

Abstract
A major event in land plant evolution is the origin of vascular tissues, which ensure the long-distance transport of water, nutrients and organic compounds. However, the molecular basis for the origin and evolution of plant vascular tissues remains largely unknown.
Here, we investigate the evolution of the land plant TAL-type transaldolase (TAL) gene and its potential function in rice (Oryza sativa) based on phylogenetic analyses and transgenic experiments, respectively.
TAL genes are only present in land plants and bacteria. Phylogenetic analyses suggest that land plant TAL genes are derived from Actinobacteria through an ancient horizontal gene transfer (HGT) event. Further evidence reveals that land plant TAL genes have undergone positive selection and gained several introns following its acquisition by the most recent common ancestor of land plants. Transgenic plant experiments show that rice TAL is specifically expressed in vascular tissues and that knockdown of TAL expression leads to changes in both the number and pattern of vascular bundles.
Our findings show that the ancient HGT of TAL from bacteria probably plays an important role in plant vascular development and adaptation to land environments.
Keywords: bacteria, horizontal gene transfer (HGT), land plants, rice (Oryza sativa), TAL-type transaldolase (TAL), vascular development
Introduction
Horizontal gene transfer (HGT), also known as lateral gene transfer (LGT), refers to the transfer of genetic material between organisms with reproductive isolation. HGT plays an important role in the adaptive evolution of recipient lineages because the acquisition of novel genes may allow the recipient organism to explore new niches or to utilize new resources (Keeling & Palmer, 2008; Andersson, 2009). HGT is a major force driving the evolution of prokaryotes and leads to the spread of certain adaptive traits, such as antibiotic resistance and virulence (Nogueira et al., 2009; Palmer et al., 2010). Traditionally, HGT is thought to be frequent in prokaryotes and unicellular eukaryotes, but rare in multicellular eukaryotes, such as animals and plants (Huang & Gogarten, 2008; Keeling & Palmer, 2008; Keeling, 2009; Huang, 2013). However, recent investigations indicate that HGT also play important roles in the evolution of animals and plants (Richards et al., 2009; Dunning Hotopp, 2011; Yang et al., 2013). A genome-wide scan of the moss Physcomitrella patens revealed that 128 nuclear genes were acquired through HGT from prokaryotes, fungi, or viruses. These genes are involved in many plant-specific activities, including xylem formation, plant defense, nitrogen recycling, and the biosynthesis of starch, polyamines, hormones, and glutathione (Yue et al., 2012, 2013).
The pentose phosphate pathway (PPP), a key series of enzymatic reactions, is a process that generates NADPH and pentoses (five-carbon sugars) (Kruger & von Schaewen, 2003). There are two distinct branches in this pathway, one being the oxidative branch that leads to the production of NADPH, and the other the nonoxidative branch with roles in the synthesis of five-carbon sugars (Caillau & Paul Quick, 2005). In the nonoxidative branch of the PPP, transaldolase (TA) is a key enzyme that catalyzes the reversible transfer of a dihydroxyacetone group from fructose-6-phosphate to erythrose-4-phosphate, yielding sedoheptulose-7-phosphate and glyceraldehyde-3-phosphate. In addition, the roles of transaldolase, together with transketolase, also include a link between the glycolytic and PPP (Takayama et al., 1997; Caillau & Paul Quick, 2005).
Based on their conserved domains, the transaldolase family is divided into three subfamilies, including the transaldolase-like (TAL, cd00955), transaldolase_FSA (cd00956), and transaldolase_TalAB (cd00957) subfamilies. Although the TA subfamilies are well conserved in biochemical properties and the architecture of active sites, their overall sequence similarities are quite low (Caillau & Paul Quick, 2005; Samland et al., 2011). Land plants possess two types of TAs, including TAL and TalAB. The TalAB-type TA was proposed as the classical type, and members of this subfamily are ubiquitous to all three domains of life (i.e. archaea, bacteria, and eukaryotes), whereas the TAL-type TA is present only in land plants and bacteria. Two types of plant TA genes appear to be differentially expressed in response to environmental factors, suggesting that the TAL and TalAB isoforms have a nonoverlapping role in plant metabolism (Caillau & Paul Quick, 2005).
The ubiquity of the TAL-type TA in land plants suggests that its functions include a wide range of selectivity. In this study, the sequence similarity, phylogenetic relationships, and taxonomic distribution of the TAL-type TA genes were used to determine its origin in land plants. Our analyses revealed that the land plant TAL genes originated from an ancient HGT event from bacteria. Further studies with transgenic rice revealed that the TAL gene is required for rice vascular patterning.
Materials and Methods
Identification of TAL genes
To identify the genes encoding TAL-type TA in the land plants, the protein sequence of the Arabidopsis gene At5g13420 was used as a query to search the Phytozome database (Goodstein et al., 2012). If a protein sequence satisfied E ≤ 10−10, it was selected as a candidate protein. Then, the CD-search tool in the Conserved Domain Database (CDD) of the National Center for Biotechnology Information (NCBI; Marchler-Bauer et al., 2013) was used to predict the transaldolase-like domain (cd00955). The newly identified TAL sequences detected in the land plants were used iteratively to search the respective sequence database. The deduced nucleotide and protein sequences of land plant TAL genes identified in this analysis were downloaded from the Phytozome database.
