

Introduction
I was born to John and Inez Frey in Plain City, Ohio, on November 14, 1935, and I received primary and secondary education in the local public schools. My father worked in seasonally complementary trades as a sheep shearer and painting contractor. He hired me in the contracting business during school breaks through junior and senior high school and college. He instilled in me the values of hard work and uncompromising honesty, which have served me well. My mother managed the local office of the General Telephone Company. I have been happily married to Carolyn Scott Frey since 1961. We have two daughters, Suzanne and Cynthia, and three granddaughters, Samantha, Carrie, and Bonnie.
I served in the United States Army (1954–1956), and I was educated in chemistry at The Ohio State University (B.S., 1959). I was a chemist in the United States Public Health Service in 1960–1963. I did graduate work at the University of Cincinnati Evening College until 1964 and then at the University of Michigan and Brandeis University. I received my Ph.D. degree in biochemistry from Brandeis University under Robert H. Abeles in 1968. I was a postdoctoral fellow in chemistry at Harvard University under Frank H. Westheimer in 1968. Professor Abeles inspired me to become a biological chemist; he and Professor Westheimer trained me to address significant problems by critical and imaginative methods.
I established my research program at The Ohio State University in 1969 as an assistant professor of chemistry and progressed to professor of chemistry in 1979. In 1981, I moved to the University of Wisconsin-Madison as a professor of biochemistry and the Institute for Enzyme Research. I retired in 2008.
Beginning at the United States Public Health Service
Before pursuing my Ph.D. degree, I conducted laboratory research at the Sanitary Engineering Center of the United States Public Health Service in Cincinnati, Ohio, from 1960 to 1963. The center later became part of the newly created Environmental Protection Agency. I worked as an immunochemist in the food chemistry laboratory. I purified the paralytic shellfish toxin, now known as saxitoxin, from mussels and linked it chemically to bovine serum albumin. I did not know the chemical structure of saxitoxin at the time, although the elemental formula was available. From this, it appeared that the molecule contained amine or guanidine groups. Applying the chemistry of the noted biochemist Heinz Fraenkel-Conrat, I used formaldehyde to create methylene linkages between saxitoxin and serum albumin. The conjugate proved to be an immunogen in rabbits, allowing saxitoxin to serve as a hapten. The rabbits responded by producing anti-saxitoxin antibodies that neutralized the paralytic properties of saxitoxin. This work introduced me to immunology and toxicology, and the results led to the publication of my first research paper (1).
To gain more control over my professional life, I decided to move on to graduate school in pursuit of a Ph.D. degree. I joined Professor Abeles' laboratory at the University of Michigan in January 1964, and we moved to Brandeis University in August 1964. I did one year of postdoctoral research at Harvard in 1968 (Fig. 1).
FIGURE 1.

Photo of the author, Perry Allen Frey.
Starting Research in Enzymology
I focused my enzymological research, first at The Ohio State University in January 1969 and later at the University of Wisconsin-Madison in 1981, on the identification and chemical characterization of transient intermediates in stepwise enzymatic reactions. Many enzymatic reactions proceed in several chemical steps by way of transient intermediates. Often, the intermediates are high in energy and chemically unstable. A thorough understanding of any chemical process requires knowledge of the structures of intermediates. Unraveling such reaction trajectories forms the basis of much chemical and biochemical research. Moreover, according to Hammond's postulate, a high-energy or transient intermediate must be regarded as closely related to the transition state for a reaction. Theory holds that enzyme catalysis arises from the stabilization of transition states through tight binding. Therefore, an enzyme tightly binds a transition state-like intermediate. A stable molecule structurally similar to a transient intermediate must also be avidly bound by an enzyme. This expectation forms the basis for the development of transition state analogs as inhibitors for enzymes identified as drug targets. For all of these reasons, the characterization of transient intermediates should be regarded as an important objective in enzymology.
2-Acetylthiamin Diphosphate
In a central step of aerobic energy metabolism, pyruvate undergoes NAD+- and CoA-dependent dehydrogenation and decarboxylation to acetyl-CoA, NADH, and CO2. The pyruvate dehydrogenase (PDH) complex catalyzes this process at the active sites of three enzymes within the complex: pyruvate dehydrogenase (E1), dihydrolipoyl acetyltransferase (E2), and dihydrolipoyl dehydrogenase (E3). E1 is thiamin diphosphate (ThDP)-dependent;
E2 has covalently bound lipoic acid (LipS2), and E3 is a flavoprotein with FAD. In the 1970s and 1980s, the overall reaction could be chemically described by Reactions 1–5, with the overall reaction being Reaction 6, the summation of Reactions 1–5.
 |
At the time, investigators addressed many questions about the PDH complexes, including the molecular weight, quaternary structure, spatial interactions among the enzymes, and regulation of activity. David Speckhard and Maria Maldonado in our group contributed to these issues for the PDH complex from Escherichia coli. Associates John Reardon and Heechung Yang synthesized alkylating and acylating derivatives of undecagold complexes. Undecagold could be visualized and masses determined by scanning transmission electron microscopy. We prepared undecagold-labeled components of the PDH complex and, in collaborations with James Hainfeld and Joseph Wall at the electron microscopy facility of the Brookhaven National Laboratory, proceeded to clarify the molecular masses of the core and reconstituted complexes (2–5). We also collaborated with my brother Professor Terrence G. Frey at San Diego State University to employ undecagold clusters as labels of thiol groups in E2 and mitochondria. The undecagold-labeled dihydrolipoyl groups appeared as pairs of bright spots in microscope images (6).
My main chemical interest in the PDH complex was in Reaction 2, the reductive transacetylation of lipoyl groups in LipS2-E2 by E1·hydroxyethyl-ThDP. This reaction involves dehydrogenation of hydroxyethyl-ThDP coupled with acetyl transfer to Lip(SH)2-E2 to form acetyl-S-Lip(SH)-E2. In NMR and kinetics experiments, Yuh-Shyong Yang in our group had shown S8-acetyllipoyl groups to be produced in this reaction (7).
The mechanistic issue was whether 2-acetyl-ThDP could be an intermediate in Reaction 2. Mechanisms not involving 2-acetyl-ThDP could be written, and no evidence on this issue had been published. 2-Acetyl-ThDP had never been synthesized and was thought to undergo hydrolysis too fast to be isolated and characterized. Our group focused on the possible intermediacy of 2-acetyl-ThDP and obtained the results outlined in Fig. 2.
FIGURE 2.

Overview of experiments identifying 2-acetyl-ThDP as an intermediate in reactions of the PDH complex.
Although Reaction 1 was not strictly irreversible, reversal could not occur in the absence of CO2; exclusion of CO2 made it irreversible. Because other steps were readily reversible, Claire CaJacob and I considered whether reversibility in those steps could lead to ThDP-dependent hydrolysis of acetyl-CoA through the intermediate formation of 2-acetyl-ThDP in Reaction 2. Accordingly, incubation of the PDH complex with NADH and acetyl-CoA in buffer led to the ThDP-dependent hydrolysis of acetyl-CoA. This could be explained by the intermediate formation of 2-acetyl-ThDP. Having no possibility to revert to pyruvate in the absence of CO2, the system allowed 2-acetyl-ThDP to undergo hydrolysis to acetate and ThDP (Fig. 2, right). Corazon Anonuevo Steginsky in our group similarly observed ThDP-dependent hydrolysis of succinyl-CoA catalyzed by the analogous 2-ketoglutarate dehydrogenase complex, presumably via the intermediate formation of 2-succinyl-ThDP (8, 9).
With Lim Leung and Douglas Flournoy, we also studied the reactions of E1 with 3-fluoropyruvate (Fig. 2, bottom). 3-Fluoropyruvate reacted in place of pyruvate to undergo E1·ThDP-catalyzed decarboxylation according to Reaction 1 to produce 2-fluoro-1-hydroxyethyl-ThDP. α,β-Elimination of fluoride produced 2-enolacetyl-ThDP, which, upon ketonization, became 2-acetyl-ThDP. We then investigated its further reactions within E1. We found that E1·ThDP catalyzed the reaction of 3-fluoropyruvate to CO2, fluoride, and acetate (10). This suggested decarboxylation, fluoride elimination, and ketonization to 2-acetyl-ThDP, followed by hydrolysis. We then also found that 3-fluoropyruvate, in the additional presence of dihydrolipoamide and CoASH with the PDH complex, reacted to form acetyl-CoA (11). Moreover, reaction of 3-fluoropyruvate with the PDH complex in the presence of dihydrolipoamide produced S-acetyldihydrolipoamide. All results pointed to the intermediate formation of 2-acetyl-ThDP from 3-fluoropyruvate. No NAD+ was required because 3-fluoropyruvate was at the appropriate oxidation level to react with dihydrolipoamide.
Our group took a more direct approach. Kenneth Gruys synthesized 2-acetyl-ThDP by chromic acid oxidation of synthetic hydroxyethyl-ThDP (Fig. 2, left). Christopher Halkides characterized 2-acetyl-ThDP in acidic aqueous solutions by NMR as a mixture of three interconvertible forms at equilibrium: the parent keto form, the keto hydrate, and the internal keto adduct with the 6-amino group of the pyrimidine ring (12). 2-Acetyl-ThDP proved to be stable in acidic solutions and prone to hydrolysis at higher pH values, displaying a unique pH-rate profile.
Brief reaction of [3-14C]pyruvate with the PDH complex in the presence of ThDP, NAD+, and CoASH followed closely by an acid quench produced a 14C-labeled compound that co-migrated with authentic 2-acetyl-ThDP upon chromatographic separation (Fig. 2, top). Yields were compatible with a transient intermediate, varying from 2 to 12% in the amount of PDH complex, depending on conditions (12). The pH-rate profile for hydrolysis of 2-[14C]acetyl-ThDP from the trapping experiments proved to be identical to that of authentic synthetic 2-acetyl-ThDP, definitively identifying 2-[14C]acetyl-ThDP as a transient intermediate in the reaction of [3-14C]pyruvate. 2-Acetyl-ThDP proved to be an intermediate in other enzymatic reactions as well (13).
Galactose Metabolism in the Leloir Pathway
The Leloir pathway catalyzes the metabolism of galactosyl and glucosyl groups at the level of UDP-sugars. Galactokinase catalyzes the phosphorylation of galactose to α-d-galactose-1-P; galactose-1-P uridylyltransferase (GalT) catalyzes the reaction of α-d-galactose-1-P with UDP-glucose to produce UDP-galactose and α-d-glucose-1-P. UDP-galactose 4-epimerase catalyzes the transformation of UDP-galactose to UDP-glucose. The pathway transforms α-d-galactose into glucose-1-P for energy metabolism. The reversibility of 4-epimerase and uridylyltransferase allows the production of UDP-galactose for galactosyltransferases from UDP-glucose.
