

Background: Phosphorylation of Sae2 by Mec1/Tel1 in S. cerevisiae has been shown, but its function was poorly understood.
Results: The conserved threonines of Sae2 have a redundant role in DNA damage response, and their phosphorylation directly interacts with Rad53, Dun1, and Xrs2 via their FHA domains.
Conclusion: Phosphorylation of Sae2 regulates its DNA repair function.
Significance: This work identifies the associated proteins of phosphorylated Sae2.
Keywords: Checkpoint Control, DNA Damage Response, DNA Repair, Protein Phosphorylation, Protein-Protein Interaction, Dun1, FHA Domains, Rad53, Sae2, Xrs2
Abstract
Saccharomyces cerevisiae Sae2 and its ortholog CtIP in higher eukaryotes have a conserved role in the initial processing of DNA lesions and influencing their subsequent repair pathways. Sae2 is phosphorylated by the ATR/ATM family kinases Mec1 and Tel1 in response to DNA damage. Among the Mec1/Tel1 consensus phosphorylation sites of Sae2, we found that mutations of Thr-90 and Thr-279 of Sae2 into alanine caused a persistent Rad53 activation in response to a transient DNA damage, similar to the loss of Sae2. To gain insight into the function of this phosphorylation of Sae2, we performed a quantitative proteomics analysis to identify its associated proteins. We found that phosphorylation of Thr-90 of Sae2 mediates its interaction with Rad53, Dun1, Xrs2, Dma1, and Dma2, whereas Rad53 and Dun1 additionally interact with phosphorylated Thr-279 of Sae2. Mutations of the ligand-binding residues of Forkhead-associated (FHA) domains of Rad53, Dun1, Xrs2, Dma1, and Dma2 abolished their interactions with Sae2, revealing the involvement of FHA-specific interactions. Mutations of Thr-90 and Thr-279 of Sae2 caused a synergistic defect when combined with sgs1Δ and exo1Δ and elevated gross chromosomal rearrangements. Likewise, mutations of RAD53 and DUN1 caused a synthetic growth defect with sgs1Δ and elevated gross chromosomal rearrangements. These findings suggest that threonine-specific phosphorylation of Sae2 by Mec1 and Tel1 contributes to DNA repair and genome maintenance via its interactions with Rad53 and Dun1.
Introduction
DNA double-stranded breaks (DSBs)3 are one of the most deleterious forms of DNA damage. If left unrepaired, DSBs can lead to aberrant chromosomal rearrangements and cell death (1). Two major pathways are known to be involved in DNA DSB repair, including nonhomologous end joining and homologous recombination (2). A critical step in the choice of these DNA DSB repair pathways is the nucleolytic processing of DSBs. Several enzymes have been identified to catalyze this nucleolytic processing in Saccharomyces cerevisiae. The Mre11-Rad50-Xrs2 complex and Sae2 act at the initial steps of DSB recognition and processing, followed by an extensive resection by the nucleases Dna2 and Exo1 together with the Sgs1-Top3-Rmi1 complex (3, 4). DNA DSB processing is tightly coupled to the DNA damage checkpoint, which becomes activated to halt the cell cycle, thus allowing time for DNA repair. Tel1 binds to Xrs2 of the Mre11-Rad50-Xrs2 complex and is thus recruited to DNA DSBs (5). Following the processing of DSBs into ssDNA, replication protein-A binds to ssDNA and recruits Mec1 (6), which has a major role in DNA damage checkpoint activation. Once recruited to the site of DNA damage, Mec1 and Tel1 phosphorylate many proteins at the sites of DNA damage, including replication protein-A, Mre11-Rad50-Xrs2, and Sae2 (7–9). Mutations of MEC1 and TEL1 are known to cause substantial increases in gross chromosomal rearrangements (GCRs) (10–12). Although telomere fusion is thought to contribute to chromosomal rearrangements observed in the mec1Δ tel1Δ mutant, loss of telomerase alone does not cause similar chromosomal rearrangements (11, 12), indicating that defective DNA repair might also be involved. Despite the fact that many substrates of Mec1 and Tel1 have been identified, a major challenge has been to identify and characterize those Mec1/Tel1 substrates whose phosphorylation specifically regulates DNA repair and genome maintenance.
