

Abstract
The routine application of Csp3-hybridized nucleophiles in cross-coupling reactions remains an unsolved challenge in organic chemistry. The sluggish transmetalation rates observed for the preferred organoboron reagents in such transformations are a consequence of the two-electron mechanism underlying the standard catalytic approach. We describe a mechanistically distinct single-electron transfer-based strategy for the activation of organoboron reagents toward transmetalation that exhibits complementary reactivity patterns. Application of an iridium photoredox catalyst in tandem with a nickel catalyst effects the cross-coupling of potassium alkoxyalkyl- and benzyltrifluoroborates with an array of aryl bromides under exceptionally mild conditions (visible light, ambient temperature, no strong base). The transformation has been extended to the asymmetric and stereoconvergent cross-coupling of a secondary benzyltrifluoroborate.
The immense impact of transition metal–catalyzed cross-coupling has been well recognized, with the Suzuki-Miyaura reaction in particular emerging as a preferred method for the construction of C-C bonds in both industrial and academic settings (1). Traditionally, cross-coupling reactions employ a three-step catalytic cycle (Fig. 1): (i) oxidative addition of an organic halide at Pd0, (ii) transmetalation of an organometallic nucleophile to an organo-palladium(II) electrophile, and (iii) reductive elimination from a diorganopalladium(II) species, releasing the coupled product and regenerating the Pd0 catalyst (1, 2). Although these methods are highly effective for Csp2-Csp2 coupling, extension to Csp3 centers has proven challenging because of lower rates of oxidative addition and transmetalation, as well as the propensity of the alkylmetallic intermediates to undergo facile β-hydride elimination (2). Recent advances in ligand technology and the use of alternative metals, such as nickel, have greatly expanded the scope of the electrophilic component, extending even to sterically hindered and unactivated alkyl substrates, and have largely succeeded in retarding problematic β-hydride elimination (3). Despite the progress achieved in advancement of the other fundamental steps, transmetalation has remained largely unchanged since the inception of cross-coupling chemistry. As such, cross-couplings conducted under the traditional mechanistic manifold typically result in transmetalations that are rate-limiting (4).
Fig. 1. Comparison of transmetalation in the palladium-catalyzed Suzuki-Miyaura cross-coupling and the proposed single-electron transmetalation in photoredox/nickel cross-coupling.

Ir = Ir[dFCF3ppy]2(bpy)PF6, R = generic organic subunit, Ar = aryl group.
To date, strategies aimed at accelerating the rate of transmetalation of Csp3-hybridized boronic acid reagents have been largely rudimentary. In most cases, excess base and high temperature are used, thereby limiting functional group tolerance and augmenting deleterious side reactions (5). Stoichiometric Ag and Cu salts have been shown to improve transmetalation efficiency in some systems (6–8), although the mechanism by which the acceleration is achieved is unclear (9), thus limiting their widespread application. Often, the only viable alternative to overcome a slow transmetalation is to abandon the readily available boronic acids and make use of more reactive organometallic reagents. Thus, alkylboranes, alkylzincs, or the corresponding Grignard reagents—all of which lack functional group tolerance and are unstable to air—are often used for alkyl cross-coupling (1).
The challenge of alkylboron transmetalation was recognized to arise directly from mechanistic limitations inherent in the two-electron nature of the conventional process, wherein reactivity is inversely proportional to heterolytic C-B bond strength, thus predisposing Csp3 nucleophiles for failure in cross-coupling reactions (10, 11). Rather than attempting to override the inherent biases of the conventional transmetalation pathway, we anticipated that development of an activation mode based on single-electron transfer (SET) chemistry would constitute a more efficient strategy for engaging this class of reagents (Fig. 1). Trends in homolytic C-B bond strength (12) dictate that such a reaction manifold would exhibit reactivity trends complementary to that of a traditional cross-coupling, with Csp3-hybridized nucleophiles now ideally primed for successful implementation.
