D–S6H). These effects could be due to subcellular mislocalization of SynIII upon overexpression in vivo, as we demonstrated in neuronal cultures (Figure S6I).
SynIII Is Phosphorylated by CDK5 at Ser404 Both In Vitro and In Vivo
Similar to the phenotype found for SynIII KD during development, CDK5 KO leads to a delay in radial migration and misoriented PNs (Ohshima et al., 2007).
By analyzing the amino acid sequence of SynIII, we observed that it carries a conserved consensus sequence for CDK5 phosphorylation, with Ser404 predicted to be the target (Songyang et al., 1996). Moreover, we found that CDK5 was expressed in rat cortices during development as early as E15 (Figures 5A and 5B) and that CDK5 and SynIII expression are highly correlated at perinatal ages (Figure 5C). Interestingly, we found that SynIII and CDK5 co-immunoprecipitated from lysates of rat primary cortical neurons grown for 4 days in vitro (DIV) (Figure 5D), demonstrating that SynIII and CDK5 physically interact. SynIII and the neuron-specific CDK5 activator p35 consistently colocalized in developing neurons in vitro (Figure 5E).
Figure 5.

SynIII Interacts with CDK5 and Is Phosphorylated on Ser404
(A) Representative western blot showing the temporal expression of CDK5 in comparison to actin in lysates of rat brain cortices.
(B) Quantification of CDK5 expression in experiments as in (A).
(C) Positive correlation between SynIII and CDK5 expression at the indicated ages. Dashed line: best fit (Pearson’s correlation: ∗∗p < 0.01). The data in (B) and (C) were normalized first to actin and then to the maximum detected optical density value; the values are expressed as means (±SEM; n = 3 animals/age).
(D) Rat cortical neurons were lysed at 4 DIV and subjected to co-immunoprecipitation with anti-SynIII and anti-CDK5-specific antibodies and the appropriate control IgG (IP). INPUT, 10% of total lysate; SUPERNATANT, unbound fraction.
(E) Confocal images of cultured neurons (3 DIV) stained for SynIII or p35. The scale bar represents 10 μm.
(F) FLAG-WT SynIII and FLAG-S404A rSynIII were expressed in COS7 cells and immunoprecipitated with an anti-FLAG affinity gel. Protein expression and efficient immunoprecipitation were verified through western blot with an anti-SynIII antibody (INPUT, 5% total lysate; SUPERNATANT, unbound fraction).
(G) The immunoprecipitated proteins (IP) were dephosphorylated and used as substrates for in vitro phosphorylation with active purified complex p35/CDK5 and radioactive [γ-32P]ATP. Radioactive phosphate incorporation in SynIII was detected through autoradiography of the SDS-PAGE gel.
(H) Quantification of band intensity from the experiments shown in (D). The data are mean percentages (±SEM) of S404A-SynIII phosphorylation normalized to total phosphorylation of WT-SynIII (Student’s t test: ∗∗p < 0.01). The number of independent experiments is shown in parentheses.
(I) Low-energy collision-induced dissociation (CID) tandem mass spectrum (MS/MS) showing phosphorylation at Ser404 of SynIII. The precursor mass measurement was collected in the Orbitrap to 1PPM of the theoretical mass. The MS/MS was collected in the linear ion trap. The site of phosphorylation was not ambiguous based on multiple diagnostic ions, particularly the y-12 and y-7 daughter ions. The high SEQUEST Xcorr value of 4.2 is given. In addition, diagnostic of Ser/Thr phosphorylation was the major peak in the spectrum corresponding to a neutral loss of phosphoric acid off the doubly charged precursor.
(J) Western blot showing the temporal expression of SynIII and P-SynIII in comparison to actin in lysates of rat brain cortices.
(K) Quantification of P-SynIII expression normalized to actin or SynIII, as in (A). The data were then normalized to the maximum detected optical density value; the values are expressed as means (±SEM; n = 3 animals/age).
See Figure S7.
To test whether SynIII is indeed phosphorylated by CDK5, we performed an in vitro phosphorylation assay (Figures 5F–5H). We first generated a non-phosphorylatable SynIII mutant in which Ser404 was replaced with an Ala. When immunoprecipitated and dephosphorylated SynIII or its S404A mutant was phosphorylated in vitro by active p35/CDK5 complex (Figure 5G), we observed that SynIII was phosphorylated by CDK5, whereas phosphorylation was significantly decreased by ∼31% in the S404A SynIII mutant (Figures 5G and 5H). This result indicates that Ser404 represents almost 1/3 of the total CDK5-mediated SynIII phosphorylation.
To test whether CDK5 phosphorylates SynIII in vivo, we performed liquid chromatography tandem mass spectrometry (LC-MS/MS) on endogenous SynIII from rat brains at P1, the peak of SynIII expression. In Figure 5I, the predicted m/z values of fragment ions are shown in blue or red, indicating identified b- and y-type ions, respectively. This experiment confirmed the presence of Ser404 phosphorylation in vivo.
