

Abstract
Tumor heterogeneity makes combination chemotherapy one of the preferred modes of treatment regimens. In this work, sequential exposure of two anticancer agents, paclitaxel (Tx) followed by topotecan (TPT), was shown to have a synergistic effect on non-small cell lung cancer (NSCLC) cell line, NCI-H460. In order to improve patient compliance, the aforementioned concept was translated into a drug delivery system comprising of poly(d,l-lactide-co-glycolide) (PLGA)–chitosan composite particles. TPT-containing chitosan micro-/nanoparticles were prepared by the facile technique of electrospraying and encapsulated within PLGA microparticles using emulsion-solvent evaporation technique for delayed release of TPT. The formulation containing Tx- and TPT-loaded composite particles demonstrated synergism when exposed to NCI-H460 cellular aggregates (tumoroids) generated in vitro. Overall, the results of this study demonstrated the potential of the formulation containing Tx and PLGA–chitosan (TPT-loaded) composite particles for the treatment of lung cancer.
Keywords: drug delivery system, solid tumor, paclitaxel, topotecan, sequential administration
Introduction
Tumor heterogeneity and adaptability have limited the application of conventional single anticancer agent treatment regimen. As a consequence, there has been a gradual shift toward combination treatment protocols that involve the use of two or more chemotherapeutic agents with different modes of action. Further, it has been demonstrated that some anticancer agents such as paclitaxel (Tx) improve response rates following combination with other anticancer agents as compared to the single-agent (only Tx) regimen.1,2 In most cases of combination treatments, the drugs act on dividing cells, wherein the cancer cell population with reduced sensitivity (or resistance) to one type of anticancer drug is acted upon by a second drug in order to obtain an improved or additive/synergistic response.3
In combination chemotherapy, anticancer agents can be administered either simultaneously or sequentially, depending on the mode of action of the two drugs and their toxicity profiles. For example, in a previous study, simultaneous administration of irinotecan (topoisomerase 1 inhibitor) and axitinib (tyrosine kinase inhibitor) demonstrated enhanced antiproliferative and proapoptotic activities on human dermal microvascular endothelial cells and pancreatic cancer cells as compared to the individual drugs.4 In another study, sequential exposure of two anticancer agents – Tx, a microtubule-interfering agent, followed by topotecan (TPT), a topoisomerase-1 inhibitor5 – demonstrated a synergistic response on a colon cancer cell line.6 However, in the aforementioned cases, these drugs were administered in free form. Administration of anticancer agents in free form to patients can be associated with certain disadvantages such as poor solubility (in case of Tx), poor stability, reduced drug half-life, dose-related toxicity, increased side effects, reduced patient compliance, and increased costs to achieve a therapeutic concentration.7,8 In order to overcome the aforementioned disadvantages, drugs are usually loaded as cargo in a delivery vehicle that would release them in a sustained manner.9–11 These therapeutic cargo carriers can enhance the stability (half-life) of the anticancer drugs and can increase patient compliance by decreasing the number of doses.8,12–14 In addition to this, drug carriers can be modulated to deliver the anticancer drugs to a specific target site (using targeting molecules or by intratumoral injection), thereby increasing bioavailability and decreasing systemic toxicity.15,16 This study involves the development of a drug delivery system (DDS) that employs TPT and Tx as the drugs for combination chemotherapy.
Among the various cancer types, lung cancer is the leading cause of cancer-related deaths throughout the world.17 Therefore, this study intended to establish a DDS for lung cancer treatment. Although a few studies have reported the application of Tx- or TPT-loaded microparticles or nanoparticles in the treatment of lung cancer as single-agent treatment vehicles,18–21 none of them have employed both Tx and TPT in a single formulation. Since it has been reported that Tx followed by TPT (sequential administration) was synergistic on colon cancer cells and lung cancer patients,6,22 we proposed to translate the aforementioned concept in the form of a core–shell-type DDS. The DDS would provide immediate release of Tx, followed by a drug-free period (or minimal TPT release period), which in turn would be followed by sustained release of TPT (Figure 1). Immediate release of Tx would be provided by dispersing Tx in a colloidal suspension of core–shell composite particles. Further, since the clinical treatment regimen of TPT follows the cycle of intravenous infusion every day for 5 days,22 it was hypothesized that the TPT release from the DDS should be sustained for a period of 5 days.
Figure 1.

Schematic representing the proposed events following administration of formulation containing Tx- and TPT-loaded PLGA–chitosan composite particles to an in vitro grown 3-D tumoroid.
Note: The up arrows indicate an increase in BcL-xL phosphorylation and topoisomerase 1 levels.
Abbreviations: 3-D, three-dimensional; PLGA, poly(d,l-lactide-co-glycolide); TPT, topotecan; Tx, paclitaxel.
The core–shell-type particulate DDS was fabricated using a pH-responsive core made from chitosan, which is a copolymer of N-acetyl-d-glucosamine and d-glucosamine units, obtained by alkaline deacetylation of chitin.23 Chitosan has a pKa of 6.2–6.8, which matches the acidic pH of solid tumors.24 Under mildly acidic pH conditions, chitosan remains protonated and hence swells, thereby allowing a faster drug release as compared to neutral/alkaline pH conditions.25 The shell of the formulation was fabricated using poly(d,l-lactide-co-glycolide) (PLGA), 50:50, which would allow for faster degradation due to relatively higher glycolic acid content.26 Further, PLGA has been reported to degrade faster under acidic conditions (seen in tumors) as compared to neutral pH.27 The degradation products of PLGA (lactic acid and glycolic acid) would further decrease the local pH, thereby enhancing drug release from chitosan particles. To the best of our knowledge, this is the first study that reports the sequential delivery of Tx followed by TPT using a single depot of Tx dispersed in a suspension of PLGA–chitosan composite particulate delivery system.
Materials and methods
Materials
TPT and Tx were received as gift samples from Cipla, India, and their stock solutions were prepared in double distilled water and dimethyl sulphoxide (DMSO), respectively. Chitosan (low viscosity) (≤200 mPa·s) and d-,l-glyceraldehyde were procured from Sigma-Aldrich, St Louis, MO, USA, and PLGA (50:50) (0.16–0.24 dL/g) was procured from Boehringer Ingelheim, Ingelheim, Germany, and used as received. Non-small cell lung cancer (NSCLC) cell line, NCI-H460, was procured from National Center for Cell Science, Pune, India, and was maintained at 37°C and 5% CO2 in RPMI-1640 media (Sigma-Aldrich). The media was supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 10 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer (Hi-Media, Mumbai, India), 100 mM sodium pyruvate (Hi-Media), 100 U/mL penicillin (Hi-Media), 100 μg/mL streptomycin (Hi-Media), and 0.25 μg/mL amphotericin B (Hi-Media).
Cytotoxicity studies with TPT and Tx
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as reported previously.28 Details of cell viability experiments are mentioned in the supplementary material. For all combination treatment experiments, the nature of drug interaction was analyzed by plotting isobolograms and determining combination index (CI) values.29,30 CI values were calculated using the formula,
where CTx, x and CTPT, x are concentrations of Tx and TPT used in combination, respectively (simultaneous or sequential), to cause x% (50% in this case) cell death; and ICx, Tx and ICx, TPT are concentrations of Tx and TPT used individually to cause the same effect, ie, 50% cell death. CI value of less than 1 indicates synergism, equal to 1 indicates additive effect, and greater than 1 indicates antagonism.