To identify the homologs of land plant TAL genes, BLAST searches against NCBI nr, dbEST, as well as JGI and other available eukaryotic genome databases (Supporting Information Table S1) were performed using land plant TAL proteins as queries. The sequences were further analyzed using an NCBI conserved domain search to confirm the presence of a transaldolase-like domain in the protein structure.
Phylogenetic analyses
Protein sequence alignment was performed with Clustal X (Larkin et al., 2007) followed by visual inspection and manual refinement. The gaps and ambiguously aligned sites were removed manually. The program Modelgenerator (Keane et al., 2006) was used to identify the optimal model of protein substitution and rate heterogeneity. Phylogenetic analyses were performed with a maximum likelihood (ML) approach using PhyML version 3.0 (Guindon et al., 2010) and a neighbor-joining method using MEGA (Tamura et al., 2013). The ML phylogenetic analyses were conducted with the following parameters: JTT model, estimated proportion of invariable sites, four rate categories, estimated gamma distribution parameter, and optimized starting BIONJ tree. The JTT model was also used to construct the NJ trees. A total of 100 nonparametric bootstrap samplings were performed to estimate the support level for each internal branch for both the ML and NJ trees. The branch lengths and topologies of all phylogenies were calculated using PhyML. The phylogenetic trees were visualized using the explorer program in MEGA.
Detection of positive selection
To test the selective pressure of the TAL genes during the long period of evolution in both land plants and Actinobacteria, the values of the dN : dS ratio (or ω) for two groups of TAL genes were calculated with the program codeml from PAML v4.4 (Yang, 2007). In this analysis, only Actinobacterial TAL genes located in the same branch as land plant homologs were used. Two phylogenetic trees for land plant TAL genes and their close homologs in Actinobacteria were reconstructed using PhyML (Guindon et al., 2010). The PAL2NAL program (Suyama et al., 2006) was used for the conversion of protein sequence alignment into the corresponding codon-based nucleotide alignment, which, in turn, was used for input into the codeml program in PAML. Here, we used three likelihood ratio tests (LRTs), M0 vs M3, M1a vs M2a, and M7 vs M8, to examine the selective pressure. The LRT for the comparison of M0 vs M3 was used to test the heterogeneity between the codon sites in ω, whereas the other two LRTs were used to detect the role of positive selection. For one LRT, twice the difference of the log likelihood of the two models was compared with the chi-squared (χ2) statistics, with the degrees of freedom equal to the difference in the number of parameters. In our analyses, the degrees of freedom are three for the M0/M3 test and two for the M1a/M2a and M7/M8 tests (Nielsen & Yang, 1998; Wong et al., 2004).
The improved branch-site model (Zhang et al., 2005) was also used to detect the role of positive selection that acted on the land plant TAL genes after HGT. In this model, the phylogeny is partitioned into foreground and background branches, with positive selection potentially occurring along the former. Here, the phylogenetic tree was generated using land plant and actinobacterial TAL genes, and the branch of land plants was used as the foreground. For this analysis, we compared the null hypothesis (ω fixed to 1) with the alternative hypothesis (free ω), to test whether positive selection acted on the evolution of land plant TAL genes. The Bayes empirical Bayes procedure in codeml (Yang et al., 2005) was used to calculate the posterior probability that each site was subject to positive selection in the foreground branch.
RNA extraction and gene expression analysis
Total RNA was extracted from different tissues during the heading stage. RNA extraction was performed according the manufacturer's protocol for the RNA prep pure plant kit (Tiangen, Beijing, China). Approximately 1 μg total RNA from each sample was used for first-strand cDNA synthesis. Quantitative PCR was performed using the following gene-specific primer pairs: 5′-AGATACGAGGCTGTGATTGA-3′ and 5′-TCTTGGCACCTTTCTTGAC-3′ for the rice (Oryza sativa L.) TAL gene and 5′-GATGACCCAGATCATGTTTG-3′ and 5′-GGGCGATGTAGGAAAGC-3′ for OsActin, which was a control. Quantitative PCR was performed on a ViiA7 Real-Time System (Applied Biosystems, Foster City, CA, USA) with the SYBR Premix Ex Taq system (TaKaRa, Kusatsu City, Japan). Each set of experiments was repeated three times. The relative amount of the TAL transcript is presented as the 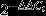 according to the ΔCt method described in the real-time PCR Applications Guide.
according to the ΔCt method described in the real-time PCR Applications Guide.
Transgenic analysis
To generate an RNAi construct, a 265 bp fragment of TAL was amplified from Nipponbare first-strand cDNA using the primers 5′-AAAGGATCCAGATACGAGGCTGTGATTGA-3′ containing a BamHI recognition site and 5′-AAAACTAGTTCTTGGCACCTTTCTTGAC-3′ containing a SpeI recognition site. A hairpin structure with two inverted repeat fragments was then constructed and transferred into the plant binary vector p1301UbiNOS and expressed under the control of the maize ubiquitin promoter (Zhou et al., 2009).
