

Abstract
Background
Sociality has many rewards, but can also be dangerous, as high population density and low genetic diversity, common in social insects, is ideal for parasite transmission. Despite this risk, honeybees and other sequenced social insects have far fewer canonical immune genes relative to solitary insects. Social protection from infection, including behavioral responses, may explain this depauperate immune repertoire. Here, based on full genome sequences, we describe the immune repertoire of two ecologically and commercially important bumblebee species that diverged approximately 18 million years ago, the North American Bombus impatiens and European Bombus terrestris.
Results
We find that the immune systems of these bumblebees, two species of honeybee, and a solitary leafcutting bee, are strikingly similar. Transcriptional assays confirm the expression of many of these genes in an immunological context and more strongly in young queens than males, affirming Bateman’s principle of greater investment in female immunity. We find evidence of positive selection in genes encoding antiviral responses, components of the Toll and JAK/STAT pathways, and serine protease inhibitors in both social and solitary bees. Finally, we detect many genes across pathways that differ in selection between bumblebees and honeybees, or between the social and solitary clades.
Conclusions
The similarity in immune complement across a gradient of sociality suggests that a reduced immune repertoire predates the evolution of sociality in bees. The differences in selection on immune genes likely reflect divergent pressures exerted by parasites across social contexts.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0628-y) contains supplementary material, which is available to authorized users.
Background
Group living confers many benefits (for some examples see [1-4]) and highly social insects such as ants - epitomes of a highly organized animal society - have risen to ecological dominance in many ecosystems of the world [5]. But group living is also associated with costs. Parasites present an enhanced risk to social animals, as large group size [6], high density, and often close relatedness among individuals increases the exposure and spread of infectious diseases (for example, [7-14]; but see [15]). On the continuum of sociality, eusocial insects are an extreme, forming dense colonies with often very highly related individuals (up to an average coefficient of relatedness of r = 0.75), where individuals perform specific functions within the group, at its simplest specializing as reproductive and worker castes. Given a generally higher risk of disease in social insect colonies, it is surprising that complete genome sequencing revealed that honeybees (Apis mellifera) had approximately only one-third as many immune genes as the two existing genomic model insect systems at the time, Drosophila melanogaster and Anopheles gambiae [16]. Honeybee biology differs from these model species in several ways, which may partly explain the striking difference in immune genome organization among these taxa. For instance, honeybees have a suite of hygienic behaviors where they groom both themselves and others, and live on food (pollen and nectar) that is also relatively clean (not withstanding the fact that food-borne diseases have been described in honeybees, for example, [17,18]). The observation that ant genomes also have few immune genes [19] indicates that this deficiency may be a more general characteristic of social hymenoptera and not primarily an artifact of honeybee breeding [20]. Sociality may instead typically allow for group-based defenses ('social immunity' [21]) that should reduce selective pressures on the evolution and maintenance of immune genes. Given the recent and dramatic declines in populations of important bee pollinators [22-24] and the role of parasites in some of these declines (for example, [23,25,26]), understanding the architecture of the immune system of bees in relation to other insects is increasingly important.
Bumblebees (genus Bombus) are essential natural and commercial pollinators and have been declining due to anthropogenic disturbances, including habitat destruction and fragmentation (reviewed in [24,27]), but also due to introduced competitors [28,29], and more recently pesticides [30,31] and parasites [24,26,32,33] have been implicated as important drivers of declines. Bumblebee declines are of both ecological and practical importance as they contribute substantially to human food crops either directly [24,27,34] or as part of a community of wild pollinators that are supplemented by managed honeybees [35]; therefore, they also aid the maintenance of plant diversity [36]. Among Bombus species, Bombus impatiens, and Bombus terrestris, both key commercial and natural pollinators, have been most extensively studied, in particular for host-parasite interactions [37-40]. These two species occupy comparable niches in North America (B. impatiens) and Europe (B. terrestris). They last shared a common ancestor approximately 18 million years ago [41].
While B. terrestris and B. impatiens share ecological factors, such as diet, with honeybees, they differ from the latter in colony organization, sociality, longevity, and mating system. Bumblebees, including B. terrestris and B. impatiens, are less advanced in their sociality than honeybees, as the physiological and morphological difference between queens and workers is not as pronounced, division of labor is weak, and colonies are much smaller (dozens or hundreds instead of thousands of workers) and very simply organized [42]. Bumblebee colonies as a whole are also shorter-lived than those of honeybees, with bumblebee queens living for one year but the colony persisting for only a few months, whereas honeybee queens and their colonies can live for several years. Like most bumblebee species, B. terrestris queens mate singly and B. impatiens queens mate singly or occasionally doubly [43], whereas Apis queens mate with between 10 and over 100 males [44-47]. This has important consequences for disease susceptibility as both multiply mated honeybees [48] and B. terrestris [49] that were artificially inseminated with sperm from multiple males produce colonies with lower parasite loads than colonies from singly mated queens.
All of these differences may have profound consequences for the evolution of their immune systems. Here, using the recently sequenced complete genomes of both the North American B. impatiens and the European B. terrestris we explore patterns of immune system evolution across a social gradient by comparing the immune repertoire and sequences of immune genes of these two species of bumblebees with those of two species of highly social honeybees and the solitary leaf-cutting bee Megachile rotundata.
Results
Immunological repertoire
Regardless of social organization, all bee species examined shared a core set of immune genes, including all members of the canonical immune pathways (Figure 1) with only minor differences in gene numbers (Figure 2). We found no relationship between the degree of sociality and the total number of canonical immune-related genes. With regard to important immune response effectors, such as anti-microbial peptides (AMPs), both Bombus species have only a single copy of defensin, which is present in two copies in A. mellifera; on the other hand, Bombus have an expanded set of serine protease inhibitors (serpins; Figure 3). We identified five, highly similar (average 75% sequence similarity), putative serpin 3/4-like genes in B. terrestris. Initial homology searches found four serpins (XP_003399186.1, XP_003399187.1, XP_003399188.1, XP_003402576.1) while a revised search using proteomic data confirmed the expression of a fifth serpin, originally described as a pseudogene (XR_132181.1; Figure 3). The proteomic data also identified two unique multiple-peptide supported isoforms of XR_132181.1 (TJ Colgan et al., unpublished data). Four serpins are clustered on genomic scaffold 11.4 while the fifth serpin (XP_003402576.1) is on an unassembled genome contig (GroupUn430). B. impatiens appears to have three novel serpins (XP_003487908.1, XP_003487890.1, and XP_003487917.1) clustered on genomic scaffold scf_0203. Homology searches for bumblebee serpins against sequences of other members of the superfamily Apoidea identified single orthologs for the eusocial honeybee A. mellifera, and the solitary leafcutter bee M. rotundata. Outside of the Hymenoptera, these serpins shared sequence similarity with serpin-1 (alaserpin) of the lepidopteran Manduca sexta.
Figure 1.
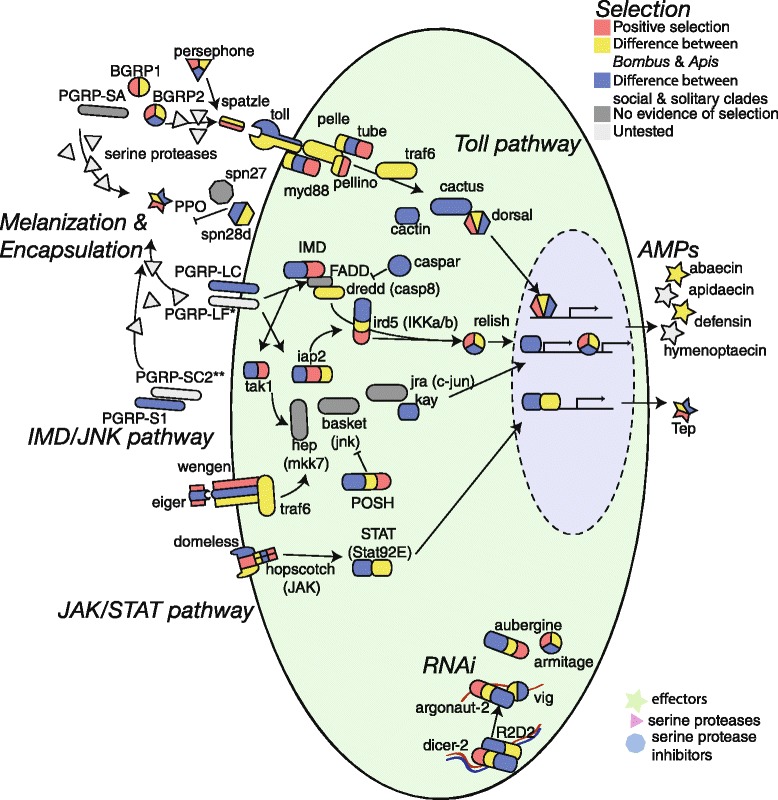
Diagram of the classical insect immune responses to parasites: Toll, IMD/JNK, JAK/STAT pathways and the melanization and antiviral RNA interference responses. Colors of the genes indicate evidence of selection as detected by either positive selection (across the four taxa phylogeny, on the branch between Bombus and Apis, the branch leading to Bombus, Apis, or Megachile) in red, or differences in selection between Bombus and Apis (yellow), or between the social and solitary clades (blue). More complete information about selection on these genes can be found in Additional files 8, 9, 10 and 11. *PGRP-LF is only found in B. impatiens. **PGRP-SC2 is not among the automated predictions for B. terrestris, although sequence in the trace archive suggests that it is present. We also detect expression of PGRP-SC2 in B. terrestris. AMP, anti-microbial peptide.
Figure 2.
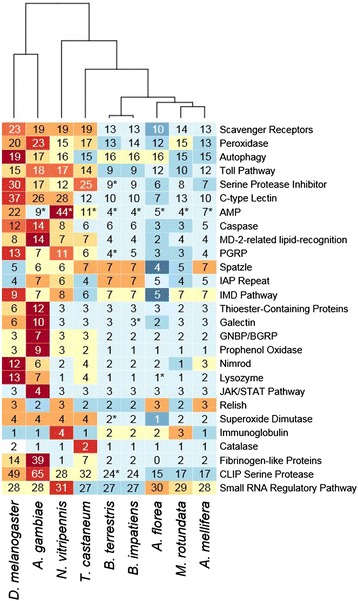
Number of genes belonging to 27 families of immune genes from OrthoDB. The colors in this heatmap reflect the number of genes in that category relative to the other species. Numbers with asterisks were manually adjusted according to our annotation efforts or the literature. The tree represents a clustering analysis using Euclidean distances based on the number of genes within these groups.
Figure 3.
