

Abstract
Aims
In cardiac muscle, Ca2+ release from sarcoplasmic reticulum (SR) is reduced with successively shorter coupling intervals of premature stimuli, a phenomenon known as SR Ca2+ release refractoriness. We recently reported that the SR luminal Ca2+ binding protein calsequestrin 2 (Casq2) contributes to release refractoriness in intact mouse hearts, but the underlying mechanisms remain unclear. Here, we further investigate the mechanisms responsible for physiological release refractoriness.
Methods and Results
Gene-targeted ablation of Casq2 (Casq2 KO) abolished SR Ca2+ release refractoriness in isolated mouse ventricular myocytes. Surprisingly, impaired Ca2+-dependent inactivation of L-type Ca2+ current (ICa), which is responsible for triggering SR Ca2+ release, significantly contributed to loss of Ca2+ release refractoriness in Casq2 KO myocytes. Recovery from Ca2+-dependent inactivation of ICa was significantly accelerated in Casq2 KO compared to wild-type (WT) myocytes. In contrast, voltage-dependent inactivation measured by using Ba2+ as charge carrier was not significantly different between WT and Casq2 KO myocytes. Ca2+-dependent inactivation of ICa was normalized by intracellular dialysis of excess apo-CaM (20 μM), which also partially restored physiological Ca2+ release refractoriness in Casq2 KO myocytes.
Conclusions
Our findings reveal that the intra-SR protein Casq2 is largely responsible for the phenomenon of SR Ca2+ release refractoriness in murine ventricular myocytes. We also report a novel mechanism of impaired Ca2+-CaM-dependent inactivation of Cav1.2, which contributes to the loss of SR Ca2+ release refractoriness in the Casq2 KO mouse model and, therefore, may further increase risk for ventricular arrhythmia in vivo.
Keywords: Calsequestrin, calmodulin, calcium release restitution, L-type calcium channel, sarcoplasmic reticulum
1. Introduction
Precise control of the release of Ca2+ from intracellular stores of sarcoplasmic reticulum (SR) in cardiac myocytes is important for normal contractility and functioning of cardiac muscle [2]. On the other hand, spontaneous Ca2+ release from ryanodine receptor (RyR2) SR Ca2+ release channels is thought to be one of the underlying mechanisms responsible for ventricular arrhythmia and sudden cardiac death. Both acquired (such as acute myocardial infarction and heart failure) and inherited (e.g. catecholaminergic polymorphic ventricular tachycardia) arrhythmia syndromes are accompanied by abnormalities in Ca2+ handling in cardiac myocytes [2]. Published reports suggest that spontaneous Ca2+ release from the SR, particularly due to alterations in RyR2 activity, may serve as a trigger for ventricular arrhythmia in heart failure [3–6]. Moreover, interventions that decrease sudden cardiac death and increase survival in patients with heart failure also normalize cardiac Ca2+ handling [7, 8]. In aggregate, these data suggest that altered myocyte Ca2+ release and ventricular arrhythmias are closely linked.
Calsequestrin 2 (Casq2) is a high-capacity Ca2+-binding protein located in the junctional sarcoplasmic reticulum (jSR) of cardiac muscle [9–11]. Casq2 binds to the RyR2, either directly or via two other jSR proteins, junctin and triadin [12, 13] which together form the SR Ca2+ release unit (CRU) [14]. This protein complex is responsible for Ca2+ release from the SR, triggered by increase in cytosolic Ca2+ concentration due to activation of L-type Ca2+ channels (Cav1.2), also known as Ca2+-induced Ca2+ release (CICR) [15, 16]. In this way, CRUs are responsible for the precise control of Ca2+ release during excitation-contraction coupling in cardiac tissue [17]. Functionally, Ca2+ entering through Cav1.2 during an action potential triggers Ca2+ release from the SR [15]. In case of two consecutive stimuli, the amplitude of the resulting Ca2+ transient depends on the diastolic interval that preceded the second stimulus – a phenomenon known as Ca2+ release refractoriness [18].
Casq2, the major intra-SR Ca2+ buffering protein [18], has been suggested to modulate activity of RyR2 Ca2+-release channels [19, 20] regulating its sensitivity to intra-SR luminal Ca2+. We recently found that Casq2, acting both as intra-SR Ca2+ buffer and as a regulator of sensitivity of RyR2 to luminal Ca2+, importantly governs Ca2+ transient restitution in intact mouse hearts [1]. Here, we further investigate the role of Casq2 in regulating SR Ca2+ release refractoriness in isolated ventricular myocytes under experimental condition that allow control of the physiological trigger of Ca2+ release, the Cav1.2 current.
2. Methods
2.1. Ethics
All experiments were approved by the institutional animal care and use committees at Animal Care and Use Committees of Vanderbilt University and performed in accordance with NIH guidelines. Mouse heart harvest was performed under general anesthesia (inhalation of 3% isoflurane vapor) and animals euthanized by exsanguination.
2.2. Restitution protocols
Restitution curves were obtained by introducing an additional stimulation pulse (S2) at different time intervals (S1–S2 coupling intervals) with respect to the regular pacing pulses (S1) as shown in Fig. 2A. The same experimental protocol was used in all the experiments studying refractoriness of either cytosolic Ca2+ transients or Cav1.2 currents. To analyze the kinetics of the recovery of cytosolic Ca2+ transients, recovery ratios were calculated from the amplitudes of S2 (extrasystolic) and S1 (regular baseline pacing) beat. To plot restitution curves, we expressed either the S2 Ca2+ release transient or the amplitude of Cav1.2 currents during S2 stimulus as a function of the S1–S2 coupling interval, using the following formula:
Fig. 2. Refractoriness of SR Ca2+ release in field-stimulated myocytes.