Preparation of chitosan micro-/nanoparticles
Chitosan micro-/nanoparticles were fabricated by a one-step electrospraying technique as reported previously.31–33 Electrospraying was chosen since it is known to provide high encapsulation efficiencies of therapeutic molecules.34,35 Details of electrospraying experiments are mentioned in the supplementary material.
Characterization of chitosan micro-/nanoparticles
Morphology of chitosan micro-/nanoparticles was characterized using scanning electron microscopy. For this, the samples deposited on a collector plate were sputter coated with gold and imaged using an FEI Quanta 200 at a working distance of 10 mm and an accelerating voltage of 20 kV.
For size and zeta potential analysis, the samples were dispersed in deionized water using a probe sonicator (VCX-750, Sonics and Materials, Newton, CT, USA), and the experiments were performed using a Zeta Sizer (ZS90, Malvern Instruments, UK).
X-ray diffraction (XRD) studies were performed on TPT-loaded and unloaded chitosan micro-/nanoparticles in order to characterize the crystallinity of TPT within the chitosan micro-/nanoparticles, with an XRD pattern of plain TPT acting as control. The experiments were performed using ARL-XTRA (Thermo Electron Corporation, Waltham, MA, USA), and data were collected over an angular range of 7°–40° (2θ) at a scanning rate of 3° per minute with a step size of 0.05°.
Coating of chitosan micro-/nanoparticles
Chitosan micro-/nanoparticles were coated with PLGA using oil-in-water emulsion-solvent evaporation technique. Prior to the coating of chitosan micro-/nanoparticles with PLGA, they were cross-linked using d-,l-glyceraldehyde as described previously.36 The details of these experiments can be found in the supplementary material.
Characterization of PLGA–chitosan composite particles
The morphology of PLGA–chitosan composite particles was characterized using scanning electron microscopy (refer “Characterization of chitosan micro-/nanoparticles”), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), and size and zeta potential (refer “Characterization of chitosan micro-/nanoparticles”). The details for TEM and FTIR studies can be found in the supplementary material.
Encapsulation efficiency and drug release
Encapsulation efficiency and drug release experiments from the chitosan micro-/nanoparticles and PLGA–chitosan composite particles were performed, the details of which are mentioned in the supplementary material.
Bioactivity of chitosan micro-/nanoparticles and PLGA–chitosan composite particles
To assess the stability of TPT in TPT-loaded chitosan micro-/nanoparticles and compatibility of chitosan micro-/nanoparticles (following exposure to high solvent concentration and high voltages during electrospraying), NCI-H460 cells were exposed to TPT-loaded and unloaded particles in separate experiments, details of which can be found in the supplementary material. In order to provide a more appropriate simulation of the tumor microenvironment, NCI-H460 tumoroids grown on chitosan–gelatin (CG) hydrogels were used for testing the bioactivity of the composite DDS.37 It has been reported previously that these tumoroids are similar to tumors grown in vivo in terms of factors involved in tumor growth, tumor cell – extracellular matrix interaction, tumor invasion and metastasis, and drug resistance.37 Therefore, these tumoroids can act as a bridging gap between the conventional two-dimensional (2-D) culture and tumors grown in vivo. The details of this study can be found in the supplementary material.
Statistical analysis
All quantitative results were expressed as mean ± standard deviation. Statistical analysis was performed using Student’s t-test. All values of P<0.05 were considered to be statistically significant.
Results and discussion
Combination of TPT and Tx on lung cancer cell lines
The first set of experiments was performed to determine the concentrations of TPT and Tx to be used in combination therapy on the lung cancer cell line, NCI-H460. Figure 2A and B show concentration-dependent death of NCI-H460 cells following treatment with TPT and Tx, respectively. The half maximal inhibitory concentration (IC50) values for TPT and Tx at the end of 24 hours were found to be 200 and 40 nM, respectively. The data indicated that NCI-H460 cells were more sensitive to Tx than to TPT.
Figure 2.

Cell viability of NCI-H460 cells.
Notes: (A and B) Cell viability (percentage of control) of NCI-H460 cells at the end of 24 hours following treatment with increasing concentrations of TPT (A) and Tx (B). n=3. (C) Cell viability (percentage of control) of NCI-H460 cells at the end of 48 hours (24-hour drug incubation, followed by 24-hour drug-free incubation) following combination (simultaneous) treatment with TPT and Tx (ratio 5:1). n=3. (D) Cell viability (percentage of control) of NCI-H460 cells at the end of 72 hours (treatment with Tx [5 hours], followed by drug-free incubation period [19 hours], followed by treatment with various concentrations of TPT [24 hours], followed by drug-free incubation period for 24 hours). *Indicates significance (P<0.05) between additive effect of Tx and TPT when treated individually vs their sequential treatment. n=3.
Abbreviations: TPT, topotecan; Tx, paclitaxel.
The combination experiments were performed on NCI-H460 cells based on the IC50 values obtained in the previous experiment. NCI-H460 cells (Figure 2C) demonstrated antagonism when exposed to a simultaneous treatment of TPT and Tx as compared to the individual treatments. This was probably because either of the drugs may be responsible for 1) decreased uptake of the second drug by cancer cells, 2) reduced binding of the second drug to its target site, 3) modulating cellular response to the second drug, or 4) modulating the ability of each other to damage cells.38
On the other hand, sequential delivery of Tx preexposure followed by a drug-free incubation period and then TPT exposure was synergistic on NCI-H460 cells (Figure 2D). In particular, treatment of NCI-H460 cells with only TPT (0.1–10 nM) led to no cell death as compared to control cells. However, when these cells were pretreated with Tx (0.1–100 nM), followed by a drug-free incubation period (19 hours), and then treated with TPT (0.1–10 nM), there was a significant increase in cell death (P<0.05) as compared to the additive effect of individual treatment groups. This was probably because of the following reasons:6 1) Tx preexposure led to phosphorylation of an antiapoptotic protein BcL-xL that makes NCI-H460 cells more vulnerable to TPT and, as a consequence, apoptosis;39,40 2) Tx preexposure also resulted in an increase in topoisomerase 1, which is the substrate for the second drug, TPT (enhanced topoisomerase 1 levels may be because of the increased stability of topoisomerase-1 enzyme or RNA due to their interaction with Tx); and/or 3) the drug-free incubation period allowed an increase in the percentage of cells in S phase of cell cycle, thereby increasing the probability of TPT–topoisomerase 1–DNA complexes and, eventually, cell death.
Further analysis of the effect of the combination of TPT and Tx on NCI-H460 cells was performed using the isobole method29,30 (Figure 3). Tx and TPT concentrations that led to 50% cell death were plotted on the X- and Y–axes, respectively, and the diagonal line joining these two points represented the zero isobole. Simultaneous treatment of Tx and TPT at concentrations that led to a 50% reduction in NCI-H460 cells as compared to control cells were on the right side of the zero isobole line. Therefore, when provided simultaneously, the concentrations of TPT and Tx that led to 50% cell death were relatively higher (37.5 and 7.5 nM, respectively) as compared to the individual treatment of TPT or Tx, thereby demonstrating that the simultaneous treatment was antagonistic. The CI values were found to be 2.5 for NCI-H460 cells, indicating antagonism, corroborating data obtained from the viability assay (Figure 2C). On the other hand, concentrations of Tx and TPT that led to 50% decline in NCI-H460 cell viability when provided sequentially were on the left side of the zero isobole line, thereby showing that the sequential treatment was synergistic. The CI value for sequential administration of Tx followed by TPT was found to be 0.04 for NCI-H460 cells, indicating synergism and corroborating data obtained from the viability assay (Figure 2D).
Figure 3.