The first challenge associated with the realization of this ideal is the oxidative profile of radical capture at a transition metal center (R• + Mn → R-Mn+1), which necessitates a subsequent reduction to maintain the redox neutrality of a traditional transmetalation. Here, application of visible-light photoredox catalysis (13, 14), was envisioned to satisfy the requirements of this unique series of SETs. Encouragement in this regard was found in the work of Sanford and Glorius, who had demonstrated that cooperative catalysis between transition metals and photoredox catalysts is indeed possible (15–17). Potassium organotrifluoroborates were identified as promising partners in this new class of cross-couplings, as previous reports have documented their ability to function as carbon radical sources upon single-electron oxidation (18, 19). Furthermore, Akita and co-workers have used Ir[dFCF3ppy]2(bpy)PF6 (dFCF3ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine; bpy = bipyridine) as a catalyst for the oxidation of activated potassium organotrifluoroborates, demonstrating the feasibility of their implementation in the proposed single-electron transmetalation manifold (20, 21).
Studies were initiated with nickel as a result of its high reactivity toward organic halides and its favorable single-electron redox potentials. We anticipated that the combination of a monomeric Ni(0) catalyst 1 and an aryl halide 2 would result in rapid oxidative addition, generating Ni(II) species 3. Concomitantly, visible-light irradiation of Ir[dFCF3ppy]2(bpy)PF6 4 would generate the excited-state complex 5, the reduction potential of which is sufficiently high [electrochemical potential of reduction Ered = +1.21 V (22, 23)] to induce single-electron oxidation of an activated alkyltrifluoroborate 6 [electrochemical potential of oxidation Eox = −1.10 V (20)], affording the desired alkyl radical 7 upon fragmentation. Subsequent capture of the alkyl radical at Ni(II) would then yield high-valent Ni(III) intermediate 9, which was expected to undergo reductive elimination to generate the desired cross-coupled product 10 and Ni(I) complex 11. From here, reduction of 11 [Ered > −1.10 V (24, 25)] by the reduced form of the photocatalyst 8 [Eox = +1.37 V (21)] would regenerate both the Ni(0) catalyst 1 and the Ir catalyst 4, closing the dual catalytic cycle.
As a proof of concept, this dual catalytic single-electron transmetalation approach was applied to the cross-coupling of benzylic trifluoroborates and aryl bromides (Fig. 2). Our efforts were quickly rewarded, as a catalytic system consisting of photocatalyst 4, Ni(COD)2 (COD = 1,5-cyclooctadiene), 4,4-di-tert-butyl-2,2′-bipyridine (dtbbpy) as ligand, and 2,6-lutidine as an additive effected the cross-coupling of potassium benzyltrifluoroborate and bromobenzonitrile in 89% yield upon exposure to visible light from a 26-W compact fluorescent light bulb at room temperature for 24 hours. Control reactions performed in the absence of photocatalyst, Ni catalyst, or light resulted in no detectable product formation, confirming the essential role of each of these components in the dual catalytic process (26).
Fig. 2. Photoredoxcross-coupling of benzylic trifluoroborates and aryl bromides.

All yields are percent isolated yield of pure material after chromatography. Reactions were performed on aryl halide (0.5 mmol). Boc = tert-butoxycarbonyl, Me = methyl, Ph = phenyl, Ac = acetyl.
We next analyzed the scope of the reaction with regard to both the benzylic trifluoroborate and aryl halide. As expected, electronic modification of the trifluoroborate component had a moderate effect on reaction yield, with more electron-rich, and thus more highly stabilized, radical precursors (16 and 18) performing better than those substituted with electron-withdrawing groups (15 and 17). Substrates possessing an ortho substituent were well tolerated, as evidenced by isolation of product 13 in 82% yield. The reaction also exhibited increased efficiency on a larger scale, as diarylmethane 12 was isolated in 97% yield on a 5.5-mmol scale with reduced catalyst loading [1 mol % 4 and 1.5 mol % of Ni (COD)2 and ligand].