Of particular interest here is Sae2, which is phosphorylated by Mec1 and Tel1 in response to DNA damage (9, 13). Mutation of five SQ/TQ sites of Sae2, sae22,5,6,8,9, which conform to the consensus phosphorylation motif of Mec1 and Tel1, eliminated the bulk of its DNA damage-induced phosphorylation and caused persistent Rad53 activation in response to transient DNA damage treatment similar to the deletion of SAE2 (9, 13). Furthermore, these sae2 mutations caused a defect in suppression of chromosomal translocation mediated by nonhomologous end joining (14). Interestingly, mammalian CtIP, ortholog of Sae2, is phosphorylated by ATR and ATM (15–17), which are orthologs of Mec1 and Tel1, respectively. In particular, phosphorylation of Thr-859 on human CtIP by ATR/ATM was shown to have a role in homologous recombination (17). Moreover, T859A mutation of CtIP caused a reduced replication protein-A focus formation and loss of viability following camptothecin (CPT) treatment. Thr-859 of human CtIP is conserved in Xenopus, and a corresponding phosphorylation of Thr-818 on Xenopus CtIP was shown to regulate CtIP association with chromatin (16). Finally, Thr-279 of Sae2, which conforms to a Mec1/Tel1 consensus phosphorylation site, corresponds to Thr-859 of human CtIP and Thr-818 of Xenopus CtIP. These findings suggested that phosphorylation of this conserved threonine of Sae2/CtIP by ATR/ATM family kinases probably has an important function in DNA repair, although the specific function of Thr-279 of Sae2 has not yet been determined.
Sae2 is also phosphorylated by cyclin-dependent kinase (CDK) (18). It was shown that phosphorylation of Sae2 on Ser-267 is required for cells to confer resistance to DNA-damaging agents, and the S267A mutation impaired resection of an irreparable DSB induced by HO endonuclease. A recent study showed that phosphorylation of Sae2 by CDK and Mec1/Tel1 appeared to alter its oligomeric state by converting Sae2 into a monomeric and active state (19). However, Ctp1, the ortholog of Sae2 in Schizosaccharomyces pombe, lacks such CDK phosphorylation. Instead, Ctp1 is phosphorylated by casein kinase (20), which was shown to mediate an interaction between Ctp1 and the Forkhead-associated (FHA) domain of Nbs1, the S. pombe ortholog of S. cerevisiae Xrs2. On the other hand, several putative CDK sites on human CtIP were shown to facilitate the interaction between CtIP and Nbs1 (17), suggesting that different kinases phosphorylate Ctp1 and CtIP to promote their association with Nbs1 in different organisms. A possible interaction between Sae2 and Xrs2 in S. cerevisiae has not been identified. It is also unclear whether phosphorylation of Sae2 by CDK, casein kinase, or possibly other kinases helps to regulate the interaction between Sae2 and Xrs2.
Here we characterized phosphorylation of Sae2 further. Through mutagenesis analysis of Mec1 and Tel1 phosphorylation sites of Sae2, we identified two conserved threonine residues, Thr-90 and Thr-279 of Sae2, to have a redundant role for its function in the DNA damage response. We further applied quantitative mass spectrometry (MS) to identify Sae2-associated proteins, which are mediated by phosphorylation of Thr-90 and Thr-279, and then examined the role of this phosphorylation of Sae2 in DNA repair and maintenance of genome integrity.
EXPERIMENTAL PROCEDURES
Plasmids and Yeast Genetic Methods
SAE2 plus 200 base pairs of upstream sequence was cloned into a pFA6a plasmid using PacI and AscI sites (21). Sae2 and Rad9 were tagged in the C terminus with a His6-3× HA (3HA) epitope from pFA6a. FHA domains of Dma1, Dma2, Rad53-FHA1, Rad53-FHA2, and Dun1 were cloned into pGEX-4T1 plasmid such that an N-terminal GST tag was fused to each FHA domain. The Xrs2-FHA domain was cloned into C-terminal His6-FLAG-TEV-Protein A (TAF) in pET21a plasmid (22). All point mutations were introduced by site-directed mutagenesis and confirmed by DNA sequencing.
The yeast strains used are isogenic with W303 or S288c, as shown in Table 1. Standard yeast genetic methods were used to introduce mutations into chromosomal loci, which were confirmed by DNA sequencing. For plate sensitivity, cells were grown to late log phase and normalized before serial dilution and spotting onto YPD and drug plates with the indicated concentrations. Cells were grown at 30 °C and imaged after 2–3 days unless otherwise noted. For the dose-dependent killing curve, cells in log phase were split and treated with indicated doses of methyl methanesulfonate (MMS) for 1 h. Cells were added to an equal volume of 10% sodium thiosulfate to quench the MMS before serial dilution and plating onto YPD. Cells were incubated for 3 days at 30 °C. The percentage viability was calculated by dividing the number of viable colonies for each strain after the MMS treatment compared with no treatment, and an average of three independent experiments was used. CPT sensitivity as measured by liquid culture was performed as described previously (23, 24). Briefly, overnight cultures were diluted to YPD containing 2% DMSO to grow for 5 h in log phase. For slow growing cultures, this pregrowth was done overnight. Cultures were then diluted to an A600 of 0.001 in YPD with 2% DMSO containing camptothecin at varying concentrations of 0 (untreated), 1, 5, 10, and 20 μm. Cultures were grown until the untreated control had reached 10 doublings before subsequent treated cultures were measured. The slope of the plot ln(A/A0) was calculated, and the ratio of SlopeC (treated) versus SlopeC = 0 (untreated) from averages of three independent experiments is shown in the graphs. Fluctuation analysis used in analyzing GCRs was described previously (25).