I initially addressed the reaction mechanism of the NAD+-dependent UDP-galactose 4-epimerase in E. coli. Up to that time, all NAD+-dependent hydride transfers in biochemistry were strictly stereospecific, and 4-epimerase raised the question of how non-stereospecificity could occur. The uridine diphosphoryl moiety of the substrate proved to play dual essential roles in the action of 4-epimerase.
My associates Timple G. Wee, Shan Wong, and Uan Kang verified UDP-4-ketoglucose as a tightly bound intermediate and devised a clear understanding of the basis for non-stereospecific action (reviewed in Ref. 14). The UDP moiety proved to be the enzyme-binding anchor, with very little binding to the sugar. This allowed the glucosyl and galactosyl moieties to undergo free rotations within the active site. The constellation of glycosyl-C3(OH), C4(H), and C4(OH) proved to be similar in reactive rotamers of galactosyl and glucosyl moieties, so NAD+ accessed C4(H) equally with either substrate.
In chemical, kinetic, and NMR spectroscopic experiments, my associates Yun-Hua Wong, George Flentke, Yijeng Liu, Janeen Vanhooke, Yaoming Wei, and James Burke showed that in a second essential role, binding of the UDP moiety chemically activated and increased the reduction potential of NAD+ in 4-epimerase (15–19). Associates Adrian Hegeman, Barbara Gerratana, and Jeffrey Gross further showed how the NAD+ and active site residues in the homologous dTDP-glucose 4,6-dehydratase catalyze dehydration of dTDP-glucose to dTDP-4-keto-6-deoxyglucose (20).
The overall 4-epimerase mechanism rationalizes how nature requires three enzymes to interconvert glucosyl and galactosyl moieties differing only in stereochemical configuration at C4. In nature, the installation of UDP as the anchor in 4-epimerase requires phosphorylation of galactose by the kinase and uridylyl transfer by the transferase (21). The Leloir pathway and 4-epimerase mechanism stand in contrast to the organic chemistry laboratory, where NaBH4 readily reduces UDP-4-ketoglucose to the mixture of UDP-galactose and UDP-glucose.
After mentioning to graduate student Lee-Jun Wong that the galactosemia-defect enzyme, GalT, should function in two steps through a uridylyl-enzyme intermediate, she verified ping-pong kinetics and isolated the covalent intermediate. Chemical degradation, site-directed mutagenesis and chemical rescue experiments in our laboratory performed by Sue-Lein Yang, Teresa Field, Frank J. Ruzicka, and Jeongmin Kim identified His166 as the uridylylation site. Sandaruwan Geeganage proved the kinetic competence of GalT His166-UMP. Details of chemical mechanisms in the Leloir pathway are reviewed elsewhere (21).
Phosphoryl- and Nucleotidyl-enzyme Intermediates and Phosphorus Stereochemistry
My group's focus on GalT broadened my attention to mechanisms of nucleotidyl- and phosphotransferases in general. Nearly all were controversial regarding covalent nucleotidyl- or phosphoryl-enzyme intermediates. In cases of ping-pong kinetics, covalent intermediates could be identified. In cases of sequential kinetics with ternary or quaternary complexes, the issue remained ambiguous. Often, covalent phosphoenzymes were reported, but the question of whether they represented artifacts remained. To address this issue, our group and those under Fritz Eckstein at the Max Planck Institute for Experimental Medicine, Jeremy R. Knowles at Harvard University, Stephen J. Benkovic at Pennsylvania State University, Wojciech Stec at the Polish Academy of Sciences, John A. Gerlt at the University of Illinois at Urbana-Champaign, and Gordon Lowe at the University of Oxford pursued definitive results through phosphorus stereochemistry. Biological phosphates, being achiral or prochiral at tetrahedral phosphorus, had to be synthesized with P-substituents such as S, 18O, and/or 17O stereospecifically placed to study stereochemistry (22).
The ping-pong mechanism of GalT implied overall retention of configuration at Pα of UDP-glucose through stereochemical inversion in two steps. Our laboratory developed methods to synthesize P-chiral substrates for this and a number of other enzymes. Stereochemical studies also required methods to determine configuration at phosphorus in P-chiral substrates and products. Our laboratory developed 31P NMR and chemical/mass spectrometric methods for assigning relative and, ultimately, absolute configurations at chiral phosphorus.
Two of our group's first methods are illustrated in Fig. 3 as examples. Kwan-Fu Sheu initially studied adenylate kinase, which catalyzes the phosphorylation of AMP by ATP. He demonstrated first that the Rp- and Sp-epimers of adenosine 5′-O-(1-thiodiphosphate) (ADPαS) displayed different 31P NMR chemical shifts. This 31P NMR property subsequently could be employed to distinguish P-epimers and to assign P-configurations. He then showed that adenylate kinase catalyzed stereospecific phosphorylation of adenosine 5′-phosphorothioate to (Sp)-ADPαS (Fig. 3A) (23). Other phosphotransferases turned out also to display this property of stereospecific phosphorylation in our and other laboratories. This property enabled us to assign configurations to phosphorus in molecules with P-chiral [18O]phosphorothioate groups.
FIGURE 3.

Syntheses of P-chiral biochemical phosphorothioates. A, adenylate kinase-catalyzed stereospecific phosphorylation of adenosine 5′-phosphorothioate. B, chemical transformation of (Sp)-[α-18O2]ADPαS into (Rp)-[γ-18O]ATPγS.
In stereochemical work on phosphotransferases, P-chiral adenosine 5′-O-(3-[3-18O]thiotriphosphate) ([γ-18O]ATPγS) would be needed. In our group, John P. Richard and Hsu-Tso Ho synthesized molecules, such as (Sp)-[γ-18O]ATPγS by the method outlined in Fig. 3B (24). This proved to be a substrate for most phosphotransferases, and the configurations of the [18O]phosphorothioate groups in the products were determined by chemical, mass spectrometric, or 31P NMR methods. R. Douglas Sammons, Thomas Meade, and Radha Iyengar devised methods for displacing sulfur from P-chiral phosphorothioate esters with 18O or 17O from water (25, 26). Our group applied these methods to elucidate the stereochemical course of phosphotransfer (Table 1).
TABLE 1.
Stereochemistry of substitution at phosphorus
ApA, adenyl 3,5′-adenosine; OAA, oxalacetate; PEP, phosphoenolpyruvate.
| Enzyme or chemical reaction | Reaction | Stereochemistry | Ref. |
|---|---|---|---|
| Galactose-1-P uridylyltransferase | UDP-Gal + Glc-1-P ⇌ UDP-Glc + Gal-1-P | Retention | 27 |
| UDP-Glc + GalT His166⇌Glc-1-P + GalT His166-UMP | Inversion | 28 | |
| UDP-glucose pyrophosphorylase | UTP + Glc-1-P ⇌ UDP-Glc + PPi | Inversion | 29 |
| Nucleoside-diphosphate kinase | ATP + NDP ⇌ NTP + ADP | Retention | 27 |
| Adenylate kinase | ATP + AMP ⇌ ADP + ADP | Inversion | 30 |
| Nucleoside phosphotransferase | AMP + nucleoside ⇌ NMP + adenosine | Retention | 31 |
| Adenosine kinase | ATP + adenosine ⇌ ADP + AMP | Inversion | 32 |
| Polynucleotide kinase | ATP + 5′-HO-ApA ⇌ ADP + 5′-PO-ApA | Inversion | 33 |
| Glycerol kinase | Glycerol + ATP ⇌ sn-glycerol-3-P | Inversion | 34 |
| Phosphoenolpyruvate carboxykinase | |||
| Mitochondrial | OAA + GTP → PEP + CO2 + GDP | Inversion | 35 |
| Cytosolic | OAA + ITP → PEP + CO2 + IDP | Inversion | 36 |
| DNA polymerase | |||
| Polymerase I | Overall | Inversion | 37 |
| T7 polymerase | Overall | Inversion | 38 |
| Mevalonate-PP decarboxylase | |||
| Mevalonate-PP + ATP⇌5-P-mevalonate-PP + ADP | Inversion | 39 | |
| Gentamicin nucleotidyltransferase | Overall | Inversion | 40 |
| Adenylate cyclase (Bordetella pertussis) | Overall | Inversion | 40 |
| FHIT (fragile histidine triad) | Overall | Retention | 41 |
| Phosphoanhydride chemical synthesis | Inversion | 42 | |
| Desulfurization of P-chiral ADPαS to P-chiral [α-18O]ADP | Inversion | 43 | |
| Catalyzed thiophosphoanhydride synthesis | Epimerization | 44 |
The outcomes of our stereochemical studies appear in Table 1. Reactions of P-chiral substrates in single-displacement mechanisms proceed with inversion of configuration at phosphorus. Overall retention implicates a double-displacement mechanism. Accordingly, the reaction of GalT proceeds with overall retention of configuration at Pα. In an improbable scenario, overall retention could result from two retention steps. To test this remote possibility, Richard S. Brody and Abolfazl Arabshahi in our group proved that the first step of the reaction, transfer of the uridylyl-αS group to His166, proceeds with inversion of Pα, proving overall retention in two inverting steps, as illustrated in Reactions 7 and 8 (27).
REACTION 7.

REACTION 8.

Phosphorus stereochemistry resolved all questions of enzymatic single or double displacement at phosphorus. The results led also to the statement of the principle of “economy in the evolution of binding sites” in enzymes. The first six entries in Table 1 exemplify the principle. In a reversible nucleotidyl transfer or phosphotransfer, there are two P-acceptors, one in the forward direction and the other in the reverse direction. When the P-acceptors are structurally and electrostatically similar, the kinetic mechanism is ping pong with overall retention at phosphorus. In these cases, the P-donor-binding site encompasses the P-acceptor subsite, with an intervening enzymatic nucleophile. Such a site is sufficient to support the overall reaction. The enzymatic nucleophile covalently mediates phosphotransfer between the two P-acceptors, which bind to the P-acceptor subsite, but not at the same time, thus the ping-pong mechanism and covalent E-P intermediate. Otherwise, when the P-acceptors are structurally and electrostatically dissimilar, more binding sites are required, the kinetics is sequential with ternary complexes, and the stereochemistry is inversion at phosphorus. In the latter cases, there is no need for an enzymatic nucleophile within a ternary complex. This principle appears to hold in other classes of group transferases as well (45).
Identification and Characterization of Radical Intermediates
In 1986, I refocused my research to the field of radical enzymology, believing that much of the new chemistry in biology might be radical chemistry. As a graduate student in the 1960s, free radicals were regarded as off-limits for enzyme intermediates. Radicals were high-energy species and regarded as too highly reactive and difficult to control to be accommodated within the active sites of enzymes. By the 1970s, a stable tyrosyl radical in the active site of E. coli ribonucleotide reductase began to change things in enzymology. For some time, it was regarded as an anomaly and an exception. However, evidence gradually appeared indicating the possibility of radicals in oxygenation reactions and in the reactions of adenosylcobalamin, the vitamin B12 coenzyme. I took an interest in S-adenosylmethionine, which I thought might react as a radical. Ultimately, many newly discovered enzymes requiring S-adenosylmethionine have been found to function through radical mechanisms.