A - Experimental protocol used to assess refractoriness of SR Ca2+ release in a cytosol of murine LV myocytes using electric field stimulation. Premature extra stimuli (S2) were applied at successively shorter S1–S2 coupling intervals following a 1 Hz conditioning train (S1 stimuli). B and C - representative examples of Ca2+ transient recorded in WT and Casq2 KO myocytes, respectively in response to the S1–S2 protocol shown in A. Note the complete loss of Ca2+ release refractoriness in the Casq2 KO myocyte. D – Average S2 Ca2+ release fraction plotted as a function of varying the S1–S2 coupling interval (n=7 for WT, open diamonds; n=6 for Casq2 KO, black diamonds; **P<0.005, *P<0.05). E - SR Ca2+ content measured by rapid caffeine application (n = 5–6 myocytes per group).
Our approach for analyzing SR Ca2+ restitution is different from the one that was used in our previous reports [1]. To better reflect that Ca2+ reuptake is not complete and hence the SR is still depleted during the very premature S2 beats, we use the value of the peak of the S2 transient rather than the S2 transient amplitude (see Fig. 1 for details). This approach takes into account the Ca2+ in the cytosol that is not yet taken up when the premature S2 stimulus is delivered and obviously cannot be released. We posit that this is a more accurate approach for measuring RyR2 release refractoriness in the intact myocyte.
Fig. 1. Calculation of S2 amplitude.
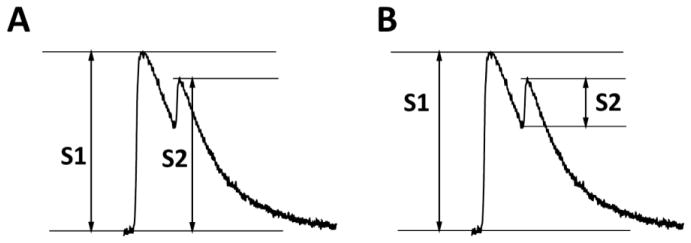
To calculate the restitution curve of Ca2+ release, we included the Ca2+ that still remains in the cytosol for measuring the amplitude of the premature S2 beat (A), because that Ca2+ has not been taken up into the SR and hence cannot be released. Previous approaches [1] measured S2 as the height of the premature S2 (B), which results in a restitution curve that mostly reflects the rate of SR Ca2+ uptake, and not the intrinsic refractoriness of the RyR2 Ca2+ release channel complex.
2.3. Myocyte isolation and cytosolic Ca2+ transient measurements
Ventricular myocytes from 12 to 16 weeks old male and female Casq2 KO or wild-type (WT) mice from the same C57BL/6 strain were isolated using a modified collagenase/protease method as previously described [21]. All experiments on field-stimulated myocytes were conducted in Tyrode’s solution (TS) containing (in mM): NaCl 134, KCl 5.4, MgCl2 1, CaCl2 2, glucose 10, HEPES 10, adjusted to pH 7.4 with NaOH. After isolation, myocytes were loaded with Fura-2 acetoxymethyl ester, Fura-2 AM as described previously [22]. Briefly, myocytes were incubated with 2 μM Fura 2 AM for 6 minutes at room temperature to load the indicator in the cytosol. Myocytes were then washed twice for 10 minutes with TS containing 250 μM probenecid to retain the indicator in the cytosol. A minimum of 30 min was allowed for de-esterification before imaging the cells.
Fura-2–loaded healthy rod-shaped isolated ventricular myocytes were placed into the experimental chamber, field stimulated, and superfused with TS. Intracellular Ca2+ transients were measured using a dual-beam excitation fluorescence photometry setup (IonOptix Corp.) utilizing the protocol shown in Fig. 2A. After that, myocytes were exposed for 4 seconds to TS containing 10 mmol/l caffeine using a rapid concentration-clamp system. The amplitude of the caffeine-induced Ca2+ transient was used as an estimate of total SR Ca2+ content [18]. All experiments were conducted at room temperature (~23°C). Ca2+ transients were analyzed using specialized data analysis software (IonWizard, IonOptixCorp.). Excitation wavelengths of 360 and 380 nm were used to monitor the fluorescence signals of Ca2+-bound and Ca2+-free fura-2, and [Ca2+]i measurements are reported as fluorescence ratios (Fratio).
For the measurements of cytosolic Ca2+ transients in voltage-clamp mode, cells were loaded with fluo-4 pentapotassium salt (final concentration 100 μM), added into pipette solution from stock. Pipette solution contained (in mM): CsCl 125, MgATP 5, MgCl2 1, glutathione (GSH) 5, cAMP 0.05, HEPES 20, adjusted to pH 7.25 with CsOH. External K-free solution contained (in mM): NaCl 134, CsCl 5, MgCl2 1, CaCl2 2, glucose 10, HEPES 10, adjusted to pH 7.4 with NaOH. The same S1–S2 stimulation protocol, only applied in voltage-clamp mode, was used. Fig. 3A demonstrates representative examples of membrane currents and corresponding [Ca2+]i transients from the cytosol in response to S1 and S2 voltage stimuli. Again, in the end of experiment each cell was exposed to TS containing 10 mmol/l caffeine to estimate total SR Ca2+ content. All chemicals, unless otherwise specified, were obtained from Sigma (St. Louis, MO).