Finally, we generated a phosphorylation-state-specific antibody recognizing phospho-Ser404 in SynIII. After testing its specificity for phospho-Ser404 (Figure S7B), we analyzed the phosphorylation of SynIII on Ser404 in vivo. We found that the absolute amount of phosphorylated SynIII present in the cortex plateaued at P1–P7 and then dropped to undetectable levels at P16–P35. However, the fraction of phospho-SynIII to total SynIII was high at very early stages of development (E15–E18) and steadily decreased thereafter to become undetectable at P16 (Figures 5J and 5K).
SynIII Phosphorylation at CDK5 Site Regulates SynIII Effects
To test whether SynIII is indeed a downstream effector of CDK5 during development in vivo, we generated a pseudo-phosphorylated SynIII mutant by replacing Ser404 with an Asp (D), which mimics the negative charge of the phosphate group (rSynIII S404D). We next verified that both S404A and S404D rSynIII expression and subcellular distribution were similar to that of the WT protein (Figures S7C–S7E).
When expressed alone, rSynIII S404A caused a delay in radial migration at E21 similar to that observed in the SynIII KD experiments (Figures 6A and 6B), indicating that this mutant may act in a dominant-negative manner on endogenous SynIII. Interestingly, when overexpressed at P7, rSynIII S404A resulted in the appearance of ectopic cells without orientation defects (Figures 6C, 6E, and 6F), suggesting that the presence of the endogenous SynIII is sufficient to guarantee proper neuronal orientation. Consistent with the dominant-negative effect of the mutant on migration, when S404A rSynIII was co-expressed with shRNA at a 0.5:1 or 1:1 ratio (as was done for WT rSynIII; Figures 2H and 2I), it did not rescue the shRNA-induced migration defects at either E21 (Figures 6A and 6B) or P7 (Figures 6C and 6E). Moreover, the co-transfection of S404A rSynIII with shRNA at either ratio did not rescue the orientation defects in PNs located in the CP (Figure 6D) or in ectopic PNs (Figure 6F). Conversely, when co-electroporated with shRNA at a 0.5:1 ratio, S404D rSynIII significantly rescued the delay in radial migration caused by SynIII KD at E21 (Figures S7F and S7G).
Figure 6.

The Non-phosphorylatable SynIII Mutant at the CDK5 Phosphorylation Site Does Not Rescue shRNA-Induced Defects
(A) Confocal images of cortical coronal sections of animals at E21 transfected with shRNAscr (control), S404A rSynIII, S404A rSynIII/shRNA, or shRNA. The scale bar represents 100 μm.
(B) Quantification of the number of PNs residing in the VZ/SVZ, IZ, or CP in shRNAscr (control)-, S404A rSynIII-, S404A rSynIII/shRNA-, or shRNA-expressing cortices. The data are mean percentages (±SEM) of the total number of fluorescent cells in the section (one-way ANOVA; post hoc Holm-Sidak test: all conditions versus control ∗∗∗p < 0.001).
(C) Confocal images of cortical coronal sections at P7 from animals transfected with shRNAscr (control), S404A rSynIII, S404A rSynIII/shRNA (0.5:1 ratio), S404A rSynIII/shRNA (1:1 ratio), or shRNA. The scale bars represent 100 μm. Arrowheads, example PNs that did not complete radial migration (black, UP neurons; gray, IN neurons).
(D) Quantification of the distribution of CP cells in the three orientation classes, as in Figure 1D (one-way ANOVA; post hoc Holm-Sidak test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). The data are mean percentages (±SEM) of the total number of CP cells.
(E) Quantification of the number of control, S404D rSynIII+, S404D rSynIII/shRNA+ (0.5:1 ratio), S404D rSynIII/shRNA+ (1:1 ratio), or shRNA+ cells that did not complete migration (one-way ANOVA; post hoc Holm-Sidak test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
(F) Quantification of the percentage of inverted cells with respect to the total ectopic cells in the control, S404D rSynIII alone, S404D rSynIII/shRNA (0.5:1 ratio), and S404D rSynIII/shRNA (1:1 ratio) conditions (one-way ANOVA; post hoc Holm-Sidak test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
The data in (E) and (F) are mean percentages (±SEM) of the total number of ectopic cells. See Figure S7.
SynIII Is Downstream of Sema3A-Signaling Cascade
CDK5 is a downstream effector in the Sema3A pathway, and alterations in this pathway at various levels all cause a phenotype similar to SynIII KD in the cortex (Polleux et al., 2000; Behar et al., 1996). To test the possibility that SynIII acts downstream of the Sema3A-signaling cascade, we investigated SynIII phosphorylation by CDK5 in 4-DIV cortical neurons after stimulation with 1 nM Sema3A. We observed that SynIII was phosphorylated on Ser404 only 5 min after the stimulus, and its phosphorylation significantly increased after 20 and 30 min in comparison to vehicle-treated cells (Figures 7A and 7B).
Figure 7.

S404D rSynIII Expression Rescues the Phenotype Caused by KD of the Sema3A Receptor NP1
(A) Representative western blot showing the phosphorylation of SynIII at Ser404 upon stimulation of 4-DIV primary cortical neurons with Sema3A.
(B) Quantification of the phosphorylation of SynIII at Ser404 (optical density values are normalized to total SynIII expression) in experiments, as in (A). The data are mean percentages (±SEM) normalized to the control values (n = 6 samples per time point). The data are shown in a scatterplot that was fit with a logarithmic regression function (one-way ANOVA; post hoc Tukey’s test: ∗∗p < 0.01).