For expression analysis, a 1138 bp genomic fragment upstream of the rice TAL gene translation start codon was PCR-amplified with the primers 5′-AAAGGATCCGGCAGATTTAGTAGAGCCATTTCTC-3′ and 5′-AAACCATGGTGACTCACATGATGGGGCTCCTG-3′. The DNA fragment was cloned into the BamHI and NcoI sites of the vector pCAMBIA1301. The resulting plasmid was transformed into rice, and three independent TAL promoter-beta-glucuronidase (GUS) transgenic plants were analyzed using the GUS staining assay, as previously described (Zhou et al., 2009). All constructs were transformed by Agrobacterium tumefaciens-mediated transformation (Hiei et al., 1994).
Morphological analysis
For histology, the fresh culm samples were removed from plants at the heading stage and then fixed in 2% glutaraldehyde, dehydrated in a graded ethanol series, and embedded in Spurr resin. Transverse sections were cut with an Ultracut EM UC7 (Leica, Solms, Germany), stained with 0.5% toluidine blue, and photographed using a DM1000 microscope system (Leica).
Leaf and stem morphometric analyses were performed at the heading stage. Leaf width and vascular pattern parameters were measured through the middle region of the flag leaf. The vascular pattern parameters of the leaves were measured in the dark-field microscopic digital images. The number of vascular bundles in the outer and inner rings of the stem was quantified in the digital microscopic images of the transverse sections through the middle region of the first and second internodes of the plants at the heading stage.
Results
TAL genes are widespread in land plants
Blast searches against the plant database revealed that each sequenced land plant genome contains at least one gene encoding TAL proteins. To explore the origin and evolutionary process of the land plant TAL genes, we characterized TAL genes from species representing the main lineages of land plants, including moss P. patens, lycophyte Selaginella moellendorffii, gymnosperm Picea sitchensis, five monocots and nine dicots (Table S2). We finally identified 19 TAL genes from the 17 genomes. Both the Linum usitatissimum and Populus trichocarpa genomes contained two TAL genes, whereas other sampled land plant genomes possessed only one TAL gene. Both the paralogous pairs were located on the terminal of the phylogenetic tree (Fig.1), indicating that they were formed through two distinct recent duplication events. We also found that both of these two paralogous pairs resulted from segmental duplication because there were highly conserved genes in the flanking regions for the two pairs of paralogous TAL genes in L. usitatissimum and P. trichocarpa.
Fig 1.

The phylogenetic tree of the land plant TAL-type transaldolase (TAL) genes and their exon/intron structures. The numbers above the branches represent the bootstrap values for the maximum likelihood and distance analyses, respectively. The asterisks indicate values < 50%. The exons are indicated by boxes, whereas introns are indicated by lines. The number above an intron indicates the phase.
Structural analysis of the TAL genes in land plants was performed by comparing the exon/intron organization. By comparing the CDS sequences with the corresponding genomic DNA sequences, we showed that the coding regions of all land plant TAL genes are interrupted by five to seven introns (Fig.1). The positions and phases of the majority of these introns in land plant TAL genes are conserved, demonstrating that the primary gene structure of this family was formed in the common ancestor of land plants. We also observed that the coding regions of TAL genes contained six introns in dicots, but only five introns in monocots. Through comparisons with homologs in P. patens and S. moellendorffii, we conclude that an intron in the middle of the monocot TAL genes was lost after the split between monocots and dicots.
Land plant TAL gene was horizontally acquired from bacteria through an ancient transfer
The origin of land plants is a key event in the history of life and has led to dramatic changes in the environment and ecological systems of Earth. Evidence revealed that land plants evolved from charophycean green algae c. 480–490 million yr ago (Sanderson et al., 2004). Therefore, the majority of land plant genes must be vertically inherited. We searched the NCBI nr and JGI databases for the homologs of land plant TAL proteins. However, no homologs were found in any other eukaryote. The blast results also revealed that homologs of the land plant TAL proteins exist in bacteria only. In addition, the NCBI CDD also showed that genes encoding the TAL type TA proteins are found in land plants and bacteria only. The taxonomic distribution suggests that the evolution of the TAL gene is a result of an ancient HGT between bacteria and the ancestor of the land plants. Similarity searches and the presence of a conserved transaldolase-like domain (cd00955) indicate that the TAL genes are distributed widely in most bacterial lineages. The universality of the distribution in bacteria also suggests that this gene may have first emerged in bacteria.
To determine the origin of the land plant TAL genes, we sampled representative taxonomical groups of cellular organisms in the nr database to build a phylogenetic tree (Fig.2). All selected proteins for the phylogenetic analyses possessed a transaldolase-like domain. Both the ML and distance phylogenetic trees showed a similar topology. Although the selected sequences cover the majority of primary bacterial lineages, in our analysis, the phylogenetic tree of the TAL proteins is not congruent with the species phylogeny, suggesting extensive gene losses, HGT, and selection within bacteria. In the phylogenetic tree, all the sampled land plant TAL genes form a single clade with high bootstrap support. The monophyly of land plant TAL orthologs strongly suggests that they have a single origin and are derived from a unique gene already present in the ancestor of land plants. In addition, we also observed that land plant TAL genes are in the clade of Actinobacteria genes, with high bootstrap support values for both methods. In addition, the close phylogenetic relationship of plant and Actinobacteria TALs is supported by the observation that land plant TALs have the highest similarity with their actinobacterial homologs. These findings demonstrate that land plant TAL genes originated from a single ancient HGT event from Actinobacteria before the separation of the land plant lineages.
