

Abstract
Observational studies have reported different effects of adiposity on cardiovascular risk factors across age and sex. Since cardiovascular risk factors are enriched in obese individuals, it has not been easy to dissect the effects of adiposity from those of other risk factors. We used a Mendelian randomization approach, applying a set of 32 genetic markers to estimate the causal effect of adiposity on blood pressure, glycemic indices, circulating lipid levels, and markers of inflammation and liver disease in up to 67,553 individuals. All analyses were stratified by age (cutoff 55 years of age) and sex. The genetic score was associated with BMI in both nonstratified analysis (P = 2.8 × 10−107) and stratified analyses (all P < 3.3 × 10−30). We found evidence of a causal effect of adiposity on blood pressure, fasting levels of insulin, C-reactive protein, interleukin-6, HDL cholesterol, and triglycerides in a nonstratified analysis and in the <55-year stratum. Further, we found evidence of a smaller causal effect on total cholesterol (P for difference = 0.015) in the ≥55-year stratum than in the <55-year stratum, a finding that could be explained by biology, survival bias, or differential medication. In conclusion, this study extends previous knowledge of the effects of adiposity by providing sex- and age-specific causal estimates on cardiovascular risk factors.
Introduction
The incidence of overweight and obesity is increasing rapidly on a global level. Adiposity constitutes an important risk factor for cardiovascular disease (CVD), which is the major cause of morbidity and mortality for both men and women (1), although CVD generally occurs later in women than in men (2,3). There is strong evidence from randomized clinical trials (4,5) that weight loss induced by dietary change reduces the levels of cardiovascular risk factors, such as blood pressure and circulating lipid levels. However, intervention studies of weight loss are often difficult to interpret because the intervention may affect several separate pathways. For example, bariatric surgery can affect glucose metabolism through pathways other than weight loss (6). We and others (7,8) have previously applied Mendelian randomization methods to assess and confirm the causal role of adiposity in cardiometabolic disease. In Mendelian randomization study designs, one or several genetic variants, usually single nucleotide polymorphisms (SNPs), associated with a modifiable risk factor are used as instrumental variables (IVs). The IVs provide unbiased estimates of the causal relation of the exposure to the risk factor (here, adiposity) with an outcome of interest (here, other cardiovascular risk factors) (9).
Age and sex differences in adiposity and body fat distribution, as well as physiology of the heart and blood vessels, are evident even before overt CVD becomes evident (10,11). As an example, men are usually less sensitive to insulin than women, given a certain BMI, which may partly be explained by differences in fat distribution (12). Moreover, the strength of association between obesity and a number of cardiovascular risk factors, such as blood pressure (13–15) and total serum cholesterol (16) and fasting glucose (17) levels, has been reported to decline with age. The role of weight loss as a health-promoting intervention in older age groups has been debated lately as large studies (18) have shown that the excess mortality from obesity markedly declines with age and that the risk of sarcopenia and osteoporosis increases with weight loss. Improved understanding of the effect of adiposity in different age and sex groups may advance prevention of CVD through the possibility of efforts directed toward groups with the largest causal effect. However, stratified observational studies suffer from the risk of confounding and reverse causation, problems that can be avoided using a Mendelian randomization design.
The objective of the current study was to use a Mendelian randomization design to assess whether adiposity causally affects known cardiovascular risk factors at a similar magnitude in men and women and before and after 55 years of age.
Research Design and Methods
Study Population
Population-based studies of European ancestry enrolled in the European Network for Genetic and Genomic Epidemiology (ENGAGE) Consortium were invited to participate in the study. In total, 30 studies contributed to the primary and/or secondary analysis in the current study (Supplementary Table 1). A standardized analysis plan was provided to all partners, and individual-level analyses were performed at each center separately. The distributions of age, BMI, and sex in the participating cohorts are shown in Supplementary Table 2.
Genetic Instruments
An individual nonweighted genetic risk score using the 32 SNPs (“lead SNPs”) reported by Speliotes et al. (19) was used as the IV in 25 studies for the main analyses (listed in Supplementary Table 3). The score was created by summing the number of BMI-increasing alleles (0, 1, 2) for each of the 32 SNPs. When direct genotype data were not available for an SNP in a particular study, we prioritized as follows: 1) imputed allele dosage data with high imputation quality (IMPUTE proper_info≥0.4 or MACH r2hat≥0.3) (20); 2) information from directly genotyped proxy SNPs from a predefined list of variants that are in high linkage disequilibrium (LD) with lead SNPs; or 3) imputation to a prespecified allele dosage (2*reported effect allele frequency from the study by Speliotes et al. [19]). Eighteen of the studies included in the genetic score analyses had directly genotyped or high-quality imputed genotype information on all SNPs, while seven studies lacked information on three SNPs each and used the prespecified allele dosage for those three SNPs.
In secondary analyses, we used one SNP as the IV to maximize the study sample (five additional ENGAGE studies had information on this SNP only, for a total of 30 studies). For this instrument, we used direct genotype information for the FTO variant rs9939609 from participating studies when possible (16 studies). If rs9939609 was not genotyped directly, we prioritized as follows: 1) the HapMap II CEU (European) reference panel imputed genetic information (hapmap.ncbi.nlm.nih.gov) for rs9939609 (six studies) or 2) genotype information from a predefined list of proxies that are in high LD with rs9939609. We used the directly genotyped proxies rs11075989 (four studies, r2 with rs9939609 = 1.0 in HapMap II), rs3751812 (one study, r2 = 1.0), rs1558902 (one study, r2 = 0.93), and rs1421085 (two studies, r2 = 0.93). We estimated the effects of the BMI-increasing A allele of rs9939609 or for the corresponding alleles from proxies (using HapMap II CEU LD data) on BMI and cardiovascular risk factors.
Individuals were excluded from analysis when the overall genotyping array sample call rate was <95%. All studies reported SNPs with Hardy-Weinberg equilibrium exact P values >0.0001 and a call rate >0.95 for genotyped SNPs.
Outcomes
We studied the following quantitative outcomes: 1) diastolic and systolic blood pressure where observed values were increased with 15 mmHg for systolic blood pressure and 10 mmHg for diastolic blood pressure in case of reported hypertension medication; 2) circulating lipid fraction (only in individuals not receiving lipid-lowering medication) (concentrations of HDL cholesterol [HDL-C], LDL cholesterol [LDL-C], total cholesterol, and triglycerides); 3) measurements of glucose homeostasis in nondiabetic subjects using the following glycemic traits: concentrations of fasting glucose, 2-h glucose from the oral glucose tolerance test (OGTT), hemoglobin A1c (HbA1c), and fasting insulin; 4) liver enzyme activity (alanine aminotransferase [ALT], γ-glutamyl transferase [GGT]); and 5) inflammation markers (concentrations of C-reactive protein [CRP] and interleukin-6 [IL-6]). The measurement methods used in each study and the respective matrix are reported in Supplementary Tables 4, 5, and 6. The following variables were transformed to the natural logarithmic scale prior to further analysis due to their skewed distribution when examining normal probability plots in preliminary analysis (levels of fasting insulin, ALT, GGT, CRP, IL-6, and triglycerides). All quantitative phenotypes including BMI were z score (SD) standardized prior to further analysis to reduce heterogeneity across studies and to make it possible to compare the adiposity effect between outcomes.
Statistical Analysis
Association Analyses
We evaluated associations of BMI and IVs with each outcome separately, as well as associations between IVs and BMI. For all analyses, we used linear regressions in each study separately, assuming an additive effect of the number of BMI-increasing alleles. The associations between BMI and outcomes are hereafter referred to as “observational.” All analyses were performed in the full study sample, as well as stratified by age at measurement (cutoff 55 years of age), and in men and women separately. If measurements were available both before and after age 55 years for the same individuals, these individuals were included in both age strata (using the respective measurement). Regression analyses were adjusted for age and sex (only in the sex-pooled analysis) by including age at measurement and sex as covariates. Individual studies were adjusted for additional study-specific covariates such as the study center when relevant. Random-effects meta-analysis (21,22) of the study-specific results was performed using the rmeta package, in R version 3.0.0.