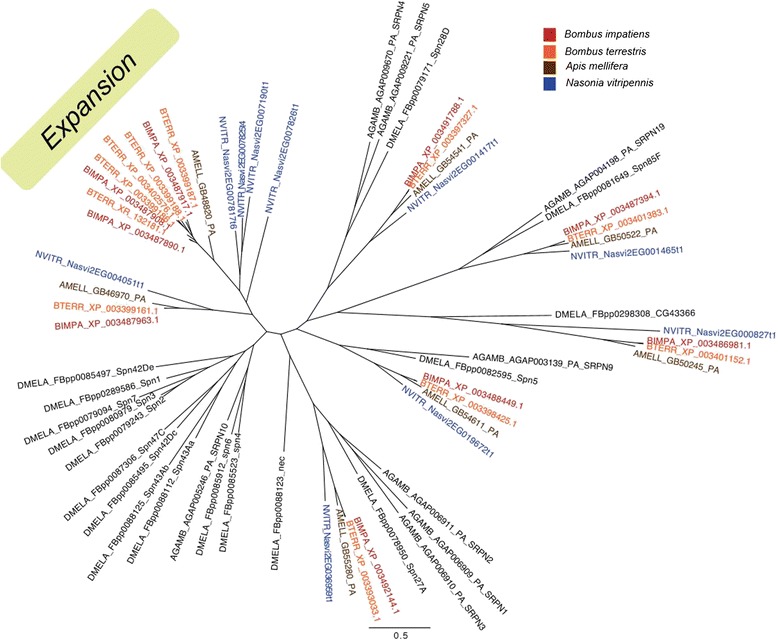
Gene tree of serine protease inhibitors showing the expansion within Bombus (green box). Hymenopteran species are labeled by color and Dipterans are labeled black.
We also find what appears to be a homolog of the apoptosis-involved caspase decay (Figure 4). There also appears to be a Hymenoptera-specific clade of caspases that share the most homology with Ice in Drosophila. We find an additional PGRP (peptidoglycan receptor protein) in B. impatiens (XP_003487752), which is missing in B. terrestris and A. mellifera. On the genomic sequence, this novel PGRP is immediately downstream of XP_003487751, which is homologous to XP_003400160 in B. terrestris and XP_392452 in A. mellifera, likely from tandem duplication.
Figure 4.
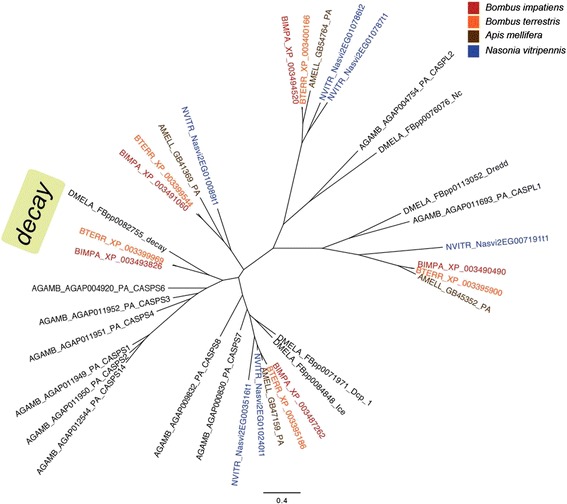
Gene tree of caspases showing the Bombus genes that appear similar to D. melanogaster decay (green box). Hymenopteran species are labeled by color and Dipterans are labeled black.
Immunological expression
We used quantitative PCR to determine whether 27 candidate immune genes (Table 1) were functionally expressed in B. terrestris, including if they were differentially expressed following exposure to Gram-negative or Gram-positive bacterial cues. We measured expression in gynes and males also to investigate sex-specific patterns. All surveyed genes were actively expressed in both gyne and male B. terrestris, including the novel serpins (serpin 3/4 A, serpin 3/4 B, alaserpin) and the decay homolog. Both sex (Tables 2 and 3; Figure 5) and treatment (Figure 6) significantly influenced expression of this battery of genes and the different sexes responded differently to the treatments as revealed by the sex*treatment interaction (Tables 2 and 3; Figure 6). The recognition receptors beta-glucan receptor protein 2 (BGRP2) and peptidoglycan receptor protein SA (PGRP-SA) were more strongly expressed in queens than males, but BGRP1 and PGRP-LB had male-biased expression. PGRP-SC2 was more strongly expressed in queens but was also upregulated in queens given the challenge whereas males downregulated this gene upon challenge. All antimicrobial peptides (AMPs) were more strongly expressed in queens than males and most were induced upon challenge and induced more dramatically in queens. The effectors lysozyme, transferrin, the signaling transducer relish, antiviral genes argonaute and aubergine, and melanization related genes PPO and punch follow a similar pattern with queen-biased expression and greater induction of expression when there was a significant treatment by sex interaction. An exception to this general pattern is the serpin 27a, which inhibits melanization. Queens had lower expression of this gene and the expression appears to be reduced upon bacterial exposure. Males did not reduce their expression of serpin 27a as intensely as the queens did.
Table 1.
Gene and primer details used for quantitative PCR
| Gene | Putative gene function | NCBI accession | Forward primer | Reverse primer | Product size (base pairs) |
|---|---|---|---|---|---|
| AK | Arginine kinase, housekeeping | AF_492888 | CTGGACTCTGGTGTCGGTAT | GTCTTTTGGTGGATGCTTGT | 129 |
| PLA2 | Phospholipase A2, housekeeping | FN_391388 | TATCTTTCAATGCCCAGGAG | GTCGTAACAAATGTCATGCG | 129 |
| ef1α | Elongation factor 1α | XM_003401944 | GCTGGTGACTCGAAGAACAATC | GGGTGGTTCAACACAATAACCTG | 74 |
| BGRP1 | Recognition receptor, Toll pathway | XM_003397996 | AACGTGGAAGTCAAAGATGG | GCGAACGATGACTTGGTATT | 206 |
| BGRP2 | Recognition receptor, Toll pathway | XM_003394713 | TAACTCCCTTTGGAAACACG | GGCGGTAAAATACTGAACGA | 249 |
| PGRP-S1 | Recognition receptor, Imd pathway | XM_003400112 | TTTCCATGTTGCTCGCTTCG | CGCGGTTTCCCTTTCGATATTAG | 77 |
| PGRP-LC | Recognition receptor, Imd pathway | XM_003396463 | CAGCCACCTACGACAGATTT | GTACATTCCGCTTGTGTCCT | 101 |
| PGRP-SA | Recognition receptor, Toll pathway | XM_003401893 | CGTGAAGGAGCTCATACCAT | CCAGGACTCATAGTGGCTGT | 200 |
| PGRP-SC2 | Recognition receptor, Imd pathway | XM_003493213 | TTGGTTGGCGAAGATGGAAAC | CGCGCTTGGATTATGACCAAC | 132 |
| pelle | Signal molecule, Toll pathway | XM_003399470 | TAAATCGACCTATGCAAGCC | GGGTATAGCTGCTTCTGCTG | 107 |
| relish | Signal molecule, Imd pathway | XM_003399472 | CAGCAGTAAAAATCCCCGAC | CAGCACGAATAAGTGAACATA | 156 |
| basket | Signal molecule, JNK pathway | XM_003402794 | GGAACAAGATAATCGAGCAACTG | CTGGCTTTCAATCGGTTGTG | 177 |
| hopscotch | Signal molecule, JAK/STAT pathway | XM_003401903 | CACAGACTGAAGCAGGTTGA | CATATGGGTAATTTGGTGCC | 353 |
| abaecin | Antimicrobial peptide (AMP) | XM_003394653 | GCCACAATATGTGGAATCCT | ATGACCAGGGTTTGGTAATG | 141 |
| apidaecin | Antimicrobial peptide (AMP) | XM_003402966 | CCCGACTAATGTACCTGCCA | GAAGGTGCGAATGTGTTGGA | 131 |
| defensin | Antimicrobial peptide (AMP) | XM_003395924 | GTCTGCCTTTGTCGCAAGAC | GACATTAGTCGCGTCTTCTTCG | 139 |
| hymenoptaecin | Antimicrobial peptide (AMP) | XR_132450 | TTCATCGTACTGGCTCTCTTCTG | AGCCGTAGTATTCTTCCACAGC | 85 |
| TEPA | Effector molecule, JAK/STAT pathway | XM_003399699 | GCGTTCTATGACCACCTGTT | TACAGGTTACTCCACAGCCC | 212 |
| lysozyme3 | Bacteriolytic effector | XM_003394052 | TATGGGCAAGAAGATTCGAC | GTGTACATCGTTCACGCATC | 219 |
| transferrin | Iron-binding protein, antibacterial | XM_003401163 | CAATTTCTTCACCGCATCCT | CCTCGTTATTTGGCTTGCAT | 131 |
| ferritin | Iron transportation protein | XM_003393332 | AAAGAATTGGACGCAAATGG | CAGCGAACTGATGTCCAAGA | 259 |
| serpin27a | Serine protease inhibitor, prophenoloxidase cascade | XM_003392985 | CCGATCATCCATTCGTATTC | ACCTGCACTTGATATCCCTG | 164 |
| PPO | Prophenoloxidase, melanin synthesis | HM142999 | AGCGGCATAATACGTTGTGT | CCGAGGGATAGAAAGTCTCC | 329 |
| punch | Enzyme, melanin synthesis | XR_131852 | ATTGCCAGGACACTTTCAAC | TACAAGCTGGAAACGGAAAC | 211 |
| kayak | JNK pathway | Bter:08277927 | ACGCAATATGGGTGGCAGAA | TGAACGAAGACGACAGACCG | 271 |
| serpin 3/4A | Novel serine protease inhibitor | XM_003399138 | GCAGAGACAAATGTTGAAGCAC | CACAGTCTGGGATAATGAAGAACC | 78 |
| serpin 3/4B | Novel serine protease inhibitor | XM_003399140 | ATGGTGCTTTGTTCATCAGTCG | GACCCAATGACAGCAGTAACAG | 97 |
| alaserpin | Novel serine protease inhibitor | XM_003399139 | TGCTGAAATGCTAGATGACACG | GCATATCGCTCGTTAACTCAGG | 104 |
| argonaute 2 | RNA-interference, possible antiviral function | XM_003398481 | AATTGCAAGATCAACCTGCC | CCTACCCAAAGACAAGGCAA | 175 |
| aubergine | RNA-interference, possible antiviral function | XM_003400641/XM_003400642 | GTCGCCCTTCTGCATATCTC | AAGATCGAACTGCTATCCGC | 190 |
| decay | Caspase mediating apoptosis | XM_003399921 | AAGAAGACCTCGGTCCTTAGAC | CAGCTGCAAATGAAGTAATGCG | 74 |
Table 2.
MANOVA results for all validated immune genes
| Df | Pillai | Approximate F | Num Df | Den Df | P | |
|---|---|---|---|---|---|---|
| Sex | 1 | 0.986 | 128.635 | 27 | 51 | <0.0001 |
| Treatment | 3 | 1.445 | 1.823 | 81 | 159 | 0.00067 |
| Sex*Treatment | 3 | 1.365 | 1.640 | 81 | 159 | 0.0042 |
| Residuals | 77 |
Table 3.