Fig 2.
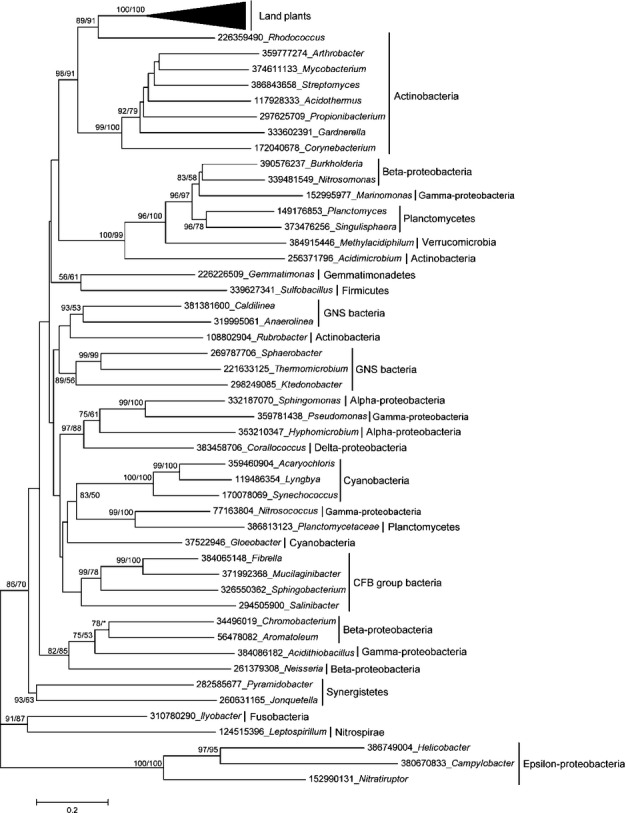
Phylogenetic analyses of TAL-type transaldolase (TAL) proteins. The numbers above the branches represent the bootstrap values for the maximum likelihood and distance analyses. All sequences were obtained from the National Center for Biotechnology Information (NCBI), except for those in the green plants, and each protein is indicated by the GI numbers in NCBI and its genus.
Positive selection facilitated the evolution of TAL genes in plants
The likelihood ratio tests of positive selection were applied using ML methods and the codon substitution models of Yang and colleagues (Nielsen & Yang, 1998; Yang et al., 2005; Yang, 2007). In this analysis, all the sampled land plant TAL genes and eight actinobacterial genes following to the same branch of plants were tested, respectively. First, we compared models M0 and M3 to determine whether there were dN : dS ratio variations for the codon positions for TAL genes both in land plants and Actinobacteria. Overall, both maximum likelihood estimates for the dN : dS values with model M0 were close to zero (Table S3), suggesting that purifying selection was the predominant force in the evolution of the TAL gene in both land plants and Actinobacteria. However, the log-likelihood differences between model M3 and M0 were statistically significant for both lineages, indicating that selective constraint level varied across amino acid positions. Secondly, the likelihood ratio tests to compare the data fit to models M2a vs M1a and M8 vs M7 were used to determine whether positive selection promoted the divergence of the TAL genes in both lineages. No amino acid site was found to be influenced by positive selection during the evolution of TAL genes either in land plants or in Actinobacteria. These results reveal that the primary constraints for the TAL gene in land plants after fixation from a horizontal gene transfer event was purifying selection.
To assess whether land plants are characterized by a different pattern of molecular evolution of the TAL genes compared with those in Actinobacteria, a new phylogeny was constructed using the TAL proteins in the branch of land plants and Actinobacteria. The branch of the new phylogeny leading to land plant TAL genes was classified as the foreground branch and those in Actinobacteria as the background branches. We found that the model that permitted a class of positively selected codons with dN : dS > 1 for the land plants branch had a significantly better fit to the data than the model in which this class of codon was restricted to dN : dS = 1 (Table 1). Because LRTs suggest that positive selection acted on the evolution of the TAL gene in land plants, the method of Bayes empirical Bayes (Yang et al., 2005) was used to evaluate the positively selected sites and their posterior probabilities. A total of 52 codons were identified with a > 50% posterior probability of dN : dS > 1 along the land plant branch. Of these, 11 amino acid sites have a 95% posterior probability of positive selection (Fig. S1). The most reasonable explanation for these results is that the TAL gene has undergone adaptive evolution in the ancestor of land plants during a short period of time after the HGT event, although the dominant force for the evolution in land plants of these amino acid sites is purifying selection.
Table 1.