(C) Representative western blot showing the temporal expression of NP1 in comparison to actin in lysates of rat brain cortices.
(D) Quantification of NP1 expression in experiments as in (C).
(E) Positive correlation between SynIII and NP1 expression at the indicated ages. Dashed line: best fit (Pearson’s correlation: ∗p < 0.05). The data in (D) and (E) were normalized first to actin and then to the maximum detected optical density value; the values are expressed as means (±SEM; n = 3 animals/age).
(F) Confocal images of cultured neurons (DIV 3) stained for SynIII (green) or NP1 (red). The scale bar represents 10 μm.
(G) Confocal images of cortical coronal sections at P7 from animals transfected with shRNAscr (control), NP1shRNA, or S404D rSynIII/NP1 shRNA. The scale bar represents 100 μm.
(H) Quantification of the number of control, NP1 shRNA+, or S404D rSynIII/NP1 shRNA+ cells that did not complete migration (one-way ANOVA; post hoc Holm-Sidak test: ∗∗∗p < 0.001).
(I) Schematic model on the involvement of SynIII in the Sema3A/CDK5 pathway. Sema3A binds to the NP1/Plexin A2 complex, which activates Fyn kinase. Active Fyn phosphorylates CDK5 on Tyr15, promoting phosphorylation of SynIII on Ser404 by p35/CDK5. Phosphorylated SynIII may interact with its cytoskeletal partners and SVs to control neuronal migration and orientation.
See Figure S7.
We next tested the causal involvement of SynIII in the Sema3A pathway in vivo. First, we investigated the expression of the Sema3A receptor neuropilin 1 (NP1) (Chen et al., 2008) in rat brain cortices. NP1 expression was highly similar to that of SynIII during development, with a maximum expression between E21 and P7, followed by a sharp decrease immediately thereafter (Figures 7C–7E). Moreover, as was seen for p35, we observed a colocalization of SynIII and NP1 in developing neurons in vitro (Figure 7F), suggesting that these three molecules may indeed be part of the same signaling cascade.
Next, to investigate the Sema3A/CDK5/SynIII pathway in vivo, we electroporated a shRNA against NP1 in rat embryos (Chen et al., 2008). As expected (Chen et al., 2008), we observed a very strong effect on the radial migration of layer II/III PNs at P7, with only ∼35% of PNs reaching the CP (Figures 7G and 7H). However, when we co-expressed S404D rSynIII and NP1 shRNA, we significantly rescued radial migration (Figures 7G and 7H), with ∼50% more PNs reaching the CP in comparison to NP1 shRNA-electroporated brains.
Discussion
During cortical development, post-mitotic PNs migrate away from the VZ along RG fibers toward the surface of the CP. On reaching the superficial layer, PNs stop migrating, detach from RG fibers, and morphologically differentiate.
In this study, we showed that SynIII, the Syn isoform that is most precociously expressed, plays a key role in cortical development. KD, KO, or upregulation of SynIII caused defects in radial migration, with the appearance of ectopic cells at later stages. Moreover, SynIII KD/KO caused significant orientation defects in ectopic PNs as well as in PNs that fully completed their radial migration. Interestingly, in misoriented PNs, the centrosome was properly located on the side of the apical dendrite. If centrosome localization ahead of the nucleus in the direction of migration is necessary for neuronal migration (Solecki et al., 2009), the question arises of how most of the SynIII-KD-misoriented neurons completed their migration. It is possible that SynIII-KD neurons migrate along glial fibers when the leading process is directed to the pial surface and stop migrating when they change orientation. Conceivably, re-orientation of migrating PNs occurs several times during migration, and—consistent with the higher levels of SynIII shRNA in ectopic PNs—the more SynIII is KD, the more migrating cells will re-orientate, resulting in a slower migration and ultimate ectopic positioning. Conversely, PNs expressing lower levels of SynIII shRNA will reach the CP, detach from RG fibers, and, without the fiber’s restriction, acquire all possible degrees of orientation before morphological maturation. SynIII KD causes the described phenotypes through a cell-autonomous mechanism, as confirmed by an intact RG scaffold in shRNA-transfected brains.
The migration and orientation defects involved the Sema3A-signaling cascade; indeed, upon Sema3A stimulation, SynIII is phosphorylated by CDK5. As shown by SynIII phospho mutant experiments, this phosphorylation is essential for SynIII activity. Thus, we propose a unifying model in which SynIII acts downstream Sema3A/CDK5 signaling to control neuronal migration and orientation (Figure 7I). Given that SynIII KD caused similar defects when performed in neuronal progenitors of both layer II/III and layer IV/V, and considering that CDK5 activity is essential for migration and dendrite development of layer V PNs (Ohshima et al., 2007), we hypothesize that the mechanism by which SynIII regulates proper cortical organization is generalizable to the development of all cortical layers. Future studies should investigate whether SynIII also regulates migration in other brain areas, as previously described for CDK5 (Jessberger et al., 2008).