High levels of versatility and functional group tolerance were observed with regard to the aryl halide partner. Substrates bearing electrophilic functional groups that would be incompatible with more highly reactive organometallic nucleophiles were well tolerated. Protic functional groups, including amide 27, sulfonamide 39, phenol 26, pyrazole 32, and -NHBoc 28, could also be used. Substrates possessing substituents ortho to the halide (25, 37) were tolerated, albeit in diminished yield. The absence of a strong base permitted the coupling of amino acid derivative 28 with no observable epimerization, demonstrating the potential utility of this method for late-stage functionalization of peptides or for use with molecules containing other base-sensitive functional groups.
A variety of nitrogen-containing heteroaryl bromides—classically challenging yet highly valued substrates because of their prevalence in biologically active compounds (27)—performed well under the optimized reaction conditions. Pyridine substrates were coupled in all possible regioisomeric configurations (29, 30, 31, 33). Other important N-heterocycles, including pyrimidine 34, indole 35, and quinoline 37, proved to be competent partners. Although five-membered heterocyclic bromides generally exhibited poor reactivity, electron-deficient thienyl bromides were coupled in moderate yields, leading to 38 and 39.
Several practical and more sustainable features derive from this approach to cross-coupling. Previous approaches to the cross-coupling of benzylboron compounds with aryl halides have required excess (3 equiv) aqueous base and temperatures no lower than 60°C (28–30). Furthermore, the present reaction makes use of air-stable and inexpensive bipyridine ligands with low loading of the Ni catalyst. A derivative of photo-catalyst 4 has recently been made commercially available and is similarly effective in promoting the desired reactivity.
The reported cross-coupling reactions generally exhibited levels of efficiency and functional group tolerance equal to or surpassing those of traditional cross-coupling reactions on similar substrates. Most reactions cleanly afforded the desired product, with the remaining mass balance consisting of only unreacted aryl halide. Competing homocoupling of the trifluoroborate to afford bibenzyl derivatives was undetectable by crude high-performance liquid chromatography analysis, allowing use of only a slight excess (1.2 equiv) of this reaction partner, which is typical in traditional Suzuki-Miyaura cross-couplings. Also of note is the compatibility of this reaction manifold with functional groups susceptible to single-electron oxidation or potentially reactive toward the radical intermediates, including phenol, anilide, and thienyl substructures, as well as five-membered nitrogen heterocycles. Remarkably, even 4-bromostyrene could be used as an electrophile without competitive radical capture or polymerization, affording diarylmethane 40 in 61% yield. The surprising compatibility of the reaction manifold with this substrate may suggest a mechanistic scenario more complex than that proposed in Fig. 1. However, the radical nature of the activation mode is strongly supported by the previous studies of Akita (20, 21) and our own experiments using chiral ligand scaffolds and racemic secondary alkyl nucleophiles (see below).
As a demonstration of the broader potential of the application of single-electron transmetalation in this dual catalytic cross-coupling, the conditions optimized for use with primary benzylic trifluoroborates were directly applied to the cross-coupling of a secondary (α-alkoxy)alkyltrifluoroborate (Fig. 3). Comparable single-electron oxidation potentials (20, 21) serve as the singular commonality between these structurally dissimilar reagents. Thus, C-C bond formation via single-electron transmetalation proceeded smoothly under these extremely mild and unoptimized conditions. The differential reactivity between the two-electron and single-electron transmetalation processes is underscored by the stark differences in conditions previously reported for (α-alkoxy)alkyltrifluoroborate cross-coupling (5 equiv CsOH, 105°C, 24 hours) versus those described herein (31). The observed reactivity also serves to demonstrate tolerance of substrates possessing β-hydrogens—a characteristic that is requisite for any general method for the cross-coupling of alkyl substructures.
Fig. 3.
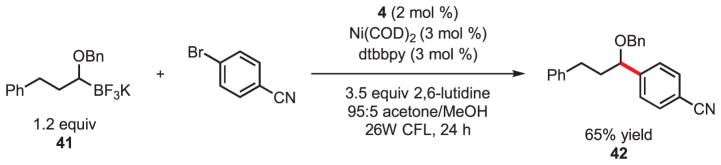
Photoredox cross-coupling of a secondary (α-alkoxy)alkyltrifluoroborate with 4-bromobenzonitrile.
