

Recently we reported improved proteome coverage and quantitation metrics for low abundance proteins within whole proteomes by incorporation of a novel digestion and depletion strategy prior to a standard shotgun proteomic analysis. Our goal was to improve the proteomic metrics of low abundance proteins by reducing proteolytic background, namely reducing the highly sampled peptides derived from high abundance proteins. Our method was successful in reducing proteolytic background. Conceptually we rationalized these gains resulted from a selective digestion and removal of abundant proteins, as peptides. Since our report, the mechanism by which our gains were achieved has been challenged in a Correspondence by Ye et al. (Nat. Methods, 2014). In response, we have reanalyzed our data in a peptide-centric manner and propose a refined kinetic mechanism consistent with established competitive substrate kinetics.
Through a simplified derivation beginning with a classical Michaelis-Menten competitive substrate model and further quantitative analysis of our data, we provide a refined depletion mechanism that more accurately describes the complex mixtures we previously analyzed. Our revised qualitative expression describing depletion of early-generated peptides from proximal fast tryptic cleavage sites with high specificity constants (V/K) (Supplementary Note 1) is illustrated by the following equation:
| (1) |
where χA,depleted is the mole fraction of substrate A after complete (tc) and depletion (td) digestion times expressed as mole fraction of total substrate cleavage sites. So expressed, tryptic sites have different specificity constants as well as abundances. Substrate cleavage results in the generation of two shorter polypeptides that can be subsequently cleaved into more substrates over time. The relative cleavage rates are governed by each site’s relative specificity constant. From this perspective, we redefine the mechanism for depletion and enrichment of the digestion and depletion of abundant proteins (DigDeAPr) method. Early-generated peptides, derived from fast substrate sites (i.e. high V/K) within ~ 100 amino acids of each other, are removed at the point of our 10K molecular weight cutoff (MWCO) spin-filter depletion step. Clearing of these early-generated peptides prior to further digestion, provides an enrichment of peptides resulting from slower tryptic sites in the subsequent complete digestion step. By use of equation 1 we illustrate the expected adjustment in peptide abundance resulting from limited digestion and depletion (Figure 1a) as driven by the relative cleavage site specificity constants (V/K). When peptide abundance is considered between control and DigDeAPr runs, the expected trend is observed (Figure 1b, Supplementary Figure 1, and Supplementary Note 2) consistent with theory. Notably, the use of 10-fold more starting material and depletion of early-generated peptides acts to equalize the measured abundance of all peptides. Since peptide abundances are used to estimate protein abundance with shotgun proteomics,1–4 the equalization of peptides acts to also equalize the measurable abundance of proteins, as we found empirically in our initial analysis.
Figure 1.
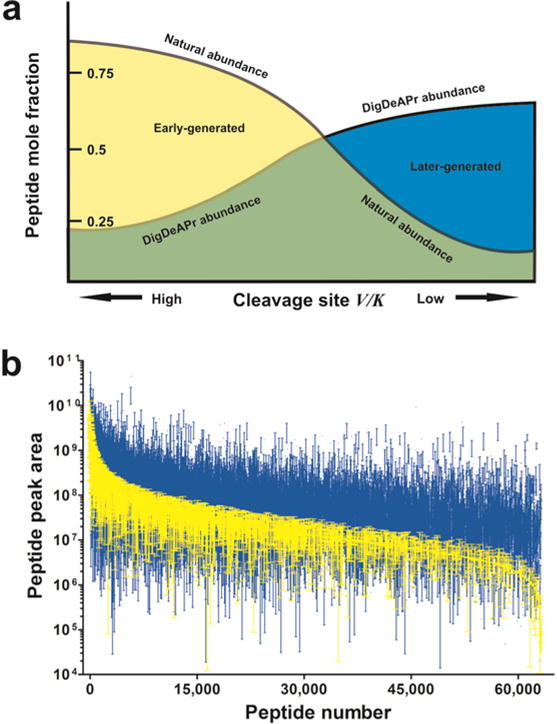
Theoretical and empirical comparison of DigDeAPr mechanism. (a) Schematic of dominant mechanism from digestion and depletion based on the cleavage site specificity constant (V/K) for a given protease. The natural abundance of peptides from a complete protease digestion are adjusted by the use of ten times as much material and depletion of early-generated peptides to enrich for later-generated peptides with lower cleavage site specificity constants. (b) Rank abundance plots of peptide chromatographic peak areas from triplicate control (yellow) and DigDeAPr (blue) runs representing early- and later-generated peptides, respectively. Standard deviations are represented by error bars.