Tight Spatial and Temporal Regulation of SynIII Controls Neuronal Migration and Orientation during Development
Although many factors have been identified in recent years to be involved in cortical migration, we are still far from understanding the entire framework, and many actors await identification. Among these, SynIII was a good candidate. First, the emergence of the Syn3 gene correlates with the appearance of the telencephalic region during evolution (Kao et al., 1999). Second, we demonstrated that SynIII is highly expressed and phosphorylated on Ser404 during development and has a well-defined spatial expression along cortical layers at distinct developmental stages. Finally, in vitro data suggest a role for SynIII in neuronal development (Ferreira et al., 2000).
Here, we showed that SynIII modulates neuronal migration and orientation in vivo. In particular, a tight regulation of SynIII expression is necessary for proper cortical development. Indeed, SynIII expression peaks in a defined temporal window, beyond which it is detrimental for cortical PN development, as proven by the following facts: (1) both SynIII KD and overexpression caused defects in radial migration and in the morphological maturation of PNs; (2) a shRNA-insensitive rSynIII completely rescued the KD phenotypes only when co-expressed with shRNA at optimal levels; and (3) the cortical phenotype caused by SynIII KD during early development was irreversible and persisted even when the expression of the protein was endogenously downregulated at later developmental stages, such as P14 (also see the Supplemental Discussion).
SynIII KO and SynIII shRNA-Transfected Brains Share Common Features
Data from the literature indicated no major brain abnormalities in SynIII KO mice (Feng et al., 2002), whereas our data demonstrated that acute KD of SynIII by RNAi profoundly affected cortical development. Interestingly, many other cases have been described where acute KD of a specific protein in a subset of neurons by IUE leads to strong defects in brain development, whereas the constitutive KO of the same protein does not lead to an apparent phenotype (e.g., DCX, Corbo et al., 2002 versus Bai et al., 2003; p75, Lee et al., 1992 versus Zuccaro et al., 2014; GABAB receptor, Schuler et al., 2001 versus Bony et al., 2013). Two main reasons could explain this finding: (1) the chronic and widespread depletion of the protein in KO mice could trigger redundancy or compensatory mechanisms by other proteins that overcome the deficits in brain development and/or (2) subtle developmental impairments in KO animals may be simply overlooked due to the complexity of the in vivo system and/or the lack of adequate methods to study mild defects. Here, we provided evidence for both possibilities. Indeed, marking developing neurons with a fluorescent reporter via IUE in SynIII KO mice allowed us to examine their cortical development with an unprecedented focus on neuronal migration and orientation. Using this technique allowed us to conclude that SynIII KO animals display the same phenotype as observed in shRNA-transfected brains, although to a milder extent.
Because of the high homology between SynIII and SynI/II, we hypothesized that they could compensate for the loss of SynIII and that this compensation could result in a mild developmental phenotype in KO animals. Nevertheless, we showed that SynII is not significantly expressed at perinatal ages in the cortex and that the overexpression of SynI did not rescue SynIII shRNA-mediated effects. It is possible that the differences between SynI and SynIII in terms of their amino acid sequence, location of phosphorylation sites, and spatio-temporal expression profile could account for their lack of involvement in neuronal migration and orientation.
In conclusion, we hypothesize that proteins other than SynI and II may compensate for SynIII loss in KO animals, and we propose the use of IUE for expressing fluorescent reporters as a means to uncover subtle developmental phenotypes in KO animals.
SynIII Is Part of the Sema3A Pathway
Our data consistently show that SynIII KD leads to a peculiar effect in the orientation of neurons located in the CP and also in the ectopic cells. Interestingly, the orientation defects in cortical neurons described in the literature are primarily associated with the faulty expression of genes involved in the Sema3A-signaling pathway (Behar et al., 1996; Chae et al., 1997; Chen et al., 2008; Ohshima et al., 2007; Polleux et al., 1998, 2000; Sasaki et al., 2002; Shelly et al., 2011). Sema3A is a secreted protein well known for its role as a guidance signal upon binding to its receptor NP1. NP1 associates with its co-receptor plexin A2 to activate Fyn kinase. Then, Fyn activation promotes CDK5 binding to plexin A2 and eventually activates CDK5 by phosphorylating it on Tyr15 (Figure 7I).
SynIII carries a highly conserved putative site for CDK5 phosphorylation (Ser404). We demonstrated that SynIII physically interacts with CDK5 and is a substrate for the kinase by showing that (1) SynIII and CDK5 are both expressed at perinatal ages in the cortex and colocalize in developing neurons in culture; (2) SynIII and CDK5 co-immunoprecipitate; and (3) SynIII is phosphorylated by CDK5 both in vitro and in vivo.
The rescue experiments performed with phospho mutants in vivo proved that Ser404 phosphorylation has a biological significance and that the phenotype we observed in SynIII-null PNs is at least partially due to a missing step in CDK5 signaling. Moreover, we showed that Ser404 phosphorylation increased upon Sema3A stimulation in vitro and that the pseudo-phosphorylated mutant S404D rSynIII partially compensated for the loss of Sema3A signaling caused by NP1 KD in vivo. Thus, our data demonstrate that SynIII is part of the Sema3A pathway (Figure 7I; also see the Supplemental Discussion).