Parameters of the branch-site models used for the detection of positive selection
| Model | Loge L | Parameters |
|---|---|---|
| Null | −17 119.9710 | p0 = 0.8158, p1 = 0.0466, p2a = 0.1302, p2b = 0.0074 |
| Background: ω0 = 0.0471, ω1 = 1.0000, ω2a = 0.0471, ω2b = 1.0000 | ||
| Foreground: ω0 = 0.0471, ω1 = 1.0000, ω2a = 1.0000, ω2b = 1.0000 | ||
| Alternative | −17 112.9058** | p0 = 0.7947, p1 = 0.0455, p2a = 0.1511, p2b = 0.0087 |
| Background: ω0 = 0.0485, ω1 = 1.0000, ω2a = 0.0485, ω2b = 1.0000 | ||
| Foreground: ω0 = 0.0485, ω1 = 1.0000, ω2a = 48.9623, ω2b = 48.9623 |
**, P < 0.01.
TAL gene is involved in rice vascular patterning
To ascertain the potential function of this horizontally acquired gene, the rice TAL gene was selected for functional detection. Quantitative PCR analysis suggested that rice TAL was expressed in organs such as the leaf, panicle, stem, knot, and root (Fig.3a). Our results also revealed that flowering panicles accumulated the TAL transcript at the highest level and also at the lowest part of the sheath at the heading stage. Furthermore, we generated transgenic rice plants expressing a fragment of the TAL promoter fused with the GUS reporter gene. Three independent TAL promoter-GUS transgenic plants were analyzed. All of them showed a similar expression pattern. TAL-GUS expression was observed with a pattern of strong tissue specificity (Fig.3b–i), and GUS activity was detected mainly in the vascular bundles of tissues, including the large and small veins of leaves (Fig.3e,f). In a cross-section of the stem at the heading stage, high GUS activities were detected in the phloem of vascular bundles (Fig.3h,i). The finding that TAL is preferentially expressed in vascular tissues indicated that this gene may be involved in rice vascular patterning.
Fig 3.
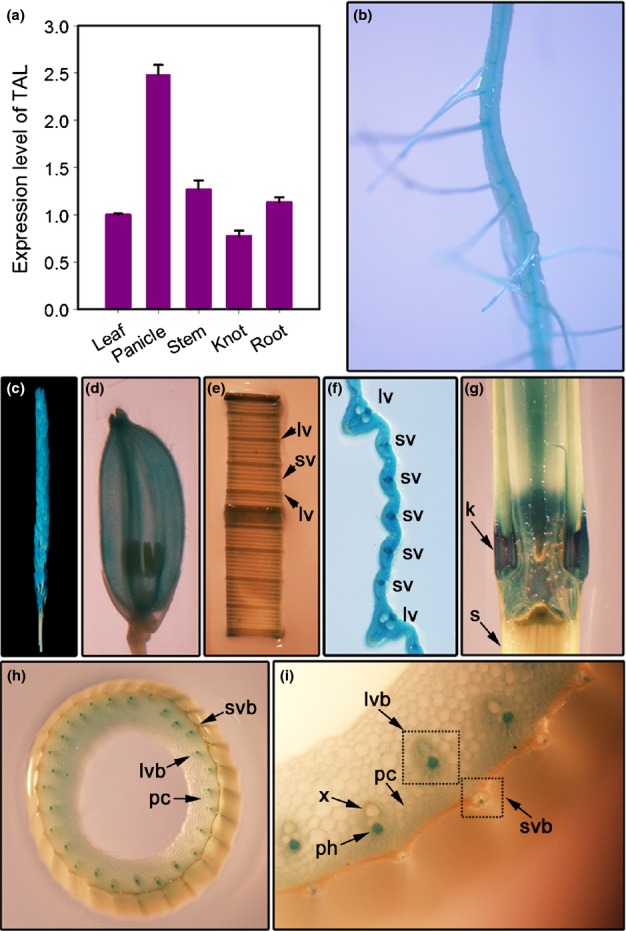
Expression analyses of the rice (Oryza sativa) TAL-type transaldolase (TAL) gene. (a) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of rice TAL expression. Total RNA was isolated from the leaf, panicle, stem, knot, and root of wildtype plants at the heading stage. Amplification of the rice Actin gene was used as a control. Error bars represent + 1SE. (b) Beta-glucuronidase (GUS) activity in root. (c) GUS activity in panicle. (d) GUS activity in glume. (e) GUS activity in leaf at the heading stage. (f) Magnified image of the leaf in (e) showing the strong expression of TAL in developing vascular bundles of leaf. (g) GUS activity in knot and stem. (h) Transverse section of a third internode at the heading stage, showing the expression of TAL promoter-GUS in the vascular bundles of the stem. (i) Magnified image of the stem in (h) showing the strong expression of TAL in the phloem of vascular bundles. lv, large vein; sv, small vein; s, stem; k, knot; lvb, large vascular bundles; svb, small vascular bundles; pc, parenchyma cells; x, xylem; ph, phloem.