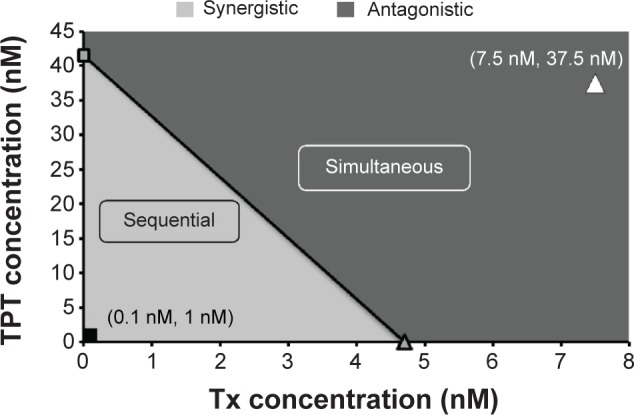
Isobologram of TPT and Tx on NCI-H460 cells demonstrating equitoxic doses of TPT and Tx either alone or in combination (simultaneous or sequential).
Notes: The diagonal line connects IC50 values of TPT and Tx and represents zero interaction isoboles. Values on the right side of the diagonal line indicate an antagonistic interaction between the drugs (simultaneous treatment), while those on the left side of the diagonal line indicate a synergistic interaction (sequential treatment).
Abbreviations: IC50, half maximal inhibitory concentration; TPT, topotecan; Tx, paclitaxel.
Taken together, these results indicated that simultaneous treatment of Tx and TPT was antagonistic, whereas sequential administration of Tx followed by TPT (with a drug-free incubation period in between) demonstrated synergism on NCI-H460 cells.
Characterization of TPT-loaded chitosan micro-/nanoparticles
Figure 4A shows a scanning electron micrograph of TPT-loaded chitosan micro-/nanoparticles demonstrating a smooth rounded morphology. The size of chitosan micro-/nanoparticles was 642.6±14.9 nm, with a polydispersity index of 0.283±0.012. Further, due to the presence of amine groups in chitosan, the zeta potential of chitosan micro-/nanoparticles was positive and determined to be 34.67±0.71 mV, indicating that the particles would be relatively stable in a suspension. XRD was performed to determine the crystallinity of TPT within TPT-loaded chitosan micro-/nanoparticles. Figure 4B shows the XRD patterns of TPT, chitosan micro-/nanoparticles, and TPT-loaded chitosan micro-/nanoparticles. Peaks in the XRD pattern corresponded to most of the characteristic peaks in crystalline form of TPT hydrochloride.41 However, no such peaks were observed in TPT-loaded chitosan micro-/nanoparticles or unloaded chitosan micro-/nanoparticles. Absence of peaks attributed to noncrystallinity of TPT in the TPT-loaded chitosan micro-/nanoparticles and indicated that TPT was homogenously dispersed and present at the molecular state within chitosan micro-/nanoparticles.42 Drug encapsulation efficiency of TPT-loaded chitosan micro-/nanoparticles was estimated to be approximately 100% for 1:20 of TPT:chitosan and 90.09% for 1:10 of TPT:chitosan. The high encapsulation efficiencies can be attributed to the hydrophilic nature of TPT and the favorable interaction between TPT and chitosan.
Figure 4.
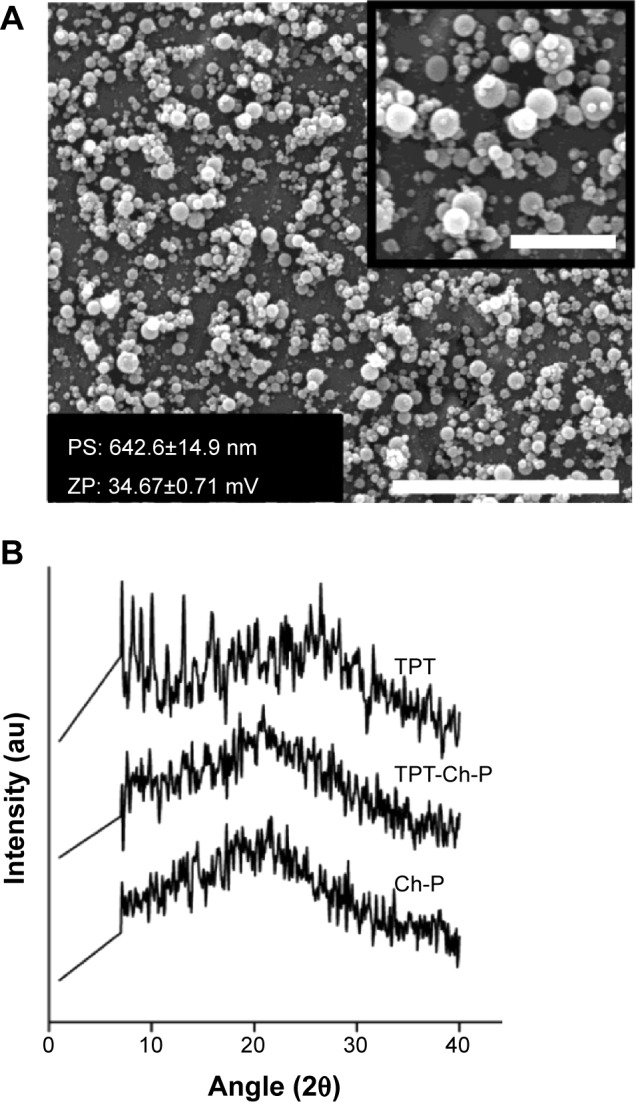
Characterization of chitosan micro-/nano-particles.
Notes: (A) Scanning electron micrograph of TPT-encapsulated Ch-P synthesized by the electrospraying technique (scale bar: 10 μm). Inset represents a higher magnification (scale bar: 2.5 μm). (B) X-ray diffraction pattern of TPT, Ch-P, and TPT-loaded Ch-P.
Abbreviations: au, arbitrary units; Ch-P, chitosan micro-/nanoparticles; PS, particle size; TPT, topotecan; ZP, zeta potential.
TPT release from chitosan micro-/nanoparticles
TPT release from chitosan micro-/nanoparticles was performed under two different pH conditions, pH 6.8 and 7.4 (due to the difference in pH between normal and tumor tissue24). Under both pH conditions studied, TPT from chitosan micro-/nanoparticles showed a burst release for the first 10 hours (Figure 5). This could be attributed to the presence of adsorbed TPT on the surface of chitosan micro-/nanoparticles that was readily released into the medium as a result of desorption. TPT release was faster in phosphate-buffered saline (PBS) (pH 6.8) as compared to PBS pH 7.4, indicating the influence of pH on TPT release from chitosan micro-/nanoparticles. This was probably because, at pH 6.8 and below, chitosan exists as a polycation (pKa for chitosan is between 6.2–6.8).43 Therefore, pH 6.8 (PBS) caused protonation of chitosan chains, whereas pH 7.4 (PBS) resulted in deprotonation of chitosan chains. The protonation in turn resulted in enhanced repulsion between amine groups of chitosan, which led to swelling of chitosan particles and faster TPT diffusion, thereby resulting in enhanced TPT release at pH 6.8 (PBS) as compared to pH 7.4 (PBS).44 It was also observed that the release profile of TPT from chitosan micro-/nanoparticles at pH 6.8 and 7.4 reached a plateau after 40 hours, which could be attributed to drug saturation or drug instability.
Figure 5.