Univariate ANOVA results for each gene tested in the MANOVA
| Sex | Treatment | Sex*Treatment | |
|---|---|---|---|
| BGRP1 | *** | ~ | – |
| BGRP2 | *** | ~ | – |
| PGRP-S1 | *** | – | – |
| PGRP-LC | – | – | – |
| PGRP-SA | ** | – | – |
| PGRP-SC2 | *** | – | ** |
| abaecin | *** | *** | ~ |
| apidaecin | *** | ~ | – |
| defensin | *** | *** | * |
| hymenoptaecin | *** | *** | – |
| lysozyme | *** | – | – |
| transferrin | *** | – | * |
| ferritin | – | – | – |
| tepA | – | – | ~ |
| relish | *** | *** | – |
| basket | – | – | – |
| hopscotch | *** | – | – |
| kayak | *** | – | – |
| punch | – | *** | * |
| serpin ¾A | ** | – | – |
| serpin ¾B | – | – | – |
| alaserpin | – | – | – |
| serpin27a | *** | *** | – |
| PPO | * | – | – |
| argonaute 2 | *** | – | – |
| aubergine | *** | – | – |
| decay | – | – | – |
Transformed expression values (dCt) were treated as dependent on the sex of the bees (male/queen) and the treatment they received (naïve, sterile Ringers solution injection, injection with Arthrobacter globiformis, or injection with Escherichia coli). –, P > 0.1; ~, P < 0.1; *P < 0.05; **P < 0.01; ***P < 0.001; full statistics can be found in Additional file 13.
Figure 5.
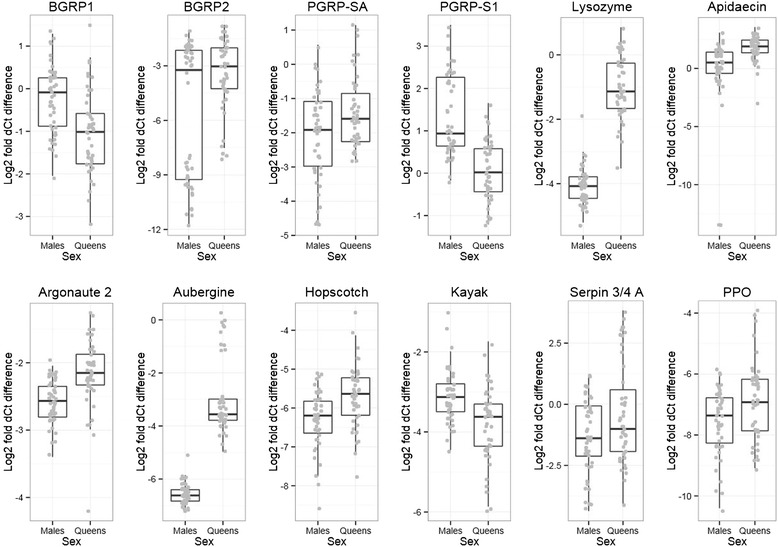
Logfold gene expression relative to invariant housekeeping genes in males and young queens (gynes). All genes shown here are significantly differentially expressed between the sexes. Full details of these statistics can be found in the supplemental materials.
Figure 6.
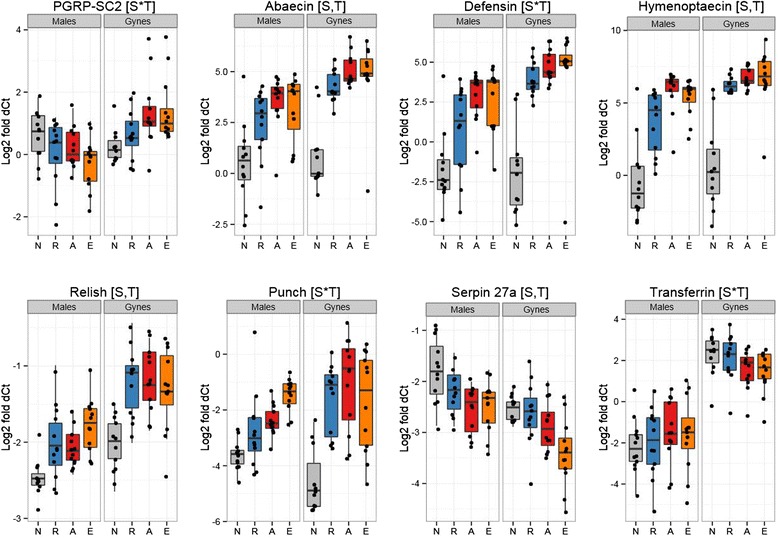
Logfold gene expression relative to invariant housekeeping genes in males and gynes according to treatment (x-axis: N, naïve; R, Ringer injection; A, Arthrobacter globiformis injection; E, Escherichia coli injection). Next to the gene name we depict whether the expression differed significantly according to sex (S), treatment (T), or the interaction between sex and treatment (S*T). Full details of these statistics can be found in the supplemental materials.
Signatures of selection
While we did not identify any pattern of immune gene numbers varying with sociality, we did find variation in the evolution of these immune genes both between the highly social Apis clade and the less social Bombus clade, and between the solitary Megachile and the broader social clade containing Apis and Bombus. Globally, the ratio of non-synonymous to synonymous substitutions was ω = 0.12 ± 0.01 (mean ± standard error of the mean) and ω = 0.10 ± 0.01 in the four- and five-taxa M0 analyses, respectively (Additional files 1 and 2), although ω differed dramatically across ortholog groups (range 0.367 to 0.0001, 0.30 to 0.0047; Additional files 3, 4, 5, 6, and 7). The orthologs of dscam (down syndrome cell adhesion molecule), three antiviral proteins (aubergine, argonaute 2, and rm62) a trypsin-like serine protease that is homologous to the scavenger receptor tequila, a CLIP serine protease orthologous to CG4998, the peroxidase cardinal, a caspase ark, and the Toll signaling protein pellino showed evidence of positive selection across the four-taxa (Apis and Bombus) phylogeny (Table 4; Figures 7 and 8). Across the whole five-taxa phylogeny we again found evidence for positive selection on argonaute 2, dscam, ark, and the CG4998 ortholog, and additionally found positive selection on a second CLIP serine protease without a clear D. melanogaster ortholog but which is similar to CG11843 and snake, which are involved in toll signaling [50] (Table 5). In the branch leading to Apis the small RNA regulatory or anti-viral gene drosha, and the RNA helicase rm62, which has been implicated in both RNA interference [51] and bacterial response [52], the bacterial recognition gene BGRP1, a serine protease inhibitor, the caspase ark and IMD of the IMD pathway, are under selection (Table 6). On the branch leading to Bombus we find evidence of selection on argonaute 2, rm62-F (which is also an RNA helicase but has not been directly linked to immune responses), and the toll-7 receptor, which has been implicated in viral defenses [53]. We also find evidence of selection on a number of members of the toll pathway, including dorsal, myd88, and BGRP1, which recognizes bacterial pathogens and initiates toll pathway signaling. Domeless, the receptor of the JAK/STAT pathway, had the most sites showing evidence of selection while dorsal showed stronger evidence of positive selection but across fewer sites. Two catalases, ark and catalase, a serpin and a scavenger receptor, snmp1, also appear to be under selection in bumblebees (Table 7; Figure 9). A number of genes show evidence of different selection between honeybees and bumblebees (Figure 9; Table 8; Additional file 8), including dorsal, spaetzle, and tube, all from the toll pathway, a nimrod gene, argonaute 2, a number of serpins, and dscam. Considerably more genes differ in selection between the social and solitary clades (Figure 10; Additional file 9) perhaps in part due to the difference in time since sharing an ancestor with both Bombus and Apis. However, genes that exhibit signs of different selection within the social clades (upper diagonal in Figure 10) are likely more robust than those showing signs of selection only in the solitary M. rotundata (lower diagonal) as the genes that appear to be evolving rapidly in the solitary group might be inflated due to the disproportionate phylogenetic distance of M. rotundata to the Apis and Bombus clades. A summary of genes for which we found evidence of selection and according to which selection model is provided in Additional file 10 (four taxa: Bombus and Apis) and Additional file 11 (five taxa: Bombus, Apis, and Megachile).
Table 4.
Genes under positive selection (using FDR < 0.05) across the whole phylogeny (4 taxa tree)
| OrthoDB group id a | Gene b | Classification sites c | Total ratio d | Likelihood | p-value q-value e | BH-corrected sites f | Positively selected |
|---|---|---|---|---|---|---|---|
| EOG66DJHX-2 | dscam | Immunoglobulin | 777 | 186.607 | 0.00000 | 0.00000 | 4L, 6R, 8S, 11D, 13G, 14D, 20Q, 22A, 24M, 26A, 30T, 35A, 37T, 43E, 44P, 52R, 54T, 56I, 58T, 60P, 63K, 65I, 66H |
| EOG6HHMH6 | serpin-23 | Scavenger receptor | 2066 | 15.366 | 0.00046 | 0.00987 | 36S, 87S, 90Q, 92K, 288P, 334S, 397N, 431S, 490D, 761P, 772R, 811K, 815T, 816Y, 824S, 877S, 1782K, 1788E |
| EOG66DJHQ | aubergine | Small RNA regulatory path-members | 787 | 15.955 | 0.00034 | 0.00858 | 219K, 269K, 287A, 342S, 348D, 359G, 397R, 410E, 415P, 431G, 436D, 621K |
| EOG64B8H5 | CLIP-A10 | CLIP serine protease | 816 | 29.218 | 0.00000 | 0.00003 | 1I, 3H, 21V, 25P, 37P, 291K, 333T, 335T, 344S, 457S |
| EOG6KKWHX | argonaute-2 | Small RNA regulatory pathway members | 896 | 20.248 | 0.00004 | 0.00150 | 24W, 27N, 43S, 48Q, 58S, 59N, 81D, 103I, 519F |
| EOG6J3TZ2 | cardinal | Peroxidase | 1203 | 14.276 | 0.00079 | 0.01489 | 35M, 46S, 538A, 711A, 742E, 743T, 882D, 931V |
| EOG6VX0M3 | ark | Caspase | 1263 | 17.732 | 0.00014 | 0.00423 | 67G, 386G, 752T, 1028G, 1078T, 1112F |
| EOG6JQ2CF | LOC100642575 (B. terr) | Scavenger receptor | 924 | 22.212 | 0.00002 | 0.00075 | 75S, 78P, 111I, 112P, 647S |
| EOG6W9GK1-3 | rm62-B1 | Small RNA regulatory pathway members | 431 | 11.766 | 0.00279 | 0.04179 | 425A, 430S, 431E |
| EOG634TNR | pellino | Toll pathway | 431 | 12.386 | 0.00204 | 0.03407 | 1P, 2S |
aGroup identifiers are from OrthoDB 6 (http://cegg.unige.ch/orthodb6).
bUnless otherwise specified, gene names are taken from the A. mellifera or D. melanogaster orthologs.
cTotal number of codons in the alignment after trimming with Gblocks.
dComparison of model M7 versus M8.
eMultiple test correction by the method of Benjamini and Hochberg to control the false discovery rate (only groups where FDR < 0.05 are shown).
fSites are classified as under positive selection if the Bayesian posterior probability > 0.75 (>0.95 in bold italic). Sites where are italic.
Reference sequence taken from A. mellifera.
Figure 7.