IV Analyses
We used IV estimators to quantify the strength of the causal associations between adiposity and other cardiovascular risk factors. The estimator (βIV) was calculated as the ratio between the two regression coefficients determined from association meta-analyses in each stratum (Eq. 1): estimated genetic score or FTO effect on the given outcome (βgenetic instrument_outcome), and estimated genetic score or FTO effect on BMI (βgenetic instrument_BMI).
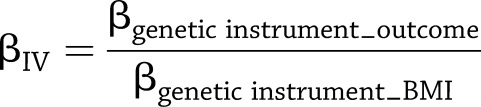 |
(1) |
For quantitative outcomes with a single instrument, the IV estimator derived by Equation 1 is identical to that derived by the widely used two-stage least squares method (23). The SEs for the IV estimators were calculated using the delta method (Eq. 2), which we have previously evaluated for this purpose (7):
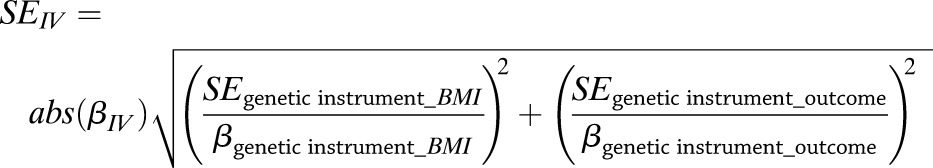 |
(2) |
For each trait, we tested the null hypothesis of no difference between the IV estimator and the observational regression-based estimator via the z test. We also tested differences between estimates for men and women and differences between the two age strata using the same approach.
Multiple Testing Correction
We used the Benjamini and Hochberg (24) procedure implemented in the STATA multproc procedure to correct the level of significance to a 5% false discovery rate (FDR). The procedure was applied to all 266 P values in the main analysis together.
Results
Association of the Genetic Score With BMI
Random-effects meta-analysis of the association between the genetic score and BMI in 25 studies (n = 81,764) showed a positive effect (0.030 SD units of BMI per additional allele [95% CI 0.028–0.033], P = 2.8 × 10−107). Stratum-specific point estimates were similar to those of the pooled data, and no differences between strata were found using the z test (Table 1).
Table 1.
Association of genetic score and FTO variant with BMI
| Genetic score |
FTO |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nstud | n | β (95% CI) | P | F | Pdiff | nstud | n | β (95% CI) | P | F | Pdiff | |
| All | 25 | 81,764 | 0.030 (0.028–0.033) | 2.80 × 10−107 | 484 | NA | 30 | 141,800 | 0.081 (0.070–0.091) | 1.40 × 10−50 | 224 | NA |
| Men | 24 | 38,704 | 0.029 (0.025–0.032) | 3.63 × 10−56 | 249 | 0.19 | 29 | 68,971 | 0.075 (0.062–0.087) | 7.60 × 10−31 | 133 | 0.28 |
| Women | 24 | 43,060 | 0.032 (0.029–0.035) | 1.06 × 10−97 | 440 | 29 | 72,829 | 0.084 (0.072–0.096) | 2.00 × 10−44 | 195 | ||
| ≥55-years-old group | 21 | 37,105 | 0.027 (0.023–0.032) | 3.28 × 10−30 | 130 | 0.14 | 25 | 52,057 | 0.076 (0.061–0.091) | 1.90 × 10−23 | 100 | 0.39 |
| <55-years-old group | 23 | 45,775 | 0.032 (0.029–0.034) | 1.60 × 10−119 | 540 | 28 | 90,855 | 0.084 (0.071–0.097) | 9.70 × 10−38 | 165 | ||
β (95% CI), per-allele effect on BMI on SD scale; n, total number of individuals in the analysis; nstud, number of studies in analysis; Pdiff, P value for test of differences between strata.
Associations of BMI With Cardiovascular Risk Factors
We applied the FDR procedure to correct for all 266 tests reported in Tables 2, 3, and 4. At a 5% FDR level, the corrected critical P value was estimated to 0.022, which should be considered as the P value threshold to denote statistical significance in this report.
Table 2.
Nonstratified observational and IV analyses based on a genetic score of the association of adiposity with cardiovascular risk factors
| Observational analyses |
IV analyses |
||||||
|---|---|---|---|---|---|---|---|
| n | β (95% CI) | P* | n | β (95% CI) | P* | PIV-obs* | |
| Diastolic blood pressure | 117,230 | 0.25 (0.23–0.27) | 2.5 × 10−136 | 66,997 | 0.15 (0.03–0.26) | 0.01 | 0.08 |
| Systolic blood pressure | 117,781 | 0.22 (0.20–0.24) | 2.3 × 10−101 | 67,553 | 0.16 (0.04–0.28) | 0.01 | 0.31 |
| Fasting glucose | 69,110 | 0.19 (0.15–0.22) | 2.9 × 10−26 | 37,181 | 0.09 (−0.01 to 0.19) | 0.07 | 0.06 |
| 2-h post-OGTT glucose | 22,204 | 0.19 (0.11–0.27) | 5.9 × 10−6 | 4,596 | 0.11 (−0.19 to 0.40) | 0.48 | 0.61 |
| HbA1c | 29,451 | 0.16 (0.11–0.21) | 5.1 × 10−10 | 26,901 | 0.19 (0.01–0.36) | 0.03 | 0.79 |
| ln fasting insulin | 40,165 | 0.48 (0.44–0.52) | 1.2 × 10−127 | 24,614 | 0.31 (0.17–0.45) | 9.6 × 10−6 | 0.02 |
| ln CRP | 71,960 | 0.33 (0.30–0.36) | 2.7 × 10−102 | 47,781 | 0.30 (0.20–0.41) | 8.8 × 10−9 | 0.56 |
| ln IL-6 | 11,878 | 0.13 (0.06–0.19) | 2.0 × 10−4 | 10,285 | 0.26 (0.08–0.45) | 0.01 | 0.18 |
| HDL-C | 93,015 | −0.28 (−0.29 to −0.26) | 5.8 × 10−211 | 58,387 | −0.33 (−0.42 to −0.24) | 1.1 × 10−12 | 0.29 |
| LDL-C | 86,793 | 0.11 (0.08–0.14) | 5.7 × 10−17 | 52,858 | 0.08 (−0.02 to 0.19) | 0.11 | 0.64 |
| Total cholesterol | 107,741 | 0.08 (0.06–0.10) | 9.7 × 10−15 | 59,329 | 0.00 (−0.11 to 0.11) | 0.97 | 0.17 |
| ln triglycerides | 101,709 | 0.31 (0.30–0.32) | <1 × 10−300 | 54,417 | 0.23 (0.12–0.34) | 2.7 × 10−5 | 0.18 |
| ln ALT | 45,473 | 0.23 (0.19–0.26) | 6.5 × 10−31 | 23,927 | 0.09 (−0.09 to 0.26) | 0.32 | 0.13 |
| ln GGT | 71,859 | 0.22 (0.20–0.24) | 4.3 × 10−87 | 26,831 | 0.15 (0.00–0.29) | 0.04 | 0.35 |
All models were adjusted for sex and age. β (95% CI), effect per SD change of BMI on trait (SD scale); PIV-obs, P value from test of difference between observational and IV analyses.
*A nominal P value of <0.022 is considered significant at a 5% FDR level.
Table 3.