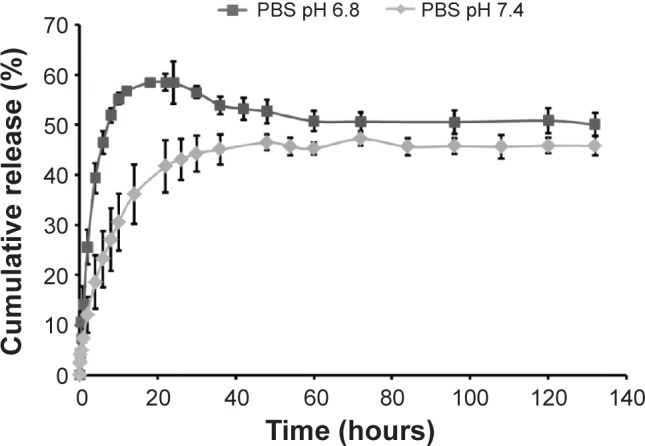
In vitro release kinetics of TPT from TPT-encapsulated chitosan micro-/nanoparticles in PBS; pH 6.8 and 7.4. n=3.
Abbreviations: PBS, phosphate-buffered saline; TPT, topotecan.
Hence, it can be concluded that chitosan micro-/nanoparticles would allow for faster drug release in tumor tissue (mildly acidic) as compared to healthy tissues.
Bioactivity of TPT released from chitosan micro-/nanoparticles
TPT was encapsulated within chitosan micro-/nanoparticles by electrospraying and thus was under the influence of high voltage and high solvent concentrations during the electrospraying process. As a result, its bioactivity could be adversely affected. Since this study was intended to develop a particulate delivery system for the treatment of lung cancer, bioactivity of TPT encapsulated in chitosan micro-/nanoparticles was tested on non-small lung cancer line, NCI-H460, as a function of time. It was observed that with increase in incubation period, there was a decrease in cell viability in NCI-H460 cells (Figure 6). Therefore, TPT released from chitosan micro-/nanoparticles was active in spite of the harsh conditions used during electrospraying and was being released in a continuous manner that resulted in increased cell death with increase in time. However, the placebo particles caused minimal cell death, indicating their cytocompatibility. Further, the active lactone form of TPT is more stable under acidic pH conditions and hence predominates under such conditions.5 Since the conditions used to synthesize chitosan micro-/nanoparticles and the tumor pH are acidic in nature, TPT is expected to be stable and retain its activity during the electrospraying process as well as in solid tumors.
Figure 6.
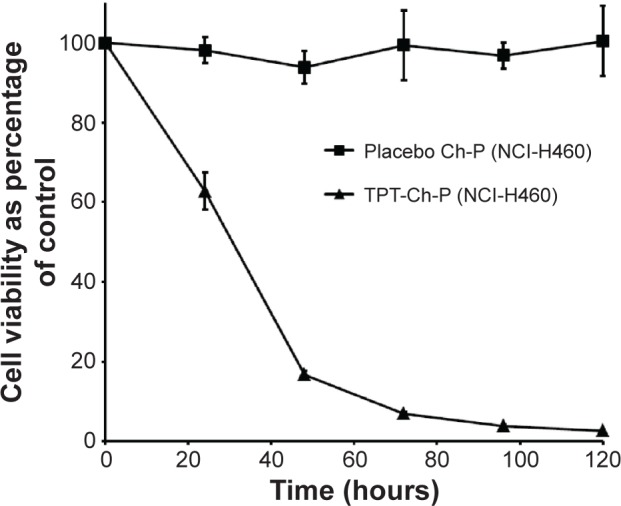
Bioactivity (cell viability) of TPT-loaded Ch-P (0.05 mg/mL) on NCI-H460 cells. n=3.
Abbreviations: Ch-P, chitosan micro-/nanoparticles; TPT, topotecan.
Fabrication of PLGA–chitosan composite particles
For the sequential delivery system to be effective, there is a need for a drug-free incubation period between the two drug treatments. The strategy employed in this work to enable the drug-free incubation time of approximately 19 hours was to coat the chitosan particles with relatively fast hydrolytically degrading polymer – PLGA (50:50). For the synthesis of PLGA-coated chitosan composite particles, chitosan micro-/nanoparticles were cross-linked with d-,l-glyceraldehyde to prevent TPT loss from chitosan particles while coating. The encapsulation efficiency of TPT in chitosan micro-/nanoparticles decreased to 27.95% following cross-linking as compared to uncross-linked particles. This was probably because of the loss of the adsorbed TPT on chitosan particle surface during cross-linking and washing procedures.
Figure 7A shows a scanning electron micrograph of PLGA-coated chitosan particles. The average size of composite particles was approximately 2,134±67.5 nm, with a polydispersity index of 0.343±0.024. The large difference in particle size between composite particles (2,134±67.5 nm) and chitosan particles (642.6±14.9 nm) indicated the possible presence of multiple chitosan particles within one PLGA microparticle. This speculation was further supported by the presence of undulations on the surface of PLGA–chitosan composite particles, which were probably due to multiple chitosan particles within one PLGA particle.45 These undulations/blebs were absent in plain PLGA (data not shown) or chitosan particles (Figure 4A). In order to confirm PLGA coating on chitosan micro-/nanoparticles, TEM of composite particles was performed. Presence of a darker shell around a lighter core indicated the presence of a PLGA shell around chitosan micro-/nanoparticles core (Figure 7B). TPT encapsulation efficiency of PLGA–chitosan composite particles was 13.96% as against 27.97% in glyceraldehyde-cross-linked chitosan particles. The decrease in TPT loading in coated particles as compared to uncoated could be attributed to the hydrophilicity of TPT that led to the loss during coating process.
Figure 7.

Characterization of PLGA-chitosan composite microparticles.
Notes: (A) Scanning electron micrograph of PLGA–Ch-P (scale bar: 2 μm). (B) Transmission electron micrograph of PLGA-Ch-P. The PLGA shell is indicated by the darker exterior due to the presence of iron oxide particles (6 nm) (scale bar: 0.5 μm). (C) FTIR analysis of PLGA-Ch-P (unloaded and TPT loaded), Ch-P, and PLGA powder. Arrow indicates peak at 1,760 cm–1 corresponding to the C=O stretch that is characteristic of PLGA. (D) Zeta potential of Ch-P and PLGA-Ch-P. n=3.
Abbreviations: Ch-P, chitosan micro-/nanoparticles; FTIR, Fourier transform infrared spectroscopy; PLGA, poly(d,l-lactide-co-glycolide); PLGA-Ch-P, poly(d,l-lactide- co-glycolide)–chitosan composite microparticles; PS, particle size; TPT, topotecan.
Further, FTIR of chitosan micro-/nanoparticles, PLGA, and PLGA–chitosan composite particles (TPT loaded and unloaded) was performed to demonstrate the presence of PLGA coating on chitosan micro-/nanoparticles (Figure 7C). The characteristic peak of the ester bond (C=O stretch) of PLGA was observed at 1,760 cm−1 46 for PLGA and PLGA-coated chitosan composite particles (TPT loaded and unloaded) and was absent in chitosan micro-/nanoparticles. This confirmed the presence of PLGA in composite particles.
Zeta potential of PLGA–chitosan composite particles decreased from a positive value of +34.6±0.7 mV to a negative value of −6.99±2.44 mV (Figure 7D), further corroborating the presence of PLGA coating on chitosan micro-/nanoparticles.
Taken together, the results of this study confirmed the presence of PLGA coating on chitosan micro-/nanoparticles resulting in the formation of PLGA–chitosan composite particles.