Hypothetical Mechanisms of Action of SynIII
How could SynIII translate Sema3A signaling into inputs for neuronal migration and orientation? Little is known regarding SynIII interactors. In general, Syn family members can bind to synaptic vesicles (SVs), actin (Benfenati et al., 1989a, b, 1992), and tubulin (Petrucci and Morrow, 1991) through the highly conserved A, C, and E domains (Candiani et al., 2010; Kao et al., 1999). These domains are also present in SynIII. Indeed, SynIII also binds SVs and regulates their trafficking within nerve terminals (Kao et al., 1998). As the regulation of cytoskeletal dynamics and SV trafficking are requisites for membrane rearrangements during cell migration (Itofusa and Kamiguchi, 2011), SynIII may participate in radial migration by the latter processes (Figure 7I). The phenotypic features of SynIII-KD or -overexpressing PNs support this hypothesis: delayed migration, ectopic positioning, and morphologically impaired leading processes are typical of neurons that lose their contact with RG fibers (Elias et al., 2007; Gupta et al., 2003; Sanada et al., 2004; Valiente et al., 2011). The dysregulation of SynIII activity could indeed interfere with the cytoskeletal dynamics and membrane rearrangements involved in the formation and disassembly of cell adhesions and, consequently, interactions between migrating neurons and RG fibers (Rivas and Hatten, 1995; also see the Supplemental Discussion).
Concluding Remarks
In this study, we showed that SynIII acts downstream Sema3A and regulates the radial migration and orientation of PNs. Neuronal migration is fundamental for the development of a fully functional brain, as demonstrated by the implication of defective neuronal migration in a number of neurodevelopmental disorders (Ayala et al., 2007). Interestingly, experimental evidence implies the Sema3A/SynIII pathway in schizophrenia. Indeed, Sema3A-signaling dysregulation in brains from individuals with schizophrenia (Eastwood et al., 2003) and a genetic linkage between SynIII mutations and schizophrenia have been reported (Hall et al., 2007; Lachman et al., 2005; Porton et al., 2004). Moreover, as we observed in SynIII KD/KO animal model brains, reduced SynIII expression (Porton and Wetsel, 2007), abnormal cytoarchitecture, and dendritic aberrations (Arnold et al., 1991; Benes et al., 1991; Glantz et al., 2000; Mirnics et al., 2000; Rajkowska et al., 1998; Rosoklija et al., 2000) have also been reported in the brains of individuals with schizophrenia. Interestingly, the altered directionality of axonal elongation in SynIII-KD neurons that we describe in the present study is consistent with the disruption of axonal fiber connectivity in schizophrenic patients (Zalesky et al., 2011). Finally, SynIII KO mice exhibit abnormal sensory motor gating in response to prepulse inhibition (Geyer et al., 2001), similar to what is observed in patients with schizophrenia and in mouse models of schizophrenia.
Thus, the clarification of the role of SynIII in the Sema3A pathway could be important in shedding light on the pathogenesis of neurodevelopmental diseases and further increase our knowledge of the complex mechanisms that regulate cortical development in health and disease.
Experimental Procedures
Generation of Constructs
For detailed cloning procedures and list of used primers, see the Supplemental Experimental Procedures.
Cell Cultures, Transfection, and Stimulation
COS7 Cell Culture and Transfection
COS7 cells were cultured in Dulbecco’s MEM (DMEM) (GIBCO) supplemented with 10% fetal calf serum (GIBCO), 1% L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Biowhittaker) and maintained at 37°C in a 5% CO2 humidified atmosphere. The cells were transfected with Fugene 6 (Roche). For detailed procedures, see the Supplemental Experimental Procedures.
Cortical Neuron Culture and Neuronal Stimulation
Primary cultures of dissociated cortical neurons were prepared from E18 rats and maintained in Neurobasal medium supplemented with 2% B-27, 0.5 mM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). The neurons were maintained at 37°C in a 5% CO2 humidified atmosphere. For stimulation experiments, the neurons were washed with Neurobasal medium and incubated for 30 min at 37°C. mSema3A-Fc (1 nM; R&D Systems) was added to the medium and, after incubation, the cells were lysed and processed for western blot.
Western Blotting
Total cell lysates were obtained from COS7 cells 48 hr after transfection or from cortical neuron cultures at 4 DIV. For detailed procedures, reagents, and a list of antibodies, see the Supplemental Experimental Procedures.
Immunoprecipitation and In Vitro Protein Dephosphorylation/Phosphorylation Assays
Following transient transfection, COS7 cells were lysed and SynIII and S404 SynIII were immunoprecipitated and dephosphorylated. The samples were then subjected to in vitro phosphorylation. For detailed procedures and reagents, see the Supplemental Experimental Procedures.
Animals and IUE
Sprague-Dawley rats and C57BL/6J mice were from Charles River Laboratories or Harlan. SynIII KO mice were generated by homologous recombination (Feng et al., 2002) and backcrossed onto the C57BL/6J background for over ten generations. All of the experiments were performed in accordance with the guidelines established by the European Communities Council (Directive 2010/63/EU of September 22nd, 2010) and were approved by the Italian Ministry of Health.
Standard IUE was performed as previously described (dal Maschio et al., 2012). For detailed procedures and reagents, see the Supplemental Experimental Procedures.