TABLE 1.
Yeast strains used in this study
All W303 strains were isogenic to HZY1077 and HZY1078. Background for all S288c strains was ura3-52 leu2Δ1 his3Δ200 lys2ΔBgl hom3-10 ade2Δ1 ade8.
| Strain | Genotype | Background | Reference/Source |
|---|---|---|---|
| HZY1077 | MATa ade2-1 can1-100 his3-1115 leu2-3,112 trp1-1 ura3-1 RAD5+ | W303 | Xiaolan Zhao |
| HZY1078 | MATα ade2-1 can1-100 his3-1115 leu2-3,112 trp1-1 ura3-1 RAD5+ | W303 | Xiaolan Zhao |
| HZY1239 | MATα sae2Δ::HIS3 | W303 | This study |
| HZY1240 | MATα sae2-T90A::kanMX6 | W303 | This study |
| HZY1241 | MATα sae2-S249A::kanMX6 | W303 | This study |
| HZY1242 | MATα sae2-T279A::kanMX6 | W303 | This study |
| HZY1243 | MATα sae2-T90A,S249A::kanMX6 | W303 | This study |
| HZY1244 | MATα sae2-T90A,T279A::kanMX6 | W303 | This study |
| HZY1235 | MATα sae2-S249A,T279A::kanMX6 | W303 | This study |
| HZY1236 | MATα sae2-T90A,S249A,T279A::kanMX6 | W303 | This study |
| JLY030 | MATa SAE2-3HA::HIS3MX6 bar1Δ::URA3 | W303 | This study |
| JLY032 | MATα sae2-T90A,T279A-3HA::kanMX6::HIS3MX6 | W303 | This study |
| JLY035 | MATα sae2-T90A,S249A,T279A-3HA::kanMX6::HIS3MX6 | W303 | This study |
| JLY048 | MATα sae2-T90A-3HA:: kanMX6::HIS3MX6 | W303 | This study |
| JLY050 | MATα sae2-S249A-3HA::kanMX6::HIS3MX6 | W303 | This study |
| JLY053 | MATα sae2-T279A-3HA::kanMX6::HIS3MX6 | W303 | This study |
| SCY249 | MATa sml1Δ::TRP1arg4Δ | S288c | Chen et al. (2010) |
| JLY089 | MATα RAD9-3HA::kanMX6 | W303 | This study |
| JLY091 | MATα RAD9-3HA::kanMX6 sae2Δ::HIS3 | W303 | This study |
| JLY093 | MATα RAD9-3HA::HIS3MX6 sae2-2AQ(T90A,T279A)::kanMX6 | W303 | This study |
| JLY158 | MATα mrc1Δ::URA3 | W303 | This study |
| JLY160 | MATα rad9Δ::URA3 | W303 | This study |
| JLY161 | MATα sae2-2AQ::kanMX6 mrc1Δ::URA3 | W303 | This study |
| JLY162 | MATα sae2-2AQ::kanMX6 mrc1Δ::URA3 | W303 | This study |
| JLY163 | MATα sae2-2AQ::kanMX6 rad9Δ::URA3 | W303 | This study |
| JLY164 | MATα sae2-2AQ::kanMX6 rad9Δ::URA3 | W303 | This study |
| JLY230 | MATa exo1Δ::HIS3 | W303 | This study |
| JLY235 | MATα sgs1Δ::URA3 | W303 | This study |
| JLY233 | MATα sae2-2AQ::kanMX6 sgs1Δ::URA3 | W303 | This study |
| JLY234 | MATa sae2-2AQ::kanMX6 exo1Δ::HIS3 | W303 | This study |
| JLY221 | MATa dun1Δ::HIS3 | W303 | This study |
| JLY223 | MATα rad53-R70A,N107A::kanMX6 | W303 | This study |
| JLY641 | MATa rad53-R70A,N107A::kanMX6 dun1Δ::HIS3 | W303 | This study |
| JLY635 | MATa sae2-2AQ(T90A,T279A)::URA3 dun1Δ::HIS3 | W303 | This study |
| JLY637 | MATα sae2-2AQ(T90A,T279A)::URA3 rad53-R70A,N107A::kanMX6 | W303 | This study |
| JLY639 | MATa sae2-2AQ(T90A,T279A)::URA3 dun1Δ::HIS3 rad53-R70A,N107A::kanMX6 | W303 | This study |
| JLY391 | MATα xrs2-R32G,S47A::kanMX6 | W303 | This study |
| JLY335 | MATα xrs2-R32G,S47A::kanMX6 sgs1Δ::URA3 | W303 | This study |
| JLY395 | MATα xrs2-R32G,S47A::kanMX6 exo1Δ::natMX4 | W303 | This study |
| JLY229 | MATa/MATα sae2-2AQ::kanMX6/SAE2 sgs1Δ::URA3/SGS1 exo1Δ::HIS3/EXO1 | W303 | This study |
| JLY540 | MATa/MATα sae2-T90A::kanMX6/SAE2 sgs1Δ::URA3/SGS1 exo1Δ::natMX4/EXO1 | W303 | This study |
| JLY541 | MATa/MATα sae2-T279A::kanMX6/SAE2 sgs1Δ::URA3/SGS1 exo1Δ::natMX4/EXO1 | W303 | This study |
| JLY171 | MATa/MATα rad53-R70A,N107A:: kanMX6/RAD53 dun1Δ::HIS3/DUN1 sgs1Δ::URA3/SGS1 | W303 | This study |
| JLY496 | MATa/MATα dun1Δ::HIS3/DUN1 sgs1Δ::URA3/SGS1 exo1Δ::natMX4/EXO1 | W303 | This study |