Fig. 4 shows the radicals 4a–4n discovered by our group. By purely chemical methods, we identified the radicals shown in blue. For example, Ded-Shih Huang and Frank J. Ruzicka found isomerization of radicals 4a and 4b to be required, by precedent in organic chemistry, to explain product formation in the action of methane monooxygenase on 1,1-dimethylcyclopropane (46). We characterized the other radicals shown in black in rapid-mixed freeze-quenched samples by EPR spectroscopy in collaboration with my Wisconsin colleague George H. Reed and his associates, who extracted the maximum structural information from isotope-edited spectra.
FIGURE 4.

Radical intermediates discovered in enzymatic active sites. Radicals 4a and 4b, methane monooxygenase; 4c, dioldehydrase; 4d, lysine 2,3-aminomutase and dioldehydrase; 4e, dioldehydrase and class II ribonucleotide reductase; 4f, glutamate 2,3-aminomutase; 4g–4j, lysine 2,3-aminomutase; and 4k–4n, lysine 5,6-aminomutase.
Dioldehydrase and Ribonucleotide Reductase
My introduction to this field began with my training for a Ph.D. degree under Professor Abeles. Dioldehydrase catalyzes the dehydration of propane-1,2-diol to propionaldehyde and proceeds by hydrogen transfer from C1 to C2. The reaction requires the coenzyme adenosylcobalamin, shown in Fig. 5A, in addition to dioldehydrase and a potassium ion.
FIGURE 5.
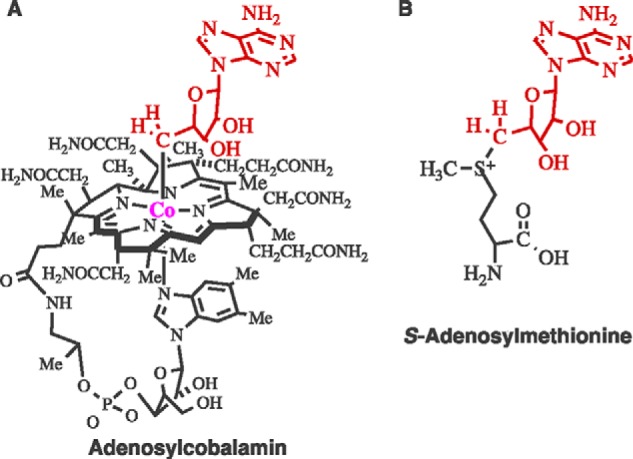
Structures of adenosylcobalamin and complex of S-adenosylmethionine and [4Fe-4S].
Adenosylcobalamin-dependent isomerization reactions follow the pattern of Reaction 9, in which a group (R) in a substrate migrates from Cβ to Cα, and a hydrogen cross-migrates from Cα to Cβ. The reaction of dioldehydrase follows this pattern in the first step of Reaction 10, producing propionaldehyde hydrate. Dehydration leads to propionaldehyde.
REACTION 9.

REACTION 10.

In my Ph.D. research on dioldehydrase, I found the following results, among others. (i) Reaction of the complex of dioldehydrase and adenosylcobalamin (E·Cob(III)-CH2Ado) with the suicide inactivator glycolaldehyde cleaved the Co–C5′ bond to cob(II)alamin and 5′-deoxyadenosine. (ii) Suicide inactivation by [2-3H]glycolaldehyde produced 5′-[3H]deoxyadenosine. (iii) Reaction of [1-3H]propane-1,2-diol as the dioldehydrase substrate produced [5′-3H]adenosylcobalamin. Reaction of [5′-3H]adenosylcobalamin with dioldehydrase and propane-1,2-diol produced [3H]propionaldehyde (47). These results led directly to the conclusion that C5′ in adenosylcobalamin mediates hydrogen transfer in reactions of dioldehydrase. This function of adenosylcobalamin proved to hold for all other adenosylcobalamin-dependent enzymes (48).
At the time, free radicals had not been discovered in enzymatic reactions and did not appear in my Ph.D. dissertation. Jack Halpern at The University of Chicago and Richard Finke at the University of Oregon later discovered the weakness of the cobalt–carbon bond in adenosylcobalamin and its tendency to undergo homolytic scission to cob(II)alamin and the 5′-deoxyadenosyl radical (49, 50). This introduced radical chemistry into the field of adenosylcobalamin, and radical intermediates were found in adenosylcobalamin-dependent reactions. This led to the generic mechanism in Reactions 11–15 for isomerizations,
 |
where •CH2Ado is the 5′-deoxyadenosyl radical, S• is the radical arising from abstraction of a hydrogen from the substrate S–H, and P• is the product-related radical.
Thirty years after completing my Ph.D. research, I returned to dioldehydrase to complete analysis of suicide inactivation by glycolaldehyde (47). Associate Andreas Abend found that suicide inactivation led to radical 4c in Fig. 4, the product of hydrogen abstraction from glycolaldehyde (51, 52).
In our group, Philip A. Schwartz showed that photolysis of adenosylcobalamin in the presence of O2 produces 5′-peroxyadenosine through the reaction of O2 with the 5′-deoxyadenosyl radical (4e) in Fig. 4 (53). Schwartz further showed that O2-dependent cleavage of adenosylcobalamin at the active site of dioldehydrase also produces 5′-peroxyadenosine through radical 4e (54).
Class II ribonucleotide reductase catalyzes the adenosylcobalamin-dependent reduction of ribonucleoside triphosphates to 2′-deoxynucleoside triphosphates by a mechanism requiring the intermediate formation of a thiyl radical at Cys408. Andreas Abend in our group synthesized 5′-(R)-[5′-2H]adenosylcobalamin and demonstrated that ribonucleoside-triphosphate reductase catalyzes epimerization of C5′ in the presence of the allosteric activator dGTP. Dawei Chen in our group carried out a detailed kinetic analysis and proved that C5′ epimerization proceeds faster than reduction of the substrate. He further proved that the inactive mutant reductases C408A and C408S also catalyze C5′ epimerization faster than the overall reduction by wild-type reductase. The results implicated the 5′-deoxyadenosyl radical (4e) as an intermediate in the generation of the Cys408 thiyl radical (55). JoAnne Stubbe at the Massachusetts Institute of Technology collaborated in this research.
Lysine 2,3-Aminomutase
In 1970, Horace A. Barker and associates at the University of California, Berkeley, discovered clostridial lysine 2,3-aminomutase in bacterial lysine metabolism (56). The transformation of l-lysine into l-β-lysine, a 2,3-amino migration, followed the pattern of adenosylcobalamin-dependent enzymes (Reaction 9) but did not involve adenosylcobalamin. Instead, the enzyme required S-adenosylmethionine and pyridoxal phosphate (PLP), and it contained a trace of iron.
Adenosylcobalamin and S-adenosylmethionine share the 5′-deoxyadenosyl moiety (Fig. 5, A and B) but are otherwise chemically different. In particular, the S+–C5′ bond in S-adenosylmethionine is >30 kcal/mol stronger than the Co–C5′ bond in adenosylcobalamin.
Graduate student Marcia Moss and I began studies of lysine 2,3-aminomutase, and I continued with Janina Baraniak, Marcus Ballinger, Bin Song, Robert M. Petrovich, Kafryn Lieder, Weiming Wu, Elham Behshad, Jennifer Miller, Dawei Chen, Frank J. Ruzicka, and Olafur Magnusson. The reaction of clostridial lysine 5,6-aminomutase, a 5,6-amino migration discovered by Thressa C. Stadtman at the National Institutes of Health (57), also followed the course of Reaction 9 and required adenosylcobalamin, not S-adenosylmethionine.
In our first work on lysine 2,3-aminomutase, Moss showed that tritium in [5′-3H]adenosylmethionine appeared in l-lysine and l-β-lysine. This implicated the 5′-deoxyadenosyl moiety of S-adenosylmethionine in hydrogen transfer, as in adenosylcobalamin-dependent reactions (58, 59). We wrote the radical mechanism in Fig. 6 to account for our results.
FIGURE 6.

Radical mechanism in the reaction of lysine 2,3-aminomutase.
Of the four radicals in Fig. 6, our group characterized three by rapid-mix/freeze-quench EPR and pulsed magnetic resonance spectroscopy. The kinetically competent C2 radical in Fig. 6 appeared in the steady state of the reaction of lysine as radical 4g (Fig. 4) (60–63). The enantiomeric radical 4h appeared in the steady state in the action of E. coli lysine 2,3-aminomutase (64). Senior associate Frank J. Ruzicka discovered clostridial glutamate 2,3-aminomutase and found intermediate radical 4f (65).
The other radicals in Fig. 6 are too high in energy to be observed by EPR. In radicals 4f–4h, the unpaired electrons are stabilized by delocalization into the carboxylate group. High-energy radicals in Fig. 6 can be stabilized by adjacent, sterically compact, functional groups that delocalize the unpaired electron. 4-Thia-l-lysine reacts as a substrate for lysine 2,3-aminomutase, and the substrate-related radical 4i appears in the steady state (66, 67). Trans-4,5-dehydro-l-lysine reacts as a suicide inactivator of lysine 2,3-aminomutase and is converted to the allylic radical 4j, with cleavage of the S+–C5′ bond and formation of 5′-deoxyadenosine (68). Dehydration between C3′ and C4′ of the adenosyl moiety in S-adenosylmethionine leads to S-3′,4′-anhydroadenosylmethionine, which functions as a coenzyme for lysine 2,3-aminomutase. In reaction with lysine 2,3-aminomutase and lysine, the EPR spectrum of 5′-deoxy-3′,4′-anhydroadenosine-5′-yl signals the transient appearance of the kinetically competent radical 4d (69). In the reaction of dioldehydrase with the analogous 3′,4-anhydroadenosylcobalamin, radical 4d appears and interacts magnetically with cob(II)alamin in a triplet spin system (70).
Our group turned its attention to the theretofore unknown mechanism by which lysine 2,3-aminomutase promotes cleavage of S-adenosylmethionine to the 5′-deoxyadenosyl radical. With graduate student Robert M. Petrovich, we found lysine 2,3-aminomutase to harbor a catalytically essential [4Fe-4S] cluster (71, 72). One iron proved to be unique in that it could be removed by controlled oxidation to form a [3Fe-4S] cluster and reductively restored with FeSO4 and a reducing agent. In addressing cleavage of the S+–C5′ bond, Squire J. Booker synthesized Se-adenosylselenomethionine and found it to activate lysine 2,3-aminomutase. We then collaborated with Robert Scott and Nathaniel Cosper at the University of Georgia, experts in selenium x-ray absorption spectroscopy, to determine the fate of selenium upon cleavage of the Se+–C5′ bond, with particular reference to whether selenium would become a ligand to the unique iron in [4Fe-4S]. Upon cleavage of Se-adenosylselenomethionine at the active site by reaction with trans-4,5-dehydro-l-lysine to form the allylic radical 3j (Fig. 4), the x-ray absorption data revealed direct Fe–Se ligation (73). We concluded that upon cleavage of Se+–C5′, selenium became ligated to the unique iron in the cluster. Subsequent work with Dawei Chen in a collaboration with Brian Hoffman and Charles Walsby at Northwestern University showed that the interaction of S-adenosylmethionine with the [4Fe-4S] cluster entailed direct ligation of the methionyl amino and carboxylate group with the unique iron (74). This interaction had been found in the pyruvate formate-lyase activase, another S-adenosylmethionine enzyme (75). Integrating the spectroscopic results, we wrote the mechanism in Reaction 16 for the cleavage of S-adenosylmethionine into the 5′-deoxyadenosyl radical (74).