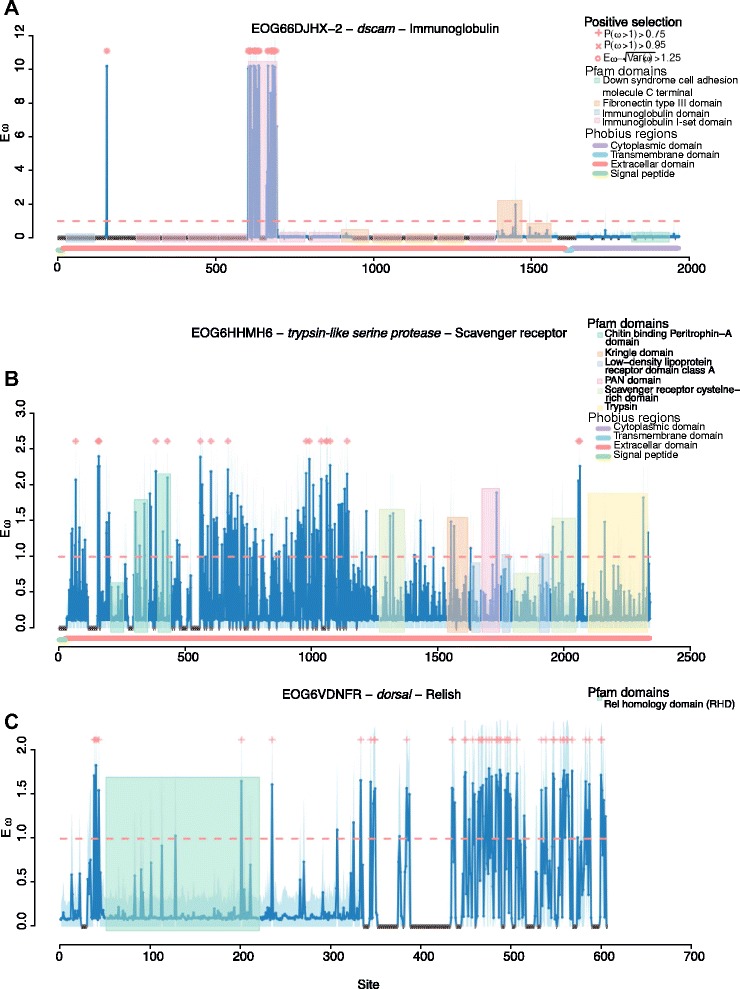
Sites under selection within the Apis, Bombus phylogeny for three genes of interest. The title for each gene presents the OrthoDB accession, the gene name, and the immune category. We only present a subset of the genes that showed an overall signature for selection highlighting codons at three different significance thresholds: Bayesian posterior probability >0.75 (plus signs along the top of each panel), >0.95 (x’s), and where Eω - sqrt(Var(ω)) > 1.25 (circles). The blue shadow indicates an estimate of error at each codon. We show Pfam domains in colored blocks and Phobius regions along the x-axis. Crosshatched regions were trimmed from the alignment.
Figure 8.
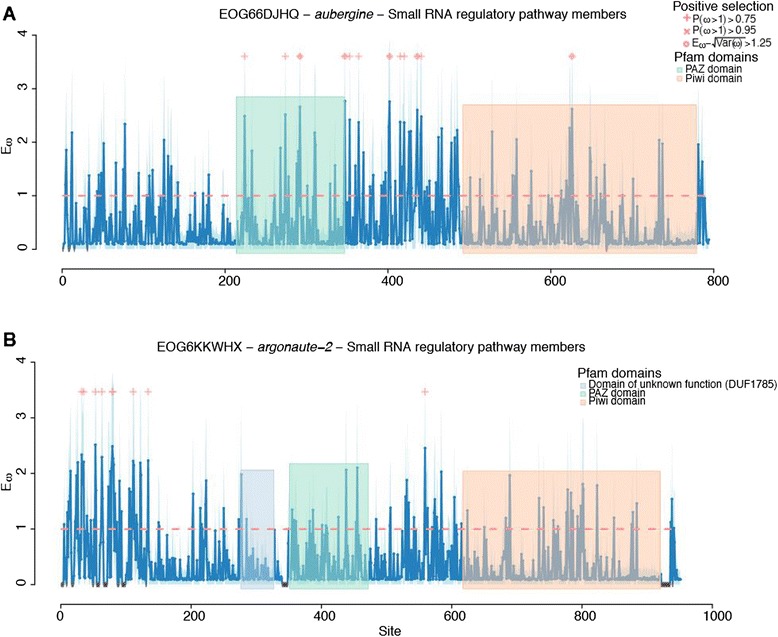
Sites under selection within the Apis, Bombus phylogeny for two viral response genes. The title for each gene presents the OrthoDB accession, the gene name, and the immune category. We only present a subset of the genes that showed an overall signature for selection highlighting codons at three different significance thresholds: Bayesian posterior probability >0.75 (plus signs along the top of each panel), >0.95 ('x's), and where Eω - sqrt(Var(ω)) > 1.25 (circles). The blue shadow indicates an estimate of error at each codon. We show Pfam domains in colored blocks and Phobius regions along the x-axis. Crosshatched regions were trimmed from the alignment.
Table 5.
Genes under positive selection (using FDR < 0.05) across the whole phylogeny (5 taxa tree)
| OrthoDB group id a | Gene b | Classification sites c | Total ratio d | Likelihood | p-value q-value e | BH-corrected sites f | Positively selected |
|---|---|---|---|---|---|---|---|
| EOG6KKWHX | argonaute-2 | Small RNA regulatory pathway members | 810 | 31.839 | 0.00000 | 0.00001 | 22W, 25N, 41S, 55S, 56N, 57S, 69L, 73D, 85D, 95I, 329S, 346N, 450F, 692L, 693R |
| EOG66DJHX-2 | dscam | Immunoglobulin | 489 | 44.704 | 0.00000 | 0.00000 | 2M, 4A, 13A, 20R, 24I, 26T, 28P, 31K, 33I, 34H, 451G, 452G |
| EOG6QRFKP | CLIP-C1B | CLIP serine protease | 330 | 13.650 | 0.00109 | 0.03063 | 14L, 15Q, 66L, 72M, 118A, 132L, 195Q, 313N |
| EOG6VX0M3 | ark | Caspase | 1128 | 17.871 | 0.00013 | 0.00619 | 495S, 629T, 904G, 954T, 988F |
| EOG64B8H5 | CLIP-A10 | CLIP serine protease | 792 | 14.259 | 0.00080 | 0.02824 | 1I, 3H, 20V, 318T |
aGroup identifiers are from OrthoDB 6 (http://cegg.unige.ch/orthodb6).
bUnless otherwise specified, gene names are taken from the A. mellifera or D. melanogaster orthologs.
cTotal number of codons in the alignment after trimming with Gblocks.
dComparison of model M7 versus M8.
eMultiple test correction by the method of Benjamini and Hochberg to control the false discovery rate (only groups where FDR < 0.05 are shown).
fSites are classified as under positive selection if the Bayesian posterior probability > 0.75 (>0.95 in bold italic). Sites where are Italic.
Reference sequence taken from A. mellifera.
Table 6.
Genes under positive selection (using FDR < 0.05) on the branch to Apis (5 taxa tree)
| OrthoDB group id a | Gene b | Classification | Total sites c | Likelihood ratio d | p-value | BH-corrected q-value e | Positively selected sites f |
|---|---|---|---|---|---|---|---|
| EOG6VX0M3 | ark | Caspase | 1128 | 9.974 | 0.00079 | 0.02812 | 412N, 484N, 593S, 862P, 941N, 953L |
| EOG66Q57J | LOC100642902 (B. terr) | Serine protease inhibitor | 1189 | 9.555 | 0.00100 | 0.02812 | 425D, 452I, 540S, 622S, 721M |
| EOG634TN0 | drosha | Small RNA regulatory pathway members | 1290 | 8.884 | 0.00144 | 0.03380 | 58A, 94N, 155M, 278V |
| EOG6XWDCW | rm62-C | Small RNA regulatory pathway members | 492 | 10.537 | 0.00058 | 0.02812 | 32S, 130I, 151S, 269S |
| EOG6DV43B | immune deficiency | IMD pathway | 249 | 9.882 | 0.00083 | 0.02812 | 139V, 141S |
| EOG6RV16R-1 | BGRP-1 | GNBP | 459 | 9.619 | 0.00096 | 0.02812 | 151L |
aGroup identifiers are from OrthoDB 6 (http://cegg.unige.ch/orthodb6).
bNumber of codons remaining in the alignment after trimming with Gblocks.
cComparison of Branch-site model A versus a constrained version with ω 2 = 1.
eMultiple test correction by the method of Benjamini and Hochberg to control the false discovery rate (only groups where FDR < 0.05 are shown).
fSites are classified as under positive selection if the Bayesian posterior probability > 0.75 (> 0.95 in bold). The reference sequence is from A. mellifera.
Table 7.
Genes under positive selection (using FDR < 0.05) on the branch to Bombus (5 taxa tree)
| OrthoDB group id a | Gene b | Classification | Total sites c | Likelihood ratio d | p-value | BH-corrected q-value e | Positively selected sites f |
|---|---|---|---|---|---|---|---|
| EOG666T1W | domeless | JAK/STAT pathway | 1435 | 9.552 | 0.00100 | 0.01951 | 24L, 102R, 224S, 526A, 770T, 799N, 838V, 942V, 952I, 954A, 959L, 960A, 992Q, 1055R, 1056W, 1312T, 1316D |
| EOG6VDNFR | dorsal | Relish | 353 | 22.813 | 0.00000 | 0.00013 | 104Q, 177S, 309R, 316K, 317I, 318S, 332S, 333Y, 334N, 336S, 347N, 350R |
| EOG66Q57J | LOC100642902 (B. terr) | Serine protease inhibitor | 1189 | 20.354 | 0.00000 | 0.00023 | 165S, 230D, 247P, 419T, 500S, 502D, 590Q, 617S |
| EOG6BG7B9 | snmp1 | Scavenger receptor | 430 | 18.183 | 0.00001 | 0.00047 | 77G, 217G, 227K, 346L, 353K, 394N, 395K |
| EOG6VX0M3 | ark | Caspase | 1128 | 7.406 | 0.00325 | 0.04167 | 156L, 668S, 992R, 1074N, 1079L |
| EOG6ZPC9T | rm62-F | Small RNA regulatory pathway members | 545 | 11.406 | 0.00037 | 0.01032 | 120R, 136G, 169Q, 542N, 543K |
| EOG6RV16R-1 | BGRP-1 | GNBP | 459 | 9.363 | 0.00111 | 0.01951 | 222R, 229E, 370P, 458W |
| EOG6X0K8Q | myd88 | Toll pathway | 209 | 9.782 | 0.00088 | 0.01951 | 45E, 83F, 133P, 199D |
| EOG6Z8WBN | catalase | Catalase | 181 | 7.431 | 0.00321 | 0.04167 | 53A, 83T, 89S |
| EOG6931ZS-1 | TLR-7 | Toll receptor | 1299 | 12.107 | 0.00025 | 0.00886 | 230T, 1190K, 1191D |
| EOG6KKWHX | argonaute-2 | Small RNA regulatory pathway members | 810 | 8.276 | 0.00201 | 0.03147 | 684A |
aGroup identifiers are from OrthoDB 6 (http://cegg.unige.ch/orthodb6).
bNumber of codons remaining in the alignment after trimming with Gblocks.
cComparison of Branch-site model A versus a constrained version with ω 2 = 1.
eMultiple test correction by the method of Benjamini and Hochberg to control the false discovery rate (only groups where FDR < 0.05 are shown).
fSites are classified as under positive selection if the Bayesian posterior probability > 0.75 (> 0.95 in bold). The reference sequence is from A. mellifera.