| JLY497 | MATa/MATα rad53-R70A,N107A::kanMX6/RAD53 sgs1Δ::URA3/SGS1 exo1Δ::natMX4/EXO1 | W303 | This study |
| HZY2672 | MATα sae2-2AQ::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2709 | MATα sae2-2AQ::natMX4 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2801 | MATa sae2-2AQ::natMX4 sgs1Δ::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2802 | MATα sae2-2AQ::natMX4 sgs1Δ::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3041 | MATa sae2-2AQ::kanMX6 exo1Δ::natMX4 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3042 | MATα sae2-2AQ::kanMX6 exo1Δ::natMX4 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2908 | MATa rad53-R70A::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2909 | MATα rad53-R70A::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2912 | MATa rad53-R70A::kanMX6 dun1Δ::HIS3 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY2913 | MATα rad53-R70A::kanMX6 dun1Δ::HIS3 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3377 | MATa rad53Δ::HIS3 sml1Δ::TRP1 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3378 | MATα rad53Δ::HIS3 sml1Δ::TRP1 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3192 | MATα exo1Δ::natMX4 sgs1Δ::TRP1 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| HZY3193 | MATα exo1Δ::natMX4 sgs1Δ::TRP1 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| JLY410 | MATa sae2-2AQ::natMX4 xrs2-R32G::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| JLY411 | MATα sae2-2AQ::natMX4 xrs2-R32G::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| JLY412 | MATa xrs2-R32G::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
| JLY413 | MATα xrs2-R32G::kanMX6 can1::hisG yel072w::CAN1/URA3 iYEL072W::hph | S288c | This study |
Biochemical Methods
For Western blot analysis, protein extracts were prepared using a trichloroacetic acid (TCA) method. To monitor Rad53 phosphorylation, we used a mouse monoclonal α-Rad53, EL7E1 serum (Dr. Marco Foiani). To detect Sae2-3HA and Rad9-3HA, we used the 3F10 antibody (α-HA; Roche Applied Science). For pull-down experiments, various Sae2-3HA cells were treated with 25 μg/ml phleomycin for 1 h. Native protein extracts from these cells were prepared using a glass bead method described previously (26). FHA-domain resins of Xrs2, Rad53-FHA1, and Dun1 were incubated with protein extracts derived from Sae2-3HA strain in TBS-N buffer (50 mm Tris-HCl, pH 7.4, 0.5% Nonidet P-40, 150 mm NaCl) for 2 h at 4 °C and washed several times. FHA-binding proteins were eluted by boiling in 1% SDS or with cleavage by tobacco etch virus protease. To dephosphorylate Sae2-3HA in cell extract, we incubated Sae2-3HA cell extract with 4000 units of λ-phosphatase for 1 h at 30 ºC in the presence of 2 mm MnCl2 without EDTA and β-glycerophosphate. To preserve phosphorylation of Sae2, 5 mm EDTA and 5 mm β-glycerophosphate were included to inhibit cellular phosphatases during the preparation of Sae2-3HA cell extract, which was not treated by λ-phosphatase. Equal amounts of both cell extracts (with and without λ-phosphatase treatment) were then used for an FHA domain pull-down experiment. To prepare unphosphorylated peptide of Sae2, Thr(P)-90- and Thr(P)-279-containing phosphopeptides of Sae2 bound to neutravidin resins were dephosphorylated by 800 units of λ-phosphatase before they were used for binding experiments with various FHA domains.