REACTION 16.

Senior associate Frank J. Ruzicka obtained diffraction-quality crystals of lysine 2,3-aminomutase in complex with S-adenosylmethionine and lysine in our laboratory. We collaborated with Professor Dagmar Ringe and Bryan Lepore at Brandeis University to obtain a molecular structure (76). The structure fully supported all that had been found by spectroscopy and providing detailed information about the molecular structure and all lysine 2,3-aminomutase contacts with Se-adenosylselenomethionine, lysine, and PLP.
With associates Glen T. Hinckley and Susan C. Wang, we addressed the energetics of one-electron reductive cleavage of S-adenosylmethionine to the 5′-deoxyadenosyl radical. The electrochemical reduction potential for the [4Fe-4S] cluster in lysine 2,3-aminomutase proved to be −0.43 V in the presence of S-adenosylmethionine (77). The reduction potential for a generic trialkyl sulfonium ion (−1.8 V) gave a difference of 1.4 V, meaning that electron transfer would not be possible unless the gap in reduction potentials could be decreased. Electrochemical data showed that the addition of a surrogate for lysine (alanine + ethylamine) lowered the reduction potential for [4Fe-4S] in lysine 2,3-aminomutase to −0.6 V. Moreover, the reduction potential for S-adenosylmethionine bound to [4Fe-4S] at the active site proved to be −0.99 V (78). Thus, binding of lysine and S-adenosylmethionine closed the gap in reduction potentials from 1.4 to 0.4 V, enabling one-electron transfer.
In 1984, Joachim Knappe at the University of Heidelberg found the activation of the pyruvate formate-lyase activase by S-adenosylmethionine to be accompanied by glycyl radical formation in pyruvate formate-lyase and production of 5′-deoxyadenosine (79). Joan B. Broderick, now at Montana State University, and associates discovered the iron-sulfur cluster in pyruvate formate-lyase activase, and in the year 2000, they showed it to be [4Fe-4S] (80). The 1990s saw the discovery of biotin synthase, lipoyl synthase, and class III ribonucleotide reductase as S-adenosylmethionine-activated [4Fe-4S] enzymes (81–87, 90). All catalyzed chemically difficult reactions involving C–H cleavage. By the turn of the century, the genes encoding these proteins and lysine 2,3-aminomutase had been cloned and sequenced. Heidi J. Sofia at the National Human Genome Research Institute and associates found the motif CxxxCxxC in common among these enzymes. This motif proved to bind the [4Fe-4S] clusters. Sofia and colleagues searched the genomic database for proteins with this motif and found >600 proteins in 2001 involved in highly diverse biological processes (88). She named them the radical SAM superfamily. By 2008, the superfamily had grown to >2800 proteins in over 40 families (89). The superfamily has grown to >48,000 proteins in nearly 70 families (Structure-Function Linkage Database) that catalyze complex reactions, including repair of thymine dimers, vitamin biosynthesis, antibiotic biosynthesis, methylation of DNA, anaerobic heme biosynthesis, carbide insertion into the FeMo cofactor of nitrogenase, methanogenesis, and other chemically difficult reactions, often presumably by radical mechanisms (91, 92).
Lysine 5,6-Aminomutase
The internal transfer of the C6 amino group to C5 of d-lysine, l-lysine, or l-β-lysine is catalyzed by lysine 5,6-aminomutase, a PLP enzyme. 5,6-Aminomutase requires adenosylcobalamin but neither S-adenosylmethionine nor iron (Fig. 5A). A chemical mechanism analogous to that in Fig. 6 for lysine 2,3-aminomutase can be written, with substitution of adenosylcobalamin as the source of the 5′-deoxyadenosyl radical. In this mechanism, the initial substrate radical would be the C5 lysyl-PLP radical, and the rearranged product-related radical would be the C6 methylene radical.
Graduate student Christopher H. Chang initiated research in our group on the 5,6-aminomutase (93). Elham Behshad and Kuo-Hsiang Tang carried the work forward. No radicals could be detected by EPR spectroscopy in the steady states of reactions of any lysine substrate. This could be attributed to the high energy of all of the putative radicals that would appear in the mechanism. However, the lysine 2,3-aminomutase substrate 4-thia-l-lysine stabilized the C3 radical in Fig. 6, and the 4-thia group could be expected to stabilize equally well an unpaired electron at C5. Accordingly, Tang discovered radicals 4k–4n in the reactions of the substrate analogs 4-thia-d- and 4-thia-l-lysine (94). Radicals 4k and 4l in Fig. 6 are analogs of C5 radical intermediates in the reactions of d- or l-lysine. The molecular structure of the resting enzyme with cobalamin and PLP bound, obtained in collaboration with Catherine Drennan and Frederick Berkovitch at the Massachusetts Institute of Technology, implies a major structural reorganization attending the binding of lysine (95).
In research on aminomutases, I expected to find S-adenosylmethionine to be an evolutionary predecessor of the more complex adenosylcobalamin, which is produced in 24 biosynthetic steps. A corollary might be that the more elegant adenosylcobalamin dominates over the “inferior” predecessor. The results on lysine 2,3-aminomutase support S-adenosylmethionine as a possible predecessor, and it is as active as any adenosylcobalamin enzyme. However, the sheer size of the radical SAM superfamily does not support adenosylcobalamin as a superior radical initiator.
Role of Low-barrier Hydrogen Bonding in Serine Protease Reactions
Low-barrier Hydrogen Bonding in Chymotrypsin
I shall explain how I came to study strong low-barrier hydrogen bonds (LBHBs), with special reference to catalysis by chymotrypsin. In beginning my research program in 1969, I resolved not to study chymotrypsin. It had been chemically and kinetically analyzed in detail, and a crystal structure identified the catalytic triad of Ser195, His57, and Asp102. His57 functioned as a base, removing the β-OH proton of Ser195 as the β-oxygen attacked the substrate carbonyl group to form a metastable tetrahedral intermediate. Collapse of the intermediate with release of the N-terminal cleavage peptide produced peptidyl chymotrypsin, which underwent hydrolysis to free chymotrypsin and the C-terminal cleavage peptide. I regarded chymotrypsin to be in good hands and did not see how I could contribute.
The above mechanism explained everything, with one exception. His57, displaying a pKa value of 7, seemed too weak a base to abstract a proton from serine. Principles of physical organic chemistry would mandate a base with a pKa value between those of the serine β-OH, pKa = 13.6 (96), and the N terminus of the leaving cleavage peptide, pKa = 9. The standard rationale was that the transition state would embody the appropriate basicities, but there was no explanation for how this would be brought about. A solution awaited experiments in 1972, 1987, and the 1990s.
My colleague W. Wallace Cleland (Fig. 7), seeking to explain low-deuterium fractionation factors (Φ < 1.0) in several enzymatic mechanisms, published a paper in 1992 suggesting the presence of LBHBs in certain enzymes (97). Deuterium fractionation factors expressed the equilibrium binding of deuterium relative to hydrogen. Low factors indicated favorable hydrogen versus deuterium binding. The cysteine β-SH displayed low-deuterium fractionation, but cysteine was absent in these systems. LBHBs also displayed low-deuterium fractionation. Simultaneously, John A. Gerlt, now at the University of Illinois, and Paul G. Gassman at the University of Minnesota provided a theoretical basis for short-strong H-bonding in certain enzymatic mechanisms (98).
FIGURE 7.

The author with W. Wallace Cleland (left) in 2007 in Waltham, Massachusetts.
Cleland presented the deuterium fractionation evidence at a conference we attended, and on the flight home, he commented on the unfavorable reception he received. I suggested that he find spectroscopic evidence. Cleland asked what kind of spectroscopy, and I suggested NMR. In that moment, I recalled the work of Robillard and Shulman in 1972. They found a downfield 1H NMR signal at 18 ppm in chymotrypsin at low pH values and assigned it to the proton bridging His57 and Asp102 (99). This proton resonated at 15 ppm at pH values above 7 and at 18 ppm at low pH values. John L. Markley, then at Purdue University, independently extended this to trypsin (100). The 18 ppm signal for protonated His57 seemed consistent with an LBHB. I immediately realized that His57 would be protonated at and above pH 7 in the metastable tetrahedral intermediates for peptide hydrolysis. Could an LBHB stabilize these intermediates and related transition states?
My Ph.D. preceptor, Professor Abeles, introduced peptidyl trifluoromethyl ketones as transition state analog inhibitors of serine proteases, including chymotrypsin. He and his associates found the methyl esters of N-acetyl-phenylalanine, N-acetyl-glycyl-phenylalanine, N-acetyl-valyl-phenylalanine, and N-acetyl-leucyl-phenylalanine to be good substrates for chymotrypsin, with the best being N-acetyl-leucyl-phenylalanine methyl ester. They synthesized analogous peptidyl trifluoromethyl ketones in which -CF3 replaced -OCH3, and they showed that they formed tetrahedral adducts with Ser195 in chymotrypsin (101–103). The compounds were excellent inhibitors and analogs of the metastable tetrahedral intermediates, with the best being N-acetyl-leucyl-phenylalanyl-CF3, with a dissociation constant of 0.2 nm for the ketonic (unhydrated) form. I thought that the adducts would include protonated His57 and display LBHBs. I called Abeles and asked if he had retained a sample of the best inhibitor and explained my thoughts. He replied that he would find out and get back to me. I then found in the library that, in 1987, Liang and Abeles (103) had already published what I had in mind. In the complex of chymotrypsin with N-acetyl-leucyl-phenylalanyl-CF3, they found a downfield proton at 18.7 ppm that persisted at pH 9. I called Abeles immediately and told him that he had already done what I had planned, and he promptly recalled his and Liang's earlier work. He also sent me a sample of the inhibitor. We repeated the work and found the downfield signal at 18.9 ppm.