Figure 9.
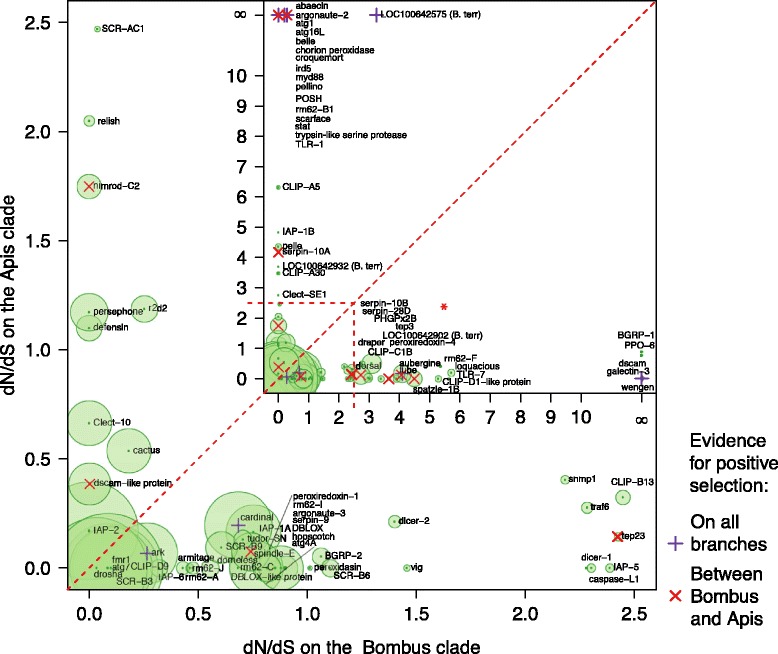
Differences in evolutionary pressure between Apis and Bombus across orthology groups. Names are taken from D. melanogaster when available. The size of the point is scaled according to the proportion of codons that are evolving under different selection in the two clades. Names were moved to improve legibility taking care to maintain x-axis position in the insert (denoted with an asterix). Full table of these results can be found in Additional file 8.
Table 8.
Genes under positive selection (using FDR < 0.05) on the branch between Bombus and Apis (4 taxa tree)
| OrthoDB group id a | Gene b | Classification | Total sites c | Likelihood ratio d | p-value | BH-corrected q-value e | Positively selected sites f |
|---|---|---|---|---|---|---|---|
| EOG6VDNFR | dorsal | Relish | 476 | 36.202 | 0.00000 | 0.00000 | 121Q, 194S, 228S, 318L, 326R, 330W, 333K, 334I, 335S, 345D, 348N, 350Q, 351N, 353A, 358Y, 359P, 363D, 367K, 368S, 369N, 372D, 373T, 375A, 376K, 377L, 380A, 384Q, 386T, 387T, 390S, 392D, 394D, 396C, 397D, 398T, 400T, 401S, 403Q, 404M, 407F, 410L, 411S, 415K, 420T, 422P, 425P, 433K, 434Q, 440V, 441P, 443E, 446Q, 447S, 448L, 453N, 454T, 458S, 462S, 463P, 465E, 467G, 468K, 471S, 472E, 473K, 474K, 476T |
| EOG6ZPC96 | nimrod-C2 | Nimrod | 1802 | 26.848 | 0.00000 | 0.00001 | 37Q, 47Q, 188M, 444H, 458K, 511M, 522V, 535V, 537E, 542Q, 550K, 576C, 582E, 599Y, 612P, 617T, 619V, 626P, 628V, 633R, 643V, 644N, 663R, 669S, 677E, 693S, 1010P |
| EOG66Q57J | LOC100642902 (B. terr) | Serine protease inhibitor | 1327 | 39.488 | 0.00000 | 0.00000 | 40G, 213S, 287E, 291D, 299V, 334V, 341S, 452S, 503T, 507H, 508S, 509D, 577G, 597D, 631T, 666D, 704K, 708S, 710A, 743S, 758K, 759W, 775Q, 778S, 820K, 856M, 858Q |
| EOG6BG79T | spindle-E | Small RNA regulatory pathway members | 1273 | 7.774 | 0.00265 | 0.03328 | 17H, 55Q, 157D, 175S, 254N, 391Q, 492G, 749T, 751S, 787I, 832F, 1026P, 1131S, 1237N, 1248T |
| EOG68SF83 | tep23 | Thioester-containing protein | 1694 | 10.554 | 0.00058 | 0.01087 | 15T, 39Y, 84S, 204P, 288G, 652A, 683S, 1070S, 1092S, 1466L, 1467S, 1470E, 1482A, 1543L |
| EOG6QNKCB | spatzle-1B | Spaetzle | 169 | 7.766 | 0.00266 | 0.03328 | 3S, 10C, 14E, 17S, 22A, 36S, 62S, 96A, 116T, 142S |
| EOG6866VT | tube | Toll pathway | 298 | 8.801 | 0.00151 | 0.02258 | 24S, 30S, 45M, 195L, 267L, 287V, 295N |
| EOG6XWDDG-1 | serpin-10A | Serine protease inhibitor | 385 | 11.507 | 0.00035 | 0.00743 | 88S, 253F, 335S, 341C, 344P |
| EOG6W3R35 | belle | Small RNA regulatory pathway members | 683 | 14.230 | 0.00008 | 0.00303 | 134T, 278I, 602S, 633Q, 664S |
| EOG6KKWHX | argonaute-2 | Small RNA regulatory pathway members | 896 | 12.187 | 0.00024 | 0.00665 | 44S, 49S, 746S |
| EOG6HHMH6 | serpin-23 | Scavenger receptor | 2066 | 10.002 | 0.00078 | 0.01303 | 1459K |
| EOG66DJHX-1 | dscam-like protein | Immunoglobulin | 1847 | 12.000 | 0.00027 | 0.00665 | None |
aGroup identifiers are from OrthoDB 6 (http://cegg.unige.ch/orthodb6).
bNumber of codons remaining in the alignment after trimming with Gblocks.
cComparison of Branch-site model A versus a constrained version with ω 2 = 1.
eMultiple test correction by the method of Benjamini and Hochberg to control the false discovery rate (only groups where FDR < 0.05 are shown).
fSites are classified as under positive selection if the Bayesian posterior probability > 0.75 (> 0.95 in bold). The reference sequence is from A. mellifera.
Figure 10.
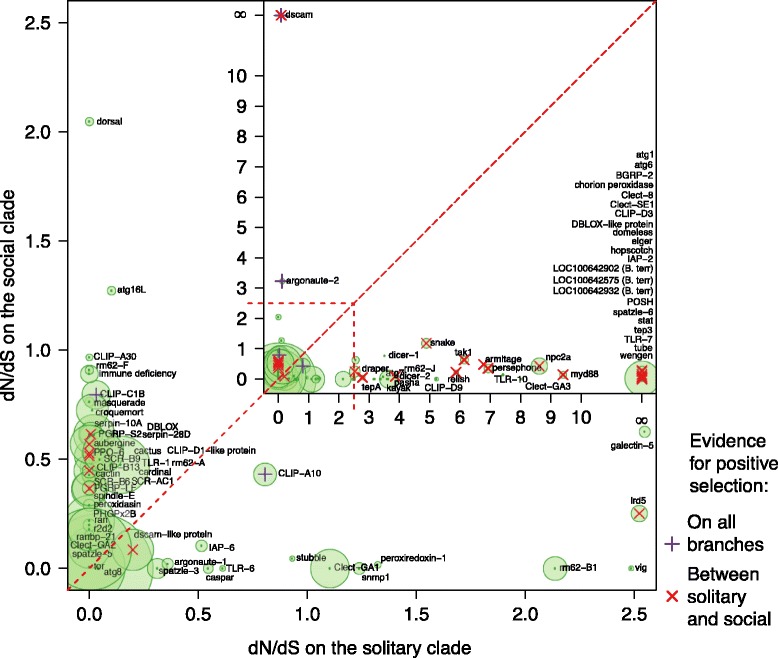
Differences in evolutionary pressure between social (Apis and Bombus) and solitary (M. rotundata) across orthology groups. Names are taken from D. melanogaster when available. The size of the point is scaled according to the proportion of codons that are evolving under different selection in the two clades Names were moved to improve legibility. A full table of these results can be found in Additional file 9.
Discussion
We find that the genomes of both species of bumblebee encode a remarkably similar repertoire of immune-related genes to the honeybee A.mellifera and solitary leafcutting bee M. rotundata (Figure 2). All the components of the major immune pathways, Toll, IMD, JAK/STAT, JNK, and the antiviral machinery are present in both Bombus species. Furthermore, the subset of these genes that we surveyed are detectibly expressed and many are induced upon immune activation. Indeed, these immune genes are expressed in a sex-specific fashion as predicted by Bateman’s principle of greater investment into maintenance for the choosier sex, usually females [54]. The sex differences in expression appear to be independent of gene dose since the expression of housekeeping genes was not significantly different between haploid males and gynes.
Overall, the number of immune genes is very consistent among the sequenced bees regardless of their degree of sociality, that is, from solitary (Megachile) to primitive (Bombus) and higher (Apis) eusociality (Figure 2). Primitive eusociality evolved about 87 million years ago in corbiculate bees [55], whereas higher sociality evolved in the Apini and Meliponi/Bombini with sociality being presumably secondarily lost in the Euglossini [55]. According to our results, the solitary bee M. rotundata, which split from the Apidae some 105 million years ago, has a comparable number of immune genes to honeybees and both Bombus species. These results suggest that the immune repertoire of A. mellifera, which was described as depauperate relative to dipterans, is probably a characteristic of bees more generally and predates the evolution of sociality and certainly existed before advanced eusociality in bees, and perhaps even as far back as before the split with ants [20]. Therefore, the relatively limited immune repertoire of honeybees does not seem to be the result of the transition to sociality and the associated behavioral adaptations for social immunity as suspected before [16]. An intriguing but purely speculative thought is that, rather than sociality reducing the need for immune genes, reduced immune complexity may have facilitated (for example, by way of easing the self/foreign distinction) or empowered (by way of allowing for social defenses) the evolution of social groups in the first place.
Both Bombus species have a small expansion of serpins (Figure 3). These serpins appear similar to the silkworm moth B. mori’s antitrypsin, which is involved in prophenoloxidase (PPO) regulation and is upregulated upon fungal infection [56]. We confirmed that these serpins are expressed in B. terrestris when challenged and are thus likely functional. The honeybee homolog seems to have a mutation within the binding region PS00284, which does not conform to the consensus pattern of this active site. It is unclear whether this gene in honeybees is a functional serpin. We also find a caspase that is similar to Decay in D. melanogaster (Figure 4), which has not been found in either A. mellifera or Nasonia vitripennis.