Our depletion runs provide a defined, limited digestion time point for consideration of the aforementioned kinetic efficiencies through analysis of early- and later-generated peptides and fast and slow tryptic cleavage sites (Supplementary Note 3). Early-generated peptides should be depleted and have lower abundances after DigDeAPr when compared to control runs, while later-generated peptides should be enriched and have higher abundances. Using label-free chromatographic peak area ratios of peptides in both control and DigDeAPr runs, we quantified 13,628 and 13,112 peptides in HEK (Figure 2a) and yeast cells, respectively, that were used to classify peptides as early- or later-generated by their relative ratios. Both distributions show defined populations of peptides that were depleted (log2 ratio ≤ −1), unchanged (–1 < log2 ratio < 1), and enriched (log2 ratio ≥ 1). Focusing on the HEK peptide distribution, motif analysis of cleaved (Figure 2b) and missed cleaved (Figure 2c) tryptic sites on depleted peptides validates that early-generated peptides from proximal fast tryptic cleavage sites (< ~100 amino acids apart) are selectively removed during the 10 kDa depletion step (Supplementary Note 4). Similarly, tryptic motifs of enriched, later-generated peptides represent slow cleavage sites (Figure 2d) that remain uncleaved within polypeptides of greater than 10 kDa at the depletion time point. Thus, consideration of tryptic sites and peptides in the digestion and depletion mechanism is essential and illustrates the depletion and enrichment of peptides from fast and slow tryptic cleavage sites, respectively.
Figure 2.
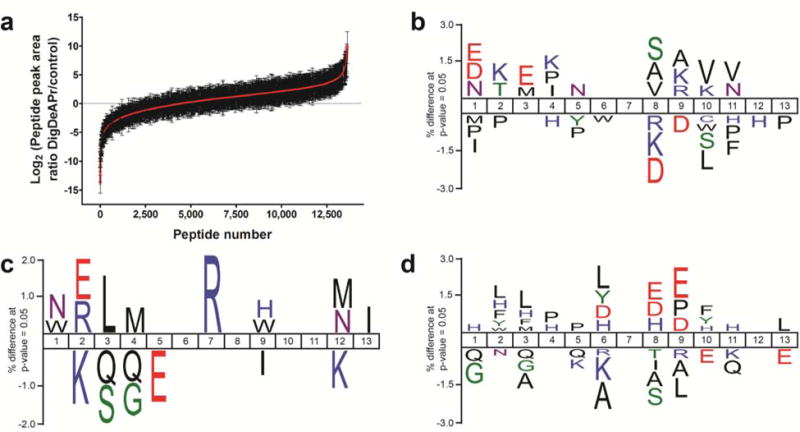
Motif analysis to support DigDeAPr mechanism. (a) Distribution of quantified HEK peptide ratios using label-free peak area measurements from Fonslow et al. Tryptic site motif analysis using IceLogo6 and peptide ratios from analysis of HEK cells categorized by peak area ratios: (b) depleted (log2 ratio ≤ 1) cleaved sites (n = 5,834) versus unchanged (−1 < log2 ratio < 1) cleaved sites (n = 11,885), (c) depleted missed cleavage sites (n = 2,846) versus unchanged missed cleavage sites (n = 5,438), and (d) enriched (log2 ≥ 1) cleaved sites (n = 22,239) versus unchanged cleaved sites (n = 11,885).