Fig. 3. Refractoriness of SR Ca2+ release in voltage-clamped myocytes.

A –voltage clamp protocol (bottom trace), corresponding membrane currents (middle trace) and [Ca2+]i (top trace) during an S1 and S2 stimuli used to assess Ca2+ release refractoriness in voltage-clamped myocytes. To maintain constant Ca2+ trigger, SR Ca2+ release was activated with ICa tail currents that elicited maximal Ca2+ release during the S1 train. B and C - representative examples of Ca2+ transients recorded in WT and Casq2 KO myocytes, respectively, in response to the S1–S2 voltage clamp protocol shown in A. D - S2 Ca2+ release fraction changes as a result of varying the S1–S2 coupling interval (n=12 for WT, open circles and Casq2 KO, black circles; **P <0.005, *P <0.05). E – SR Ca2+ content measured by rapid caffeine application (n = 8 myocytes per group).
2.4. Measurements of Cav1.2 current
For measurements of Cav1.2 current, freshly isolated murine ventricular myocytes were whole-cell patched in Tyrode’s solution and then solution was changed to K+-free solution (described above) containing either 2 mM CaCl2 or 2mM BaCl2. In all experiments, myocytes were pre-incubated for 30 min in 50 μM Ryanodine + 10 μM Thapsigargin + 30 μM TTX to eliminate SR Ca2+ release and block sodium currents. The pipette solution contained (in mM): CsCl 110, MgCl2 1, MgATP 5, cAMP 0.2; EGTA 14; Fluo-4 0.1, Hepes 20; pH=7.25 (CsOH). For experiments testing effect of calmodulin (CaM) on inactivation of ICa, CaM purified from bovine testes (Sigma) was added into pipette solution for a final concentration of 20 μM. All experiments were carried out at room temperature.
2.5. Western Blot
Ventricular cardiomyocytes obtained from wild-type and Casq2 KO mice were homogenized and centrifuged at 100 g for 1 min at 4°C to eliminate the cellular debris. The supernatants were used for immunoblotting. The extracted proteins were separated on SDS-PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% milk and incubated overnight with primary antibodies: Calmodulin (Cell signaling antibodies) and GAPDH (Ambion). Specific bands were detected using Pierce ECL protein detection system. Western blot analyses were performed using ImageJ software (NIH).
2.6. Statistical analysis
Differences between groups were assessed using a one-way analysis of variance (ANOVA). If statistically significant differences were found, individual groups were compared with Student’s two-sided t test. Results were considered statistically significant if the P value was <0.05. Unless otherwise indicated, results are expressed as arithmetic means ± SE.
3. Results
3.1. Refractoriness of SR Ca2+ release is eliminated in ventricular myocytes lacking Casq2
Previous studies have shown that ventricular myocytes isolated from Casq2 KO mice exhibit elevated rates of premature spontaneous Ca2+ releases, delayed after depolarizations and triggered beats, and catecholamine-induced ventricular arrhythmias in vivo [21, 22]. We hypothesized that the arrhythmogenic potential of loss of Casq2 may be related to how quickly a secondary Ca2+ release can be elicited in cardiac myocyte. To investigate this hypothesis, we compared refractoriness of SR Ca2+ release in intact ventricular myocytes from WT and Casq2 KO mice using a field stimulation protocol (Fig. 2A). Under these conditions, myocytes from WT mice exhibit robust time-dependent refractoriness of Ca2+ release from the SR, i.e., the cytosolic Ca2+ transients in response to premature S2 stimuli were significantly smaller compared to the transients elicited during regular S1 pacing, especially at the shortest S1–S2 interval (Fig. 2B). Unlike WT, myocytes from Casq2 KO animals exhibited near complete absence of SR Ca2+ release refractoriness, even at very short S1–S2 coupling intervals (Fig. 2C).
Average restitution curves were plotted for each group (Fig. 2D), demonstrating an almost complete lack of SR Ca2+ release refractoriness in Casq2 KO myocytes. At the same time, SR Ca2+ content, estimated by the amplitude of Ca2+ transient as response to rapid application of caffeine (10 mM/L), was similar for both models (Fig. 2E). Our results indicate that Casq2 protein is responsible for SR Ca2+ refractoriness observed in isolated ventricular myocytes. Our data are in agreement with our previous report, where dramatic acceleration of Ca2+ release recovery was found at the whole heart level [1].
3.2. Accelerated recovery of Cav1.2 current contributes to the loss of SR Ca2+ release refractoriness in Casq2 KO myocytes
There are several factors that may contribute to the refractoriness of SR Ca2+ release in cardiac muscle. Since Cav1.2 current serves as the trigger for Ca2+ release from the SR during the cardiac action potential, refractoriness of Cav1.2 channels can be one of potential contributors to SR Ca2+ release refractoriness. To test this hypothesis, we next used an experimental protocol in which voltage clamp was used instead of field stimulation (Fig. 3A). Cav1.2 tail currents were used to activate Ca2+ release from the SR. This approach allows maintaining the Ca2+ current trigger essentially constant and hence eliminating possible refractoriness of Cav1.2 channels. Fig. 3B and C demonstrate representative recordings from voltage-clamped WT and Casq2 KO myocytes in response to the S1–S2 protocol shown above. Comparison of respective Ca2+ transient records from two approaches (field stimulation vs. voltage clamp) demonstrates the importance of Cav1.2 refractoriness for SR Ca2+ release, because refractoriness of SR Ca2+ release was accelerated substantially in voltage-clamped compared to field-stimulated cells for both WT and Casq2 KO myocytes.