Age-stratified observational and IV analysis based on a genetic score of the association of adiposity with cardiovascular risk factors
| <55 years |
≥55 years | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| n | β (95% CI) | P* | PIV-obs* | n | β (95% CI) | P* | PIV-obs* | Pdiff* | |
| Observational analyses | |||||||||
| Diastolic blood pressure | 71,588 | 0.29 (0.26–0.32) | 2.7 × 10−98 | 48,210 | 0.23 (0.19–0.27) | 4.4 × 10−35 | 0.01 | ||
| Systolic blood pressure | 71,753 | 0.25 (0.22–0.27) | 2.3 × 10−85 | 48,596 | 0.22 (0.18–0.26) | 2.0 × 10−26 | 0.19 | ||
| Fasting glucose | 45,047 | 0.16 (0.13–0.20) | 7.3 × 10−21 | 25,089 | 0.24 (0.20–0.27) | 4.7 × 10−37 | 0.005 | ||
| 2-h post-OGTT glucose | 13,778 | 0.18 (0.10–0.25) | 3.1 × 10−6 | 8,426 | 0.21 (0.09–0.32) | 4.6 × 10−4 | 0.66 | ||
| HbA1c | 14,824 | 0.13 (0.07–0.20) | 1.1 × 10−4 | 14,397 | 0.18 (0.12–0.24) | 8.8 × 10−9 | 0.35 | ||
| ln fasting insulin | 26,322 | 0.48 (0.44–0.52) | 1.8 × 10−118 | 14,815 | 0.50 (0.45–0.54) | 1.6 × 10−97 | 0.57 | ||
| ln CRP | 41,854 | 0.40 (0.37–0.43) | 2.5 × 10−175 | 30,090 | 0.30 (0.26–0.33) | 2.4 × 10−68 | 2.51 × 10−6 | ||
| ln IL-6 | 6,719 | 0.16 (0.06–0.26) | 0.001 | 5,159 | 0.14 (0.08–0.21) | 1.9 × 10−5 | 0.73 | ||
| HDL-C | 57,672 | −0.28 (−0.30 to −0.27) | 1.1 × 10−304 | 36,171 | −0.28 (−0.31 to −0.25) | 9.1 × 10−99 | 0.83 | ||
| LDL-C | 56,392 | 0.16 (0.13–0.18) | 4.4 × 10−33 | 31,499 | 0.02 (0.00–0.04) | 0.08 | 9.73 × 10−16 | ||
| Total cholesterol | 69,736 | 0.12 (0.10–0.15) | 9.8 × 10−23 | 39,112 | 0.00 (−0.02 to 0.02) | 0.96 | 1.0 × 10−12 | ||
| ln triglycerides | 68,777 | 0.33 (0.31–0.35) | 4.3 × 10−224 | 34,061 | 0.27 (0.25–0.29) | 9.4 × 10−181 | 2.4 × 10−5 | ||
| ln ALT | 27,956 | 0.24 (0.18–0.30) | 1.1 × 10−13 | 17,517 | 0.19 (0.17–0.21) | 3.1 × 10−69 | 0.2 | ||
| ln GGT | 47,202 | 0.25 (0.22–0.28) | 4.5 × 10−50 | 24,657 | 0.18 (0.16–0.20) | 4.5 × 10−65 | 5.5 × 10−4 | ||
| IV analyses | |||||||||
| Diastolic blood pressure | 35,518 | 0.21 (0.09–0.33) | 8.0 × 10−4 | 0.20 | 32,596 | 0.01 (−0.16 to 0.18) | 0.93 | 0.01 | 0.06 |
| Systolic blood pressure | 35,681 | 0.21 (0.12–0.30) | 2.5 × 10−6 | 0.41 | 32,989 | 0.06 (−0.14 to 0.26) | 0.56 | 0.13 | 0.17 |
| Fasting glucose | 20,742 | 0.10 (−0.01 to 0.22) | 0.09 | 0.29 | 17,380 | 0.04 (−0.11 to 0.19) | 0.58 | 0.01 | 0.54 |
| 2-h post-OGTT glucose | 2,077 | −0.01 (−0.41 to 0.39) | 0.97 | 0.38 | 2,519 | 0.13 (−0.27 to 0.53) | 0.53 | 0.71 | 0.64 |
| HbA1c | 13,614 | 0.10 (−0.05 to 0.24) | 0.19 | 0.65 | 13,287 | 0.25 (0.01–0.48) | 0.04 | 0.58 | 0.29 |
| ln fasting insulin | 15,985 | 0.26 (0.10–0.42) | 0.001 | 0.01 | 9,526 | 0.32 (0.12–0.52) | 0.002 | 0.09 | 0.64 |
| ln CRP | 25,822 | 0.35 (0.23–0.48) | 7.5 × 10−8 | 0.48 | 21,943 | 0.18 (0.01–0.35) | 0.04 | 0.17 | 0.10 |
| ln IL-6 | 5,368 | 0.37 (0.12–0.61) | 0.004 | 0.13 | 4,917 | 0.13 (−0.13 to 0.40) | 0.32 | 0.96 | 0.21 |
| HDL-C | 32,977 | −0.36 (−0.47 to −0.25) | 3.4 × 10−10 | 0.20 | 26,425 | −0.28 (−0.40 to −0.17) | 1.2 × 10−6 | 0.97 | 0.36 |
| LDL-C | 32,016 | 0.15 (0.05–0.25) | 0.004 | 0.92 | 21,851 | −0.10 (−0.32 to 0.12) | 0.38 | 0.30 | 0.04 |
| Total cholesterol | 33,002 | 0.10 (−0.05 to 0.24) | 0.20 | 0.73 | 27,344 | −0.19 (−0.36 to −0.01) | 0.04 | 0.04 | 0.015 |
| ln triglycerides | 33,018 | 0.28 (0.16–0.41) | 1.3 × 10−5 | 0.45 | 22,438 | 0.12 (0.00–0.25) | 0.05 | 0.02 | 0.09 |
| ln ALT | 11,215 | 0.22 (0.05–0.39) | 0.01 | 0.84 | 12,712 | −0.10 (−0.39 to 0.19) | 0.50 | 0.05 | 0.06 |
| ln GGT | 13,915 | 0.24 (0.10–0.38) | 0.001 | 0.87 | 12,916 | −0.01 (−0.24 to 0.21) | 0.92 | 0.10 | 0.07 |
All models were adjusted for sex and age. β (95% CI), effect per SD change of BMI on trait (SD scale); Pdiff, difference between the two strata (only significant P values are shown); PIV-obs, P value from test of difference between observational and IV analyses.
*A nominal P value of <0.022 is considered significant at a 5% FDR level.
Table 4.