I and John Tobin proceeded to assign the downfield protons in protonated His57 of chymotrypsin and the tetrahedral complex of chymotrypsin with N-acetyl-leucyl-phenylalanyl-CF3 as LBHBs, the only LBHBs assigned in a protein at the time (104). Simultaneously, W. Wallace Cleland and Maurice M. Kreevoy at the University of Minnesota elaborated on their theory of catalysis by LBHBs (105). With Tobin, we put forward in Fig. 8A a mechanism for acylation of chymotrypsin and postulated that the LBHB stabilized the metastable tetrahedral intermediate and, by Hammond's postulate, the transition state (104). We noted the similarity of this tetrahedral intermediate to the Ser195 adduct of N-acetyl-leucyl-phenylalanyl-CF3 (Fig. 8B) and emphasized that Liang and Abeles had found the downfield proton in this complex and shown it to persist above pH 9. We further pointed out that Richard Henderson at the Medical Research Council had methylated His57 in chymotrypsin, decreasing activity by 2 × 105 (106). This chemical mutation disrupted the LBHB, meaning that it could account for 105 of the rate enhancement in chymotrypsin, corresponding to transition state stabilization of 7 kcal/mol. We further noted the downfield proton in cis-urocanic acid and suggested it as a “first approximation” to a model for protonated His57 (104).
FIGURE 8.

LBHBs in chymotrypsin. A, role for the His57-Asp102 LBHB in the acylation of Ser195 in chymotrypsin. B, LBHB in the tetrahedral adduct of chymotrypsin with N-acetyl-leucyl-phenylalanyl-CF3 (N-AcLF-CF3).
In Fig. 8A, we assigned partial charges y+ to His57 and y− to Asp102, where 0.5 ≤ y < 1.0 (104). We did not know the exact position of the proton in the LBHB and defined y accordingly. A value of 0.5 for y would correspond to a symmetrical H-bond, which was unlikely. A value between 0.5 and 1.0 in an unsymmetrical LBHB seemed more likely. In the absence of a paramagnetic metal or a ring current effect, we thought the downfield proton to be magnetically deshielded because of elongation of the Nδ1–H bond due to the LBHB. Elongation corresponded to pulling the proton away from the imidazole ring, leaving increased electron density in the ring and increased basicity at the His57 Nϵ3. In this way, the basicity of His57 would increase in the approach to the transition state, making it a suitable base to catalyze proton transfer from Ser195.
The excellent review by Hibbert and Emsley (107) on strong H-bonds in small molecules guided us in our assignment. They defined three classes of H-bonds: conventional weak H-bonds (<10 kcal/mol), intermediate strong H-bonds (10–20 kcal/mol), and very strong H-bonds (>24 kcal/mol). The authors pointed out that the intermediate strong H-bonds could form between heteroatoms with similar proton affinities, and they displayed downfield proton chemical shifts (δH 18–20 ppm), low-deuterium fractionation factors, deuterium isotope effects on NMR chemical shifts, and linear H-bonding with less than van der Waals contact distances separating heteroatoms. We adopted these properties as distinguishing features of LBHBs.
With associates Constance S. Cassidy and Jing Lin and Wisconsin colleague John L. Markley, we found corroborative evidence for the LBHB in peptidyl trifluoromethyl ketone complexes of chymotrypsin (108–112). The downfield proton chemical shifts were 18.6–18.9 ppm depending on peptidyl structure. The deuterium fractionation factors were 0.3–0.4. Activation enthalpies for chemical exchange of the LBHBs with water were 14.7–19.4 kcal/mol, >10 kcal/mol higher than for conventional H-bonds. The LBHBs displayed deuterium and tritium isotope effects on chemical shift. These were the expected properties for the intermediate strong H-bonds defined by Hibbert and Emsley (107).
Catalytic Significance of the LBHB
As evidence of the significance of LBHBs, the chymotrypsin adducts with N-acetyl-phenylalanyl-CF3, N-acetyl-glycyl-phenylalanyl-CF3, N-acetyl-valyl-phenylalanyl-CF3, and N-acetyl-leucyl-phenylalanyl-CF3 displayed pKa values for His57 from 10.7 for N-acetyl-phenylalanyl-CF3 to 12.1 for N-acetyl-leucyl-phenylalanyl-CF3. In linear free energy correlations, plots of literature values of log(kcat/Km) for peptidyl methyl esters against pKa or δH for the corresponding peptidyl trifluoromethyl ketones proved to be linear with positive slopes. Plots of log Ki for peptidyl trifluoromethyl ketones against δH or pKa for the same inhibitors proved to be linear with negative slopes.
The difference in pKa for His57 in the tetrahedral complex of chymotrypsin·N-acetyl-leucyl-phenylalanyl-CF3 (pKa = 12) and free chymotrypsin (pKa = 7) corresponds to a free energy difference of 7 kcal/mol, the same as the difference in activation energies for free chymotrypsin and N-methylated His57 chymotrypsin. Thus, the physicochemical properties of peptidyl trifluoromethyl ketone adducts support the proposition of the importance of the LBHB in catalysis. All evidence points to the role of the LBHB in elevating the pKa of His57 to between 9 and 13 at the tetrahedral intermediate, making it the ideal base catalyst for serine protease action.
Analogous evidence for complexes of peptidyl trifluoromethyl ketones with the serine protease subtilisin included downfield δH and low-deuterium fractionation for the His-Asp contact (113). Further evidence showed the contact distance between the His57 Nδ1 and Asp102 Oδ1 of the complex of chymotrypsin with N-acetyl-leucyl-phenylalanyl-CF3 to be less than the van der Waals contact distance (114). In addition, the crystal structure of subtilisin at atomic resolution clearly showed a lengthening of the His57 Nδ1H–H bond (115).
LBHBs in Small Molecules
Constance S. Cassidy, John Tobin, Jing Lin, and Mary Cloninger in our group undertook studies of LBHBs in small molecules. Physical and physical organic chemists had defined the physicochemical properties of LBHBs in small molecules (107).
Fig. 9 illustrates energy profiles for weak, intermediate strength, and very strong H-bonds. Weak H-bonds, as in water, display double-minimum energy profiles separated by high barriers. Intermediate strength H-bonds, or LBHBs, are shorter than allowed by van der Waals radii, and the barrier is lowered to near the zero-point vibrational energy, as in the complex of N-methylimidazole (N-MeIm) with 2,2-dichloropropionate. This complex (δH = 18 ppm) displays an anti-symmetrical C–O vibrational frequency lying between those of analogous ion pairs (N-MeImH+–−OOC-R) formed with stronger acids and those of neutral complexes (N-MeIm–HOOC-R formed with weaker acids) (116). Both ion paired and neutral complexes display chemical shifts upfield from 18 ppm. The spectroscopic properties of the 18 ppm complex are as expected for an LBHB.
FIGURE 9.

Three classes of hydrogen bonds: weak, low-barrier, and single-well.
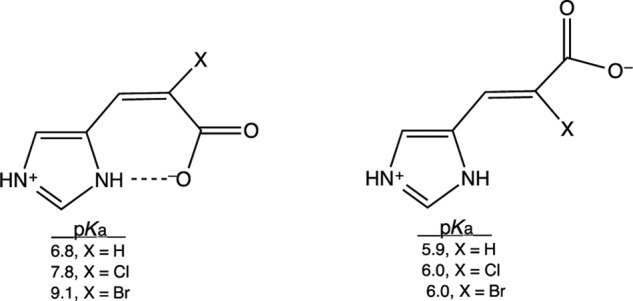
Associate Mary Cloninger showed that the downfield intramolecular H-bond in cis-urocanic acid can be altered by introducing steric bulk at C2. The His57-Asp102 contact in chymotrypsin is sterically compressed in x-ray crystal structures of peptidyl trifluoromethyl ketone complexes (114). By the foregoing hypothesis, the basicity of His57 (pKa = 12) can be attributed to this contact. The imidazole basicities of 2-substituted cis- and trans-urocanates in water support this hypothesis, as shown in Structure 1 (117). The increasing sizes of the 2-chloro and 2-bromo substituents increase the pKa in cis-urocanates but not in the trans-isomers. The electronic effects of chlorine and bromine cannot account for the results, so the steric effects must be invoked. The increased basicity can be ascribed to the increased strength of the H-bond and alternatively by increased electrostatic interaction in the contact. In either case, the closer contacts in the cis-2-chloro- and 2-bromourocanates lead to increased base strength in the imidazole ring. This likely occurs in the chymotrypsin transition states.
STRUCTURE 1.
Strong H-bonds in Aqueous/Organic Media
Our group addressed widespread reservations as to whether LBHBs could be present in aqueous media (118). We chose to study intramolecular LBHBs in sterically enforced H-bonding within molecules, such as hydrogen maleate monoanion (H-maleate) in Fig. 9B, perhaps the most thoroughly studied strong H-bond in organic chemistry. Typical research of this molecule involved crystal structures or strictly anhydrous, aprotic solvents in solution. Under these conditions, H-maleate served as an example of a single-well H-bond. Crystal structures showed the H-bond to be nearly symmetrical, and perfectly symmetrical in the imidazolium salt (119). The downfield proton in anhydrous solvents appeared at >20 ppm.
Theoretical calculations put the energy of the H-bond variously at >24 kcal/mol in vacuum. McAllister (120) studied the effect of medium polarity and found the H-bond energy to be ∼20 kcal/mol in “a dielectric continuum” similar to water. In aqueous solution, the difference in pKa values of 1.9 and 6.2 for maleic acid could not be accounted for on purely electrostatic grounds, but an energy of 12–15 kcal/mol for the intramolecular H-bond could explain the difference (118).
Why then would the LBHB in H-maleate not appear in aqueous solutions? We reasoned that it might undergo exchange too quickly with protons in water to appear in the NMR spectrum. Therefore, Constance S. Cassidy, John Tobin, and Jing Lin in our group studied the NMR spectra of H-maleate in 90:10 (v/v) acetone-d6/H2O at −50 °C to decrease the exchange rate (121). These solutions contained 0.4 mol fractions of water molecules (40%). We also studied other sterically strained dicarboxylate monoanions, H-2,2-dimethyl malonate, and H-cis-cyclohexane 1,2-dicarboxylate in the same solvent, and we found the downfield signals at 20.2, 19.0, and 19.2 ppm, respectively. Therefore, intermediate strong intramolecular H-bonds exist in organic/aqueous media. Substrate-binding sites in enzymes clearly are not aqueous, although water has access at the surface. The acetone-d6/H2O likely allows more access to water than a binding site in a protein. In further work on the chemical properties of intramolecular H-bonding, Lin in our group showed the activation energy for exchange with medium protons and deuterium fractionation to be compatible with an LBHB (122)
Nearly all studies of ultra strong H-bonds in chemistry employed non-biological molecules. A recent study showed that all of the acid-base groups in proteins can form ultra strong H-bonds (123).
At the time we made our assignments of LBHBs (104), I told my associates that the worst reaction we could expect would be for other investigators simply to accept our claim. This would signal the end of research on LBHBs in serine proteases. I need not have been concerned. Many objections followed, including the theory that strong H-bonds could not exist in water. This made it necessary to carry out much more detailed and critical tests. The results outlined above fully corroborated our original claim.
Acknowledgments
I am exceedingly grateful to my graduate and postdoctoral students for sharing the thrill of discovery with me. I am also grateful to senior investigators in Asia, Europe, and the Americas for valuable collaborations. I am equally grateful to the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of General Medical Sciences of the National Institutes of Health for continuously supporting my research through predoctoral fellowships from 1964 to 1967, a postdoctoral fellowship in 1968, and individual research grants from 1969 to my retirement in 2008.