TPT release from PLGA–chitosan composite particles
Since the goal of this study was to devise a DDS that enables delayed release of TPT from PLGA–chitosan composite particles, a drug release study was performed in PBS at two pH values, pH 6.8 (similar to tumor tissue pH) and 7.4 (physiological pH) (Figure 8), as explained in “TPT release from chitosan micro-/nanoparticles” section. It was observed that only 23.7%±4.1% of TPT was released during the first 24 hours at pH 6.8 (pH of tumor tissue) and 16.4%±3.1% at pH 7.4 (physiological pH), thereby preventing the initial burst release as observed in uncoated particles (Figure 5). With increase in time of incubation, there was an increase in TPT release from PLGA-coated chitosan composite particles over a period of 144 hours, probably attributed to the erosion process. As observed for chitosan micro-/nanoparticles (Figure 5), the drug release from composite particles was also faster at pH 6.8 as compared to pH 7.4, due to increased protonation of chitosan at pH 6.8 as against de-protonation at pH 7.4 (Figure 8). Higher drug release at pH 6.8 can also be attributed to faster degradation of PLGA, whose degradation is known to be catalyzed in an acidic environment.27 The sensitivity of PLGA and chitosan to an acidic environment led to an overall increase in TPT release kinetics at pH 6.8 as compared to pH 7.4. This was clearly evident from the cumulative release at the 144-hour time point, which was 56.4%±5.9% at pH 7.4 and 90.66%±8.9% at pH 6.8. Further, during the first 6 hours when Tx would be administered, approximately 12.5%±3.5% TPT was released at pH 6.8 and 7.2%±2.1% TPT was released at pH 7.4, indicating minimal interaction between Tx and TPT, which would otherwise lead to antagonism.
Figure 8.
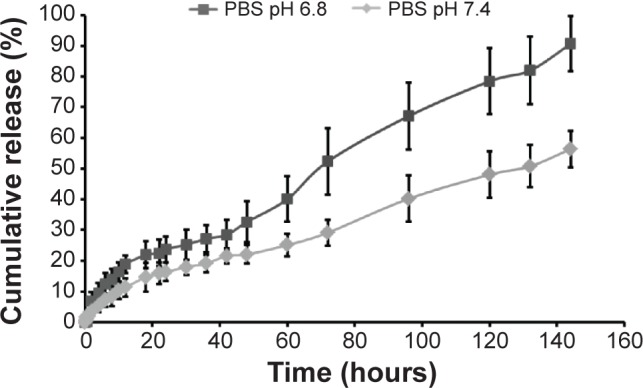
In vitro release kinetics of TPT from PLGA–chitosan composite microparticles in PBS; pH 6.8 and 7.4. n=3.
Abbreviations: PLGA, poly(d,l-lactide-co-glycolide); PBS, phosphate-buffered saline; TPT, topotecan.
Taken together, the results of this study indicated that the PLGA coating led to a decrease in burst release of TPT (irrespective of the pH) and that TPT released from coated particles was higher at pH 6.8 than at pH 7.4.
Bioactivity of formulation (Tx + TPT-loaded PLGA–chitosan composite particles)
Bioactivity of the formulation on NCI-H460 tumoroids generated on three-dimensional (3-D) CG scaffolds (Figure S1) was analyzed. In the first set of experiments, these tumoroids were exposed to a simultaneous treatment of TPT and Tx (ratio 5:1) and compared with individual TPT and Tx treatment. It was observed that the simultaneous treatment was antagonistic (Figure S2), thereby corroborating the results obtained on 2-D TCPS surfaces (Figure 2C). In the next set of experiments, the tumoroids grown on CG scaffolds were exposed to sequential treatment of Tx, followed by TPT (5 hours of Tx preexposure, followed by a drug-free incubation period of 19 hours, followed by 24-hour treatment with TPT), and compared to individual treatment of Tx and TPT (matched for concentration and duration). Tx exposure followed by TPT treatment was demonstrated to be synergistic on 3-D tumoroids (Figure 9) and corroborated the results obtained on 2-D TCPS (Figure 2D). After demonstrating synergism using free drugs, the same study was performed using the developed formulation (Tx + TPT-loaded PLGA–chitosan composite particles). It was observed that the NCI-H460 tumoroids demonstrated enhanced cell death following exposure to the formulation as compared to the additive effect of Tx (free drug) and TPT (PLGA-coated TPT-loaded chitosan composite particles) when provided individually (P<0.05) (Figure 9). It should be noted that the formulation was not able to provide a completely drug-free incubation period in between, leading to 23.7%±4.1% TPT release at pH 6.8. This could have adversely affected the synergism between Tx and TPT since their simultaneous treatment is antagonistic (Figure 2C). However, the formulation demonstrated synergism probably due to minimal interaction between Tx and TPT during the first 5 hours of Tx preexposure. Further, even though the decline in tumoroid viability following treatment with sequential administration of free drugs or drugs in formulation was insignificant, it should be noted that the formulation would be administered as a single dose that can significantly increase patient compliance. More specifically, the formulation could possibly avoid six intravenous injections (one for Tx and five for TPT) for the same dose. Further, since this formulation is being proposed for loco-regional therapy (intratumoral administration), it is expected that the systemic toxicity caused by intravenous administration of free drugs will be significantly minimized. This would lead to increased bioavailability at tumor site and decreased side effects. This DDS can especially find applications in inoperable obstructive NSCLC (ie, tracheal malignant neoplasms that cannot be resected by surgery)47 in the form of intratumoral injections that could result in a decrease in tumor size thereby rendering them operable. Further, in another study, it has been reported that tumor priming through apoptosis-inducing agents such as Tx and docetaxel has resulted in altered tumor architecture (in terms of interstitial space) and drug transport (convective transport and diffusion), thereby resulting in enhanced subsequent delivery of drug/nanoparticle.48 Similar tumor priming could possibly play a role in enhanced cell death using the developed formulation. These advantages, combined with increased patient compliance, demonstrated the clinical potential of the developed formulation for chemotherapy-based treatment of lung cancer.
Figure 9.
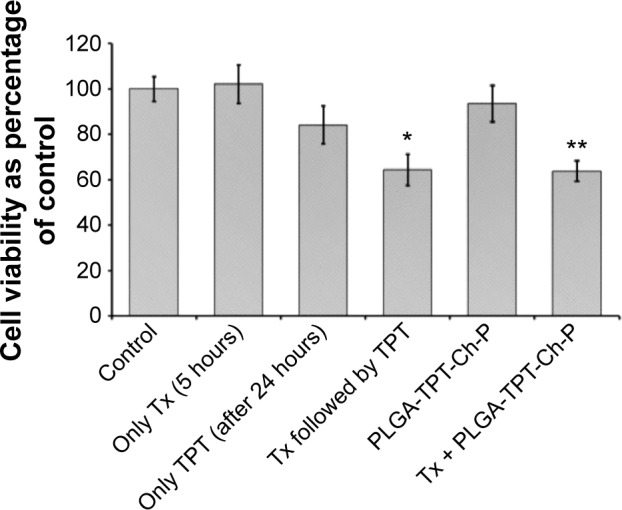
Cell viability (percentage of control) of NCI-H460 tumoroids generated on 3-D chitosan–gelatin scaffolds following treatment with Tx (5 hours), TPT (after 24 hours), Tx followed by TPT, TPT-loaded PLGA–chitosan composite microparticles, and combination of Tx- and TPT-loaded PLGA–chitosan composite particles.
Notes: Tx and TPT concentrations were 100 and 500 nM, respectively. *Indicates significance (P<0.05) between additive effect of Tx and TPT when treated individually vs their sequential treatment. **Indicates significance (P<0.05) between additive effect of Tx- and TPT-loaded PLGA–chitosan composite particles (Tx + PLGA-TPT-Ch-P) when treated individually vs their combination on NCI-H460 tumoroids. n=3.