Sex-stratified observational and IV analysis based on a genetic score of the association of adiposity with cardiovascular risk factors
| Women |
Men |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| n | β (95%CI) | P* | PIV-obs* | n | β (95% CI) | P* | PIV-obs* | Pdiff* | |
| Observational analyses | |||||||||
| Diastolic blood pressure | 59,738 | 0.22 (0.20–0.24) | 2.5 × 10−101 | 57,492 | 0.29 (0.27–0.32) | 8.8 × 10−106 | 3.3 × 10−5 | ||
| Systolic blood pressure | 59,859 | 0.21 (0.18–0.23) | 3.4 × 10−77 | 57,922 | 0.24 (0.22–0.27) | 1.76 × 10−94 | 0.01 | ||
| Fasting glucose | 35,823 | 0.18 (0.15–0.21) | 2.9 × 10−26 | 33,288 | 0.20 (0.15–0.24) | 1.2 × 10−17 | 0.48 | ||
| 2-h post-OGTT glucose | 10,248 | 0.16 (0.05–0.28) | 0.01 | 11,956 | 0.22 (0.15–0.28) | 9.51 × 10−11 | 0.41 | ||
| HbA1c | 15,676 | 0.17 (0.12–0.21) | 9.1 × 10−14 | 13,775 | 0.18 (0.09–0.27) | 5.6 × 10−5 | 0.76 | ||
| ln fasting insulin | 22,137 | 0.42 (0.38–0.46) | 7.6 × 10−98 | 18,027 | 0.58 (0.52–0.65) | 1.1 × 10−73 | 2.0 × 10−5 | ||
| ln CRP | 40,072 | 0.37 (0.34–0.40) | 5.0 × 10−107 | 31,888 | 0.30 (0.27–0.33) | 9.3 × 10−71 | 0.002 | ||
| ln IL-6 | 6,652 | 0.15 (0.08–0.22) | 8.0 × 10−5 | 5,226 | 0.10 (0.05–0.16) | 2.0 × 10−4 | 0.31 | ||
| HDL-C | 49,949 | −0.27 (−0.30 to −0.25) | 5.2 × 10−150 | 43,039 | −0.29 (−0.31 to −0.27) | 1.4 × 10−194 | 0.36 | ||
| LDL-C | 47,619 | 0.11 (0.08–0.13) | 2.5 × 10−20 | 39,174 | 0.11 (0.07–0.15) | 1.8 × 10−8 | 0.73 | ||
| Total cholesterol | 55,223 | 0.06 (0.04–0.08) | 3.6 × 10−10 | 52,518 | 0.11 (0.08–0.14) | 1.5 × 10−13 | 2.6 × 10−16 | ||
| ln triglycerides | 52,873 | 0.26 (0.24–0.28) | 4.5 × 10−182 | 48,836 | 0.39 (0.36–0.41) | 4.2 × 10−204 | 0.005 | ||
| ln ALT | 24,013 | 0.16 (0.13–0.19) | 1.9 × 10−28 | 21,460 | 0.32 (0.28–0.36) | 1.6 × 10−55 | 5.9 × 10−11 | ||
| ln GGT | 38,127 | 0.17 (0.15–0.20) | 2.3 × 10−48 | 33,732 | 0.31 (0.28–0.33) | 2.2 × 10−96 | 2.16 × 10−12 | ||
| IV analyses | |||||||||
| Diastolic blood pressure | 34,333 | 0.07 (−0.04 to 0.18) | 0.24 | 0.01 | 32,664 | 0.23 (0.06–0.40) | 0.008 | 0.44 | 0.11 |
| Systolic blood pressure | 34,459 | 0.10 (−0.01 to 0.20) | 0.06 | 0.05 | 33,094 | 0.21 (0.04–0.37) | 0.02 | 0.65 | 0.29 |
| Fasting glucose | 22,066 | 0.09 (−0.03 to 0.21) | 0.14 | 0.15 | 15,104 | 0.10 (−0.06 to 0.26) | 0.23 | 0.24 | 0.93 |
| 2-h post-OGTT glucose | 2,360 | 0.18 (−0.22 to 0.57) | 0.39 | 0.95 | 2,225 | 0.08 (−0.55 to 0.72) | 0.80 | 0.68 | 0.81 |
| HbA1c | 14,332 | 0.13 (−0.01 to 0.28) | 0.06 | 0.69 | 12,569 | 0.28 (0.06–0.51) | 0.01 | 0.41 | 0.28 |
| ln fasting insulin | 14,085 | 0.29 (0.14–0.45) | 3.0 × 10−4 | 0.12 | 10,518 | 0.36 (0.02–0.70) | 0.04 | 0.19 | 0.73 |
| ln CRP | 27,107 | 0.29 (0.09–0.49) | 0.004 | 0.45 | 20,674 | 0.26 (0.12–0.40) | 3.0 × 10−4 | 0.59 | 0.79 |
| ln IL-6 | 5,840 | 0.38 (0.14–0.62) | 0.002 | 0.07 | 4,445 | 0.08 (−0.22 to 0.37) | 0.62 | 0.86 | 0.11 |
| HDL-C | 31,201 | −0.34 (−0.45 to −0.23) | 7.9 × 10−10 | 0.22 | 27,146 | −0.31 (−0.46 to −0.16) | 4.0 × 10−5 | 0.75 | 0.74 |
| LDL-C | 29,250 | 0.10 (−0.01 to 0.22) | 0.08 | 0.96 | 23,595 | 0.04 (−0.09 to 0.17) | 0.60 | 0.26 | 0.45 |
| Total cholesterol | 31,515 | −0.02 (−0.14 to 0.09) | 0.68 | 0.96 | 27,796 | 0.03 (−0.13 to 0.18) | 0.74 | 0.11 | 0.88 |
| ln triglycerides | 29,744 | 0.25 (0.14–0.37) | 2.8 × 10−5 | 0.16 | 24,660 | 0.24 (0.06–0.42) | 0.01 | 0.30 | 0.61 |
| ln ALT | 14,701 | 0.11 (−0.04 to 0.26) | 0.14 | 0.51 | 9,226 | 0.09 (−0.14 to 0.32) | 0.43 | 0.05 | 0.90 |
| ln GGT | 16,794 | 0.13 (−0.06 to 0.32) | 0.18 | 0.64 | 10,037 | 0.17 (−0.09 to 0.43) | 0.20 | 0.32 | 0.80 |
All models were adjusted for age. Pdiff, difference between men and women (only significant P values are shown); PIV-obs, P value from test of difference between observational and IV analyses; β (95% CI), effect per SD change of BMI on trait (SD scale); ln, transformed to natural logarithm scale.
*A nominal P value of <0.022 is considered significant at a 5% FDR level.
The random-effects meta-analysis showed that BMI was associated with all quantitative phenotypes in all strata (Tables 2, 3, and 4, observational analysis) with the exception of the ≥55-year stratum, where no association was found with LDL-C and total cholesterol levels (Table 3).
Instrumental Variable Analysis Using the Genetic Score
Nonstratified IV analysis using the genetic score showed evidence for causal effects of adiposity on the following: 1) diastolic and systolic blood pressure (after adjustment for blood pressure medication); 2) levels of HDL-C and triglycerides (in individuals not receiving lipid-lowering medication); 3) fasting levels of insulin; and 4) levels of CRP and IL-6 (Table 2).
Stratified IV analysis showed consistent evidence for causal effects of adiposity in all age and sex strata for HDL-C. For the other outcomes, we found evidence of causality in up to three of the strata (Tables 3 and 4).
Differences Between Age Strata for the Observational and IV Analyses of the Association of Adiposity With Outcomes
We observed significantly larger regression coefficients in the observational analysis for BMI in the <55-year stratum than in the ≥55-year stratum for 6 of the 14 outcomes, as follows: diastolic blood pressure and levels of LDL-C, total cholesterol, triglycerides, GGT, and CRP. We found a smaller regression coefficient for the association of BMI with fasting glucose levels in the <55-year stratum (Table 3).
In line with the observational analysis, for these six outcomes all IV point estimates were higher in the <55-year stratum than in the ≥55-year stratum. However, these differences were nonsignificant, except for the total cholesterol analysis (P = 0.015).
Differences Between Sex Strata for the Observational and IV Analyses of the Association of Adiposity With Outcomes
In observational analyses, we observed larger regression coefficients in men than in women for associations of BMI with systolic and diastolic blood pressure and levels of total cholesterol, triglycerides, ALT, GGT, and fasting insulin. In line with the observational analysis for these seven outcomes, all IV point estimates were consistently higher (but not significantly different) in men than in women, with the exception of triglycerides and ALT. There were larger effects of adiposity on CRP levels in women than in men in the observational analysis, and, in line with this, the IV point estimate was higher for women, but again was not significantly different (Table 4).
Differences Between Observational Analysis and IV Analysis
We found the IV estimates for diastolic blood pressure and fasting glucose level to be significantly reduced or abolished compared with the observational estimates in the ≥55-year stratum (Table 3). This finding implies the presence of confounding of the observational results for these outcomes in the old stratum, which was not as prominent in the young stratum. We also found that the IV was significantly smaller than the observational estimate for fasting insulin levels, both in nonstratified analysis and for <55-year stratum. Further, the IV estimates in women were found to be significantly smaller than the observational estimates for diastolic blood pressure.
Instrumental Variable Analysis Using FTO
In secondary analyses, we performed random-effects meta-analysis of the association between FTO variant and BMI in the 30 studies (n = 141,800) that showed a positive effect of the A-allele of rs9939609 on BMI (0.081 SD units per additional A-allele [95% CI 0.070–0.091], P = 1.4 × 10−50) (Table 1). The results using FTO as an IV are presented in Supplementary Tables 7, 8, and 9. Overall, the results were similar in directions and effect sizes to those using the genetic score. Because the sample size was much larger for the FTO instrument for some outcomes, the FTO IV estimates were in some instances more precise (smaller SEs) with respect to the estimates using the genetic score. In addition to the findings presented above, the FTO analysis provided evidence of a causal effect of adiposity on ALT activity. Further, the IV estimate using FTO was significantly higher for men than for women for fasting insulin level (P = 0.01), a result that is consistent with the observational estimate (Supplementary Table 9).