Next, we compared the restitution kinetics of Cav1.2 currents in WT vs Casq2 KO myocytes (Fig. 4 A). Myocytes were pre-incubated with ryanodine (50 μM) and thapsigargin (10 μM) for 30 min to eliminate SR Ca2+ release. 200 μM cAMP was present in the pipette solution to fully phosphorylate the channels and to model conditions of adrenergic stimulation under which CPVT-related arrhythmias usually occur. Both groups had decreased ICa at short S1–S2 intervals, but surprisingly, ICa recovered significantly faster in Casq2 KO vs WT myocytes. At the same time, current-voltage relationships were not significantly different between the two groups (Fig. 4 B). Thus, recovery of Cav1.2 in mice lacking Casq2 is accelerated and could contribute to loss of SR Ca2+ release refractoriness in this model.
Fig. 4. Restitution of Cav1.2 current is accelerated in Casq2 KO myocytes.

A – Average Cav1.2 current restitution curve for WT (white circles, n=12) and Casq2 KO (black circles, n=12) myocytes. Inset shows representative examples of Cav1.2 current records for each group and the voltage protocol. Cav1.2 currents were elicited by a 50-ms depolarizing steps to 0mV from holding potential −70 mV. Myocytes were pre-incubated in 50 μM Ryanodine + 10 μM Thapsigargin + 30 μM TTX for 30 min to deplete the SR and eliminate INa. **P <0.005, *P<0.05. B – current-voltage relationships of Cav1.2 peak currents of WT (white circles, n=6) and Casq2 KO (black circles, n=6). There was no significant difference in voltage dependence or Cav1.2 current density between the two mouse models.
3.3. Ca2+-dependent inactivation of Cav1.2 to cytosolic Ca2+ is suppressed in Casq2 KO myocytes
Cav1.2 channels exhibit both voltage- and Ca2+-dependent inactivation [23, 24]. A plausible explanation for the accelerated recovery of ICa in Casq2 KO myocytes is that Cav1.2 channel inactivation is impaired. Therefore, we next investigated the rate of decay of ICa using a longer depolarizing step of 200 ms. The declining phase of ICa (=inactivation) was well fitted by a single-exponential function. The rate of inactivation was substantially slower in Casq2 KO myocytes (τ=93±11 ms, n=10) compared to WT (τ=45±4 ms, n=10, p<0.005), as shown in Fig. 5A. To distinguish between voltage- and Ca2+-dependent inactivation, we next replaced Ca2+ in the external solution with equimolar Ba2+. Ba2+ currents are nearly insensitive to Ba2+-dependent inactivation of Cav1.2 channels [25]. Consequently, inactivation of IBa was much slower than that of ICa (compare Fig. 5A and 5B). Importantly, the difference in τ between WT and Casq2 KO myocytes was eliminated (WT: τ=145±19 ms, Casq2 KO: τ=145±20 ms, n=10 for each group). Moreover, substituting Ba2+ for Ca2+ also abolished the differences in the Cav1.2 current restitution curves in Casq2 KO and WT myocytes (Fig. 5C). Taken together, these results indicate that Ca2+-dependent inactivation of Cav1.2 is defective in Casq2 KO myocytes, which is responsible for the difference between Cav1.2 current restitution curves of WT and Casq2 KO myocytes.
Fig. 5. Ca2+-dependent inactivation of Cav1.2 current is defective in Casq2 KO myocytes.

A – Representative Cav1.2 current for WT (light gray) and Casq2 KO (black) myocytes elicited by 200-ms depolarizing step to 0 mV from holding potential −70 mV in bath solution containing 2 mM Ca2+. To illustrate the difference in Cav1.2 current inactivation, current traces were normalized to peak amplitude value for each myocyte. B – Cav1.2 currents recorded in bath solution containing 2 mM Ba2+. Note that substituting Ca2+ with Ba2+ completely abolished the difference in Cav1.2 current inactivation. C – Average restitution curves and representative records (inset) of Ba2+ current for WT (white circles, n=13) and Casq2 KO (black circles, n=13) myocytes. The same experimental conditions and voltage protocol as in Fig. 4 were used.
3.4. Apo-Calmodulin rescues the inactivation defect of Cav1.2 current and partially recovers refractoriness of SR Ca2+ release in Casq2 KO myocytes
The Ca2+-free form of CaM (apo-CaM) is tightly bound to the C-terminus of Cav1.2 channels and serves as Ca2+ sensor for Ca2+-dependent inactivation [26, 27]. To test if CaM-dependent regulation of Cav1.2 channels was altered, we added apo-CaM (20 μM) to the pipette solution and repeated the ICa measurements. Apo-CaM significantly accelerated inactivation of ICa in Casq2 KO myocytes (Fig. 6A), rendering the ICa inactivation kinetics almost identical to that of WT myocytes (Casq2 KO + CaM: τ=46±3 ms, n=7, vs. WT: τ=45±4 ms for WT, n=10, p=0.8). In contrast, adding CaM had no significant effect on the rate of ICa inactivation in WT myocytes (Fig. 6B). Fig. 6C and Table 1 compare tau values for all the experiments. Addition of CaM also normalized the ICa restitution curve of Casq2 KO (Fig. 6D). At the same time, the total amount of CaM protein was not different between WT and Casq2 KO myocytes (Supplemental Fig. 1).