Abbreviations: 3-D, three-dimensional; Ch-P, chitosan micro-/nanoparticles; PLGA, poly(d,l-lactide-co-glycolide); TPT, topotecan; Tx, paclitaxel.
Conclusion
This work involved the development of a drug delivery formulation that allows for combination treatment with Tx and TPT in a sequential manner. It was shown that independent exposure of the two drugs (free-state) demonstrated a concentration-dependent toxicity on a NSCLC line, NCI-H460. Further, simultaneous exposure of Tx and TPT on NCI-H460 cells demonstrated antagonism. On the other hand, sequential administration involving preexposure of Tx followed by a drug-free incubation period and then TPT exposure showed synergism.
This concept was then translated into a drug delivery formulation that allowed for Tx preexposure followed by a delayed release of TPT. For this, TPT was successfully encapsulated within a chitosan core by electrospraying technique. Since chitosan is pH responsive and swells under acidic conditions, chitosan micro-/nanoparticles demonstrated higher TPT release at acidic pH (tumor microenvironment) as compared to neutral conditions. TPT-loaded chitosan micro-/nanoparticles further demonstrated a decrease in NCI-H460 cell viability with an increase in incubation period, thereby indicating that TPT bioactivity remained unaltered in spite of high solvent/voltage conditions used during the process of electrospraying. TPT-loaded chitosan micro-/nanoparticles were successfully coated with PLGA (to enable a drug-free incubation period) by emulsion-solvent evaporation technique. The size of PLGA-coated chitosan composite particles was approximately 2,134±67.5 nm as against 642.6±14.9 nm for chitosan micro-/nanoparticles, indicating the possible presence of multiple chitosan particles within a single PLGA particle. Presence of a dark shell around the chitosan core in TEM, C=O stretch in FTIR study, and drop in surface charge of coated particles (from +34.6 to −6.99 mV) after coating together confirmed the presence of PLGA coating. Cumulative TPT release from the coated particles was restricted to 23.7%±4.1% (as against 58.5%±4.3% in uncoated particles) for the first 24 hours and gradually increased to 90.66%±8.9% at the end of 144 hours. The developed formulation comprising Tx- and TPT-loaded composite particles, when tested on an in vitro 3-D tumor model, demonstrated synergism as compared to the additive effect of Tx (free-state) and TPT (PLGA-coated TPT-loaded chitosan composite particle) when treated individually. Although a drug-free incubation period would have been desirable in the first 24 hours, the developed formulation showed a synergistic effect in spite of the TPT release (approximately 23%) during the first 24 hours. Such a formulation that provides a sequential delivery of Tx, followed by TPT at the target site, would increase drug half-life and patient compliance and improve treatment (ie, reduced side effects, improved efficiency, reduced cost to patient, reduced time in hospital for patient, and better utilization of hospital resources). Taken together, the formulation (Tx and PLGA-coated TPT-loaded chitosan composite particles) demonstrates potential to be used as a single depot combination therapy for lung cancer treatment. Although in our studies we demonstrate this combination treatment modality for lung cancer, it can be extrapolated to all other cancer types that use Tx and TPT in their treatment protocol.49
Supplementary materials
Cell viability – MTT assay
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which involves the conversion of MTT dye (Sigma-Aldrich, St Louis, USA) to a purple-colored formazan product, an indicator of cell metabolic activity/viability. Briefly, 5×103 cells were treated with increasing concentrations of topotecan (TPT) and paclitaxel (Tx). Following 24 hours of drug treatment, cell culture medium was removed and the cells were washed with 1× phosphate-buffered saline (PBS) (pH 7.4). They were then incubated with MTT dye (0.5 mg/mL in serum-free media) for 4 hours at 37°C. Following incubation, the media were removed and insoluble formazan crystals were dissolved in DMSO. The absorbance was then recorded using a microplate reader (Tecan, Sunrise) at 570 nm, with a reference wavelength of 650 nm. Cell viability of drug-treated samples was normalized and represented as percentage with respect to untreated control. The half maximal inhibitory concentration (IC50) values (drug concentrations that resulted in 50% decline in cell viability) obtained using the toxicity data of TPT and Tx on lung cancer cell lines were the ratios used for simultaneous treatment experiments. For sequential exposure of Tx and TPT, NCI-H460 cells were exposed to Tx for 5 hours, followed by a drug-free incubation period for 19 hours and then incubation with increasing concentrations of TPT for 24 hours. For all combination experiments (Tx and TPT for simultaneous, and TPT for sequential experiments), cells were treated with the drugs for 24 hours, following which they were incubated in drug-free medium for another 24 hours before performing the MTT assay.
Preparation of chitosan micro-/nanoparticles
Briefly, chitosan was dissolved in acetic acid (Merck, India) and was transferred to a glass syringe, connected to a syringe pump (Harvard Apparatus, 11 Plus), and subsequently electrosprayed to form micro-/nanoparticles under the influence of high voltage (gamma high voltage). For the synthesis of TPT-loaded chitosan micro-/nanoparticles, the drug was added to the polymer solution prior to electrospraying (1:10 and 1:20 ratio of TPT:polymer) and then electrosprayed. The optimized parameters for the synthesis of drug-loaded chitosan micro-/nanoparticles were chitosan concentration, 1.5% (w/v); acetic acid concentration, 90% (v/v); electrospraying distance, 12 cm; electrospraying voltage, 25 kV; needle gauge, 26 G; and flow rate, 0.1 mL/h.
Coating of chitosan micro-/nanoparticles
For coating of chitosan micro-/nanoparticles with PLGA, it was speculated that while PLGA solution forms particles during the oil-in-water emulsification, it entraps chitosan micro-/nanoparticles within, thereby resulting in the formation of PLGA–chitosan composite particles. For this, PLGA was dissolved in chloroform, and chitosan micro-/nanoparticles were dispersed in chloroform. Prior to dispersion in poly(vinyl alcohol) (PVA) which was dissolved in water, the aforementioned solutions were mixed together and emulsified in PVA for different rotation speeds and time periods. The emulsions were then stirred to allow for chloroform evaporation, centrifuged, washed twice with distilled water to remove PVA, and then freeze-dried to obtain powdered PLGA–chitosan composite particles. Various parameters for PLGA coating on chitosan micro-/nanoparticles were optimized by one-at-a-time optimization method. The parameters that were optimized were PLGA concentration (1%–2% w/v), homogenization speed (5,000–15,000 rpm) (IKA, T25 Ultra Turrax), PVA concentration (1%–5% w/v), and time of homogenization (5–15 minutes). The optimized parameters were 2% PVA (w/v) and 2% PLGA (w/v) homogenized for 5 minutes at 10,000 rpm.
For cross-linking of chitosan particles prior to coating, 0.6 mg of glyceraldehyde per milligram of chitosan micro-/nanoparticles was added to the chitosan micro-/nanoparticle suspension under magnetic stirring for 2 hours at 5°C. Following this, the particles were centrifuged at 10,000 rpm for 10 minutes, washed with acetone (precooled at 5°C), and then vacuum dried.
Characterization of PLGA-coated chitosan micro-/nanoparticles
For TEM, chitosan micro-/nanoparticles were emulsified along with PLGA in a solution of 2% PVA containing iron oxide nanoparticles (~6 nm). The samples were placed on a TEM grid (Tedpella, USA) following solvent evaporation and subsequent washing steps (to remove PVA) and imaged using a Tecnai 20 U Twin transmission electron microscope. For FTIR studies, chitosan micro-/nanoparticles, PLGA, and PLGA-coated chitosan particles (TPT loaded and unloaded) were thoroughly milled with KBr and pressed to form pellets, and spectra were then obtained using a Bruker FTIR Vertex-70.