Despite having simpler colony organization and shorter colony lifespan, both bumblebee species nevertheless appear largely like honeybees in their immune-gene characteristics. Indeed, they also appear similar to the solitary leaf-cutting bee M. rotundata. While the complement of canonical immune genes may be consistent, it is important to recognize that our understanding of immunity is largely based on the known repertoire of non-social insects, and in particular the fruit fly D. melanogaster. As such, we are limited in being able to identify only known immune genes that have been functionally characterized in model systems. Bees may have further unexplored immune genes, novel defenses, and social behaviors that aid disease control and are unavailable to solitary species [21]. These adaptations are also genetically controlled, but the genes behind these traits are less well defined than the canonical immune response genes. Thus, while the Apoidea may appear to have consistent immune genomic profiles at the level of genes shared, they may differ considerably in the genetic underpinning of other key aspects of disease control in a social context, such as grooming, nest hygiene, and other behaviors. As a class, immune genes are rapidly evolving [57-60]. Here we explored which, among these immune genes, show particularly rapid evolution, or differences in selection among the different clades investigated. We found that some genes are under stronger selection in Bombus compared with Apis (genes below the diagonal in Figure 9), and a number of genes are under stronger positive selection in the social clade (upper diagonal in Figure 9) than in M. rotundata. While it is likely that clades with ω > 1 are under positive selection, these results should also be interpreted cautiously because without population data it is not possible to distinguish positive selection from relaxed constraints on selection [61]. Interestingly, we found a strong signature of selection on dscam, a gene primarily important for neuronal self-avoidance, but that is increasingly of interest in host-parasite interactions because alternative splicing of this gene can theoretically produce over 150,000 isoforms in D. melanogaster [62]. As such, dscam is hypothesized to be important for host-parasite specificity in susceptibility, and for specific immune memory [63]. The region under selection in dscam is limited to the beginning of the aligned protein (Figure 7A). This region corresponds to the fifth immunoglobulin I-set domain (sixth immunoglobulin domain overall). All of the previous immunoglobulin domains (1 to 5) were trimmed because they were not present in the A. mellifera gene. This gene appears to be under selection at least in the fifth immunoglobulin I-set domain but may also be variable in earlier domains. A previous study that examined the sequence of alternatively splicing exon cassettes did not detect selection in the crustacean Daphnia magna and several Drosophila species, at immunoglobulin (Ig) 2, 3 and 7 [64]. Our domain, however, likely corresponds to Ig4 or 5 in [64] and thus was not directly tested in their analysis. Nevertheless, our analysis is suggestive of differences in selective pressures among bee species. Among the other genes that show evidence of selection are a number of antiviral genes, including argonaute 2, aubergine (Figure 8A, B), and dicer 2, all of which have been found in other systems to be under selection [60,65]. We also detect evidence of selection on two AMPs, abaecin and defensin (Additional files 8, 10, and 11), both of which appear to be under stronger selection in the Apis clade (Figure 9). Our results corroborate those of Erler et al. [66], who also found positive selection on AMPs across several European bumblebee species. Interestingly, we find that dorsal appears to be under different selection in bumblebees than in honeybees, where Harpur and Zayed [61] found that dorsal was under purifying selection. We also find that all but one of the sites under selection in dorsal are outside of the relish domain (Figure 7C). Population level studies of the genes that appear to be evolving under different pressures in honeybees and bumblebees, and in the social and solitary clades will be instrumental in determining which of these genes are evolving under positive, relaxed or balancing selection [61].
Conclusions
Social insects have a suite of adaptations that have been hypothesized to reduce the pressure on immune system evolution, to the point of widespread gene losses, or inversely failing to produce or maintain duplicates. However, we find no evidence of great variation in immune gene complement, or in terms of the total number of immune-related genes across a gradient of sociality (highly social Apis > primitively social Bombus > solitary M. rotundata). Instead, we find a more nuanced pattern of immune system evolution, with variation in signatures of selection among these taxa. The different selective pressures that drive the evolution of these immune genes may in turn reflect the different parasite pressures and life history characteristics of different bee species. The depauperate immune repertoire of honeybees relative to model species thus appears to be ancestral to the evolution of bee sociality and not a consequence of sociality.
Materials and methods
Survey for immunological repertoire and annotation
The genomes of haploid males from a single colony of B. terrestris and of B. impatiens were sequenced by the Bumblebee Genome Consortium and the details of the sequencing, assembly, and automated annotation can be found in [67]. Using OrthoDB [68,69] orthologous groups, we identified orthologs from the two bumblebees, as well as from Apis florea, M. rotunda, and N. vitripennis, Tribolium castaneum, of previously characterized immune genes from D. melanogaster, A. gambiae, and A. mellifera that comprise 27 immune-related gene families or pathways. To complement the orthology searches, we searched for homologs of known immune proteins from the two bumblebees using blastp [70,71] against the official gene sets (NCBI RefSeqs). To confirm the absence of any proteins that appeared to be missing, we searched the genome assemblies and Short Reads Archive using tblastn.
Immunological expression
To confirm the relevance of these genes to immune activation and the validity of novel genes revealed in our annotation we challenged 2- to 3-week-old unmated male and gyne (that is, daughter queen) B. terrestris by injecting them with 2 μl of a suspension of either heat-killed E. coli (Gram-negative) or Arthrobacter globiformis (Gram-positive) at a concentration of 108 cfu/ml, or with sterile Ringer solution under the tergites of the abdomen, or as naïve controls handled them in the same way without any injection. We used 12 replicates for each treatment/caste combination (total N = 96). These experimental bees were the granddaughters and grandsons of queens collected in northern Switzerland in spring 2012 that had established colonies in the lab. We confirmed that these colonies were free of common parasites such as Crithidia bombi and Nosema bombi through visual inspection. All experimental bees were immobilized on ice for 30 minutes before treatment, including the naïve controls. After treatment we housed the bees singly with ad libitum pollen and 50% (w/w) sugar water. Eight hours after treatment we snap froze the bees in liquid nitrogen. We homogenized the abdomens before extraction with 0.5 g Zirkonium beads at 0°C to −4°C using an Omni Bead Ruptor 24 Homogenizer (OMNI International, Kennesaw, GA, USA). We then extracted total RNA using Qiagen RNeasy Plus Mini extraction kits (Qiagen, Hilden, Germany) according to the manufacturer's instructions. We confirmed RNA integrity of every sample with 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) with the RNA 6000 Nano Kits. We transcribed the RNA to cDNA using Quantitect reverse transcription kits (Qiagen) including controls without reverse transcriptase (no-RT controls) to test for genomic contamination. All samples were checked using quantitative PCR for our housekeeping genes to ensure that the no-RT controls amplified at least 10 cycles later, and thus contain less than 0.1% of the transcripts found in the RT samples.
Based on the full genomic sequences, we selected 27 candidate genes to represent various components of the immune response of insects, including the Toll, JAK/STAT, IMD and JNK pathways; recognition genes, melanization responses, various effectors and antiviral genes. We explored the expression of these genes upon immune stimulation relative to the geometric mean of three housekeeping genes (pla2, ak, ef1a). The full list of genes, their accession numbers and primers can be found in Table 1. All primers were designed using QuantPrime [72], based on the GenBank sequences (Table 1), except those for relish, which were published in [73]. The primers for kayak were designed based on a manually annotated gene given the temporary identifier (Bter:08277927). All primers were tested and have minimal dimer and high amplification efficiency (1.9 to 2.1).
We measured expression on a Fluidigm 96.96 Dynamic array on the BioMark system (Biomark Inc., Pueblo, CO, USA) using EvaGreen as a reporter according to the manufacturer’s protocol (Advanced Development Protocol 14; PN 100–1208 B). We eventually measured expression of 95 samples (12 replicates for each treatment in males and in queens with one naïve queen randomly dropped to make room for the negative control). We ran the samples with three technical replicates and used the average cycle threshold (Ct) of these technical replicates for further analysis.
We standardized expression of all genes of interest relative to the geometric mean of our three housekeeping genes (yielding deltaCt (dCt) values; all dCT values first transformed with Yeo-Johnson power transformations to improve normality and homoscedasticity, 'car' package in R) after confirming that the composite housekeeping value did not vary with sex (F1,87 = 0.09, P = 0.77), treatment (F3,87 = 0.29, P = 0.83), or their interaction (F3,87 = 0.70, P = 0.56) by ANOVA. We analyzed these dCt values using MANOVA with sex (gyne and male) and treatment (naïve, injected with Ringer’s solution, heat-killed E. coli, or heat-killed A. globiformis) as fixed, fully crossed factors (base package in R). We used MANOVA for these analyses, since the expression of any of these genes is not independent of the expression of other genes and because MANOVA accounts for multiple testing and is thus robust to type I error. When MANOVA effects were significant, we subsequently explored the univariate individual gene effects.
Building gene family phylogenies
We retrieved protein sequences of selected gene families from OrthoDB [68,69] and aligned them using ClustalW [74] and adjusted the alignments manually or with Gblocks [75] before tree-building using MrBayes [76] with the mixed model. We ran MrBayes for as long as was necessary (typically for 20,000 to 400,000 generations) to reduce the average deviation of split frequencies below 0.01 or until the split frequency approached 0.01 but did not improve further. We discarded the initial 25% of the trees as a burn-in.
Testing for signatures of selection
We extracted orthologous groups of immune-related genes from OrthoDB6 [68,69]. From the 130 orthologous groups with sequences from B. terrestris, B. impatiens, A. mellifera and A. florea we extracted 148 multiple sequence alignments containing exactly one sequence from each species. We use these 148 alignments for comparisons between the Bombus and Apis clades. From the 122 orthologous groups that contain M. rotundata sequences we further extracted 139 alignments that also contain a M. rotundata ortholog, which we use as an outgroup to compare social with solitary (non-social) bees. In six of the alignments (abaecin, basket, cactus, defensin, kayak and tak1) one or more orthologs were not present in OrthoDB6 and had to be retrieved from an alternative source (that is, NCBI). Protein sequences were aligned independently for the four-taxa (Bombus and Apis) or five-taxa trees (with Megachile) with ProGraphMSA [77] and trimmed using Gblocks with the stringent settings as described in [75]. Where orthologous groups contained multiple sequences for some species the most closely related sets of sequences were aligned. In the 12 orthologous groups that contained more than one sequence for each species we extracted the maximum number of alignments, such that each alignment contains only one sequence from each species. We retrieved cDNA sequences for the alignments from the official gene sets (A. mellifera v.4.5, A. florea v.1.0, B. impatiens v.2.0, B. terrestris v.1.0, M. rotundata v.1.0) using a custom written Python script.
We used likelihood-based codon models implemented in the PAML package [78] to analyze differences in the rate of evolution and to test for signals of selection. We tested hypotheses by using likelihood ratio tests to select the best fitting model among pairs of nested models that differ only in their representation of ω, the ratio of non-synonymous to synonymous substitutions (ω = dN/dS). We make the assumption that ω > 1 indicates positive selection, while ω < 1 and ω = 1 indicates negative and neutral selection.
The average rate of evolution was determined using the M0 [79] model, which assumes a constant ω across all sites and branches. The average ω is not a good indicator for the presence of positive selection, since functional and structural constraints ensure that most sites in functional genes are conserved [80]. Hence, we used the M7 and M8 models to test for the presence of positively selected sites. [79]. Both models allow ω to vary from site-to-site according to a Beta distribution, but the M8 model additionally allows some sites to evolve with ω > 1, to account for sites under positive selection.