Fig. 6. Apo-CaM accelerates inactivation kinetics of Cav1.2 current only in Casq2 KO myocytes.

A– Addition of apo-calmodulin (CaM, 20 μM) to the intracellular dialysis solution accelerated inactivation of Cav1.2 Ca2+ currents in Casq2 KO myocytes. B – Addition of CaM had no effect on inactivation kinetics of Cav1.2 in WT myocytes. Voltage protocol is shown below. The declining phase of Cav1.2 current was well fitted by a single exponential function. C – Average inactivation time constant (tau) for all 4 groups (n=7–10 cells per group, **P<0.005). D – Average restitution curves of Cav1.2 currents for WT (white circles, n=12) and Casq2 KO myocytes in the presence (black/white circles, n=6) and absence (black circles, n=12) of CaM (20μM) in the pipette solution. Myocytes were pre-incubated in 50 μM Ryanodine + 10 μM Thapsigargin + 30 μM TTX for 30 min. **P <0.005, *P <0.05 for Casq2 KO compared to Casq2 KO + CaM.
Table 1.
Tau values for ICa and IBa inactivation.
| N | Current density (pA/pF) | Tau (ms) | P-value (for tau vs WT) | |
|---|---|---|---|---|
| WT | 10 | 15.8 | 45 | |
| Casq2 KO | 10 | 14.8 | 93 | 0.002 (vs. WT) |
| Casq2 KO + CaM | 7 | 17.9 | 46 |
0.002 (vs. Casq2 KO) 0.8 (vs. WT) |
| WT + CaM | 7 | 15.4 | 41 | 0.4 (vs. WT) |
| WT (Ba2+) | 10 | 11.2 | 145 | |
| Casq2 KO (Ba2+) | 10 | 9.2 | 145 | 0.99 (vs. WT (Ba2+)) |
Finally, we studied the effect of apo-CaM on refractoriness of SR Ca2+ release in Casq2 KO and WT myocytes. For these experiments, we used the S1–S2 protocol in voltage-clamp mode, similar to that used in Fig. 3, but L-type Ca2+ currents were elicited by 30-ms depolarizing step to 0mV from holding potential of −70 mV rather than using tail currents to activate SR Ca2+ release (to allow physiological Ca2+-induced inactivation of L-type Ca2+ channels). AIP (1 μM) was added to intracellular solution to prevent CaMKII activation by CaM, because activated CaMKII will phosphorylate RyR2 and could accelerate SR Ca2+ release refractoriness [28]. Under these conditions, addition of CaM resulted in a partial restoration of SR Ca2+ release refractoriness in Casq2 KO myocytes (Fig. 7). Ca2+ transient decay during the S1 beat was significantly slower in Casq2 KO compared to WT myocytes, which was completely normalized by adding CaM in Casq2KO myocytes (WT + CaM: τ=226±25 ms; Casq2 KO + CaM: τ=201±11 ms; Casq2 KO: τ=425±78 ms, p< 0.05 vs. both CaM groups; n=9–14 cells per group). This result suggests that impaired CaM-dependent Cav1.2 inactivation contributes to excess Ca2+ influx in Casq2 KO myocytes. Furthermore, because CaM addition completely normalizes LTCC Ca2+ refractoriness in Casq2 KO myocytes but has no effect on WT myocytes (Fig. 6), the results of Fig. 7D also indicate that the Cav1.2 inactivation defect is responsible for approximately 50% of the loss of Ca2+ release refractoriness found in Casq2 KO myocytes.
Fig. 7. Apo-CaM partly restores SR Ca2+ release refractoriness in Casq2 KO myocytes.

Representative examples of Ca2+ transient restitution in Casq2 KO myocytes in the absence (A) and in the presence of CaM (20μM) in the pipette solution (B), as well as in WT myocytes in the presence of CaM (C). D - S2 Ca2+ release fraction plotted as a function of the S1–S2 coupling interval (n=8–10; Casq2 KO w/o CaM - black circles; Casq2 KO+CaM - grey circles, *P <0.05 (vs. Casq2 KO w/o CaM); WT+CaM – light-grey diamonds, #P <0.05 (vs. Casq2 KO+CaM)). E - No significant difference in SR Ca2+ content was detected between three experimental groups.