Encapsulation efficiency and drug release
Chitosan micro-/nanoparticles (TPT-loaded [ratio 1:20] and unloaded) were dissolved in acetic acid, whereas chitosan micro-/nanoparticles (TPT loaded [ratio 1:10] and unloaded), cross-linked micro-/nanoparticles, and PLGA-coated chitosan composite particles were dissolved in HCl (as the cross-linked particles were insoluble in acetic acid) for encapsulation efficiency and drug-loading experiments. Using a calibration curve of TPT in acetic acid and HCl, the amount of TPT in each type of particle was measured and was represented using the following formula:
Since HCl was used only for calculating the encapsulation efficiency of TPT in cross-linked particles, its effect on drug stability was not evaluated.
For drug release experiments, release kinetics of TPT from Ch-P and PLGA-coated chitosan composite particles was studied in 0.1 M PBS (pH 6.8) and 0.1 M PBS (pH 7.4) (which are tumor tissue and physiological pH respectively) at 37°C in a water bath shaker at 50 rpm. Drug release experiments were performed in sink conditions, wherein the sample was withdrawn at regular time intervals and was replenished with fresh buffer. Absorbance of the sample was measured using a UV spectrophotometer (Thermo Electron Corporation, Helios UV 1). The amount of TPT was determined using previously plotted calibration curves of TPT in 0.1 M PBS (pH 6.8) and 0.1 M PBS (pH 7.4). TPT release was plotted as percentage cumulative release (%) as a function of time (h).
Bioactivity of chitosan micro-/nanoparticles and PLGA-coated chitosan composite particles
To study the bioactivity of chitosan micro-/nanoparticles, 5×103 cells were treated with 0.05 mg/mL chitosan micro-/nanoparticles (TPT loaded and unloaded in separate experiments) following 24 hours of seeding in a 96-well plate. MTT assay was performed at the end of 24, 48, 72, 96, and 120 hours (see “Cytotoxicity studies with TPT and Tx” section), and the data of cell viability were represented as percentage of control. For the bioactivity of composite particles, 1×105 cells suspended in 10 μL volume were seeded on 3-D chitosan–gelatin (CG) scaffolds. Cells were allowed to adhere on CG scaffolds for 2–3 hours, following which 500 μL of cell culture media was added to each well. The cells were allowed to grow for a period of 6 days on the CG scaffolds (Figure S1), following which the bioactivity of PLGA-coated chitosan composite particles were studied on them. In the first set, NCI-H460 tumoroids were treated with free Tx (100 nM), free TPT (500 nM), and combination of free Tx and free TPT (ratio 1:5) (Figure S2). In the second set, NCI-H460 tumoroids were treated with Tx (5 hours of preexposure), free TPT, free Tx followed by free TPT (Tx exposure for 5 hours, followed by drug-free incubation period of 19 hours, followed by free TPT exposure for 24 hours), PLGA-coated TPT-loaded chitosan composite particles, and combination of Tx and PLGA-coated TPT-loaded chitosan composite particles. MTT assay was performed at the end of 48 hours of incubation, and the values of cell viability were represented as percentage of control.
NCI-H460 cell spheroid/tumoroid generated on chitosan–gelatin 3-D scaffolds at the end of day 6.
Note: Scale bar: 50 μm.
Abbreviation: 3-D, three-dimensional.
Cell viability (percentage of control) of NCI-H460 tumoroids generated on 3-D chitosan–gelatin scaffolds following treatment with TPT, Tx, and Tx + TPT.
Notes: Concentrations of Tx and TPT were 100 and 500 nM, respectively. *Indicates significance (P<0.05) between additive effect of Tx and TPT when treated individually vs their simultaneous treatment on NCI-H460 tumoroids (n=3).
Abbreviations: 3-D, three-dimensional; TPT, topotecan; Tx, paclitaxel.
Acknowledgments
NA acknowledges financial support from the Council of Scientific and Industrial Research (CSIR), India. DSK is a “Class-of-1970 Research Fellow” and thanks the Department of Biotechnology (DBT), India, for research funding and Cipla, India, for the gift sample of anticancer drugs. DSK and NA also thank Professor Gajbhiye, Department of Chemistry, IIT Kanpur, for iron oxide nanoparticles.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Socinski MA. Single-agent paclitaxel in the treatment of advanced non-small cell lung cancer. Oncologist. 1999;4(5):408–416. [PubMed] [Google Scholar]
- 2.Belani CP. Paclitaxel and docetaxel combinations in non-small cell lung cancer. Chest. 2000;117(4 Suppl 1):144S–151S. doi: 10.1378/chest.117.4_suppl_1.144s. [DOI] [PubMed] [Google Scholar]
- 3.Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv. 2007;7(4):216–223. doi: 10.1124/mi.7.4.8. [DOI] [PubMed] [Google Scholar]
- 4.Canu B, Fioravanti A, Orlandi P, et al. Irinotecan synergistically enhances the antiproliferative and proapoptotic effects of axitinib in vitro and improves its anticancer activity in vivo. Neoplasia. 2011;13(3):217–229. doi: 10.1593/neo.101334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 6.Bahadori HR, Greem MR, Catapano CV. Synergistic interaction between topotecan and microtubule-interfering agents. Cancer Chemother Pharmacol. 2001;48:188–196. doi: 10.1007/s002800100327. [DOI] [PubMed] [Google Scholar]
- 7.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 8.Cho K, Wang X, Nie S, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 9.Langer R. New methods of drug delivery. Science. 1990;249(4976):1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 10.Moghimi SM, Kissel T. Particulate nanomedicines. Adv Drug Deliv Rev. 2006;58(14):1451–1455. doi: 10.1016/j.addr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream. Science. 2004;303(5665):1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 12.Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. doi: 10.1016/j.addr.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Moorthi C, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci. 2011;14(1):67–77. doi: 10.18433/j30c7d. [DOI] [PubMed] [Google Scholar]
- 14.Swami A, Shi J, Gadde S, Votruba A, Kolishetti N, Farokhzad O. Nanoparticles for targeted and temporally controlled drug delivery. In: Svenson S, Prud’homme RK, editors. Multifunctional Nanoparticles for Drug Delivery Applications. New York, NY: Springer; 2012. pp. 9–29. [Google Scholar]
- 15.Parveen S, Sahoo SK. Polymeric nanoparticles for cancer therapy. J Drug Target. 2008;16(2):108–123. doi: 10.1080/10611860701794353. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011. CA Cancer J Clin. 2011;61(4):212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 18.Fonseca C, Simões S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 19.Alipour S, Montaseri H, Tafaghodi M. Preparation and characterization of biodegradable paclitaxel loaded alginate microparticles for pulmonary delivery. Colloids Surf B Biointerfaces. 2010;81(2):521–529. doi: 10.1016/j.colsurfb.2010.07.050. [DOI] [PubMed] [Google Scholar]