Slice Histology and Immunostaining
Slice histology and immunostaining were performed as previously described (Bony et al., 2013). For detailed procedures, antibodies, and reagents, see the Supplemental Experimental Procedures.
Images Acquisition and Analysis
The images were acquired using a confocal laser-scanning microscope (TCS SP5; Leica Microsystems) or with an epifluorescence microscope equipped with Neurolucida (MicroBrightField) software. For detailed procedures on acquisition and analysis, see the Supplemental Experimental Procedures.
Mass Spectrometry
Endogenous SynIII was immunoprecipitated from P1 brains. For detailed procedures, see the Supplemental Experimental Procedures. Immunoprecipitated SynIII was then processed for mass spectrometry analysis. In-gel tryptic digests were performed as previously described (Ballif et al., 2006), and extracted peptides were subjected to LC-MS/MS analysis in a linear ion trap-orbitrap mass spectrometer, as previously described (Ballif et al., 2008; Buel et al., 2010) and detailed in Supplemental Experimental Procedures.
Statistics
If not indicated otherwise in the legend, the numbers in parentheses on the histograms indicate the number of processed animals. One slice per animal was analyzed. Statistical analysis between control and experimental groups was performed with Student’s t test or, in the case of more than two experimental groups, with one-way ANOVA followed by post hoc multiple comparisons. For non-normally distributed data, ANOVA on ranks test and non-parametric post hoc comparisons were used. Outliers were excluded with the Grubb’s test. We used GraphPad (GraphPad Software) and SigmaStat (Systat Software) software. All of the data are presented as the means ± SEM.
Author Contributions
L.E.P. participated in the design of the experiments and performed the molecular biology and biochemistry experiments, IUE, immunocytochemistry, immunohistochemistry, image acquisition, and data analysis. L.E.P. also wrote the manuscript and made the figures. J.S. performed IUE, immunohistochemistry, and image acquisition and participated in biochemistry experiments. A.P. and S.G. performed the in vitro phosphorylation, CO-IP experiment, and neuronal cell culture experiments. S.S. performed the electrophysiology experiments. B.A.B. performed the mass spectrometry experiment. L.C. and F.B. designed the experiments, wrote the manuscript, and funded the project. All of the authors revised the manuscript.
Acknowledgments
We thank Drs. Hung-Teh Kao (Brown University) and Paul Greengard (The Rockefeller University) for providing us with the SynIII mutant mouse strain and with the anti-SynIII antibodies. This study was supported by research grants from the Italian Ministry of University and Research (PRIN 2010JFYFY2 to F.B. and FIRB 2010 “Futuro in Ricerca” to S.G.), the EU FP7 Integrating Project “Desire” (grant agreement no. 602531), and the Cure Epilepsy “Advanced Innovator Award” (to F.B.). The support of Telethon-Italy (grant GGP13033 to F.B.) and CARIPLO foundation (grant 2013 0879 to L.C.) is also acknowledged. Funding for the mass spectrometry analysis was provided by the Vermont Genetics Network/NIH grant 8P20GM103449 from the INBRE program of the NIGMS.
Published: April 2, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Results, Supplemental Discussion, Supplemental Experimental Procedures, and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.03.022.
Supplemental Information
References
- Arnold S.E., Lee V.M., Gur R.E., Trojanowski J.Q. Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc. Natl. Acad. Sci. USA. 1991;88:10850–10854. doi: 10.1073/pnas.88.23.10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R., Shu T., Tsai L.H. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Bai J., Ramos R.L., Ackman J.B., Thomas A.M., Lee R.V., LoTurco J.J. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Ballif B.A., Cao Z., Schwartz D., Carraway K.L., 3rd, Gygi S.P. Identification of 14-3-3epsilon substrates from embryonic murine brain. J. Proteome Res. 2006;5:2372–2379. doi: 10.1021/pr060206k. [DOI] [PubMed] [Google Scholar]
- Ballif B.A., Carey G.R., Sunyaev S.R., Gygi S.P. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J. Proteome Res. 2008;7:311–318. doi: 10.1021/pr0701254. [DOI] [PubMed] [Google Scholar]
- Behar O., Golden J.A., Mashimo H., Schoen F.J., Fishman M.C. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature. 1996;383:525–528. doi: 10.1038/383525a0. [DOI] [PubMed] [Google Scholar]
- Benes F.M., Sorensen I., Bird E.D. Reduced neuronal size in posterior hippocampus of schizophrenic patients. Schizophr. Bull. 1991;17:597–608. doi: 10.1093/schbul/17.4.597. [DOI] [PubMed] [Google Scholar]
- Benfenati F., Bähler M., Jahn R., Greengard P. Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J. Cell Biol. 1989;108:1863–1872. doi: 10.1083/jcb.108.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F., Valtorta F., Bähler M., Greengard P. Synapsin I, a neuron-specific phosphoprotein interacting with small synaptic vesicles and F-actin. Cell Biol. Int. Rep. 1989;13:1007–1021. doi: 10.1016/0309-1651(89)90016-7. [DOI] [PubMed] [Google Scholar]