To prepare phosphopeptide resins, Thr(P)-90- and Thr(P)-279-containing phosphopeptides of Sae2 (Chi Scientific) with an N-terminal biotin were first immobilized on neutravidin resins (Thermo Scientific). Phosphopeptide resins or blank resins were incubated with native protein extracts, which were pretreated with blank neutravidin resins to deplete endogenous biotinylated proteins. The native cell lysates were prepared using a glass bead method, and cells were grown using the SILAC (stable isotope labeling via amino acid in culture) method (27). After incubation, the phosphopeptide resins and blank resins were washed by the same TBS-N binding buffer. Phosphopeptide-binding proteins were eluted by incubation with 8 m urea at 37 °C for 30 min. Eluted proteins were reduced by 10 mm DTT and alkylated with 30 mm iodoacetamide before dilution and proteolysis with trypsin (Promega).
Methods Used in Quantitative Mass Spectrometry Analyses
Trypsin-digested peptides were desalted using a Sep-Pac C18 cartridge and then resuspended in 80% acetonitrile, 20% water to be fractionated on a TSKgel Amide-80 (1.0-mm inner diameter) column (TOSOH Bioscience) (28). A total of 14 fractions were collected for LC-MS/MS analysis using an Orbitrap-LTQ mass spectrometer. MS data were searched using Sorcerer-SEQUEST as described previously (26). The identified peptides were quantified using XPRESS. In cases where peptides in the mock sample were not identified, a minimal ion intensity of 1.0E4 was used to calculate the abundance ratio. Each protein was identified and quantified with at least three unique peptides. The average of their abundance ratios was used to calculate the abundance ratio for each protein for both replicate experiments.
RESULTS
Mutation of Both Thr-90 and Thr-279 of Sae2 Causes an Elevated Sensitivity to Genotoxic Agents
Sae2 has five serine and threonine residues that follow the consensus phosphorylation motif, (S/T)Q, of Mec1 and Tel1 (9). To identify the specific (S/T)Q sites of Sae2 important for its function, we performed a sequence alignment of Sae2 orthologs in fungi and human and found that Thr-90 and Thr-279 of Sae2 are conserved among fungi (Fig. 1A). In particular, Thr-279 is located in a conserved C-terminal region of Sae2, which is also present in CtIP. To study their functions, we changed Thr-90, Ser-249, and Thr-279 of Sae2 into alanine and integrated these mutations in the SAE2 chromosomal locus. First, we analyzed the effect of these sae2 mutations on the electrophoretic mobility shift of Sae2 using Sae2-3HA strain. Treatment of Sae2-3HA strain by either phleomycin or CPT caused an accumulation of slower migrating bands, representing phosphorylated species of Sae2. As shown in Fig. 1B, sae2-T90A mutation partially reduced the slowest migrating species of Sae2 caused by phleomycin treatment, whereas sae2-T279A mutation caused an accumulation of more slower migrating species of Sae2. The sae2-2AQ mutation, containing both T90A and T279A, caused an intermediate effect on the electrophoretic mobility shift of Sae2. On the other hand, sae2-S249A mutation caused a reduction of slower migrating species of Sae2, indicating that Ser-249 is a major DNA damage-induced phosphorylation site. The sae2-3AQ mutation, containing S249A, T90A, and T279A, eliminated the bulk of slower migrating bands of Sae2. Thus, Ser-249, Thr-90, and Thr-279 are the major phleomycin-induced phosphorylation sites of Sae2 (Fig. 1B, bottom, lane 7). CPT-induced Sae2 electrophoretic shift (Fig. 1B, top) appears to be somewhat less pronounced compared with phleomycin treatment, but the overall changes are similar.
FIGURE 1.

Thr-90 and Thr-279 of Sae2 are specifically involved in its function in the DNA damage response. A, sequence alignment of Sae2 orthologues revealed that Thr-90 and Thr-279 of Sae2 are conserved among various fungi, whereas Thr-279 is also conserved in human CtIP as Thr-859. B, cell lysates derived from an untagged and the Sae2-3HA strain with various sae2 (2AQ-T90A,T279A; 3AQ-T90A,T279A,S249A) mutants treated by 25 μg/ml phleomycin or 20 μm CPT for 1 h and analyzed by anti-HA immunoblotting. Ponceau staining shows equal loading. *, DNA damage-induced gel bands of Sae2-3HA. Strains used were HZY1078, JLY030, JLY032, JLY035, JLY048, JLY050, and JLY053. C, a 10-fold serial dilution of wild-type strain and various sae2 phosphorylation-defective mutants were plated on YPD plates containing no drug, 0.02% MMS, DMSO, 30 μm CPT, or 3 μg/ml phleomycin for 3 days at 30 °C. Strains used were HZY1078, HZY1235, HZY1236, HZY1239, HZY1240, HZY1241, HZY1242, HZY1243, and HZY1244. D, camptothecin sensitivity of wild-type strain (open circle; HZY1077/HZY1078), the sae2Δ mutant (open square; HZY1239), and sae2-2AQ mutant (open triangle; HZY1244) were examined in liquid culture at various concentrations over 10 doubling times. The ratio of the slope of the curve between treated (SlopeC) and untreated (SlopeC = 0) is plotted for each concentration (see “Experimental Procedures”). The average result of three independent experiments is shown. E, dose-dependent viability curve of sae2 was analyzed using the same strains as in D. Cells were treated for 1 h at the indicated MMS concentrations and then plated on YPD plates to measure the number of viable cells. The average result of three independent experiments is shown. Error bars, S.D.