- 20.Souza LG, Silva EJ, Martins AL, et al. Development of topotecan loaded lipid nanoparticles for chemical stabilization and prolonged release. Eur J Pharm Biopharm. 2011;79(1):189–196. doi: 10.1016/j.ejpb.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Nsereko S, Amiji M. Localized delivery of paclitaxel in solid tumors from biodegradable chitin microparticle formulations. Biomaterials. 2002;23(13):2723–2731. doi: 10.1016/s0142-9612(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 22.Felip E, Rosell R, Domine M, et al. Sequential dose-dense paclitaxel followed by topotecan in untreated extensive-stage small-cell lung cancer: a Spanish Lung Cancer Group phase II study. Ann Oncol. 2003;14(10):1549–1554. doi: 10.1093/annonc/mdg405. [DOI] [PubMed] [Google Scholar]
- 23.Muzzarelli R. Chitosan. In: Muzzarelli R, editor. Natural chelating polymers. Oxford, UK: Pergamon Press; 1973. pp. 144–176. [Google Scholar]
- 24.Zhang X, Lin Y, Gillies RJ. Tumor pH and its measurement. J Nucl Med. 2010;51(8):1167–1170. doi: 10.2967/jnumed.109.068981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiǧli Aydin RS, Pulat M. 5-fluorouracil encapsulated chitosan nanoparticles for pH-stimulated drug delivery: evaluation of controlled release kinetics. J Nanomater. 2012:313961. [Google Scholar]
- 26.Makadia HK, Siegel SJ. Poly lactic-co-glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011;3(3):1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Ding JX, Tang ZH, et al. In vitro evaluation of anticancer nanomedicines based on doxorubicin and amphiphilic Y-shaped copolymers. Int J Nanomedicine. 2012;7(1):2867–2697. doi: 10.2147/IJN.S30687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arya N, Arora A, Vasu K, Sood AK, Katti DS. Combination of single walled carbon nanotubes/graphene oxide with paclitaxel: a reactive oxygen species mediated synergism for treatment of lung cancer. Nanoscale. 2013;5:2818–2829. doi: 10.1039/c3nr33190c. [DOI] [PubMed] [Google Scholar]
- 29.Bovill JG. Analysis of drug interactions. Bailliere’s Clin Anaesthesiol. 1998;12(2):153–168. [Google Scholar]
- 30.Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001;298(3):865–872. [PubMed] [Google Scholar]
- 31.Arya N, Chakraborty S, Dube N, Katti DS. Electrospraying: a facile technique for synthesis of chitosan-based micro/nanospheres for drug delivery applications. J Biomed Mater Res B Appl Biomater. 2009;88(1):17–31. doi: 10.1002/jbm.b.31085. [DOI] [PubMed] [Google Scholar]
- 32.Seth A, Katti DS. A one-step electrospray-based technique for modulating morphology and surface properties of poly (lactide-co-glycolide) microparticles using Pluronics®. Int J Nanomedicine. 2012;7:5129–5136. doi: 10.2147/IJN.S34185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain AK, Sood V, Bora M, Vasita R, Katti DS. Electrosprayed inulin microparticles for microbiota triggered targeting of colon. Carbohydr Polym. 2014;112:225–234. doi: 10.1016/j.carbpol.2014.05.087. [DOI] [PubMed] [Google Scholar]
- 34.Bock N, Dargaville TR, Woodruff MA. Electrospraying of polymers with therapeutic molecules: state of the art. Prog Polym Sci. 2012;37(11):1510–1551. [Google Scholar]
- 35.Almería B, Fahmy TM, Gomez A. A multiplexed electrospray process for single-step synthesis of stabilized polymer particles for drug delivery. J Control Release. 2011;154(2):203–210. doi: 10.1016/j.jconrel.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Oliveira BF, Santana MHA, Ré MI. Spray-dried chitosan microspheres cross-linked with d, l-glyceraldehyde as a potential drug delivery system: preparation and characterization. Braz J Chem Eng. 2005;22(3):353–360. [Google Scholar]
- 37.Arya N, Sardana V, Saxena M, Rangarajan A, Katti DS. Recapitulating tumor microenvironment in chitosan-gelatin 3-D scaffolds: an improved in vitro tumor model. J R Soc Interface. 2012;9(77):3288–3302. doi: 10.1098/rsif.2012.0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaufmann SH, Peereboom D, Buckwalter CA, et al. Cytotoxic effects of topotecan combined with various anticancer agents in human cancer cell lines. J Natl Cancer Inst. 1996;88(11):734–741. doi: 10.1093/jnci/88.11.734. [DOI] [PubMed] [Google Scholar]
- 39.Poruchynsky MS, Wang EE, Rudin CM, Blagosklonny MV, Fojo T. Bcl-xL Is phosphorylated in malignant cells following microtubule disruption. Cancer Res. 1998;58(15):3331–3338. [PubMed] [Google Scholar]
- 40.Haldar S, Basu A, Croce CM. Bcl2 Is the guardian of microtubule integrity. Cancer Res. 1997;57(2):229–233. [PubMed] [Google Scholar]
- 41.Pathi SL, Shashi R, Gangrade MG, Kankan RN, Rao DR, Inventors, Cipla Limited assignee Novel crystalline forms. 2007 Apr 19; WO 2007/042799 A1. [Google Scholar]
- 42.Gómez-Gaete C, Tsapis N, Besnard M, Bochot A, Fattal E. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int J Pharm. 2007;331(2):153–159. doi: 10.1016/j.ijpharm.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 43.Lin S, Chang C-C, Lin C-W. A reversible optical sensor based on chitosan film for the selective detecton of copper ions. Biomed Eng-App Bas C. 2012;24(5):453–459. [Google Scholar]
- 44.Fan L, Wu H, Zhang H, et al. Novel super pH-sensitive nanoparticles responsive to tumor extracellular pH. Carbohydr Polym. 2008;73(3):390–400. [Google Scholar]
- 45.Bazzo GC, Lemos-Senna E, Pires ATN. Poly(3-hydroxybutyrate)/chitosan/ketoprofen or piroxicam composite microparticles: preparation and controlled drug release evaluation. Carbohydr Polym. 2009;77(4):839–844. [Google Scholar]
- 46.Sutton D, Durand R, Shuai X, Gao J. Poly(d, l-lactide-co-glycolide)/poly(ethylenimine) blend matrix system for pH sensitive drug delivery. J Appl Polym Sci. 2006;100(1):89–96. [Google Scholar]
- 47.Celikoglu F, Celikoglu SI, York AM, Goldberg EP. Intratumoral administration of cisplatin through a bronchoscope followed by irradiation for treatment of inoperable non-small-cell obstructive lung cancer. Lung Cancer. 2006;51(2):225–236. doi: 10.1016/j.lungcan.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Lu Z, Gao Y, Wientjes MG, Au JL. Improving delivery and efficacy of nanomedicines in solid tumors: role of tumor priming. Nanomedicine (Lond) 2011;6(9):1605–1620. doi: 10.2217/nnm.2211.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tiersten AD, Selleck MJ, Hershman DL, et al. Phase II study of topotecan and paclitaxel for recurrent, persistent, or metastatic cervical carcinoma. Gynecol Oncol. 2004;92(2):635–638. doi: 10.1016/j.ygyno.2003.11.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NCI-H460 cell spheroid/tumoroid generated on chitosan–gelatin 3-D scaffolds at the end of day 6.
Note: Scale bar: 50 μm.
Abbreviation: 3-D, three-dimensional.
Cell viability (percentage of control) of NCI-H460 tumoroids generated on 3-D chitosan–gelatin scaffolds following treatment with TPT, Tx, and Tx + TPT.
Notes: Concentrations of Tx and TPT were 100 and 500 nM, respectively. *Indicates significance (P<0.05) between additive effect of Tx and TPT when treated individually vs their simultaneous treatment on NCI-H460 tumoroids (n=3).
Abbreviations: 3-D, three-dimensional; TPT, topotecan; Tx, paclitaxel.