To further characterize the function of the rice TAL gene, we developed knockdown plants for the TAL gene in rice by introducing gene-specific RNA interference (RNAi) construct. We established 21 independent lines and grew them under normal conditions in the field. One line, TAL-RNAi, was used for further analysis. Quantitative PCR showed that the TAL transcript in leaves of the TAL-RNAi plant was significantly suppressed (Fig.4a). Leaves of the TAL-RNAi plants were shorter and narrower than those of the wildtype. The length and width of the TAL-RNAi flag leaves were c. 80.5 and 81.3% of the wildtype, respectively (Table S4, Fig.4f). In addition to the leaf shape, the TAL-RNAi plants also showed an altered culm elongation pattern. At maturity, the plant height of the TAL-RNAi was c. 71.0% of the wildtype (Fig.4b–e). We also compared the culm width between the TAL-RNAi and wildtype plants. As shown in Fig.4, the culm diameter of the uppermost four internodes (designated internodes I, II, III, and IV) of the TAL-RNAi was significantly smaller than the wildtype (Fig.4g,h). However, no significant difference in the culm width of the fifth internode was observed (Fig.4g,h).
Fig 4.

Phenotypic analysis of TAL-type transaldolase (TAL)-RNA interference (RNAi) in rice (Oryza sativa) plants. (a) Rice TAL gene expression levels in the leaves of wildtype (WT) and TAL-RNAi (TAL-Ri) plants during the heading stage. The Actin gene was amplified as the control. (b) Comparison of plant height between the WT and TAL-RNAi plants. (c, d) Plant phenotype of WT and TAL-RNAi plants. (e) Panicles and culms of WT and TAL-RNAi plants. (f) Flag leaves of WT and TAL-RNAi plants. (g) The diameter of axes from the first internode (I, panicle-neck internode) to the fifth internode (V, basal internode) of the main culms between the WT (green bars) and TAL-RNAi (purple bars) plants. (h) Cross-sections of the culm from the first internode to the fifth internode. (i) Transverse sections of the middle part of internode I of the WT and TAL-RNAi plants at the mature stage. (j) Transverse sections of the middle part of internode II of the WT and TAL-RNAi plants at the mature stage. lvb, large vascular bundles; pc, parenchyma cells; svb, small vascular bundles. The asterisks indicate the significance of differences as determined by Student's t-test as follows: **, P < 0.01; *, P < 0.05; ns, not significant. All data are means ± SE.
A constant relationship between the leaf blade width and the longitudinal vein quantity in rice was observed in previous studies (Qi et al., 2008). A comparison of the flag leaves revealed that the total number of large veins and small veins was reduced in the TAL-RNAi plant (Table S4). This observation indicates that the rice TAL gene affects the formation of leaf veins, which may contribute to the narrow leaf morphology of the TAL-RNAi plant. The finding that TAL affects leaf venation prompted us to examine the vascular system in the culms of the TAL-RNAi plant. Although the vascular bundles were uniformly arranged in cross-sections of the TAL-RNAi internode I and II, the number of vascular bundles, particularly in the outer ring, significantly decreased (Table S4). In addition to the reduced number of vascular bundles, the TAL-RNAi plant also contained smaller and immature vascular bundles (Fig.4i,j). Furthermore, the average number of parenchyma cells in the region between the two adjacent large vascular bundles was reduced from approximately five in the wildtype to approximately three in the TAL-RNAi plant (Fig.4i,j). These findings reveal that TAL affects the vascular system in rice and that knockdown of TAL expression alters both the number and pattern of the vascular bundles.
Discussion
Horizontal gene transfer is a major force driving the evolution of prokaryotes. Recent studies revealed that HGT also played a critical role in the transition of plants from aquatic to terrestrial environments (Yue et al., 2012, 2013). Plant genes with cyanobacterial and plastid-containing eukaryotic homologs as top hits are primarily derived from plastids, and many mitochondria-derived genes often have alpha-proteobacterial and other eukaryotic homologs as top hits (Huang & Gogarten, 2008). In this analysis, we found that no other eukaryotes contained genes encoding the TAL type TA and that land plant TAL showed the highest sequence percent identity with homologs from the Actinobacteria rather than alpha-proteobacteria and cyanobacteria. Therefore, under the assumption that the chance of the same gene being repeatedly transferred among different organismal groups is relatively low, the most parsimonious explanation is that the origin of the land plant TAL gene was the result of an ancient HGT event from Actinobacteria.
Generally, prokaryotic genes contain no introns. Although the TAL genes in bacteria have no introns, their homologs in land plants contain five to seven introns, and the majority of the intron/exon borders are highly conserved. It is postulated that the majority of these introns in land plant TAL genes arose through insertions shortly after the HGT event and before the divergence of the land plant lineages. Several other horizontally acquired eukaryotic genes were also found to have evolved introns through insertion events (Marcet-Houben & Gabaldon, 2010; Yang et al., 2013). In our analysis, we found that the dominant driving force for TAL gene evolution in both land plants and Actinobacteria was purifying selection, which contributes to functional stabilization. However, when we used the bacterial genes as a background, positive selection was found to significantly contribute to the evolution of the land plant TAL genes. Because positive selection is a major force underlying the adaptation of species to a new environment, the most reasonable explanation for these results is that the land plant TAL gene may have acquired some functional innovations through positive selection shortly after the HGT event and before the separation of major land plant lineages.