Discussion
In this Mendelian randomization study, we extended previous efforts to assess causal effects of adiposity on cardiovascular risk factors by examining this relation across age and sex groups. We based our estimations on associations of genetic instruments with 14 cardiovascular risk factors in up to 67,553 individuals in primary analyses and 116,443 individuals in secondary analyses. We provide evidence for causal effects of adiposity on blood pressure (systolic and diastolic), circulating levels of lipids (HDL-C, triglycerides), glucose homeostasis (fasting insulin levels), and markers of inflammation (CRP, IL-6). Similar to what was seen in the observational analysis, causal estimates for the effect of adiposity on markers of inflammation, liver damage, lipid levels, and blood pressure were consistently larger in the <55-year stratum than in the ≥55-year stratum, although most of these differences were not statistically significant in the IV analyses. Further, in IV analyses, the point estimates of the effect of adiposity on systolic and diastolic blood pressure were larger in men than in women. For markers of inflammation, we observed the reverse (i.e., larger causal estimates in women than in men). We observed a larger causal effect of adiposity on fasting insulin levels in men than in women when using the FTO instrument. We found that several estimates from the observational analyses were significantly different from the causal estimates in the old stratum.
Causal Effects of Adiposity on Cardiovascular Risk Factors
We confirm previous evidence that adiposity causally affects a broad range of cardiovascular risk factors. Adipose tissue has been shown to secrete >50 hormones and signaling molecules, many of them being proinflammatory or anti-inflammatory, and thereby exerting effects on insulin sensitivity (25). Adipose cells from obese individuals have higher secretion of proinflammatory cytokines such as tumor necrosis factor-α and IL-6 than cells from lean individuals (26), as well as increased secretion of adiponectin, which has been shown to increase insulin sensitivity (27). Decreased insulin sensitivity (i.e., insulin resistance) has been suggested to be a major link between obesity and cardiovascular risk factors such as hypertension, dyslipidemia, nonalcoholic fatty liver disease, and type 2 diabetes (28–30). Nevertheless, the metabolic consequences of obesity are complex. For example, nonalcoholic fatty liver disease is known to increase insulin resistance regardless of obesity, and thereby to increase the risk of diabetes. The results of the current study do not elucidate the precise mechanisms of how adiposity leads to the various metabolic phenotypes but, rather, clarify and quantify the causal role of adiposity and delineate its role in different age and sex groups.
Sex Differences in the Effects of Adiposity on Cardiovascular Risk Factors
Men and women show a sexual dimorphism in body fat proportion and distribution, and for a given BMI, women have more adipose tissue than men. However, men are more likely to deposit visceral fat in the abdominal region, while women tend to deposit their fat subcutaneously and in their lower extremities. Excess adipose tissue in the abdominal region, especially visceral fat, is associated with many of the cardiovascular risk factors (31). We found a stronger association of BMI with blood pressure, insulin, liver markers, and circulating lipids in men than in women in the observational analyses. Also, our results from the causal analyses using the FTO as an instrument suggest that a given increase in overall adiposity causes larger detrimental effects on insulin sensitivity in men than in women. This observation is consistent with previous observational studies and extends these results by demonstrating the difference in causal (IV) estimates (12,32). Moreover, in the observational analyses, we observed larger effect estimates of adiposity on CRP in women than in men, which is consistent with previous findings (33). It has been speculated that this sex heterogeneity could originate from differences in adipose endocrine function (34–36).
Age Differences in the Effects of Adiposity on Cardiovascular Risk Factors
The proportion of visceral adipose tissue generally increases with age, particularly among men and postmenopausal women, leading to increased insulin resistance (11,37). Weight loss in older people has been shown to reduce inflammatory markers but also to reduce muscle mass and bone mass density. This has led to a debate about whether weight loss interventions should be recommended at all to elderly obese individuals (38). Because of this apparent differential effect of weight changes in older and younger people, we carried out the analyses after stratification of participants according to age (<55 or ≥55 years). We chose the age cutoff of 55 years for the following reasons: 1) to achieve comparable numbers of studies with information available for the two age strata and 2) most women have entered menopause by 55 years of age. For power reasons, we could not stratify on more than two age groups. We found associations between adiposity and markers of inflammation, liver damage, blood pressure, and lipid concentrations to be attenuated by age and only the association with fasting glucose to be augmented. Differences in causal point estimates between the two age strata were more extreme than the differences in observational estimates, with an almost abolished effect of adiposity on several traits such as blood pressure in the ≥55-year stratum using IV analysis, although formal statistical tests for differences were, in general, nonsignificant (likely due to the inherent lower precision of causal than observational estimates). Our genetic score instrument was stronger in the <55-year age group than in the ≥55-year age group, as can be seen from a fourfold difference in F statistic values. This difference yielded slightly larger SEs for the IV estimates in the ≥55-year-old group, but should not bias the point estimates. The observed age-declining effects of adiposity on nonglycemic traits, such as blood pressure and circulating lipid levels, are in line with the limited prior literature (13,15). We speculate that these results could be caused by age-related changes in vascular biology and lipid metabolism, or could be explained by survival bias or a greater prescription of lipid-lowering therapy in older obese individuals. Further, we found that several of the estimates derived from cross-sectional observational analysis were significantly larger than IV estimates in the ≥55-year stratum. We believe that the IV estimates are better estimates of the causal effect of adiposity on cardiovascular risk factors, and that observational estimates in this age group should be interpreted with caution. We speculate that the observed error in the observational analysis is caused by confounders, such as the presence of other diseases or subclinical CVD, not accounted for in the models that can affect both BMI and CVD risk factors.
Strengths and Limitations
The main strengths of the present investigation include the large study size, allowing for age and sex stratification and the examination of a wide range of cardiovascular risk factors. The limitations are mainly related to the validity of the assumptions underlying causal interpretation within Mendelian randomization studies, as follows: 1) independence between the instrument and confounders (i.e., genotypes are randomized); 2) a reliable association between the genetic variant and intermediate phenotype; and 3) conditional independence between the genetic variant and the outcome, given the intermediate phenotype and the confounders (i.e., lack of pleiotropy) (9).
Possible violations of the first and the third assumptions include population stratification, pleiotropic effects, canalization, epigenetic effects, and the presence of genes associated with confounders and outcomes in LD with the genetic variants used. Neither the first nor the third assumption can be tested statistically, and conclusions about the validity of these assumptions in a given study have to be based on previous biological knowledge. In the current study, all association analyses were performed within each study separately (including individuals from a similar genetic background), and all studies included only individuals of European ancestry. Hence, bias from population stratification is deemed unlikely. With regard to the possibility of pleiotropic effects by genes included in the genetic score, we acknowledge that for most loci, neither the causal genes nor their precise effects are known. A combined genetic score, such as the one used in our study, has been suggested to balance the possible pleiotropic effects of some included SNPs (39), but there is controversy about this notion (40). Some of the SNPs with the largest effect on BMI, such as for variants in the close vicinity to FTO and MC4R, also come up in genome-wide association studies for other traits, such as HDL-C. The effects on non-BMI traits are usually proportional to their effects on BMI itself, which is a sign of mediation rather than a sign of these SNPs being pleiotropic (41). Finally, we found similar results when using the genetic score compared with those using the single SNP instrument, which argues against strong pleiotropic effects as the explanation of our results.
Another limitation is that, despite the very large sample size and strong associations of the instruments with BMI, our analysis still obtained results with wide CIs for the IV estimators, highlighting modest power. The sample sizes in IV analyses varied with phenotype and strata from 2,077 individuals (age <55 years, 2-h post-OGTT glucose levels) to 67,553 individuals (nonstratified analysis, systolic blood pressure), yielding larger SEs in the smaller groups, and lower statistical power to detect causal effects and differences between strata. This should be taken into consideration when interpreting the results. Based on a pilot study (data not shown), we chose to use a nonweighted genetic score. The use of such a nonweighted score compared with a weighted score in IV analysis has been evaluated by Burgess and Thompson (40) under different settings, and was found to yield less power but unbiased IV estimates. They further conclude that mis-specifications of the genetic model, such as nonlinear genetic effects, or effect modifications by variant-variant or variant-environment interactions do not lead to significant bias.