- Benfenati F., Valtorta F., Chieregatti E., Greengard P. Interaction of free and synaptic vesicle-bound synapsin I with F-actin. Neuron. 1992;8:377–386. doi: 10.1016/0896-6273(92)90303-u. [DOI] [PubMed] [Google Scholar]
- Bony G., Szczurkowska J., Tamagno I., Shelly M., Contestabile A., Cancedda L. Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/LKB1. Nat. Commun. 2013;4:1800. doi: 10.1038/ncomms2820. [DOI] [PubMed] [Google Scholar]
- Buel G.R., Rush J., Ballif B.A. Fyn promotes phosphorylation of collapsin response mediator protein 1 at tyrosine 504, a novel, isoform-specific regulatory site. J. Cell. Biochem. 2010;111:20–28. doi: 10.1002/jcb.22659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiani S., Moronti L., Pennati R., De Bernardi F., Benfenati F., Pestarino M. The synapsin gene family in basal chordates: evolutionary perspectives in metazoans. BMC Evol. Biol. 2010;10:32. doi: 10.1186/1471-2148-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F., Baldelli P., Valtorta F., Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chae T., Kwon Y.T., Bronson R., Dikkes P., Li E., Tsai L.H. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- Chen G., Sima J., Jin M., Wang K.Y., Xue X.J., Zheng W., Ding Y.Q., Yuan X.B. Semaphorin-3A guides radial migration of cortical neurons during development. Nat. Neurosci. 2008;11:36–44. doi: 10.1038/nn2018. [DOI] [PubMed] [Google Scholar]
- Chen Q., Che R., Wang X., O’Neill F.A., Walsh D., Tang W., Shi Y., He L., Kendler K.S., Chen X. Association and expression study of synapsin III and schizophrenia. Neurosci. Lett. 2009;465:248–251. doi: 10.1016/j.neulet.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo J.C., Deuel T.A., Long J.M., LaPorte P., Tsai E., Wynshaw-Boris A., Walsh C.A. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dal Maschio M., Ghezzi D., Bony G., Alabastri A., Deidda G., Brondi M., Sato S.S., Zaccaria R.P., Di Fabrizio E., Ratto G.M., Cancedda L. High-performance and site-directed in utero electroporation by a triple-electrode probe. Nat. Commun. 2012;3:960. doi: 10.1038/ncomms1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paula D., Bentley M.V., Mahato R.I. Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting. RNA. 2007;13:431–456. doi: 10.1261/rna.459807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood S.L., Law A.J., Everall I.P., Harrison P.J. The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol. Psychiatry. 2003;8:148–155. doi: 10.1038/sj.mp.4001233. [DOI] [PubMed] [Google Scholar]
- Elias L.A., Wang D.D., Kriegstein A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Feng J., Chi P., Blanpied T.A., Xu Y., Magarinos A.M., Ferreira A., Takahashi R.H., Kao H.T., McEwen B.S., Ryan T.A. Regulation of neurotransmitter release by synapsin III. J. Neurosci. 2002;22:4372–4380. doi: 10.1523/JNEUROSCI.22-11-04372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A., Kao H.T., Feng J., Rapoport M., Greengard P. Synapsin III: developmental expression, subcellular localization, and role in axon formation. J. Neurosci. 2000;20:3736–3744. doi: 10.1523/JNEUROSCI.20-10-03736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M.A., Krebs-Thomson K., Braff D.L., Swerdlow N.R. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl.) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Glantz L.A., Austin M.C., Lewis D.A. Normal cellular levels of synaptophysin mRNA expression in the prefrontal cortex of subjects with schizophrenia. Biol. Psychiatry. 2000;48:389–397. doi: 10.1016/s0006-3223(00)00923-9. [DOI] [PubMed] [Google Scholar]
- Gupta A., Sanada K., Miyamoto D.T., Rovelstad S., Nadarajah B., Pearlman A.L., Brunstrom J., Tsai L.H. Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat. Neurosci. 2003;6:1284–1291. doi: 10.1038/nn1151. [DOI] [PubMed] [Google Scholar]
- Hall H., Lawyer G., Sillén A., Jönsson E.G., Agartz I., Terenius L., Arnborg S. Potential genetic variants in schizophrenia: a Bayesian analysis. World J. Biol. Psychiatry. 2007;8:12–22. doi: 10.1080/15622970600892004. [DOI] [PubMed] [Google Scholar]
- Itofusa R., Kamiguchi H. Polarizing membrane dynamics and adhesion for growth cone navigation. Mol. Cell. Neurosci. 2011;48:332–338. doi: 10.1016/j.mcn.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Jessberger S., Aigner S., Clemenson G.D., Jr., Toni N., Lie D.C., Karalay O., Overall R., Kempermann G., Gage F.H. Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol. 2008;6:e272. doi: 10.1371/journal.pbio.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao H.T., Porton B., Czernik A.J., Feng J., Yiu G., Häring M., Benfenati F., Greengard P. A third member of the synapsin gene family. Proc. Natl. Acad. Sci. USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao H.T., Porton B., Hilfiker S., Stefani G., Pieribone V.A., DeSalle R., Greengard P. Molecular evolution of the synapsin gene family. J. Exp. Zool. 1999;285:360–377. [PubMed] [Google Scholar]