We next examined growth of these sae2 mutants on plates containing MMS, CPT, and phleomycin. Compared with a wild-type strain, the sae2-T90A, sae2-S249A, and sae2-T279A mutants did not show appreciable elevated sensitivity to these drugs (Fig. 1C). Interestingly, sae2-T90A,T279A (sae2-2AQ) mutant showed a significantly elevated sensitivity to MMS, CPT, and, to a lesser extent, phleomycin (Fig. 1C), albeit not to the same extent as sae2Δ mutant. Although the sae2-S249A mutation removes the majority of Sae2 electrophoretic shift (Fig. 1B), it does not alter MMS, CPT, or phleomycin sensitivity of the sae2-T90A, sae2-T279A, or sae2-2AQ mutant. Thus, Thr-90 and Thr-279 of Sae2 redundantly confer resistance to chronic treatments of MMS and CPT, whereas phosphorylation of Ser-249 of Sae2 does not contribute to this function of Sae2. To further examine CPT sensitivity of sae2-2AQ and sae2Δ mutants, we examined their growth in liquid culture in the presence of varying concentrations of CPT using a previously published method (23). We found that the sae2-2AQ mutant is less sensitive than the sae2Δ mutant at lower CPT concentrations (Fig. 1D). However, at a higher concentration, such as 20 μm CPT, the sae2-2AQ mutant grew similarly to the sae2Δ mutant (Fig. 1D). The elevated sensitivity of the sae2-2AQ mutant to chronic MMS and CPT treatments could be due to a cell cycle arrest, loss of viability, or both. To test for a loss of viability, wild-type strain and the sae2-2AQ and sae2Δ mutants were treated with increasing amounts of MMS for 1 h and quantified for surviving colonies. MMS treatment caused a dosage-dependent and increased loss of viability of the sae2-2AQ mutant, although not to the same extent as the sae2Δ mutant (Fig. 1E).
MMS Treatment Caused Persistent Rad53 Activation in sae2-2AQ Mutant
Previous studies showed that mutations of all five Mec1/Tel1 consensus phosphorylation sites of Sae2 caused persistent Rad53 phosphorylation in response to DNA damage (9, 13). Here we investigated whether Thr-90 and Thr-279 of Sae2 are specifically involved. Following MMS treatment, Rad53 became hyperphosphorylated, as was evident by its slower electrophoretic mobility (Fig. 2A). Rapid Rad53 dephosphorylation ensued within 30 min after wild-type cells were released into fresh media, and it was essentially complete within 2 h. As expected, Rad53 in the sae2Δ mutant remained phosphorylated for up to 2 h following its release into fresh media (Fig. 2A). Interestingly, Rad53 remained phosphorylated for up to 2 h after being released in fresh media in the sae2-2AQ mutant, whereas sae2-T90A and sae2-T279A mutations did not appreciably alter the kinetics of Rad53 dephosphorylation compared with wild-type strain (Fig. 2A). Thus, Thr-90 and Thr-279 of Sae2 have a redundant role in allowing Rad53 dephosphorylation to occur during recovery from a transient MMS treatment.
FIGURE 2.

Persistent Rad53 phosphorylated in the sae2-2AQ mutant following a transient DNA damage treatment. A, DNA damage checkpoint inactivation was examined using anti-Rad53 monoclonal antibody in wild-type and sae2 mutants. Cells were treated for 1 h with 0.01% MMS and subsequently resuspended into fresh YPD medium. Cells were harvested at the indicated time points to examine Rad53 phosphorylation. Representative loading control was by Ponceau stain. Strains used were HZY1077, HZY1239, HZY1240, HZY1242, and HZY1244. B, inactivation of Rad53 is correlated with dephosphorylation of HA-tagged RAD9. Rad9 phosphorylation was monitored at the indicated time points similarly as in A with wild-type, sae2-2AQ, and sae2Δ cells. Strains used were JLY089, JLY091, and JLY093. C and D, effect of combining the rad9Δ or mrc1Δ with sae2-2AQ mutations in causing DNA damage sensitivity. C, camptothecin sensitivity at various concentrations. The ratio of the slope of the curve between treated (SlopeC) and untreated (SlopeC = 0) is plotted for each concentration (see “Experimental Procedures”). The average result of three independent experiments is shown with S.D. represented by error bars. D, hydroxyurea sensitivity. Strains used for C and D were HZY1077/HZY1078, HZY1244, JLY158, JLY160, JLY161, JLY162, JLY163, and JLY164.