During their long period of evolution, land plants have adapted to terrestrial environments. To overcome the challenges of a dry environment and the lack of support for upright growth, plants evolved adaptive features, including cuticles, stomata, vascular tissue, gametangia, and seeds (Ligrone et al., 2012; Yue et al., 2012). The vascular system of plants consists of a network of continuous strands, the vascular bundles, which ensure the long-distance transport of water, nutrients, and organic compounds produced by photosynthesis in leaves (Ye, 2002). As a result of adaptive evolution, the origin of vascular tissues solved the problem of the long-distance transport of water and nutrients, thus enabling plants to gradually colonize the land. Studies revealed that HGT played important roles in the origin of genes associated with vascular development. It was proposed that the gene VEP1 functions as a positive element required for vascular strand development in land plants (Jun et al., 2002). Systematic analysis found that the ancestor of land plants acquired the VEP1 gene through an ancient HGT from bacteria (Tarrio et al., 2011). Lignin occupies the spaces in the cell wall between the cellulose, hemicellulose, and pectin components, particularly in the xylem tracheids, vessel elements, and sclereid cells. Phenylalanine ammonia-lyase (PAL), a critical enzyme of the general phenylpropanoid pathway that provides precursors for lignin monomer biosynthesis, was probably transferred from soil bacteria to fungi, and then secondarily routed to land plants (Emiliani et al., 2009). In this analysis, we not only demonstrated that the land plant TAL gene was acquired through an ancient HGT event, but also that this gene plays a significant role in the development of the plant vascular system using expression analysis and transgenic rice experiments.
Plant TA was first isolated from the potato, and further evidence shows that the plant TA is encoded by two distinct genes that evolved independently (Moehs et al., 1996; Caillau & Paul Quick, 2005). The detection of conserved domains revealed that one isoform is a TAL-type TA, such as PoTAL1 in tomato and At5g13420 in Arabidopsis, and the other is a TalAB-type, such as PoTAL2 and At1g12230. Only the TAL-type land plant TA is catalytically active when expressed as an Escherichia coli lysate (Caillau & Paul Quick, 2005). In addition, to cooperate with other OPPP enzymes to fine-tune the production of nonphotosynthetic redox power during the light phase, TA also has a specific role in plant defense mechanisms (Caillau & Paul Quick, 2005). However, green algae only possess TalAB-type TA, and no other eukaryotes contain genes encoding TAL-type TA, except land plants. In addition, different origination patterns were suggested for TAL and TalAB-type TAs because the low degree of sequence similarity between them. In this analysis, we found that the ancestor of land plants acquired the TAL-type TA from Actinobacteria through an ancient HGT event. This transfer probably confers an adaptive advantage for land plants because our transgenic experiments indicate that the rice TAL gene is involved in vascular patterning. Previous investigation of tomato TAL (ToTal1) abundance by western blot revealed that this enzyme localizes almost exclusively within vascular tissues (Caillau & Paul Quick, 2005), suggesting that the function of plant TAL gene in vascular patterning should be conserved in vascular plants, at least in angiosperm. However, we also found that the TAL gene is present in the genome P. patens, suggesting that the TAL-type TA may also have other functions in land plants in addition to its involvement in vascular patterning.
Acknowledgments
This study was supported by grants from the National Program on the Development of Basic Research (2011CB100100, 2013CBA01405), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the National Natural Science Foundation of China (31391632, 31200943, 31171187 and 31328003), the Natural Science Foundation of Jiangsu Province (BK2012261), the Natural Science Foundation of the Jiangsu Higher Education Institutions (14KJA210005), the Innovative Research Team of Universities in Jiangsu Province, NSF Assembling the Tree of Life program (DEB 0830024), and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Supporting Information
Additional supporting information may be found in the online version of this article.
Please note: Wiley Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 An alignment of the selected TAL protein sequences in land plants and Actinobacteria, showing the positively selected sites in land plants.