To highlight further the differences between the activation mode reported here and that of traditional cross-coupling, we performed a competition experiment between potassium benzyltrifluoroborate and potassium phenyltrifluoroborate. Exposure of these two nucleophiles to the photoredox cross-coupling conditions resulted in isolation of diarylmethane product 12 in 91% isolated yield, with no observable biaryl formation (Fig. 4). The ability to engage a Csp3-hybridized organo-metallic reagent selectively in a transition metal–catalyzed C-C bond-forming reaction in the presence of an equivalent Csp2-hybridized organometallic represents a striking reversal of the reactivity hierarchy of previously reported cross-coupling reactions. This effectively demonstrates the complementary reactivity patterns observed between the single-electron and two-electron transmetalation modes.
Fig. 4. Probing chemo- and stereoselectivity.

(A) Competition experiment between potassium benzyltrifluoroborate and potassium phenyltrifluoroborate under photoredox cross-coupling conditions. (B) Stereoconvergent cross-coupling of a racemic trifluoroborate 44 and aryl bromide to afford an enantioenriched product. Reactions were performed on aryl bromide (0.5 mmol). *Determined by chiral supercritical fluid chromatography (SFC). †Absolute configuration was assigned as (S) on the basis of data reported in the literature. er = enantiomeric ratio.
Another important implication of the single-electron transmetalation manifold is related to the stereochemical outcome of the alkyl transfer. Nearly all cross-coupling reactions of stereo-defined nucleophiles reported heretofore have demonstrated the transmetalation event to be stereospecific (32). Thus, enantioenriched products may only be accessed from nonracemic and configurationally stable organometallic reagents, which are often difficult to access. Isolated examples of stereoconvergence in transmetalation exist, specifically in the context of secondary benzylmagnesium reagents, a pyrrolidine-based organozinc, and a diastereoconvergent cross-coupling of substituted cyclohexylzinc reagents (33–36). Stereoconvergence in the former is thought to be enabled by dynamic kinetic resolution of the configurationally unstable Grignard reagent; the origin of selectivity in the latter is not fully understood. None of these approaches constitute a general strategy for stereoconvergent transmetalation beyond the scope of the directly explored reagents.
In contrast, the stereochemical outcome of the single-electron transmetalation is dictated by facial selectivity of the addition of a prochiral alkyl radical to a ligated Ni center. Thus, application of a chiral ligand framework renders this process asymmetric and provides a general reaction manifold in which stereoconvergent transmetalation can be achieved. Well-known stereoconvergent cross-couplings of alkyl halides, which putatively undergo a similar mechanistic step, provide guidance for selection of appropriate ligand scaffolds to maximize the stereo-selectivity of the radical capture (37). Indeed, when we used commercially available ligand L1 under slightly modified conditions, racemic trifluoroborate 44 was engaged in stereoconvergent cross-coupling with methyl 3-bromobenzoate, affording 1,1-diarylethane product 45 in 52% yield and a promising enantiomeric ratio of 75:25 (Fig. 4). The observed stereoconvergence serves as an effective mechanistic probe that supports the role of the organotrifluoroborate as a carbon radical precursor, provides evidence that the radical is intercepted by the ligated Ni complex, and suggests that C-C bond formation occurs via reductive elimination from Ni.
This preliminary result strongly implies that high levels of stereoselectivity are possible in the photoredox cross-coupling of secondary alkyl nucleophiles with appropriate modification of reaction conditions and ligand structure. Refinement of this approach to asymmetric cross-coupling will provide a powerful advancement to the field by alleviating the need for synthesis of enantio-enriched organometallic reagents. Taken together, our findings effectively validate the single-electron transmetalation manifold and dual photoredox/cross-coupling cycle as a viable alternative to conventional cross-coupling of Csp3-hybridized nucleophiles.
Supplementary Material
Acknowledgments
Supported by NIH (grant R01 GM-081376) and Pfizer. We thank Frontier Scientific for providing all of the organoboron precursors used in our work.