Activation of Rad53 requires two adaptor proteins, Rad9 and Mrc1, which are phosphorylated by Mec1 and Tel1 and control Rad53 activation in both redundant and distinct manners, depending on the types of genotoxic stresses (29–31). In particular, Rad9 has a more important role for Rad53 activation in response to DNA damage. Like Rad53, Rad9 remains hyperphosphorylated in the sae2-2AQ and sae2Δ mutants following a transient MMS treatment (Fig. 2B), indicating that the defect of Rad53 dephosphorylation in the sae2-2AQ mutant occurs at an earlier step. One possibility is that sae2-2AQ mutant suffers from a DNA repair defect. To examine this further, we examined the effect of deleting RAD9 and MRC1 on the growth of sae2-2AQ mutant in the presence of CPT, which causes covalent DNA-protein adducts. Deleting RAD9 modestly improves growth of the sae2-2AQ mutant at a lower concentration of CPT (10 μm) but not at a higher concentration of CPT (20 μm) (Fig. 2C). This finding suggests that the accumulation of CPT-induced DNA lesion may require the function of Sae2 in DNA repair via its phosphorylation at Thr-90 and Thr-279, and cell growth cannot be fully rescued by the loss of Rad9 to reduce Rad53 activity. On the other hand, deleting MRC1 does not have an appreciable effect on the growth of sae2-2AQ mutant (Fig. 2D), indicating that Mrc1 does not contribute to the CPT sensitivity of sae2-2AQ mutant.
Deletion of MRC1 is known to cause defects in DNA replication and compromise Rad53 activation in response to hydroxyurea (HU) treatment (31, 32). Interestingly, the sae2-2AQ mrc1Δ double mutant showed significantly improved growth compared with the mrc1Δ mutant in the presence of HU (Fig. 2E), whereas deleting RAD9 had little effect on the HU sensitivity of the sae2-2AQ mutant. Considering that the sae2-2AQ mutant has an elevated Rad53 activity, whereas the mrc1Δ mutant has a defect in Rad53 activation in response to HU treatment (31, 32), we reason that the sae2-2AQ mutation would elevate Rad53 activation in the sae2-2AQ mrc1Δ double mutant, thus allowing it to better cope with HU treatment than mrc1Δ mutant (Fig. 2E). Taken together, these findings suggest that the sae2-2AQ mutant may suffer from both DNA repair and hyperactivated Rad53 defects, which cause different outcomes in combination with the loss of Rad9 and Mrc1, depending on the types of genotoxic agents used.
Genetic Interactions between SAE2, SGS1, and EXO1
Sgs1 and Exo1 are known to function in DNA DSB processing (3). To explore the role of Sae2 phosphorylation in DNA resection, we first examined effect of combining various sae2 mutations with sgs1Δ and exo1Δ mutations. Interestingly, we found that the sae2-2AQ sgs1Δ exo1Δ triple mutant is lethal (Fig. 3A), whereas the sae2-T90A sgs1Δ exo1Δ and sae2-T279A sgs1Δ exo1Δ triple mutants are viable (Fig. 3A). This finding again supports a redundant role of Thr-90 and Thr-279 of Sae2 for its DNA repair function. Although the sae2Δ sgs1Δ double mutant is known to be lethal (33, 34), the sae2-2AQ sgs1Δ double mutant is viable and lacks any obvious growth defect. However, the sae2-2AQ sgs1Δ double mutant did show a significantly elevated sensitivity to CPT compared with sae2-2AQ and sgs1Δ single mutants (Fig. 3B). To further study the role of Sae2 phosphorylation in genome maintenance, we used the yel072w::CAN1/URA3 assay, which measures GCRs formed by both segmental duplication and single-copy sequences (25). Compared with wild-type strain, there is a 4-fold increase in the rate of accumulating GCRs in the sae2-2AQ mutant (Fig. 3C and Table 2), similar to that in sae2Δ mutant and a phosphorylation-defective allele where all five putative Mec1/Tel1 phosphorylation sites of SAE2 are mutated (35). The rate of accumulating GCRs in the sae2-2AQ sgs1Δ double mutant is modestly higher than that in the sgs1Δ mutant.
FIGURE 3.

Genetic interactions between the sae2-2AQ mutation and mutations that affect DNA end resection, sgs1Δ and exo1Δ. A, tetrad dissection of diploids containing heterozygous sae2-2AQ, exo1Δ, and sgs1Δ mutations. Strains used were JLY229, JLY540, and JLY541. B, camptothecin sensitivity of sae2-2AQ and sgs1Δ mutations at various concentrations. The ratio of the slope of the curve between treated (SlopeC) and untreated (SlopeC = 0) is plotted for each concentration (see “Experimental Procedures”). The average result of three independent experiments is shown with S.D. represented by error bars. Strains used were HZY1077, HZY1244, JLY233, and JLY235. C, rates of accumulating GCRs in the sae2 and sgs1Δ mutants using the yel072w::CAN1/URA3 assay measured by fluctuation analysis (see Table 2). Strains used were HZY2672, HZY2709, HZY2801, and HZY2802. D, camptothecin sensitivity shown the same as in B except with sae2-2AQ and exo1Δ mutations. Strains used were HZY1077, HZY1244, JLY230, and JLY234. E, graph of the rate of accumulating GCRs shown the same as in C except with sae2 and exo1Δ mutants. Strains used were HZY2672, HZY2709, HZY3041, and HZY3042.