4. Discussion
4.1. Casq2 – a critical determinant for physiological SR Ca2+ release refractoriness
Structurally, the CRU of cardiac myocyte includes Cav1.2 channels located in the sarcolemma and juxtaposed RyR2 SR Ca2+ release channels in the junctional SR. RyR2 channels are associated with other SR proteins like triadin and junctin, and through them also with Casq2. During action potential, Ca2+ influx via Cav1.2 causes much bigger amount of Ca2+ being released from the SR. The amplitude of the resulting cytosolic Ca2+ transient depends on the diastolic interval between two stimuli (normally, the amplitude of the second Ca2+ release is lower when the diastolic interval is short – Fig. 2 and 3), a phenomenon described as Ca2+ release restitution. Release restitution is regulated and affected by number of different voltage-, time- and Ca2+-dependent processes such as refractoriness (recovery from inactivation) of Cav1.2 channels, recovery of RyR2 channels and SR Ca2+ uptake kinetics [29]. Two previous studies have reported that accelerated restitution of SR Ca2+ release is associated with human arrhythmia diseases: Our earlier report demonstrating accelerated Ca2+ transient recovery in intact Casq2 KO hearts [1], which are a model of a rare genetic form of catecholaminergic polymorphic ventricular tachycardia (CPVT, discussed in more detail below), and a study in the canine model of post-myocardial infarction ventricular fibrillation [30]. The later study by Belevych et al. demonstrated that acceleration of Ca2+ release restitution of post-MI myocytes was attributable to RyR2 phosphorylation and oxidation. In current study, we investigate in more detail the role that Casq2 plays in Ca2+ release restitution in isolated ventricular myocytes, and also test Cav1.2 as one of potential contributors to the restitution of SR Ca2+ release. We show that not only is Casq2 a critical determinant for the restitution of Ca2+ release from the SR but also recovery of ICa is accelerated in mice lacking Casq2 and this, along with loss of intra-SR Ca2+ buffering and altered sensitivity of RyR2 to luminal Ca2+ [1], contributes to the loss of SR Ca2+ release restitution in Casq2 KO mice. We further demonstrate that accelerated recovery from inactivation of ICa in Casq2 KO model is due to impaired sensitivity of Cav1.2 channel to intracellular Ca2+ and this altered Ca2+ sensitivity can be recovered by application of excess apo-CaM.
4.2. SR Ca2+ release refractoriness and ventricular arrhythmia
CPVT is a highly lethal form of inherited arrhythmogenic disease characterized by adrenergically mediated polymorphic ventricular tachycardia caused by mutations in genes that encode CRU-composing proteins [31, 32]. The most common form is caused by mutations in RyR2 [31, 33]. Much less common, CPVT can be caused by mutations in Casq2 [32, 34], triadin or calmodulin [35]. Unlike CPVT caused by RyR2 mutations, CPVT linked to Casq2 mutations is usually autosomal recessive, with complete absence of Casq2 predicted for patients homozygous for Casq2 nonsense mutations. Despite this fact, they display surprisingly normal cardiac contractile function [34]. Ex vivo, isolated ventricular myocytes from Casq2 KO mice display normal SR Ca2+ release and contractile function under basal conditions, but exposure to catecholamines causes increased diastolic SR Ca2+ leak, resulting in premature spontaneous SR Ca2+ releases and triggered beats [21], which are considered major hallmarks of CPVT phenotype and a culprit for arrhythmia onset in vivo. Even modest reduction in Casq2 is able to increase diastolic SR Ca2+ leak and cause spontaneous SR Ca2+ release [22]. Unlike Casq2 KO [21], heterozygous Casq2 +/− mice have normal level of all the other CRU-composing SR proteins (namely, triadin-1 and junctin), which makes Casq2 the prime candidate responsible for arrhythmogenic phenotype in this model. On the other hand, triadin is crucially important for maintaining the structural and functional integrity of the cardiac CRU, and its ablation reduces SR Ca2+ release and impairs negative feedback of SR Ca2+ release on Cav1.2 currents [36]. Recently, junctin itself, without (or in addition to) involvement of Casq2, was reported as one of important modulators of RyR2 sensitivity to luminal Ca2+ [37].
Studies have shown that altered Ca2+ handling and, in particular, premature spontaneous Ca2+ releases from the SR serve as a culprit for CPVT [38]. Our data demonstrate that in Casq2 KO murine ventricular myocytes refractoriness of Ca2+ release from the SR is accelerated compared to myocytes from WT animals, and this acceleration is not accompanied by alterations in SR Ca2+ content (Fig. 2 E). These results confirm on a cellular level the accelerated recovery of Ca2+ transients previously reported from the same mouse model on a whole-heart level [1]. Unchanged SR Ca2+ content despite dramatic loss of SR Ca2+ buffering might be explained by substantial increase in SR volume of Casq2 KO myocytes, which effectively compensates for the absence of Casq2 [21].
4.3. Accelerated Cav1.2 recovery from inactivation importantly contributes to loss of Ca2+ release refractoriness in Casq2 KO myocytes
Our study shows that, surprisingly, recovery from inactivation of Cav1.2 current is accelerated in Casq2 KO mouse model (Fig. 4A), providing enhanced triggering for CICR during premature stimulation. Along with faster rise in free intra-SR Ca2+ [1], this creates a potential for much more massive, compared to WT, SR Ca2+ release at very short S1–S2 intervals. What are the possible mechanisms for the accelerated recovery of Cav1.2 currents produced by ablation of Casq2? We found no difference in Cav1.2 current density (Fig. 4B), hence, it is unlikely that the difference in recovery from inactivation is due to changes in Cav1.2 expression level. On the other hand, substituting Ca2+ with Ba2+ as charge carrier abolished the differences in Cav1.2 current inactivation and restitution (Fig. 5), suggesting that difference in Ca2+-dependent inactivation (CDI) of the channel is the most likely culprit.
We reported previously that inactivation of Cav1.2 is also impaired in ventricular myocytes lacking triadin [36]. Since expression of triadin is decreased in Casq2 KO mice [21], one could suggest that loss of triadin contributed to the accelerated recovery of Cav1.2 from inactivation. However, in triadin null myocytes, the difference in inactivation rate of Cav1.2 current was abolished by blocking RyR2 Ca2+ release with ryanodine [36]. Since all our Cav1.2 measurements in Casq2 KO myocytes were done in presence of ryanodine and thapsigargin, it seems unlikely that decrease in triadin can explain the accelerated recovery from inactivation of Cav1.2 in Casq2 KO myocytes.