In order to detect episodes of positive selection on the connecting branches between clades we used the branch-site model [81,82]. Some branches are assigned a priori to the foreground, where some sites are allowed to evolve with ω > 1, while all sites on background branches are constrained to ω ≤ 1. The branch-site model is compared to a null model where there is no difference between foreground and background branches. We used Clade model D [83] to test for more general differences between clades. This model allows ω to differ between clades in some sites. It is compared to a null model where there are no differences in ω between clades.
To ensure that the PAML optimization did not get stuck in local optima we used six different initial estimates for ω in all analyses and initialized branch lengths to values calculated with PhyML [84]. We corrected for multiple testing by controlling the false discovery rate using the method of Benjamini and Hochberg [85]. To calculate the posterior probabilities of sites being under positive selection in the M8 and Branch-site models we used the Bayes Empirical Bayes (BEB) approach implemented in PAML [86].
We repeated the analyses using Probcons [87] for aligning sequences. However, we only report the results from alignments produced by ProGraphMSA, as these alignments give more conservative estimates and hence a smaller chance of falsely reporting positive selection. Similarly, we do not report results from using Gblocks with the relaxed settings, as described in [75], or no trimming at all. These results are available from the authors.
Data
Sequence data can be found on NCBI (B. impatiens BIMP_2.0 RefSeq Assembly GCF_000188095.1, B. terrestris Bter_1.0 GCF_000214255.1, A. mellifera Amel_4.5 GCF_000002195.4, A. florea Aflo_1.0 GCF_000184785.1, M. rotundata MROT_1.0 GCF_000220905.1). Alignments used in this manuscript can be found in Additional files 12.
Acknowledgements
Some of these data were generated at the Genetic Diversity Centre of ETH Zürich. We thank the Bumblebee Genome Consortium (http://hymenopteragenome.org/beebase/) for providing genomic resources that were used for this study. This work was supported by an ERC Advanced Grant (no. 268853 RESIST) to PSH. RMW supported by a Marie Curie International Outgoing Fellowship and a Swiss National Science Foundation award 31003A-143936 to EMZ.
Abbreviations
- AMP
antimicrobial peptide
- NCBI
National Center for Biotechnology Information
- PCR
polymerase chain reaction
- RT
reverse transcriptase
Additional files
Statistics for the global ω ratio obtained by the M0 model (4 taxa tree).
Statistics for the global ω ratio obtained by the M0 model (5 taxa tree).
The 30 fastest evolving genes as determined by the M0 model (4 taxa tree).
The 30 slowest evolving genes as determined by the M0 model (4 taxa tree).
The 30 fastest evolving genes as determined by the M0 model (5 taxa tree).
The 30 slowest evolving genes as determined by the M0 model (5 taxa tree).
Genes under positive selection (using FDR < 0.05) on the branch to Megachile (5 taxa tree).
Genes that tested significant for evolving under differenct selective pressures (FDR < 0.05) between Bombus and Apis according to Clade model D (4 taxa tree).
Genes that tested significant for evolving under different selective pressures (FDR < 0.05) between the social and solitary clades according to Clade model D (5 taxa tree).
Summary of models on the 4 taxa tree.
Summary of models on the 5 taxa tree.
Amino acid alignments used for PAML analyses.
Statistical results for MANOVA and subsequent ANOVA of gene expression data.
Footnotes
Competing interests
The authors declare no competing interests.
Authors’ contributions
SMB was the group leader for this project. Genes were manually annotated by SMB, BMS, MJFB, SDB, KC, JCC, TJC, SE, JE, SH, HMGL, MM, IM, KN, RSH, GS, NY, and PSH. RMW and EMZ produced gene lists based on OrthoDB and bioinformatics analyses. SMB produced phylogenetic analyses, conducted the gene expression experiment and analysis with technical assistance from EK. LdP analyzed the signatures of selection. SMB, BMS, and PSH prepared the manuscript with input from RMW, MJFB, LdP, JCC, TJC, SE, HMGL, IM, RSH, and GS. All authors have read and approved the final version of the manuscript.
Contributor Information
Seth M Barribeau, Email: barribeaus14@ecu.edu.
Ben M Sadd, Email: bmsadd@ilstu.edu.
Louis du Plessis, Email: louis.duplessis@env.ethz.ch.
Mark JF Brown, Email: mark.brown@rhul.ac.uk.
Severine D Buechel, Email: severine.buechel@env.ethz.ch.
Kaat Cappelle, Email: kaat.cappelle@ugent.be.
James C Carolan, Email: james.carolan@nuim.ie.
Olivier Christiaens, Email: olchrist.christiaens@ugent.be.
Thomas J Colgan, Email: tcolgan@tcd.ie.
Silvio Erler, Email: silvio.erler@zoologie.uni-halle.de.
Jay Evans, Email: jay.evans@ars.usda.gov.
Sophie Helbing, Email: sophie.helbing@zoologie.uni-halle.de.
Elke Karaus, Email: elke.karaus@env.ethz.ch.
H Michael G Lattorff, Email: lattorff@zoologie.uni-halle.de.
Monika Marxer, Email: monika.marxer@env.ethz.ch.
Ivan Meeus, Email: ivan.meeus@ugent.be.
Kathrin Näpflin, Email: kathrin.naepflin@env.ethz.ch.
Jinzhi Niu, Email: jinzhi.niu@ugent.be.
Regula Schmid-Hempel, Email: regula.schmid@env.ethz.ch.
Guy Smagghe, Email: guy.smagghe@ugent.be.
Robert M Waterhouse, Email: robert.waterhouse@unige.ch.
Na Yu, Email: na.yu@ugent.be.
Evgeny M Zdobnov, Email: evgeny.zdobnov@unige.ch.
Paul Schmid-Hempel, Email: paul.schmid-hempel@env.ethz.ch.
References
- 1.Alexander RD. The evolution of social behavior. Annu Rev Ecol Systemat. 1974;5:325–83. doi: 10.1146/annurev.es.05.110174.001545. [DOI] [Google Scholar]
- 2.Hamilton WD. Geometry for the selfish herd. J Theor Biol. 1971;31:295–311. doi: 10.1016/0022-5193(71)90189-5. [DOI] [PubMed] [Google Scholar]
- 3.Sherman PW. Nepotism and the evolution of alarm calls. Science. 1977;197:1246–53. doi: 10.1126/science.197.4310.1246. [DOI] [PubMed] [Google Scholar]
- 4.Foster SA. Group foraging by a coral-reef fish - a mechanism for gaining access to defended resources. Anim Behav. 1985;33:782–92. doi: 10.1016/S0003-3472(85)80011-7. [DOI] [Google Scholar]
- 5.Wilson EO. Success and dominance in ecosystems: the case of the social insects. Ecology Institute Oldendorf/Luhe: Oldendorf, Germany; 1990. [Google Scholar]
- 6.Côté IM, Poulin R. Parasitism and group size in social animals: a meta-analysis. Behav Ecol. 1995;6:159–65. doi: 10.1093/beheco/6.2.159. [DOI] [Google Scholar]
- 7.Shykoff JA, Schmidhempel P. Parasites and the advantage of genetic-variability within social insect colonies. Proc R Soc B Biol Sci. 1991;243:55–8. doi: 10.1098/rspb.1991.0009. [DOI] [Google Scholar]
- 8.Hughes WOH, Boomsma JJ. Genetic diversity and disease resistance in leaf-cutting ant societies. Evolution. 2004;58:1251–60. doi: 10.1111/j.0014-3820.2004.tb01704.x. [DOI] [PubMed] [Google Scholar]
- 9.Tarpy DR. Genetic diversity within honeybee colonies prevents severe infections and promotes colony growth. Proc R Biol Sci. 2003;270:99–103. doi: 10.1098/rspb.2002.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reber A, Castella G, Christe P, Chapuisat M. Experimentally increased group diversity improves disease resistance in an ant species. Ecol Lett. 2008;11:682–9. doi: 10.1111/j.1461-0248.2008.01177.x. [DOI] [PubMed] [Google Scholar]
- 11.Altermatt F, Ebert D. Genetic diversity of Daphnia magna populations enhances resistance to parasites. Ecol Lett. 2008;11:918–28. doi: 10.1111/j.1461-0248.2008.01203.x. [DOI] [PubMed] [Google Scholar]
- 12.Schmid HP. Parasites in social insects. Princeton NJ: Princeton University Press; 1998. [Google Scholar]
- 13.Liersch S, Schmid-Hempel P. Genetic variation within social insect colonies reduces parasite load. Proc R Soc B Biol Sci. 1998;265:221–5. doi: 10.1098/rspb.1998.0285. [DOI] [Google Scholar]
- 14.Boomsma JJ, Schmid-Hempel P, Hughes WOH. Life histories and parasite pressure across the major groups of social insects. In: Fellowes MDE, Holloway GJ, Rolff J, editors. Insect Evolutionary Ecology. Oxon: CABI Publishing; 2005. pp. 139–75. [Google Scholar]
- 15.Hughes WOH, Eilenberg J, Boomsma JJ. Trade-offs in group living: transmission and disease resistance in leaf-cutting ants. Proc R Soc B Biol Sci. 2002;269:1811–9. doi: 10.1098/rspb.2002.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans JD, Aronstein K, Chen YP, Hetru C, Imler JL, Jiang H, et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol Biol. 2006;15:645–56. doi: 10.1111/j.1365-2583.2006.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YP, Siede R. Honey bee viruses. Adv Virus Res. 2007;70:33–80. doi: 10.1016/S0065-3527(07)70002-7. [DOI] [PubMed] [Google Scholar]
- 18.Singh R, Levitt AL, Rajotte EG, Holmes EC, Ostiguy N, Vanengelsdorp D, et al. RNA viruses in hymenopteran pollinators: evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS One. 2010;5 doi: 10.1371/journal.pone.0014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadau J, Helmkampf M, Nygaard S, Roux J, Simola DF, Smith CR, et al. The genomic impact of 100 million years of social evolution in seven ant species. Trends Genet. 2012;28:14–21. doi: 10.1016/j.tig.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simola DF, Wissler L, Donahue G, Waterhouse RM, Helmkampf M, Roux J, et al. Social insect genomes exhibit dramatic evolution in gene composition and regulation while preserving regulatory features linked to sociality. Genome Res. 2013;23:1235–47. doi: 10.1101/gr.155408.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cremer S, Armitage S, Schmid-Hempel P. Social immunity. Curr Biol. 2007;17:R693–702. doi: 10.1016/j.cub.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Oldroyd BP. What's killing American honeybees? PLoS Biol. 2007;5:1195–9. doi: 10.1371/journal.pbio.0050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cameron SA, Lozier JD, Strange JP, Koch JB, Cordes N, Solter LF, et al. Patterns of widespread decline in North American bumble bees. Proc Natl Acad Sci U S A. 2011;108:662. doi: 10.1073/pnas.1014743108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goulson D, Lye GC, Darvill B. Decline and conservation of bumble bees. Annu Rev Entomol. 2008;53:191–208. doi: 10.1146/annurev.ento.53.103106.093454. [DOI] [PubMed] [Google Scholar]
- 25.Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–7. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 26.Furst MA, McMahon DP, Osborne JL, Paxton RJ, Brown MJF. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature. 2014;506:364. doi: 10.1038/nature12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goulson D, Darvill B. Conservation. In: Bumblebees: Behaviour, Ecology, and Conservation. 2nd ed. Oxford: Oxford University Press; 2010. p. 177–217
- 28.Inoue M, Yokoyama J, Washitani I: Displacement of Japanese native bumblebees by the recently introduced Bombus terrestris (L.)(Hymenoptera: Apidae). J Insect Conservation 2008; 135–146.