Table S1 List of the 50 eukaryotic complete genome sequences or ESTs used in this study, in addition to the NCBI nr database
Table S2 List of TAL genes in 17 representative land plant genomes
Table S3 The site-specific model parameters
Table S4 Morphometric analysis of wildtype and TAL-RNAi plants
References
- Andersson JO. Gene transfer and diversification of microbial eukaryotes. Annual Review of Microbiology. 2009;63:177–193. doi: 10.1146/annurev.micro.091208.073203. [DOI] [PubMed] [Google Scholar]
- Caillau M, Paul Quick W. New insights into plant transaldolase. Plant Journal. 2005;43:1–16. doi: 10.1111/j.1365-313X.2005.02427.x. [DOI] [PubMed] [Google Scholar]
- Dunning Hotopp JC. Horizontal gene transfer between bacteria and animals. Trends in Genetics. 2011;27:157–163. doi: 10.1016/j.tig.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emiliani G, Fondi M, Fani R, Gribaldo S. A horizontal gene transfer at the origin of phenylpropanoid metabolism: a key adaptation of plants to land. Biology Direct. 2009;4:7. doi: 10.1186/1745-6150-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Research. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hiei Y, Ohta S, Komari T, Kumashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant Journal. 1994;6:271–282. doi: 10.1046/j.1365-313x.1994.6020271.x. [DOI] [PubMed] [Google Scholar]
- Huang J. Horizontal gene transfer in eukaryotes: the weak-link model. BioEssays. 2013;35:868–875. doi: 10.1002/bies.201300007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gogarten JP. Concerted gene recruitment in early plant evolution. Genome Biology. 2008;9:R109. doi: 10.1186/gb-2008-9-7-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JH, Ha CM, Nam HG. Involvement of the VEP1 gene in vascular strand development in Arabidopsis thaliana. Plant and Cell Physiology. 2002;43:323–330. doi: 10.1093/pcp/pcf042. [DOI] [PubMed] [Google Scholar]
- Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evolutionary Biology. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ. Functional and ecological impacts of horizontal gene transfer in eukaryotes. Current Opinion in Genetics & Development. 2009;19:613–619. doi: 10.1016/j.gde.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nature Reviews Genetics. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Kruger NJ, von Schaewen A. The oxidative pentose phosphate pathway: structure and organisation. Current Opinion in Plant Biology. 2003;6:236–246. doi: 10.1016/s1369-5266(03)00039-6. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Ligrone R, Duckett JG, Renzaglia KS. Major transitions in the evolution of early land plants: a bryological perspective. Annals of Botany. 2012;109:851–871. doi: 10.1093/aob/mcs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldon T. Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics. 2010;26:5–8. doi: 10.1016/j.tig.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Research. 2013;41:D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehs CP, Allen PV, Friedman M, Belknap WR. Cloning and expression of transaldolase from potato. Plant Molecular Biology. 1996;32:447–452. doi: 10.1007/BF00019096. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira T, Rankin DJ, Touchon M, Taddei F, Brown SP, Rocha EP. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Current Biology. 2009;19:1683–1691. doi: 10.1016/j.cub.2009.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer KL, Kos VN, Gilmore MS. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Current Opinion in Microbiology. 2010;13:632–639. doi: 10.1016/j.mib.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Qian Q, Bu Q, Li S, Chen Q, Sun J, Liang W, Zhou Y, Chu C, Li X, et al. Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiology. 2008;147:1947–1959. doi: 10.1104/pp.108.118778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TA, Soanes DM, Foster PG, Leonard G, Thornton CR, Talbot NJ. Phylogenomic analysis demonstrates a pattern of rare and ancient horizontal gene transfer between plants and fungi. Plant Cell. 2009;21:1897–1911. doi: 10.1105/tpc.109.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samland AK, Rale M, Sprenger GA, Fessner WD. The transaldolase family: new synthetic opportunities from an ancient enzyme scaffold. ChemBioChem. 2011;12:1454–1474. doi: 10.1002/cbic.201100072. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Thorne JL, Wikstrom N, Bremer K. Molecular evidence on plant divergence times. American Journal of Botany. 2004;91:1656–1665. doi: 10.3732/ajb.91.10.1656. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S, McGarvey GJ, Wong CH. Microbial aldolases and transketolases: new biocatalytic approaches to simple and complex sugars. Annual Review of Microbiology. 1997;51:285–310. doi: 10.1146/annurev.micro.51.1.285. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrio R, Ayala FJ, Rodriguez-Trelles F. The Vein Patterning 1 (VEP1) gene family laterally spread through an ecological network. PLoS ONE. 2011;6:e22279. doi: 10.1371/journal.pone.0022279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WS, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang Y, Zhou Y, Gao Q, Zhang E, Zhu L, Hu Y, Xu C. Evolution of land plant genes encoding L-Ala-D/L-Glu epimerases (AEEs) via horizontal gene transfer and positive selection. BMC Plant Biology. 2013;13:34. doi: 10.1186/1471-2229-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Molecular Biology and Evolution. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Ye ZH. Vascular tissue differentiation and pattern formation in plants. Annual Review of Plant Biology. 2002;53:183–202. doi: 10.1146/annurev.arplant.53.100301.135245. [DOI] [PubMed] [Google Scholar]
- Yue J, Hu X, Huang J. Horizontal gene transfer in the innovation and adaptation of land plants. Plant Signaling & Behavior. 2013;8:e24130. doi: 10.4161/psb.24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Hu X, Sun H, Yang Y, Huang J. Widespread impact of horizontal gene transfer on plant colonization of land. Nature Communications. 2012;3:1152. doi: 10.1038/ncomms2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhu J, Li Z, Yi C, Liu J, Zhang H, Tang S, Gu M, Liang G. Deletion in a quantitative trait gene qPE9-1 associated with panicle erectness improves plant architecture during rice domestication. Genetics. 2009;183:315–324. doi: 10.1534/genetics.109.102681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 An alignment of the selected TAL protein sequences in land plants and Actinobacteria, showing the positively selected sites in land plants.
Table S1 List of the 50 eukaryotic complete genome sequences or ESTs used in this study, in addition to the NCBI nr database
Table S2 List of TAL genes in 17 representative land plant genomes
Table S3 The site-specific model parameters
Table S4 Morphometric analysis of wildtype and TAL-RNAi plants