With these peptide-centric considerations of abundance, when early- and later-generated peptides are now considered in our protein abundance analyses we notably still observe an abundance-based depletion and enrichment trend in both yeast and HEK cells: more abundant proteins have more early-generated peptides identified and less abundant proteins have more later-generated peptides identified (Supplementary Figure 5 and Note 5). Based on this data and our understanding of peptide sampling in shotgun proteomics,1 we conclude that our gains originate from analysis of a different population of enriched, later-generated peptides. That is, depletion of early-generated peptides from high abundance proteins removes enough proteolytic background to unmask and identify more later-generated peptides from low abundance proteins. Although we may not have explicitly depleted abundant proteins through digestion, in our reanalysis we find that depletion or enrichment of single peptides account for ~ 30% (1/slope = 0.298) of the observed protein abundance depletion or enrichment, respectively, explained by ~ 60% (R2 = 0.57) of the protein abundance measurements (Supplementary Figure 6 and Note 6). Additionally, we found a notable overlap in depleted, early-generated yeast peptides and “proteotypic” yeast peptides (Supplementary Figure 7 and Note 5). While “proteotypic” peptides can be used to robustly identify and quantify many proteins, they can also act as proteolytic background for other less abundant or less sampled proteins and peptides.5 These results collectively indicate that depletion of highly-sampled, abundant, easily-identified, “proteotypic” peptides has a similar effect as depleting abundant proteins to improve identification and quantification of peptides from low abundance proteins.
With our refined view of peptide abundance changes and their correlation to protein changes, we propose a dominant mechanism by which our proteome coverage and quantitation gains are realized through digestion and depletion: depletion of early-generated peptides and enrichment of later-generated peptides equalizes measurable peptide abundances and unmasks less “proteotypic” peptides for improvements in low abundance protein identification and quantification. Based on these new contributing mechanisms, DigDeAPr instead represents digestion and depletion of abundantly sampled peptides and proteins through enrichment of less easily digested and identifiable proteins and peptides. Nonetheless, the combination of ten-fold more starting material with limited digestion and depletion remains a robust and promising method to remove the most easily and repeatedly detected peptides, clearing chromatographic, electrospray ionization, and mass spectrometer space for improvements in identification coverage and quantification of low abundance proteins. These new mechanistic insights suggest that varying limited digestion times in combination with the use of other proteases with different site specificity constants (V/K) and different MWCO filter sizes may hold the most potential to further improve coverage and quantitation of whole proteomes. We are excited about the future possibilities of similar methods and mechanistic investigations to further improve proteomic coverage and quantitation in shotgun proteomics.
Supplementary Material
Acknowledgments
This project was supported by the US National Center for Research Resources (5P41RR011823-17), National Institute of General Medical Sciences (8P41GM103533-17), National Institute of Diabetes and Digestive and Kidney Diseases (R01DK074798), National Heart, Lung, and Blood Institute (RFP-NHLBI-HV-10-5) and National Institute of Mental Health (R01MH067880). We thank Daniel Schwartz for help with motif alignments and James Moresco, Jeff Savas, and Antonio Pinto for comments on the manuscript.
Footnotes
Editor’s note: These additional analyses and derivations were performed by B.R.F and M.S.H, respectively. M.S.H. was added as a co-author for his assistance with understanding enzyme kinetics of these complex mixtures.
References
- 1.Liu H, Sadygov RG, Yates JR., 3rd Analytical chemistry. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 2.Zybailov B, et al. Journal of proteome research. 2006;5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 3.Griffin NM, et al. Nature biotechnology. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwanhausser B, et al. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 5.Kuster B, Schirle M, Mallick P, Aebersold R. Nature reviews. Molecular cell biology. 2005;6:577–583. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- 6.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Nature methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.