REFERENCES
- 1. Johnson H. M., Frey P. A., Angelotti R., Campbell J. E., Lewis K. H. (1964) Haptenic properties of paralytic shellfish poison conjugated to protein by formaldehyde treatment. Proc. Soc. Exp. Biol. Med. 117, 425–430 [DOI] [PubMed] [Google Scholar]
- 2. CaJacob C. A., Frey P. A., Hainfeld J. F., Wall J. S., Yang H. (1985) Escherichia coli pyruvate dehydrogenase complex: particle masses of the complex and component enzymes measured by scanning transmission electron microscopy. Biochemistry 24, 2425–2431 [DOI] [PubMed] [Google Scholar]
- 3. Yang H. C., Hainfeld J. F., Wall J. S., Frey P. A. (1985) Quaternary structure of pyruvate dehydrogenase complex from E. coli. J. Biol. Chem. 260, 16049–16051 [PubMed] [Google Scholar]
- 4. Yang H., Frey P. A., Hainfeld J. F., Wall J. S. (1986) Pyruvate dehydrogenase complex of Escherichia coli: radial mass analysis of subcomplexes by scanning transmission electron microscopy. Biophys. J. 49, 56–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y.-S., Datta A., Hainfeld J. F., Furuya F. R., Wall J. S., Frey P. A. (1994) Mapping the lipoyl groups of the pyruvate dehydrogenase complex by use of gold cluster-labels and scanning transmission electron microscopy. Biochemistry 33, 9428–9437 [DOI] [PubMed] [Google Scholar]
- 6. Frey P. A., Frey T. G. (1999) Synthesis of undecagold labeling compounds and their applications in electron microscopic analysis of multiprotein complexes. J. Struct. Biol. 127, 94–100 [DOI] [PubMed] [Google Scholar]
- 7. Yang Y.-S., Frey P. A. (1986) Dihydrolipoyl transacetylase of Escherichia coli. Formation of 8-S-acetyldihydrolipoamide. Biochemistry 25, 8173–8178 [DOI] [PubMed] [Google Scholar]
- 8. CaJacob C. A., Gavino G. R., Frey P. A. (1985) Pyruvate dehydrogenase complex of Escherichia coli. thiamin pyrophosphate and NADH-dependent hydrolysis of acetyl-CoA. J. Biol. Chem. 260, 14610–14615 [PubMed] [Google Scholar]
- 9. Steginsky C. A., Frey P. A. (1984) Escherichia coli α-ketoglutarate dehydrogenase complex. Thiamin pyrophosphate-dependent hydrolysis of succinyl coenzyme A. J. Biol. Chem. 259, 4023–4026 [PubMed] [Google Scholar]
- 10. Leung L. S., Frey P. A. (1978) Fluoropyruvate: an unusual substrate for Escherichia coli pyruvate dehydrogenase. Biochem. Biophys. Res. Commun. 81, 274–279 [DOI] [PubMed] [Google Scholar]
- 11. Flournoy D. S., Frey P. A. (1986) Pyruvate dehydrogenase and 3-fluoropyruvate: chemical competence of acetylthiamin pyrophosphate as an acetyl group donor to dihydrolipoamide. Biochemistry 25, 6036–6043 [DOI] [PubMed] [Google Scholar]
- 12. Gruys K. J., Halkides C. J., Frey P. A. (1987) Synthesis and properties of 2-acetylthiamin pyrophosphate: an enzymatic reaction intermediate. Biochemistry 26, 7575–7585 [DOI] [PubMed] [Google Scholar]
- 13. Tittmann K., Wille G., Golbik R., Weidner A., Ghisla S., Hübner G. (2005) Radical phosphate transfer mechanism for the thiamin diphosphate- and FAD-dependent pyruvate oxidase from Lactobacillus plantarum. Kinetic coupling of intercofactor electron transfer with phosphate transfer of acetyl thiamin diphosphate via a transient semiquinone/hydroxyethyl-ThDP radical pair. Biochemistry 44, 13291–13303 [DOI] [PubMed] [Google Scholar]
- 14. Frey P. A., Hegeman A. D. (2013) Chemical and stereochemical actions of UDP-galactose 4-epimerase. Acc. Chem. Res. 46, 1417–1426 [DOI] [PubMed] [Google Scholar]
- 15. Wong Y.-H., Frey P. A. (1979) Uridine diphosphate galactose 4-epimerase. Alkylation of enzyme-bound diphosphopyridine nucleotide by p-(bromoacetamido)phenyl uridyl pyrophosphate, an active-site-directed irreversible inhibitor. Biochemistry 18, 5337–5341 [DOI] [PubMed] [Google Scholar]
- 16. Flentke G. R., Frey P. A. (1990) Reaction of uridine diphosphate galactose-4-epimerase with a suicide inactivator. Biochemistry 29, 2430–2436 [DOI] [PubMed] [Google Scholar]
- 17. Burke J. R., Frey P. A. (1993) The importance of binding energy in catalysis of hydride transfer by UDP-galactose 4-epimerase: a 13C and 15N NMR and kinetic study. Biochemistry 32, 13220–13230 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., Vanhooke J. L., Frey P. A. (1996) UDP-galactose 4-epimerase: NAD+ content and a charge transfer band associated with the substrate-induced conformational transition. Biochemistry 35, 7615–7620 [DOI] [PubMed] [Google Scholar]
- 19. Wei Y., Lin J., Frey P. A. (2001) 13C NMR analysis of electrostatic interactions between NAD+ and active site residues of UDP-galactose 4-epimerase: implications for the activation induced by uridine nucleotides. Biochemistry 40, 11279–11287 [DOI] [PubMed] [Google Scholar]
- 20. Gross J. W., Hegeman A. D., Gerratana B., Frey P. A. (2001) Dehydration is catalyzed by glutamate-136 and aspartic acid-135 active site residues in Escherichia coli dTDP-glucose 4,6-dehydratase. Biochemistry 40, 12497–12504 [DOI] [PubMed] [Google Scholar]
- 21. Frey P. A. (1996) The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 10, 461–470 [PubMed] [Google Scholar]
- 22. Frey P. A. (2010) Sulfur as a mechanistic probe in enzymatic and non-enzymatic substitution at phosphorus. New J. Chem. 34, 820–828 [Google Scholar]
- 23. Sheu K.-F., Frey P. A. (1977) Enzymatic and 31P nuclear magnetic resonance study of adenylate kinase-catalyzed stereospecific phosphorylation of adenosine 5′-phosphorothioate. J. Biol. Chem. 252, 4445–4448 [PubMed] [Google Scholar]
- 24. Richard J. P., Ho H.-T., Frey P. A. (1978) Synthesis of nucleoside [18O]pyrophosphorothioates with chiral [18O]phosphorothioate groups of known configuration. Stereochemical orientations of enzymatic phosphorylations of chiral [18O]phosphorothioates. J. Am. Chem. Soc. 100, 7756–7757 [Google Scholar]
- 25. Sammons R. D., Ho H.-T., Frey P. A. (1982) Evidence implicating cyclo-diphosphates as intermediates in reactions of nucleoside phosphorothioates with cyanogen bromide. J. Am. Chem. Soc. 104, 5841–5842 [Google Scholar]
- 26. Meade T. J., Iyengar R., Frey P. A. (1985) Synthesis and rearrangements of alkyl phosphorothioates. J. Org. Chem. 50, 936–940 [Google Scholar]
- 27. Sheu K.-F., Richard J. P., Frey P. A. (1979) Stereochemical courses of nucleotidyltransferase and phosphotransferase action. Uridine diphosphate glucose pyrophosphorylase, galactose-1-phosphate uridylyltransferase, adenylate kinase and nucleoside diphosphate kinase. Biochemistry 18, 5548–5556 [DOI] [PubMed] [Google Scholar]
- 28. Arabshahi A., Brody R. S., Smallwood A., Tsai T.-C., Frey P. A. (1986) Galactose-1-phosphate uridylyltransferase. Purification of the enzyme and stereochemical course of each step of the double-displacement mechanism. Biochemistry 25, 5583–5589 [DOI] [PubMed] [Google Scholar]
- 29. Sheu K.-F., Frey P. A. (1978) UDP-glucose pyrophosphorylase. Stereochemical course of the reaction of glucose 1-phosphate with uridine-5′[1-thiotriphosphate]. J. Biol. Chem. 253, 3378–3380 [PubMed] [Google Scholar]
- 30. Richard J. P., Frey P. A. (1978) Stereochemical course of thiophosphoryl group transfer catalyzed by adenylate kinase. J. Am. Chem. Soc. 100, 7757–7758 [Google Scholar]
- 31. Richard J. P., Prasher D. C., Ives D. H., Frey P. A. (1979) Chiral [18O]phosphorothioates. The stereochemical course of thiophosphoryl group transfer catalyzed by nucleoside phosphotransferase. J. Biol. Chem. 254, 4339–4341 [PubMed] [Google Scholar]
- 32. Richard J. P., Carr M. C., Ives D. H., Frey P. A. (1980) The stereochemical course of thiophosphoryl group transfer catalyzed by adenosine kinase. Biochem. Biophys. Res. Commun. 94, 1052–1056 [DOI] [PubMed] [Google Scholar]
- 33. Bryant F. R., Benkovic S. J., Sammons D., Frey P. A. (1981) The stereochemical course of thiophosphoryl group transfer catalyzed by T4 polynucleotide kinase. J. Biol. Chem. 256, 5965–5966 [PubMed] [Google Scholar]
- 34. Pliura D. H., Schomburg D., Richard J. P., Frey P. A., Knowles J. R. (1980) Stereochemical course of a phosphokinase using a chiral [18O] phosphorothioate: comparison with the transfer of a chiral [16O,17O,18O]phosphoryl group. Biochemistry 19, 325–329 [DOI] [PubMed] [Google Scholar]
- 35. Sheu K.-F., Ho H.-T., Nolan L. D., Markovitz P., Richard J.P., Utter M. F., Frey P. A. (1984) Stereochemical course of thiophosphoryl group transfer catalyzed by mitochondrial phosphoenolpyruvate carboxykinase. Biochemistry 23, 1779–1783 [DOI] [PubMed] [Google Scholar]
- 36. Konopka J. M., Lardy H. A., Frey P. A. (1986) Stereochemical course of thiophosphoryl transfer catalyzed by cytosolic phosphoenolpyruvate carboxykinase. Biochemistry 25, 5571–5575 [DOI] [PubMed] [Google Scholar]
- 37. Brody R. S., Frey P. A. (1981) Unambiguous determination of the stereochemistry of nucleotidyl transfer catalyzed by DNA polymerase I from Escherichia coli. Biochemistry 20, 1245–1252 [DOI] [PubMed] [Google Scholar]
- 38. Brody R. S., Adler S., Modrich P., Stec W. J., Leznikowski Z. J., Frey P. A. (1982) Stereochemical course of nucleotidyl transfer catalyzed by bacteriophage T7-induced DNA polymerase. Biochemistry 21, 2570–2572 [DOI] [PubMed] [Google Scholar]
- 39. Iyengar R., Cardemil E., Frey P. A. (1986) Mevalonate-5-diphosphate decarboxylase: stereochemical course of ATP-dependent phosphorylation of mevalonate 5-diphosphate. Biochemistry 25, 4693–4698 [DOI] [PubMed] [Google Scholar]