- Lachman H.M., Stopkova P., Rafael M.A., Saito T. Association of schizophrenia in African Americans to polymorphism in synapsin III gene. Psychiatr. Genet. 2005;15:127–132. doi: 10.1097/00041444-200506000-00009. [DOI] [PubMed] [Google Scholar]
- Lee K.F., Li E., Huber L.J., Landis S.C., Sharpe A.H., Chao M.V., Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Mirnics K., Middleton F.A., Marquez A., Lewis D.A., Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Ohshima T., Hirasawa M., Tabata H., Mutoh T., Adachi T., Suzuki H., Saruta K., Iwasato T., Itohara S., Hashimoto M. Cdk5 is required for multipolar-to-bipolar transition during radial neuronal migration and proper dendrite development of pyramidal neurons in the cerebral cortex. Development. 2007;134:2273–2282. doi: 10.1242/dev.02854. [DOI] [PubMed] [Google Scholar]
- Petrucci T.C., Morrow J.S. Actin and tubulin binding domains of synapsins Ia and Ib. Biochemistry. 1991;30:413–422. doi: 10.1021/bi00216a016. [DOI] [PubMed] [Google Scholar]
- Polleux F., Giger R.J., Ginty D.D., Kolodkin A.L., Ghosh A. Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science. 1998;282:1904–1906. doi: 10.1126/science.282.5395.1904. [DOI] [PubMed] [Google Scholar]
- Polleux F., Morrow T., Ghosh A. Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature. 2000;404:567–573. doi: 10.1038/35007001. [DOI] [PubMed] [Google Scholar]
- Porton B., Wetsel W.C. Reduction of synapsin III in the prefrontal cortex of individuals with schizophrenia. Schizophr. Res. 2007;94:366–370. doi: 10.1016/j.schres.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Porton B., Kao H.T., Greengard P. Cloning of cDNAs encoding human synapsins IIa and IIb. DNA Seq. 1999;10:49–54. doi: 10.3109/10425179909033936. [DOI] [PubMed] [Google Scholar]
- Porton B., Ferreira A., DeLisi L.E., Kao H.T. A rare polymorphism affects a mitogen-activated protein kinase site in synapsin III: possible relationship to schizophrenia. Biol. Psychiatry. 2004;55:118–125. doi: 10.1016/j.biopsych.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Porton B., Rodriguiz R.M., Phillips L.E., Gilbert J.W., 4th, Feng J., Greengard P., Kao H.T., Wetsel W.C. Mice lacking synapsin III show abnormalities in explicit memory and conditioned fear. Genes Brain Behav. 2010;9:257–268. doi: 10.1111/j.1601-183X.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G., Selemon L.D., Goldman-Rakic P.S. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch. Gen. Psychiatry. 1998;55:215–224. doi: 10.1001/archpsyc.55.3.215. [DOI] [PubMed] [Google Scholar]
- Rivas R.J., Hatten M.E. Motility and cytoskeletal organization of migrating cerebellar granule neurons. J. Neurosci. 1995;15:981–989. doi: 10.1523/JNEUROSCI.15-02-00981.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosoklija G., Toomayan G., Ellis S.P., Keilp J., Mann J.J., Latov N., Hays A.P., Dwork A.J. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch. Gen. Psychiatry. 2000;57:349–356. doi: 10.1001/archpsyc.57.4.349. [DOI] [PubMed] [Google Scholar]
- Sanada K., Gupta A., Tsai L.H. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Cheng C., Uchida Y., Nakajima O., Ohshima T., Yagi T., Taniguchi M., Nakayama T., Kishida R., Kudo Y. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron. 2002;35:907–920. doi: 10.1016/s0896-6273(02)00857-7. [DOI] [PubMed] [Google Scholar]
- Schuler V., Lüscher C., Blanchet C., Klix N., Sansig G., Klebs K., Schmutz M., Heid J., Gentry C., Urban L. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)) Neuron. 2001;31:47–58. doi: 10.1016/s0896-6273(01)00345-2. [DOI] [PubMed] [Google Scholar]
- Shelly M., Cancedda L., Lim B.K., Popescu A.T., Cheng P.L., Gao H., Poo M.M. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71:433–446. doi: 10.1016/j.neuron.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki D.J., Trivedi N., Govek E.E., Kerekes R.A., Gleason S.S., Hatten M.E. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron. 2009;63:63–80. doi: 10.1016/j.neuron.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z., Lu K.P., Kwon Y.T., Tsai L.H., Filhol O., Cochet C., Brickey D.A., Soderling T.R., Bartleson C., Graves D.J. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Ciceri G., Rico B., Marín O. Focal adhesion kinase modulates radial glia-dependent neuronal migration through connexin-26. J. Neurosci. 2011;31:11678–11691. doi: 10.1523/JNEUROSCI.2678-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalesky A., Fornito A., Seal M.L., Cocchi L., Westin C.F., Bullmore E.T., Egan G.F., Pantelis C. Disrupted axonal fiber connectivity in schizophrenia. Biol. Psychiatry. 2011;69:80–89. doi: 10.1016/j.biopsych.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccaro E., Bergami M., Vignoli B., Bony G., Pierchala B.A., Santi S., Cancedda L., Canossa M. Polarized expression of p75NTR specifies axons during development and adult neurogenesis. Cell Rep. 2014;7:138–152. doi: 10.1016/j.celrep.2014.02.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.