Conclusion
For the first time in a large Mendelian randomization study, we applied stratified analysis to assess differences in the causal effect of adiposity on cardiovascular risk factors across age and sex groups. For total cholesterol levels, causal and observational estimates were larger in the <55-year stratum than in the ≥55-year stratum, a finding that could be explained by biology, survival bias, or differential medication in older obese subjects.
Supplementary Material
Article Information
Acknowledgments. The authors thank V. Soo and other personnel (EGCUT) and the staff from the Genotyping Facilities at the Wellcome Trust Sanger Institute for genotyping the data (MORGAM). The authors also thank the late Professor Paula Rantakallio (for the launch of NFBC1966 and the initial data collection), Sarianna Vaara (for data collection), Tuula Ylitalo (for administration), Markku Koiranen (for data management), and Outi Tornwall and Minttu Jussila (for DNA biobanking). The authors thank Tomas Axelsson, Ann-Christine Wiman, and Caisa Pöntinen for their excellent assistance with genotyping (GOSH, PIVUS, ULSAM studies). Sites and key personnel of the contributing MORGAM Centres: Finland: MORGAM Data Centre, National Institute for Health and Welfare, Helsinki: K. Kuulasmaa (Responsible Person), Z. Cepaitis, A. Haukijärvi, B. Joseph, J. Karvanen, S. Kulathinal, M. Niemelä, and O. Saarela. MORGAM Central Laboratory, National Institute for Health and Welfare, Helsinki: M. Perola (Responsible Person), P. Laiho, M. Sauramo. France: National Coordinating Centre, National Institute of Health and Medical Research (U258), Paris: P. Ducimetière (National Coordinator), A. Bingham. PRIME/Strasbourg, Department of Epidemiology and Public Health, EA 3430, University of Strasbourg, Faculty of Medicine, Strasbourg: D. Arveiler (Principal Investigator), B. Haas, A. Wagner. PRIME/Toulouse, Department of Epidemiology, Toulouse University School of Medicine, Toulouse: J. Ferrières (Principal Investigator), J.-B. Ruidavets, V. Bongard, D. Deckers, C. Saulet, and S. Barrere. PRIME/Lille, Department of Epidemiology and Public Health, INSERM U744-Université Lille Nord de France–Institut Pasteur de Lille: P. Amouyel (Principal Investigator), M. Montaye, B. Lemaire, S. Beauchant, D. Cottel, C. Graux, N. Marecaux, C. Steclebout, and S. Szeremeta. MORGAM Laboratory, INSERM U937, Paris: F. Cambien (Responsible Person), L. Tiret, V. Nicaud. The authors thank G. Luc, J.-M. Bard, I. Juhan-Vague, P. Morange, B. Perret, F. Gey, J. Woodside, and I. Young for their help in laboratory analyses. Italy: Research Center on Epidemiology and Preventive Medicine, Department of Clinical and Experimental Medicine, University of Insubria, Varese: M.M. Ferrario (Principal Investigator) and G. Veronesi. Research Centre on Public Health, University of Milano-Bicocca, Monza, Italy: Giancarlo Cesana and Stefano Signorini from Laboratory Medicine, Desio Hospital, are acknowledged for laboratory analysis and biobanking. U.K.: The Belfast PRIME Study, Queen's University Belfast, Belfast, Northern Ireland, U.K.: F. Kee (Principal Investigator) A. Evans (Former Principal Investigator), J. Yarnell, E. Gardner. MORGAM Coordinating Centre, Queen's University Belfast, Belfast, Northern Ireland, U.K.: A. Evans (MORGAM Coordinator), S. Cashman, F Kee. MORGAM Management Group: A. Evans (Chair, Belfast, U.K.), S. Blankenberg (Hamburg, Germany), F. Cambien (Paris, France), M. Ferrario (Varese, Italy), K. Kuulasmaa (Helsinki, Finland), A. Palotie (Cambridge, U.K.), M. Perola (Helsinki, Finland), A. Peters (Neuherberg, Germany), V. Salomaa (Helsinki, Finland), H. Tunstall-Pedoe (Dundee, Scotland, U.K.), F. Kee (Belfast, U.K.), P.G. Wiklund (Umeå, Sweden); previous members: K. Asplund (Stockholm, Sweden), L. Peltonen (Helsinki, Finland), D. Shields (Dublin, Ireland), and B. Stegmayr (Umeå, Sweden).
Funding. The study was funded by the ENGAGE (European Network for Genetic and Genomic Epidemiology) Consortium through the European Community Seventh Framework Programme grant FP7-HEALTH-F4-2007 (201413). The B58C Study was conducted in part using phenotype and genotype data from the British 1958 Birth Cohort DNA collection, which was funded by Medical Research Council grant G0000934 and Wellcome Trust grant 068545/Z/02 (http://www.b58cgene.sgul.ac.uk/). Genotyping for the B58C-WTCCC subset was funded by Wellcome Trust grant 076113/B/04/Z. The B58C-T1DGC genotyping used resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, the National Human Genome Research Institute, the National Institute of Child Health and Human Development, and the Juvenile Diabetes Research Foundation International, and was supported by National Institutes of Health (NIH) grant U01-DK-062418. B58C-T1DGC genome-wide association study data were deposited by the Diabetes and Inflammation Laboratory, Cambridge Institute for Medical Research (CIMR), University of Cambridge, which is funded by the Juvenile Diabetes Research Foundation International, the Wellcome Trust, and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre; the CIMR is in receipt of a Wellcome Trust Strategic Award (079895). The B58C-GABRIEL genotyping was supported by a contract from the European Commission Framework Programme 6 (018996) and grants from the French Ministry of Research. The EGCUT study was supported by FP7 grants (278913, 306031, 313010), the Center of Excellence in Genomics (EXCEGEN), the University of Tartu (SP1GVARENG), Estonian Research Council grant IUT20-60, the Estonian Research Roadmap through the Estonian Ministry of Education and Research, and the Estonian Science Foundation (grant ETF9353). The FINRISK study was supported by the Academy of Finland Center of Excellence in Complex Disease Genetics (grants 213506 and 129680), the Academy of Finland (grants 251217 and 139635), the Finnish Foundation for Cardiovascular Research, and the Sigrid Juselius Foundation. The FTC/Twinfat study was supported by Global Research Awards for Nicotine Dependence (GRAND) to J.K., the Wellcome Trust Sanger Institute, ENGAGE grant FP7-HEALTH-F4-2007, grant agreement number 201413, and the Academy of Finland (grants 265240 and 263278 to J.K.). The Twinfat study was supported by the Academy of Finland (grants 266286, 272376), the Novo Nordisk Foundation, Helsinki University Hospital Research Funds, the Diabetes Research Foundation, and the Finnish Foundation for Cardiovascular Research. Recruitment for the GRAPHIC Study was funded by the British Heart Foundation; and genotyping was funded by the British Heart Foundation, the Wellcome Trust, and the NIHR. The HELENA Study received funding from the European Union (EU) Sixth RTD Framework Program (contract FOOD-CT-2005-007034). The KORA research platform was initiated and financed by the Helmholtz Zentrum München, German Research Center for Environmental Health (Neuherberg, Germany), which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Part of this work was financed by the German National Genome Research Network (NGFNPlus, project number 01GS0834); the German Research Foundation (grants TH-784/2-1 and TH-784/2-2); the European Foundation for the Study of Diabetes; and, through additional funds from the Helmholtz Zentrum München, the German Diabetes Center, and the University of Ulm. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC Health) and Ludwig-Maximilians-Universität München as part of LMUinnovativ. The diabetes part of the KORA F4 study was funded by a grant from the German Research Foundation (DFG; grant RA 459/3-1). This work was supported by the Ministry of Science and Research of the State of North Rhine-Westphalia (MIWF NRW) and the German Federal Ministry of Health (BMG). This study was supported in part by a grant from the BMBF to the German Center for Diabetes Research (DZD e.V.) and to the German Center for Cardiovascular Disease Research (DZHK e.V.). The World Health Organization-MONICA population study that was developed in the north of France was supported by grants from the Conseil Régional du Nord-Pas de Calais, the Fondation pour la Recherche Médicale, ONIVINS, the Parke-Davis Laboratory, the Mutuelle Générale de l'Education Nationale (MGEN), the Réseau National de Santé Publique, the Direction Générale de La Santé, INSERM, the Institut Pasteur de Lille, and the Unité d'Evaluation du Centre Hospitalier et Universitaire de Lille. The MORGAM study was funded in part through the European Community Sixth Framework Programme Cardiogenics project (grant agreement LSHM-CT-2006-037593) and the Seventh Framework Programme ENGAGE project (grant agreement HEALTH-F4-2007-201413). The ATBC Study was supported by U.S. Public Health Service contracts N01-CN-45165, N01-RC-45035, and N01-RC-37004 from the National Cancer Institute. The PRIME Study is organized under an agreement between INSERM and the Merck Sharp & Dohme-Chibret Laboratory for the baseline examinations and the first 5 years of follow-up. The Brianza cohort study activities were supported by the Health Administration of Regione Lombardia (grants 9783/1986, 41795/1993, 31737/1997, 17155/2004, and 10800/2009). The MPP study was supported by grants from the Heart and Lung Foundation (grant 2006-01699), Sweden; and from the Region Skåne county council, Sweden. The Netherlands Study of Depression and Anxiety (NESDA [www.nesda.nl]) was funded by the Netherlands Organization for Scientific Research (NWO) (Geestkracht program grant 10-000-1002), participating institutes (VU University, Leiden University, and University of Groningen), the Neuroscience Campus Amsterdam (NCA), the Center for Medical Systems Biology (CMSB; NWO Genomics), and Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL). Genotyping was funded by the U.S. National Institute of Mental Health (NIMH) (RC2MH089951) as part of the American Recovery and Reinvestment Act of 2009. The NFBC study was supported by the Academy of Finland (project grants 104781, 120315, 129418, Center of Excellence in Complex Disease Genetics and Public Health Challenges Research Program [SALVE]); University Hospital Oulu, Biocenter; University of Oulu, Finland (75617); the European Commission (EUROBLCS, Framework 5 award QLG1-CT-2000-01643); the National Heart, Lung, and Blood Institute (grant 5R01HL087679-02) through the SNP Typing for Association with Multiple Phenotypes from Existing Epidemiologic Data (STAMPEED) programme (1RL1MH083268-01); NIH/NIMH (grant 5R01MH63706:02); the ENGAGE project and grant agreement (HEALTH-F4-2007-201413); the Medical Research Council, U.K. (grants G0500539, G0600705); the PrevMetSyn/Public Health Challenges Research Program [SALVE]); and the European Union Framework Programme 7 small-scale focused research collaborative project EurHEALTHAgeing (grant 277849). The NTR study was conducted with funding from the NWO (MagW/ZonMW grants 904-61-090, 985-10-002, 904-61-193, 480-04-004, 400-05-717, Addiction-31160008 Middelgroot-911-09-032, Spinozapremie 56-464-14192), Center for Medical Systems Biology (CMSB, NWO Genomics), NBIC/BioAssist/RK (2008.024), Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL, 184.021.007), the VU University Institute for Health and Care Research (EMGO+) and Neuroscience Campus Amsterdam (NCA), the European Science Foundation (grant EU/QLRT-2001-01254), the European Community Seventh Framework Program (grant FP7/2007-2013), ENGAGE (HEALTH-F4-2007-201413), the European Science Council (ERC Advanced, grant 230374), Rutgers University Cell and DNA Repository (NIMH, U24-MH-068457-06), the Avera Institute (Sioux Falls, SD), and the NIH (grant R01-D0042157-01A). Part of the genotyping and analyses was funded by the Genetic Association Information Network (GAIN) of the Foundation for the U.S. NIH, the U.S. NIMH (grant MH-081802), and Grand Opportunity grants 1RC2-MH-089951-01 and 1RC2-MH-089995-01 from the NIMH. The Botnia PPP study was supported by grants from the Signe and Ane Gyllenberg Foundation, the Swedish Cultural Foundation in Finland, the Finnish Diabetes Research Society, the Sigrid Juselius Foundation, the Folkhälsan Research Foundation, the Foundation for Life and Health in Finland, Jakobstad Hospital, the Medical Society of Finland, the Närpes Research Foundation and the Vasa and Närpes Health Centers, the European Community Seventh Framework Programme (FP7/2007-2013), the ENGAGE, the Collaborative European Effort to Develop Diabetes Diagnostics (grant CEED/2008-2012), and the Swedish Research Council, including Linné grant 31475113580. The QIMR-Australia study was conducted with funding from the Australian National Health and Medical Research Council (NHMRC; grants 241944, 389875, 389891, 389892, 389938, 442915, 442981, 496739, and 552485), U.S. NIH (grants AA-07535, AA-10248, and AA-014041), and the Australian Research Council (ARC; grant DP0770096). The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University Rotterdam; the NWO; the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Netherlands Heart Foundation; the Ministry of Education, Culture and Science; the Ministry of Health, Welfare, and Sports; the European Commission; and the Municipality of Rotterdam. Support for genotyping was provided by the NWO Investments (grants 175.010.2005.011, 911-03-012), the Research Institute for Diseases in the Elderly (grant 014-93-015, RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Consortium for Healthy Aging (NCHA) project 050-060-810. A.D. is supported by the NWO (veni grant 916.12.154) and the EUR Fellowship. The TwinsUK study was funded by the Wellcome Trust and the European Community Seventh Framework Programme (grant FP7/2007-2013). The study also receives support from the NIHR BioResource Clinical Research Facility and Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. Single nucleotide polymorphism genotyping was performed by the Wellcome Trust Sanger Institute and National Eye Institute via NIH/CIDR. The ULSAM/PIVUS/GOSH/TWINGENE studies were conducted using funding by the ENGAGE Consortium, European Community Seventh Framework Programme grant FP7-HEALTH-F4-2007 (201413); Swedish Research Council grants 521-2013-8689, M-2005-1112, and 2009-2298; the Swedish Research Council for Health, Working Life and Welfare (grant 2013-2292); the Swedish Heart-Lung Foundation; the Swedish Foundation for Strategic Research (SSF, ICA08-0047); the Royal Swedish Academy of Sciences; the Swedish Diabetes Foundation; the Swedish Society of Medicine; Novo Nordisk Foundation; Uppsala University; Uppsala University Hospital; and the Swedish Research Council for Infrastructures. Recruitment for the WTCCC-CAD Study was funded by the British Heart Foundation, and genotyping was funded by the Wellcome Trust. C.P.N. is supported by the NIHR Leicester Cardiovascular Biomedical Research Unit. K.K. was supported by the Orion-Farmos Research Foundation and the Academy of Finland (grant 250207). M.D.T. holds a Medical Research Council Senior Clinical Fellowship. C.G. was supported by the Helmholtz-Russia Joint Research Group. N.J.S. holds a Chair funded by the British Heart Foundation and is an NIHR Senior Investigator. G.W.M. is supported by the NHMRC Fellowship Scheme. T.D.S. is the holder of an ERC Advanced Principal Investigator award. M.I.M. is a Wellcome Trust Senior Investigator, and is supported by Wellcome trust grants 090532 and 098381.