- 29.Kanbe Y, Okada I, Yoneda M, Goka K, Tsuchida K. Interspecific mating of the introduced bumblebee Bombus terrestris and the native Japanese bumblebee Bombus hypocrita sapporoensis results in inviable hybrids. Naturwissenschaften. 2008;95:1003–8. doi: 10.1007/s00114-008-0415-7. [DOI] [PubMed] [Google Scholar]
- 30.Gill RJ, Ramos-Rodriguez O, Raine NE. Combined pesticide exposure severely affects individual- and colony-level traits in bees. Nature. 2012;491:105–9. doi: 10.1038/nature11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitehorn PR, Apos O, Connor S, Wackers FL, Goulson D. Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science. 2012;336:351–2. doi: 10.1126/science.1215025. [DOI] [PubMed] [Google Scholar]
- 32.Colla SR, Otterstatter MC, Gegear RJ, Thomson JD: Plight of the bumble bee: pathogen spillover from commercial to wild populations. Biol Conservation. 206;129:461–7.
- 33.Arbetman MP, Meeus I, Morales CL, Aizen MA, Smagghe G. Alien parasite hitchhikes to Patagonia on invasive bumblebee. Biol Invasions. 2013;15:489–94. doi: 10.1007/s10530-012-0311-0. [DOI] [Google Scholar]
- 34.Velthuis HHW, van Doorn A. A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie. 2006;37:421–51. doi: 10.1051/apido:2006019. [DOI] [Google Scholar]
- 35.Garibaldi LA, Steffan-Dewenter I, Winfree R, Aizen MA, Bommarco R, Cunningham SA, et al. Wild pollinators enhance fruit set of crops regardless of honey bee abundance. Science. 2013;339:1608–11. doi: 10.1126/science.1230200. [DOI] [PubMed] [Google Scholar]
- 36.Memmott J, Waser NM, Price MV. Tolerance of pollination networks to species extinctions. Proc R Soc B Biol Sci. 2004;271:2605–11. doi: 10.1098/rspb.2004.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid-Hempel P. On the evolutionary ecology of host-parasite interactions: addressing the question with regard to bumblebees and their parasites. Naturwissenschaften. 2001;88:147–58. doi: 10.1007/s001140100222. [DOI] [PubMed] [Google Scholar]
- 38.Otterstatter MC, Thomson JD. Contact networks and transmission of an intestinal pathogen in bumble bee (Bombus impatiens) colonies. Oecologia. 2007;154:411–21. doi: 10.1007/s00442-007-0834-8. [DOI] [PubMed] [Google Scholar]
- 39.Schmid-Hempel P. Variation in immune defence as a question of evolutionary ecology. Proc R Soc B Biol Sci. 2003;270:357–66. doi: 10.1098/rspb.2002.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadd BM, Barribeau SM. Heterogeneity in infection outcome: lessons from a bumblebee-trypanosome system. Parasite Immunol. 2013;35:339–49. doi: 10.1111/pim.12043. [DOI] [PubMed] [Google Scholar]
- 41.Hines HM. Historical biogeography, divergence times, and diversification patterns of bumble bees (Hymenoptera : Apidae : Bombus) Syst Biol. 2008;57:58–75. doi: 10.1080/10635150801898912. [DOI] [PubMed] [Google Scholar]
- 42.Goulson D. Bumblebees: Behaviour, ecology, and conservation. 2. Oxford: Oxford University Press; 2010. [Google Scholar]
- 43.Cnaani J, Schmid-Hempel R, Schmidt JO. Colony development, larval development and worker reproduction in Bombus impatiens Cresson. Insectes Sociaux. 2002;49:164–70. doi: 10.1007/s00040-002-8297-8. [DOI] [Google Scholar]
- 44.Estoup A, Solignac M, Cornuet JM. Precise assessment of the number of patrilines and of genetic relatedness in honeybee colonies. Proc R Soc B Biol Sci. 1994;258:1–7. doi: 10.1098/rspb.1994.0133. [DOI] [Google Scholar]
- 45.Schluns H, Moritz RFA, Lattorff HMG, Koeniger G. Paternity skew in seven species of honeybees (Hymenoptera: Apidae: Apis) Apidologie. 2005;36:201–9. doi: 10.1051/apido:2005006. [DOI] [Google Scholar]
- 46.Wattanachaiyingcharoen W, Oldroyd BP, Wongsiri S, Palmer K, Paar R. A scientific note on the mating frequency of Apis dorsata. Apidologie. 2003;34:85–6. doi: 10.1051/apido:2002044. [DOI] [Google Scholar]
- 47.Palmer KA, Oldroyd BP. Mating frequency in Apis florea revisited (Hymenoptera, Apidae) Insectes Sociaux. 2001;48:40–3. doi: 10.1007/PL00001743. [DOI] [Google Scholar]
- 48.Seeley TD, Tarpy DR. Queen promiscuity lowers disease within honeybee colonies. Proc R Soc B Biol Sci. 2007;274:67–72. doi: 10.1098/rspb.2006.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baer B, Schmid-Hempel P. Experimental variation in polyandry affects parasite loads and fitness in a bumble-bee. Nature. 1999;397:151–4. doi: 10.1038/16451. [DOI] [Google Scholar]
- 50.Grosshans J, Schnorrer F, Nusslein-Volhard C. Oligomerisation of Tube and Pelle leads to nuclear localisation of Dorsal. Mech Dev. 1999;81:127–38. doi: 10.1016/S0925-4773(98)00236-6. [DOI] [PubMed] [Google Scholar]
- 51.Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002;16:2497–508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleino A, Valanne S, Ulvila J, Kallio J, Myllymaki H, Enwald H, et al. Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. EMBO J. 2005;24:3423–34. doi: 10.1038/sj.emboj.7600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamoto M, Moy RH, Xu J, Bambina S, Yasunaga A, Shelly SS, et al. Virus Recognition by Toll-7 Activates Antiviral Autophagy in Drosophila. Immunity. 2012;36:658–67. doi: 10.1016/j.immuni.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rolff J. Bateman's principle and immunity. Proc Biol Sci. 2002;269:867–72. doi: 10.1098/rspb.2002.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardinal S, Danforth BN: The antiquity and evolutionary history of social behavior in bees. PLoS One. 2011;. 6(6): e21086. doi:10.1371/journal.pone.0021086 [DOI] [PMC free article] [PubMed]
- 56.Liu HF, Li YN, Jia R, Cui WZ, Mu ZM, Zhang ZF. Alternative splicing of the antitrypsin gene in the silkworm. Bombyx mori. Mol Biol Rep. 2011;38:2793–9. doi: 10.1007/s11033-010-0424-4. [DOI] [PubMed] [Google Scholar]
- 57.Jiggins FM, Kim KW. A screen for immunity genes evolving under positive selection in Drosophila. J Evol Biol. 2007;20:965–70. doi: 10.1111/j.1420-9101.2007.01305.x. [DOI] [PubMed] [Google Scholar]
- 58.Schlenke TA, Begun DJ. Natural selection drives Drosophila immune system evolution. Genetics. 2003;164:1471–80. doi: 10.1093/genetics/164.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes AL, Ota T, Nei M. Positive Darwinian selection promotes charge profile diversity in the antigen-binding cleft of class I major-histocompatibility-complex molecules. Mol Biol Evol. 1990;7:515–24. doi: 10.1093/oxfordjournals.molbev.a040626. [DOI] [PubMed] [Google Scholar]
- 60.Lazzaro B, Clark A. Rapid evolution of innate immune response genes. In: Singh R, Xu J, Kulathinal R, editors. Rapidly evolving genes and genetic systems. Oxford: Oxford University Press; 2012. pp. 203–22. [Google Scholar]
- 61.Harpur BA, Zayed A. Accelerated evolution of innate immunity proteins in social insects: adaptive evolution or relaxed constraint? Mol Biol Evol. 2013;30:1665–74. doi: 10.1093/molbev/mst061. [DOI] [PubMed] [Google Scholar]
- 62.Yu HH, Yang JS, Wang J, Huang Y, Lee T. Endodoamin diversity in the Drosophila Dscam and its roles in neuronal morphogenesis. J Neurosci. 2009;29:1904–14. doi: 10.1523/JNEUROSCI.5743-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Armitage SAO, Peuß R, Kurtz J: Dscam and pancrustacean immune memory - a review of the evidence. Dev Comparative Immunol. 2014; in press. [DOI] [PubMed]
- 64.Brites D, Encinas-Viso F, Ebert D, Du Pasquier L, Haag CR. Population genetics of duplicated alternatively spliced exons of the Dscam gene in Daphnia and Drosophila. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helbing S, Lattorff HMG 2015. Adaptive evolution of anti-viral siRNAi genes in bumblebees. bioRxiv doi.org/10.1101/017681.
- 66.Erler S, Lhomme P, Rasmont P, Lattorff HM. Rapid evolution of antimicrobial peptide genes in an insect host-social parasite system. Infect Genet Evol. 2014;23:129–37. doi: 10.1016/j.meegid.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 67.Sadd, B, Barribeau, SM, Bloch, G, Graaf, D, Dearden, P, Elsik, C, et. al The genomes of two key bumblebee species with primitive eusocial organization. Genome Biology. (in press) [DOI] [PMC free article] [PubMed]
- 68.Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2010;39:D283–8. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–65. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 71.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL: BLAST plus : architecture and applications. BMC Bioinformatics. 2009;10 [DOI] [PMC free article] [PubMed]
- 72.Arvidsson S, Kwasniewski M, Riano-Pachon DM, Mueller-Roeber B. QuantPrime - a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics. 2008;9:465. doi: 10.1186/1471-2105-9-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schlüns H, Sadd BM, Schmid-Hempel P, Crozier RH. Infection with the trypanosome Crithidia bombi and expression of immune-related genes in the bumblebee Bombus terrestris. Dev Comp Immunol. 2010;34:705–9. doi: 10.1016/j.dci.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 74.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. ClustalW and ClustalX version 2. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 75.Talavera G, Casteresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 76.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szalkowski AM, Anisimova M. Graph-based modeling of tandem repeats improves global multiple sequence alignment. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gkt628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang ZH. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 79.Yang ZH, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 81.Zhang JZ, Nielsen R, Yang ZH. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–9. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 82.Yang ZH, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–17. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 83.Bielawski JP, Yang ZH. A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J Mol Evol. 2004;59:121–32. doi: 10.1007/s00239-004-2597-8. [DOI] [PubMed] [Google Scholar]
- 84.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 85.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B Method. 1995;57:289–300. [Google Scholar]
- 86.Yang Z, Wong WSW, Nielsen R. Bayes Empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 87.Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–40. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]