4.4. Defective regulation by CaM mediates accelerated recovery of Cav1.2 in Casq2 KO myocytes
Studies have shown that Ca2+ binding to CaM is responsible for CDI [26, 27]. Acute overexpression of engineered, Ca2+-insensitive mutant CaM in rat ventricular myocytes can efficiently displace endogenous CaM on native Cav1.2 channels, resulting in strong inhibition of CDI without perturbation of other gating functions, such as voltage-dependent inactivation [27]. Since apo-CaM, the Ca2+-free form of CaM pre-associates with target molecules [39] such as L-type Ca2+ channels, whose function is subsequently modulated when an elevation of Ca2+ calcifies the associated CaM [26, 40, 41], any reduced availability of apo-CaM could contribute to impaired CDI, as has been shown experimentally for Cav1.3, where lack of pre-associated apo-CaM rendered Cav1.3 channels incapable of CDI while still being able to open normally [42]. Since there is extensive competition for CaM in the intracellular milieu (only one out of 100 is free), it seems possible that due to the increased SR Ca2+ leak and elevated diastolic Ca2+ in Casq2 KO myocytes [21, 22] available apo-CaM is reduced. Moreover, in Xenopus oocytes, injection of excess purified CaM protein was able to reduce dramatically the inhibitory effect of mutant, Ca2+ -insensitive CaM1234 on CDI of Cav1.2 channels [43]. This effect presumably is based on the competition for the same binding site located in the proximal C terminus of the α1C subunit of Cav1.2 that both “normal” CaM and mutant CaM1234 share. In brain, there is another Ca2+ sensor protein known to have overlapping binding sites with CaM in pCT of Cav1.2 – Ca-binding protein 1 (CaBP1) [43]. CaBP1 is structurally related to CaM but its effect on Cav1.2 is opposite to that of CaM: Addition of CaBP1 slows down CDI [44] and CaBP1 competes with CaM for aforementioned binding sites in Cav1.2 [43]. However, CaBP1 is not expressed in the heart [45], hence its involvement in attenuation of CDI in Casq2 KO ventricular myocytes seems doubtful. Another possibility would be that CaM protein expression is reduced in Casq2 KO myocytes. But our experiments show no difference in CaM protein between Casq2 KO and WT myocytes (suppl. Fig. 1).
4.5. Implication of defective Cav1.2 regulation by CaM for arrhythmogenesis
How could the impaired Ca2+-dependent inactivation of Cav1.2 and faster recovery of Ca2+ current contribute to the arrhythmogenesis in Casq2 KO mice? The impaired Ca/CaM-dependent inactivation of Cav1.2 will result in greater Ca2+ influx during each beat, leading to further Ca2+ loading of the myocytes especially during catecholaminergc stimulation, which, in turn, increases the probability of spontaneous diastolic Ca2+ releases. Premature spontaneous Ca2+ release from the SR is generally accepted as the culprit for CPVT [38, 48]. Interestingly, it was reported that Ca2+ channel blocker verapamil provides added benefit in CPVT patients when administered along with β-blockers [46, 47], a finding that is consistent with our hypothesis that enhanced Ca2+ influx via Cav1.2 contributes to excess myocyte Ca loading and arrhythmogenesis in Casq2 KO myocytes. Analogous to CPVT, increased SR Ca2+ leak and DADs have also been observed in animal models of heart failure [48, 49]. It is intriguing to speculate that impaired CaM dependent Cav1.2 may also contribute to arrhythmogenesis in heart failure and other heart diseases associated with increased SR Ca2+ leak, a hypothesis that should be tested in future studies.
One could argue that excess CaM used in our voltage clamp experiments can also inhibit RyR2 and in this way restore refractoriness of SR Ca2+release. While CaM is a physiological inhibitor of RyR2, it binds with high affinity to RyR2 and the CaM inhibitory action on RyR2 is saturated at physiological free concentrations of 100nM. As such, excess CaM will not further inhibit RyR2 channels and hence is not expected to contribute to the normalization of Ca2+ release refractoriness.
5. Summary and Conclusions
In the presented study we investigated mechanisms responsible for SR Ca2+ release refractoriness in cardiac muscle. We find that gene-targeted ablation of Casq2 results in complete loss of SR Ca2+ release refractoriness (Fig. 2&3). Surprisingly, accelerated recovery of ICa (Fig. 4) also contributed to the loss of SR Ca2+ release restitution in mice lacking Casq2. We found that the faster recovery of ICa in Casq2 KO mice is due to impaired Ca2+-dependent inactivation of Cav1.2 (Fig. 5), which could be reversed by excess apo-CaM (Fig. 6). Because excess CaM also partially restored release refractoriness in Casq2 KO myocytes (Fig. 7), these results suggest that impaired Ca2+-CaM-dependent inactivation of Cav1.2 independently contributes to loss of SR Ca2+ release refractoriness in Casq2 KO mice. We conclude that impaired CaM-dependent Cav1.2 regulation may independently contribute to arrhythmogenesis in CPVT, a new concept that could also be relevant in other heart diseases (e.g. heart failure) associated with increased SR Ca2+ leak.
Supplementary Material
Highlights.
Ablation of Casq2 results in complete loss of SR Ca2+ release restitution.
Accelerated recovery of ICa contributes to this phenomenon in Casq2 KO mice.
ICa recovers faster in Casq2 KO cells due to impaired Ca2+-dependent inactivation of Cav1.2.