Footnotes
www.sciencemag.org/content/345/6195/430/suppl/DC1
Materials and Methods
REFERENCES AND NOTES
- 1.de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]
- 2.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science; Sausalito, CA: 2010. [Google Scholar]
- 3.Rudolph A, Lautens M. Angew Chem Int Ed. 2009;48:2656–2670. doi: 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]
- 4.Hegedus LS, Soderberg BCG. Transition Metals in the Synthesis of Complex Organic Molecules. 3. University Science; Sausalito, CA: 2010. [Google Scholar]
- 5.Doucet H. Eur J Org Chem. 2008;2008:2013–2030. [Google Scholar]
- 6.Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024–5025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 7.Zou G, Reddy YK, Falck JR. Tetrahedron Lett. 2001;42:7213–7215. [Google Scholar]
- 8.Deng JZ, et al. Org Lett. 2009;11:345–347. doi: 10.1021/ol802556f. [DOI] [PubMed] [Google Scholar]
- 9.Aufiero M, Proutiere F, Schoenebeck F. Angew Chem Int Ed. 2012;51:7226–7230. doi: 10.1002/anie.201202504. [DOI] [PubMed] [Google Scholar]
- 10.Transmetalation rates of organoboron compounds adhere to trends established for the corresponding organostannanes.
- 11.Labadie JW, Stille JK. J Am Chem Soc. 1983;105:6129–6137. [Google Scholar]
- 12.Finch A, Gardner PJ, Pearn EJ, Watts GB. Trans Faraday Soc. 1967;63:1880–1888. [Google Scholar]
- 13.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034–9037. doi: 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahoo B, Hopkinson MN, Glorius F. J Am Chem Soc. 2013;135:5505–5508. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]
- 18.Lockner JW, Dixon DD, Risgaard R, Baran PS. Org Lett. 2011;13:5628–5631. doi: 10.1021/ol2023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasu Y, Koike T, Akita M. Adv Synth Catal. 2012;354:3414–3420. [Google Scholar]
- 21.Miyazawa K, Yasu Y, Koike T, Akita M. Chem Commun. 2013;49:7249–7251. doi: 10.1039/c3cc42695e. [DOI] [PubMed] [Google Scholar]
- 22.All electrochemical potentials are calculated versus the saturated calomel electrode (SCE).
- 23.Lowry M, et al. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 24.Cannes C, Labbé E, Durandetti M, Devaud M, Nédélec JY. J Electroanal Chem. 1996;412:85–93. [Google Scholar]
- 25.The reduction of (bpy)NiBr2 is observed as a two-electron wave at −1.1 V, implying that reduction of (bpy)NiBr to (bpy)Ni0 occurs at a more positive potential.
- 26.See supplementary materials on Science Online.
- 27.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain P, Yi S, Flaherty PT. J Heterocycl Chem. 2013;50(S1):E166–E173. [Google Scholar]
- 29.Flaherty A, Trunkfield A, Barton W. Org Lett. 2005;7:4975–4978. doi: 10.1021/ol051929x. [DOI] [PubMed] [Google Scholar]
- 30.Molander GA, Ito T. Org Lett. 2001;3:393–396. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]
- 31.Molander GA, Wisniewski SR. J Am Chem Soc. 2012;134:16856–16868. doi: 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Wang CY, Huang R, Biscoe MR. Nat Chem. 2013;5:607–612. doi: 10.1038/nchem.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi T, Tajika M, Tamao K, Kumada M. J Am Chem Soc. 1976;98:3718–3719. [Google Scholar]
- 34.Hayashi T, et al. J Am Chem Soc. 1982;104:180–186. [Google Scholar]
- 35.Cordier CJ, Lundgren RJ, Fu GC. J Am Chem Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriya K, Knochel P. Org Lett. 2014;16:924–927. doi: 10.1021/ol403673e. [DOI] [PubMed] [Google Scholar]
- 37.Do HQ, Chandrashekar ERR, Fu GC. J Am Chem Soc. 2013;135:16288–16291. doi: 10.1021/ja408561b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.