- 40. Van Pelt J. E., Iyengar R., Frey P. A. (1986) Gentamicin nucleotidyltransferase. Stereochemical inversion at phosphorus in enzymatic 2′-deoxyadenylyl transfer to tobramycin. J. Biol. Chem. 261, 15995–15999 [PubMed] [Google Scholar]
- 41. Abend A., Garrison P. N., Barnes L. D., Frey P. A. (1999) Stereochemical retention of the configuration in the action of Fhit on phosphorus-chiral substrates. Biochemistry 38, 3668–3676 [DOI] [PubMed] [Google Scholar]
- 42. Richard J. P., Frey P. A. (1983) Stereochemical course of phosphoanhydride synthesis. J. Am. Chem. Soc. 105, 6605–6609 [Google Scholar]
- 43. Sammons R. D., Frey P. A. (1982) Synthesis of Rp and Sp [α-18O]ADP from Sp and Rp β-cyanoethyl-adenosine 5′-[1-thiodiphosphate]. J. Biol. Chem. 257, 1138–1141 [PubMed] [Google Scholar]
- 44. Iyengar R., Frey P. A. (1988) Epimerization in the synthesis of thiophosphoanhydrides. Bioorg. Chem. 16, 52–61 [Google Scholar]
- 45. Frey P. A. (1992) Nucleotidyltransferases and phosphotransferases. Stereochemistry and covalent intermediates. in The Enzymes (Boyer P. D., Sigman D. S., eds) 3rd Ed., Vol. 20, pp. 141–186, Academic Press, New York [Google Scholar]
- 46. Ruzicka F., Huang D.-S., Donnelly M. I., Frey P. A. (1990) Methane monooxygenase catalyzed-oxygenation of 1,1-dimethylcyclopropane. Evidence for radical and carbocationic intermediates. Biochemistry 29, 1696–1700 [DOI] [PubMed] [Google Scholar]
- 47. Frey P. A. (2014) Travels with radicals. 5′-Deoxyadenosine and 5′-deoxyadenosine-5′-yl in radical enzymology. Acc. Chem. Res. 47, 540–549 [DOI] [PubMed] [Google Scholar]
- 48. Frey P. A. (2010) Cobalamin coenzymes in enzymology. in Comprehensive Natural Products II, Chemistry and Biology (Mander L., Liu H.-W., eds) Vol. 7, pp. 501–546, Elsevier, Oxford [Google Scholar]
- 49. Halpern J. (1985) Mechanisms of coenzyme B12-dependent rearrangements. Science 227, 869–875 [DOI] [PubMed] [Google Scholar]
- 50. Finke R. G., Hay B. P. (1984) Thermolysis of adenosylcobalamin: a product, kinetic, and cobalt-carbon (C5′) bond dissociation energy study. Inorg. Chem. 23, 3041–3043 [Google Scholar]
- 51. Abend A., Bandarian V., Reed G. H., Frey P. A. (2000) Identification of cis-ethanesemidione as the organic radical derived from glycolaldehyde in the suicide inactivation of dioldehydrase and of ethanolamine ammonia-lyase. Biochemistry 39, 6250–6257 [DOI] [PubMed] [Google Scholar]
- 52. Sandala G. M., Smith D. M., Coote M. L., Radom L. (2004) Suicide inactivation of dioldehydrase by glycolaldehyde and chloroacetaldehyde: an examination of the reaction mechanism. J. Am. Chem. Soc. 126, 12206–12207 [DOI] [PubMed] [Google Scholar]
- 53. Schwartz P. A., Frey P. A. (2007) 5′-Peroxyadenosine and 5′-peroxyadenosyl-cobalamin as intermediates in the aerobic photolysis of adenosylcobalamin. Biochemistry 46, 7284–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwartz P. A., Frey P. A. (2007) Dioldehydrase: an essential role for potassium ion in the homolytic cleavage of the cobalt-carbon bond in adenosylcobalamin. Biochemistry 46, 7293–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen D., Abend A., Stubbe J., Frey P. A. (2003) Epimerization at carbon-5′ of (5′R)-[5′-2H]adenosylcobalamin by ribonucleoside triphosphate reductase: cysteine 408-independent cleavage of the Co-C5′ bond. Biochemistry 42, 4578–4584 [DOI] [PubMed] [Google Scholar]
- 56. Chirpich T. P., Zappia V., Costilow R. N., Barker H. A. (1970) Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J. Biol. Chem. 245, 1778–1789 [PubMed] [Google Scholar]
- 57. Baker J. J., van der Drift C., Stadtman T. C. (1973) Purification and properties of β-lysine mutase, a pyridoxal phosphate and B12 coenzyme dependent enzyme. Biochemistry 12, 1054–1063 [DOI] [PubMed] [Google Scholar]
- 58. Moss M., Frey P. A. (1987) The role of S-adenosylmethionine in the lysine 2,3,-aminomutase reaction. J. Biol. Chem. 262, 14859–14862 [PubMed] [Google Scholar]
- 59. Baraniak J., Moss M. L., Frey P. A. (1989) Lysine 2,3-aminomutase. Support for a mechanism of hydrogen transfer involving S-adenosylmethionine. J. Biol. Chem. 264, 1357–1360 [PubMed] [Google Scholar]
- 60. Ballinger M. D., Reed G. H., Frey P. A. (1992) An organic radical in the lysine 2,3-aminomutase reaction. Biochemistry 31, 949–953 [DOI] [PubMed] [Google Scholar]
- 61. Ballinger M. D., Frey P. A., Reed G. H. (1992) Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 31, 10782–10789 [DOI] [PubMed] [Google Scholar]
- 62. Ballinger M. D., Frey P. A., Reed G. H., LoBrutto R. (1995) Pulsed electron paramagnetic resonance studies of the lysine 2,3-aminomutase substrate radical: evidence for participation of pyridoxal 5′-phosphate in a radical rearrangement. Biochemistry 34, 10086–10093 [DOI] [PubMed] [Google Scholar]
- 63. Chang C. H., Ballinger M. D., Reed G. H., Frey P. A. (1996) Lysine 2,3-aminomutase: rapid mix-freeze-quench electron paramagnetic resonance studies establishing the kinetic competence of a substrate-based radical intermediate. Biochemistry 35, 11081–11084 [DOI] [PubMed] [Google Scholar]
- 64. Behshad E., Ruzicka F. J., Mansoorabadi S. O., Chen D., Reed G. H., Frey P. A. (2006) Enantiomeric free radicals and enzymatic control of stereochemistry in a radical mechanism: the case of lysine 2,3-aminomutases. Biochemistry 45, 12639–12646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ruzicka F. J., Frey P. A. (2007) Glutamate 2,3-aminomutase: a new member of the radical SAM superfamily of enzymes. Biochim. Biophys. Acta 1774, 286–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu W., Lieder K. W., Reed G. H., Frey P. A. (1995) Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase: a radical centered on the β-carbon of the alternative substrate, 4-thia-l-lysine. Biochemistry 34, 10532–10537 [DOI] [PubMed] [Google Scholar]
- 67. Miller J., Bandarian V., Reed G. H., Frey P. A. (2001) Inhibition of lysine 2,3-aminomutase by the alternative substrate 4-thialysine and characterization of the 4-thialysyl radical intermediate. Arch. Biochem. Biophys. 387, 281–288 [DOI] [PubMed] [Google Scholar]
- 68. Wu W., Booker S., Lieder K. W., Bandarian V., Reed G. H., Frey P. A. (2000) Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry 39, 9561–9570 [DOI] [PubMed] [Google Scholar]
- 69. Magnusson O. T., Reed G. H., Frey P. A. (2001) Characterization of an allylic analogue of the 5′-deoxyadenosyl radical: an intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 40, 7773–7782 [DOI] [PubMed] [Google Scholar]
- 70. Mansoorabadi S. O., Magnusson O. T., Poyner R. R., Frey P. A., Reed G. H. (2006) Analysis of the cob(II)alamin-5′-deoxy-3′,4′-anhydroadenosyl radical triplet spin system in the active site of diol dehydrase. Biochemistry 45, 14362–14370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Petrovich R. M., Ruzicka F. J., Reed G. H., Frey P. A. (1991) Metal cofactors of lysine-2,3-aminomutase. J. Biol. Chem. 266, 7656–7660 [PubMed] [Google Scholar]
- 72. Petrovich R. M., Ruzicka F. J., Reed G. H., Frey P. A. (1992) Characterization of iron-sulfur clusters in lysine 2,3-aminomutase by electron paramagnetic resonance spectroscopy. Biochemistry 31, 10774–10781 [DOI] [PubMed] [Google Scholar]
- 73. Cosper N. J., Booker S. J., Ruzicka F., Frey P. A., Scott R. A. (2000) Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry 39, 15668–15673 [DOI] [PubMed] [Google Scholar]
- 74. Chen D., Walsby C., Hoffman B. M., Frey P. A. (2003) Coordination and mechanism of reversible cleavage of S-adenosylmethionine by the 4Fe-4S] center in lysine 2,3-aminomutase. J. Am. Chem. Soc. 125, 11788–11789 [DOI] [PubMed] [Google Scholar]
- 75. Walsby C. J., Ortillo D., Broderick W. E., Broderick J. B., Hoffman B. M. (2002) An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 124, 11270–11271 [DOI] [PubMed] [Google Scholar]
- 76. Lepore B. W., Ruzicka F. J., Frey P. A., Ringe D. (2005) The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc. Natl. Acad. Sci. U.S.A. 102, 13819–13824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hinckley G. T., Frey P. A. (2006) Cofactor-dependence in reduction potentials for [4Fe-4S]2+/1+ in lysine 2,3-aminomutase. Biochemistry 45, 3219–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang S. C., Frey P. A. (2007) Binding energy in the one-electron reductive cleavage of S-adenosylmethionine in lysine 2,3-aminomutase, a radical SAM enzyme. Biochemistry 46, 12889–12895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Conradt H., Hohmann-Berger M., Hohmann H. P., Blaschkowski H. P., Knappe J. (1984) Pyruvate formate-lyase (inactive form) and pyruvate formate-lyase activating enzyme of Escherichia coli: isolation and structural properties. Arch. Biochem. Biophys. 228, 133–142 [DOI] [PubMed] [Google Scholar]
- 80. Broderick J. B., Henshaw T. F., Cheek J., Wojtuszewski K., Smith S. R., Trojan M. R, McGhan R. M., Kopf A., Kibbey M., Broderick W. E. (2000) Pyruvate formate-lyase-activating enzyme: strictly anaerobic isolation yields active enzyme containing a [3Fe-4S]+ cluster. Biochem. Biophys. Res. Commun. 269, 451–456 [DOI] [PubMed] [Google Scholar]