Accelerated recovery of ICa in Casq2 KO mice can be reversed by excess apo-CaM.
Excess apo-CaM partially restores SR Ca2+ release restitution in Casq2 KO myocytes.
Acknowledgments
Funding
This work was partly supported by the United States National Institutes of Health [HL88635, HL71670 & HL108173 to B.C.K.]; by the American Heart Association [13IRG13680003 to B.C.K., 12POST12080080 to D.K.], and by the Australian National Health & Medical Research Council [APP1005974 to B.C.K.].
Footnotes
Conflict of Interest
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca(2)(+) release and reduces rate-dependent Ca(2)(+)-alternans in intact mouse hearts. J Mol Cell Cardiol. 2012;52:21–31. doi: 10.1016/j.yjmcc.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Gyorke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plank DM, Yatani A, Ritsu H, Witt S, Glascock B, Lalli MJ, Periasamy M, Fiset C, Benkusky N, Valdivia HH, Sussman MA. Calcium dynamics in the failing heart: restoration by beta-adrenergic receptor blockade. Am J Physiol Heart Circ Physiol. 2003;285:H305–315. doi: 10.1152/ajpheart.00425.2002. [DOI] [PubMed] [Google Scholar]
- 8.Terracciano CM, Hardy J, Birks EJ, Khaghani A, Banner NR, Yacoub MH. Clinical recovery from end-stage heart failure using left-ventricular assist device and pharmacological therapy correlates with increased sarcoplasmic reticulum calcium content but not with regression of cellular hypertrophy. Circulation. 2004;109:2263–2265. doi: 10.1161/01.CIR.0000129233.51320.92. [DOI] [PubMed] [Google Scholar]
- 9.Jones LR, Cala SE. Biochemical evidence for functional heterogeneity of cardiac sarcoplasmic reticulum vesicles. J Biol Chem. 1981;256:11809–11818. [PubMed] [Google Scholar]
- 10.Cala SE, Jones LR. Rapid purification of calsequestrin from cardiac and skeletal muscle sarcoplasmic reticulum vesicles by Ca2+-dependent elution from phenyl-sepharose. J Biol Chem. 1983;258:11932–11936. [PubMed] [Google Scholar]
- 11.Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J Biol Chem. 1983;258:1197–1204. [PubMed] [Google Scholar]
- 12.Kobayashi YM, Jones LR. Identification of triadin 1 as the predominant triadin isoform expressed in mammalian myocardium. J Biol Chem. 1999;274:28660–28668. doi: 10.1074/jbc.274.40.28660. [DOI] [PubMed] [Google Scholar]
- 13.Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270:30787–30796. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 15.Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228:34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- 16.Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flucher BE, Franzini-Armstrong C. Formation of junctions involved in excitation-contraction coupling in skeletal and cardiac muscle. Proc Natl Acad Sci U S A. 1996;93:8101–8106. doi: 10.1073/pnas.93.15.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bers DM. Excitation-contraction coupling and cardiac contractile force. Dordrecht; Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 19.Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. doi: 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- 20.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC. Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res. 2007;101:617–626. doi: 10.1161/CIRCRESAHA.107.157552. [DOI] [PubMed] [Google Scholar]
- 23.Kass RS, Sanguinetti MC. Inactivation of calcium channel current in the calf cardiac Purkinje fiber. Evidence for voltage- and calcium-mediated mechanisms. J Gen Physiol. 1984;84:705–726. doi: 10.1085/jgp.84.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira G, Yi J, Rios E, Shirokov R. Ion-dependent inactivation of barium current through L-type calcium channels. J Gen Physiol. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 27.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polakova E, Illaste A, Niggli E, Sobie EA. Maximal acceleration of Ca2+ release refractoriness by beta-adrenergic stimulation requires dual activation of kinases PKA and CaMKII in mouse ventricular myocytes. J Physiol. 2014 doi: 10.1113/jphysiol.2014.278051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu OZ, Lederer WJ, Sobie EA. Does the Goldilocks Principle apply to calcium release restitution in heart cells? J Mol Cell Cardiol. 52:3–6. doi: 10.1016/j.yjmcc.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 32.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 34.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 35.Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M, Borglum AD. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschafl BA, Arvanitis DA, Fuentes O, Yuan Q, Kranias EG, Valdivia HH. Dual role of junctin in the regulation of ryanodine receptors and calcium release in cardiac ventricular myocytes. J Physiol. 589:6063–6080. doi: 10.1113/jphysiol.2011.215988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe H, Knollmann BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol. 44:650–655. doi: 10.1016/j.jelectrocard.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 39.Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. 1999;79:661–682. doi: 10.1152/physrev.1999.79.3.661. [DOI] [PubMed] [Google Scholar]
- 40.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 41.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 42.Ben Johny M, Yang PS, Bazzazi H, Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun. 4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oz S, Benmocha A, Sasson Y, Sachyani D, Almagor L, Lee A, Hirsch JA, Dascal N. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+-binding protein 1. J Biol Chem. 288:12680–12691. doi: 10.1074/jbc.M113.460949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1. 2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 45.Haeseleer F, Sokal I, Verlinde CL, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca(2+)-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swan H, Laitinen P, Kontula K, Toivonen L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005;16:162–166. doi: 10.1046/j.1540-8167.2005.40516.x. [DOI] [PubMed] [Google Scholar]
- 47.Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, Belhassen B, Viskin S. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149–1154. doi: 10.1016/j.hrthm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 48.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 49.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.