Duality of Interest. The MONA LISA Study was made possible by an unrestricted grant from Pfizer and a grant from the French Agence Nationale de la Recherche (ANR-05-PNRA-018). The PRIME Study is organized under an agreement between INSERM and the Merck Sharp & Dohme-Chibret Laboratory for the baseline examinations and the first 5 years of follow-up. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. T.F., S.H., A.Pl., R.M., K.F., N.L.P., I.P., M.I.M., and E.I. wrote the first draft of the manuscript. D.P.S. (B58C), R.M., K.F., M.Kal., T.E. (EGCUT_metabo, EGCUT_omni), A.-P.S. (FR92, FR97, FR02, FR07, H2000), K.H. (FTC), C.P.N. (GRAPHIC, WTCCC-CAD), A.Mei. (HELENA, MONA LISA, MONICA), H.G. (KORAF3), M.Kob. (KORAF4), K.Kr., M.-L.N. (MORGAM), C.L., M.Å. (MPP, PPP), M.Kaa., V.H. (NFBC1966, NFBC1986), H.H.M.D. (NTR, NESDA), B.B. (QIMR), A.D., M.Kun., P.S.d.V., R.F.A.G.d.B., S.M.W. (Rotterdam), T.F. (TWINGENE, ULSAM, PIVUS, GOSH), and M.M. (TWINSUK) performed study-level analyses. A.Met. (EGCUT_metabo, EGCUT_omni), D.P.S. (B58C), A.S.Hav., V.S., S.R. (FR92, FR97, FR02, FR07), L.A.M., M.G.-G. (HELENA), K.H., K.Si., J.K. (FTC), N.L.P., E.I. (GOSH), M.D.T., N.J.S. (GRAPHIC), A.J., S.R., A.Pa. (H2000), A.Pe., K.St., W.K., (KORAF3), P.L., C.H., C.G. (KORAF4), L.Go., J.D., P.A. (MONA LISA), V.Le., D.C., P.A. (MONICA), K.Ku., M.P., A.E., J.F., J.V., J.W.G.Y., D.-A.T., D.A., P.A., G.V., P.B. (MORGAM), P.M.N., V.Ly. (MPP, PPP), B.W.P. (NESDA), M.-R.J. (NFBC1966, NFBC1986), D.I.B. (NTR), L.L., A.-C.S., E.I. (PIVUS), L.Gr., G.W.M., N.G.M., J.B.W. (QIMR), A.G.U., B.H.S., M.A.I., A.S.H., O.H.F., C.M.v.D. (Rotterdam), P.K.M., N.L.P. (TWINGENE), C.M., T.D.S. (TWINSUK), V.G., A.-C.S., E.I. (ULSAM), A.S.Hal., and N.J.S. (WTCCC-CAD) researched and provided the data; and reviewed, revised, and approved the manuscript. E.I. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0988/-/DC1.
References
- 1.Gersh BJ, Sliwa K, Mayosi BM, Yusuf S. Novel therapeutic concepts: the epidemic of cardiovascular disease in the developing world: global implications. Eur Heart J 2010;31:642–648 [DOI] [PubMed] [Google Scholar]
- 2.Wenger NK. Gender disparity in cardiovascular disease: bias or biology? Expert Rev Cardiovasc Ther 2012;10:1401–1411 [DOI] [PubMed] [Google Scholar]
- 3.Anand SS, Islam S, Rosengren A, et al.; INTERHEART Investigators . Risk factors for myocardial infarction in women and men: insights from the INTERHEART study. Eur Heart J 2008;29:932–940 [DOI] [PubMed] [Google Scholar]
- 4.Dow CA, Thomson CA, Flatt SW, Sherwood NE, Pakiz B, Rock CL. Predictors of improvement in cardiometabolic risk factors with weight loss in women. J Am Heart Assoc 2013;2:e000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siebenhofer A, Jeitler K, Berghold A, et al. Long-term effects of weight-reducing diets in hypertensive patients. Cochrane Database Syst Rev 2011;CD008274. [DOI] [PubMed] [Google Scholar]
- 6.Madsbad S, Dirksen C, Holst JJ. Mechanisms of changes in glucose metabolism and bodyweight after bariatric surgery. Lancet Diabetes Endocrinol 2014;2:152–164 [DOI] [PubMed] [Google Scholar]
- 7.Fall T, Hägg S, Mägi R, et al.; European Network for Genetic and Genomic Epidemiology (ENGAGE) consortium . The role of adiposity in cardiometabolic traits: a Mendelian randomization analysis. PLoS Med 2013;10:e1001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordestgaard BG, Palmer TM, Benn M, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med 2012;9:e1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med 2008;5:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science 2005;308:1583–1587 [DOI] [PubMed] [Google Scholar]
- 11.Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev 2013;93:359–404 [DOI] [PubMed] [Google Scholar]
- 12.Geer EB, Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med 2009;6(Suppl. 1):60–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pikilidou MI, Scuteri A, Morrell C, Lakatta EG. The burden of obesity on blood pressure is reduced in older persons: the SardiNIA study. Obesity (Silver Spring) 2013;21:E10–E13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitlock G, Lewington S, Sherliker P, et al.; Prospective Studies Collaboration . Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 2009;373:1083–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Rennie DC, Reeder BA. Age-related association between body mass index and blood pressure: the Humboldt Study. Int J Obes Relat Metab Disord 1995;19:825–831 [PubMed] [Google Scholar]
- 16.Wakabayashi I. Relationships of body mass index with blood pressure and serum cholesterol concentrations at different ages. Aging Clin Exp Res 2004;16:461–466 [DOI] [PubMed] [Google Scholar]
- 17.Wakabayashi I, Daimon T. Age-dependent decline of association between obesity and hyperglycemia in men and women. Diabetes Care 2012;35:175–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berrington de Gonzalez A, Hartge P, Cerhan JR, et al. Body-mass index and mortality among 1.46 million white adults. N Engl J Med 2010;363:2211–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speliotes EK, Willer CJ, Berndt SI, et al.; MAGIC; Procardis Consortium . Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 2008;17:R122–R128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org, 2013
- 22.Lumley T. rmeta: Meta-analysis. R package version 2.16. http://CRAN.R-project.org/package=rmeta, 2012
- 23.Palmer TM, Sterne JA, Harbord RM, et al. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol 2011;173:1392–1403 [DOI] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;57:289–300 [Google Scholar]
- 25.Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm 2013;2013:139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011;11:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao H, Fall T, van Dam RM, et al. Evidence of a causal relationship between adiponectin levels and insulin sensitivity: a Mendelian randomization study. Diabetes 2013;62:1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landsberg L, Aronne LJ, Beilin LJ, et al. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension. J Clin Hypertens (Greenwich) 2013;15:14–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am 2011;95:875–892 [DOI] [PubMed] [Google Scholar]
- 30.Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol 2013;218:R25–R36 [DOI] [PubMed] [Google Scholar]
- 31.Racette SB, Evans EM, Weiss EP, Hagberg JM, Holloszy JO. Abdominal adiposity is a stronger predictor of insulin resistance than fitness among 50-95 year olds. Diabetes Care 2006;29:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sierra-Johnson J, Johnson BD, Bailey KR, Turner ST. Relationships between insulin sensitivity and measures of body fat in asymptomatic men and women. Obes Res 2004;12:2070–2077 [DOI] [PubMed] [Google Scholar]
- 33.Thorand B, Baumert J, Döring A, et al.; KORA Group . Sex differences in the relation of body composition to markers of inflammation. Atherosclerosis 2006;184:216–224 [DOI] [PubMed] [Google Scholar]
- 34.Lear SA, Chen MM, Birmingham CL, Frohlich JJ. The relationship between simple anthropometric indices and C-reactive protein: ethnic and gender differences. Metabolism 2003;52:1542–1546 [DOI] [PubMed] [Google Scholar]
- 35.Rossi IA, Bochud M, Bovet P, et al. Sex difference and the role of leptin in the association between high-sensitivity C-reactive protein and adiposity in two different populations. Eur J Epidemiol 2012;27:379–384 [DOI] [PubMed] [Google Scholar]
- 36.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA 1999;282:2131–2135 [DOI] [PubMed] [Google Scholar]
- 37.Pascot A, Lemieux S, Lemieux I, et al. Age-related increase in visceral adipose tissue and body fat and the metabolic risk profile of premenopausal women. Diabetes Care 1999;22:1471–1478 [DOI] [PubMed] [Google Scholar]
- 38.Darmon P. Intentional weight loss in older adults: useful or wasting disease generating strategy? Curr Opin Clin Nutr Metab Care 2013;16:284–289 [DOI] [PubMed] [Google Scholar]
- 39.Davey Smith G. Random allocation in observational data: how small but robust effects could facilitate hypothesis-free causal inference. Epidemiology 2011;22:460–463 [DOI] [PubMed] [Google Scholar]
- 40.Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013;42:1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Willer CJ, Schmidt EM, Sengupta S, et al.; Global Lipids Genetics Consortium . Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.