- 81. Reed K. E., Cronan J. E. (1993) Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J. Bacteriol. 175, 1325–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Miller J. R., Busby R. W., Jordan S. W., Cheek J., Henshaw T. F., Ashley G. W., Broderick J. B., Cronan J. E., Jr., Marletta M. A. (2000) Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry 39, 15166–15178 [DOI] [PubMed] [Google Scholar]
- 83. Sanyal I., Cohen G., Flint D. H. (1994) Biotin synthase: purification, characterization as a [2Fe-2S]cluster protein, and in vitro activity of the Escherichia coli bioB gene product. Biochemistry 33, 3625–3631 [DOI] [PubMed] [Google Scholar]
- 84. Ugulava N. B., Gibney B. R., Jarrett J. T. (2000) Iron-sulfur interconversions in biotin synthase: dissociation and reassociation of iron during conversion of [2Fe-2S] to [4Fe-4S] clusters. Biochemistry 39, 5206–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eliasson R., Fontecave M., Jörnvall H., Krook M., Pontis E., Reichard P. (1990) The anaerobic ribonucleotide reductase from Escherichia coli requires S-adenosylmethionine as a cofactor. Proc. Natl. Acad. Sci. U.S.A. 87, 3314–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Harder J., Eliasson R., Pontis E., Ballinger M. D., Reichard P. (1992) Activation of the anaerobic ribonucleotide reductase from Escherichia coli by S-adenosylmethionine. J. Biol. Chem. 267, 25548–25552 [PubMed] [Google Scholar]
- 87. Mulliez E., Fontecave M., Gaillard J., Reichard P. (1993) An iron-sulfur center and a free radical in the active anaerobic ribonucleotide reductase of Escherichia coli. J. Biol. Chem. 268, 2296–2299 [PubMed] [Google Scholar]
- 88. Sofia H. J., Chen G., Hetzler B. G., Reyes-Spindola J. F., Miller N. E. (2001) Radical SAM. A novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 29, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Frey P. A., Hegeman A. D., Ruzicka F. J. (2008) The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88 [DOI] [PubMed] [Google Scholar]
- 90. Florentin D., Bui B. T., Marquet A., Ohshiro T., Izumi Y. (1994) On the mechanism of biotin synthase of Bacillus sphaericus. C. R. Acad. Sci. III 317, 485–488 [PubMed] [Google Scholar]
- 91. Stich T. A., Myers W. K., Britt R. D. (2014) Paramagnetic intermediates generated by radical S-adenosylmethionine (SAM) enzymes. Acc. Chem. Res. 47, 2235–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Broderick J. B., Duffus B. R., Duschene K. S., Shepard E. M. (2014) Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang C. H., Frey P. A. (2000) Cloning, sequencing, heterologous expression, purification, and characterization of adenosylcobalamin-dependent d-lysine 5,6-aminomutase from Clostridium sticklandii. J. Biol. Chem. 275, 106–114 [DOI] [PubMed] [Google Scholar]
- 94. Tang K.-H., Mansoorabadi S. O., Reed G. H., Frey P. A. (2009) Radical triplets and suicide inhibition in reactions of 4-thia-d- and 4-thia-l-lysine with lysine 5,6-aminomutase. Biochemistry 48, 8151–8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Berkovitch F., Behshad E., Tang K.-H., Ennus E., Frey P. A., Drennan C. L. (2004) A locking mechanism preventing radical damage in the absence of substrate, as revealed by the x-ray structure of lysine 5,6-aminomutase. Proc. Natl. Acad. Sci. U.S.A. 101, 15870–15875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bruice T. C., Fife T. H., Bruno J. J., Brandon N. E. (1962) Hydroxyl group catalysis. II. The reactivity of the hydroxyl group of serine. The nucleophilicity of alcohols and the ease of hydrolysis of their acetyl esters as related to their pKa. Biochemistry 1, 7–12 [DOI] [PubMed] [Google Scholar]
- 97. Cleland W. W. (1992) Low-barrier hydrogen bonds and low fractionation factor bases in enzymatic reactions. Biochemistry 31, 317–319 [DOI] [PubMed] [Google Scholar]
- 98. Gerlt J. A., Gassman P. G. (1993) Understanding the rates of certain enzymatic reactions: proton abstraction from carbon acids, acyl-transfer reactions, and displacement reactions of phosphodiesters. Biochemistry 32, 11943–11952 [DOI] [PubMed] [Google Scholar]
- 99. Robillard G., Shulman R. G. (1972) High resolution nuclear magnetic resonance study of the histidine-aspartate hydrogen bond in chymotrypsin and chymotrypsinogen. J. Mol. Biol. 71, 507–511 [DOI] [PubMed] [Google Scholar]
- 100. Markley J. L. (1978) Hydrogen bonds in serine proteinases and their complexes with protein proteinase inhibitors. Proton nuclear magnetic resonance studies. Biochemistry 17, 4648–4656 [DOI] [PubMed] [Google Scholar]
- 101. Imperiali B., Abeles R. H. (1986) Inhibition of serine proteases by peptidyl fluoromethyl ketones. Biochemistry 25, 3760–3767 [DOI] [PubMed] [Google Scholar]
- 102. Brady K., Abeles R. H. (1990) Inhibition of chymotrypsin by peptidyl trifluoromethyl ketones: determinants of slow-binding kinetics, Biochemistry 29, 7608–7617 [DOI] [PubMed] [Google Scholar]
- 103. Liang T. C., Abeles R. H. (1987) Complex of α-chymotrypsin and N-acetyl-l-leucyl-l-phenylalanyl trifluoromethyl ketone: structural studies with NMR spectroscopy. Biochemistry 26, 7603–7608 [DOI] [PubMed] [Google Scholar]
- 104. Frey P. A., Whitt S. A., Tobin J. B. (1994) A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 264, 1927–1930 [DOI] [PubMed] [Google Scholar]
- 105. Cleland W. W., Kreevoy M. M. (1994) Low-barrier hydrogen bonds and enzymic catalysis. Science 264, 1887–1890 [DOI] [PubMed] [Google Scholar]
- 106. Henderson R. (1971) Catalytic activity of α-chymotrypsin in which histidine-57 has been methylated. Biochem. J. 124, 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hibbert F., Emsley J. (1991) Hydrogen bonding and chemical reactivity. Adv. Phys. Org. Chem. 26, 255–379 [Google Scholar]
- 108. Cassidy C. S., Lin J., Frey P. A. (1997) A new concept for the mechanism of action of chymotrypsin: the role of the low-barrier hydrogen bond. Biochemistry 36, 4576–4584 [DOI] [PubMed] [Google Scholar]
- 109. Lin J., Cassidy C. S., Frey P. A. (1998) Correlations of the basicity of His 57 with transition state analogue binding, substrate reactivity, and the strength of the low-barrier hydrogen bond in chymotrypsin. Biochemistry 37, 11940–11948 [DOI] [PubMed] [Google Scholar]
- 110. Lin J., Westler W. M., Cleland W. W., Markley J. L., Frey P. A. (1998) Fractionation factors and activation energies for exchange of the low barrier hydrogen bonding proton in peptidyl trifluoromethyl ketone complexes of chymotrypsin. Proc. Natl. Acad. Sci. U.S.A. 95, 14664–14668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cassidy C. S., Lin J., Frey P. A. (2000) The deuterium isotope effect on the NMR signal of the low-barrier hydrogen bond in a transition-state analog complex of chymotrypsin. Biochem. Biophys. Res. Commun. 273, 789–792 [DOI] [PubMed] [Google Scholar]
- 112. Westler W. M., Frey P. A., Lin J., Wemmer D. E., Morimoto H., Williams P. G., Markley J. L. (2002) Evidence for a strong hydrogen bond in the catalytic dyad of transition-state analogue inhibitor complexes of chymotrypsin from proton-Triton NMR isotope shifts. J. Am. Chem. Soc. 124, 4196–4197 [DOI] [PubMed] [Google Scholar]
- 113. Halkides C. J., Wu Y. Q., Murray C. J. (1996) A low-barrier hydrogen bond in subtilisin: 1H and 15N NMR studies with peptidyl trifluoromethyl ketones. Biochemistry 35, 15941–15948 [DOI] [PubMed] [Google Scholar]
- 114. Brady K., Wei A. Z., Ringe D., Abeles R. H. (1990) Structure of chymotrypsin-trifluoromethyl ketone inhibitor complexes: comparison of slowly and rapidly equilibrating inhibitors. Biochemistry 29, 7600–7607 [DOI] [PubMed] [Google Scholar]
- 115. Kuhn P., Knapp M., Soltis S. M., Ganshaw G., Thoene M., Bott R. (1998) The 0.78 Å structure of a serine protease: Bacillus lentus subtilisin. Biochemistry, 37, 13446–13452 [DOI] [PubMed] [Google Scholar]
- 116. Tobin J. B., Whitt S. A., Cassidy C. S., Frey P. A. (1995) Low-barrier hydrogen bonding in molecular complexes analogous to histidine and aspartate in the catalytic triad of serine proteases. Biochemistry 34, 6919–6924 [DOI] [PubMed] [Google Scholar]
- 117. Cloninger M. J., Frey P. A. (1998) Steric enhancement of imidazole basicity in cis-urocanic acid derivatives: models for the action of chymotrypsin. Bioorg. Chem. 26, 323–333 [Google Scholar]
- 118. Frey P. A., Cleland W. W. (1998) Are there strong hydrogen bonds in aqueous solutions? Bioorg. Chem. 26, 175–192 [Google Scholar]
- 119. Jeffrey G. A. (1997) An Introduction to Hydrogen Bonding, p. 41, Oxford University Press, New York [Google Scholar]
- 120. McAllister M. A. (1997) Characterization of low-barrier hydrogen bonds. 3. Hydrogen maleate. An ab initio and DFT investigation. Can. J. Chem. 75, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 121. Cassidy C. S., Lin J., Tobin J. B., Frey P. A. (1998) Low barrier hydrogen bonding in aqueous and aprotic solutions of dicarboxylic acids: spectroscopic characterization. Bioorg. Chem. 26, 213–219 [Google Scholar]
- 122. Lin J., Frey P. A. (2000) Strong hydrogen bonds in aqueous and aqueous-acetone solutions of dicarboxylic acids: activation energies for exchange and deuterium fractionation factors. J. Am. Chem. Soc. 122, 11258–11259 [Google Scholar]
- 123. Graham J. D., Buytendyk A. M., Wang D., Bowen K. H., Collins K. D. (2014) Strong, low-barrier hydrogen bonds may be available to enzymes. Biochemistry 53, 344–349 [DOI] [PubMed] [Google Scholar]