TABLE 2.
Rate of accumulating GCRs for mutations to SAE2, RAD53, DUN1, XRS2, EXO1, and SGS1
| Genotype | yel072w::CAN1/URA3 GCR ratea | Source/Reference |
|---|---|---|
| Wild typeb | 1.97 × 10−8 (8.7) | Putnam et al. (38) |
| sae2Δb | 1.65 × 10−7 (73) | Putnam et al. (38) |
| rad53Δ sml1Δ | 2.8 × 10−7 (123) | This study |
| rad53-R70A | 7.94 × 10−8 (35) | This study |
| dun1Δb | 1.61 × 10−7 (71) | Putnam et al. (38) |
| rad53-R70A dun1Δ | 1.2 × 10−7 (53) | This study |
| sae2-2AQ | 9.1 × 10−8 (40) | This study |
| sgs1Δb | 1.93 × 10−6 (850) | Putnam et al. (38) |
| sae2-2AQ sgs1Δ | 5.2 × 10−6 (2291) | This study |
| exo1Δb | 8.44 × 10−8 (37) | Putnam et al. (38) |
| sae2-2AQ exo1Δ | 9.4 × 10−8 (41) | This study |
| exo1Δsgs1Δ | 8.31 × 10−7 (41) | This study |
| xrs2-R32G | 2.14 × 10−8 (9.4) | This study |
| sae2-2AQ xrs2-R32G | 3.61 × 10−8 (16) | This study |
a Rate of accumulating Can 5-FOA progeny. The number in parenthesis is the -fold increase relative to wild-type yel068c::CAN1/URA3 strain (2.27 × 10−9).
b Rates taken from Putnam et al. (38).
Next, we examined possible genetic interactions between Sae2 and Exo1. The sae2-2AQ exo1Δ double mutant has a CPT sensitivity comparable with that of sae2-2AQ mutant (Fig. 3D). The rate of accumulating GCRs in the sae2-2AQ exo1Δ double mutant is comparable with that in the sae2-2AQ and exo1Δ mutants (Fig. 3E). These findings indicate either a lack of genetic interaction between these mutations or an epistasis relationship between them. Taken together, these findings reveal an essential role of Sae2 phosphorylation in cells lacking both Sgs1 and Exo1 for cell survival, and Sae2 phosphorylation is important in coping with CPT-induced DNA legions in cells lacking Sgs1 but not Exo1.
Quantitative MS Identified the Proteins Associated with Phosphorylated Sae2
A possible function of phosphorylated Thr-90 and Thr-279 of Sae2 is to mediate specific protein-protein interactions. To test this, we used biotinylated phosphopeptides of Sae2 containing either Thr(P)-90 or Thr(P)-279 that were immobilized on streptavidin resins to purify their associated proteins and then identified them using quantitative MS (Fig. 4A). Two separate experiments were performed to identify the associated proteins of Sae2 using Thr(P)-90-containing peptide. As shown in Fig. 4B, those proteins whose abundance was significantly enriched using Thr(P)-90-containing peptide relative to mock-purified sample are Xrs2, Rad53, Dun1, Dma1, and Dma2. The same approach was used to identify Thr(P)-279-associated proteins. Interestingly, only Rad53 and Dun1 were enriched by both Thr(P)-90- and Thr(P)-279-containing peptides of Sae2 (Fig. 4C), whereas Xrs2, Dma1, and Dma2 were only enriched by Thr(P)-90-containing peptide of Sae2. These MS findings are summarized in Fig. 4D.
FIGURE 4.

Phosphorylated Thr-90- and Thr-279-containing peptides of Sae2 interact with multiple FHA domain-containing proteins, including Rad53, Dun1, Xrs2, Dma1, and Dma2. A, experimental method to identify the associated proteins of Thr(P)-90- or Thr(P)-279-containing phosphopeptide of Sae2 by SILAC-based quantitative MS. B, relative abundance of proteins enriched by Thr(P)-90 phosphopeptide resins compared with blank resins in two independent experiments. The abundance ratio of each protein was calculated using minimally three unique peptides per protein and shown in a log2 scale. The proteins clustered in the top right area were greatly enriched in both replicate experiments and thus considered as candidate binding proteins. C, same as in B except comparing the relative abundance of proteins enriched by Thr(P)-90 phosphopeptide resins or Thr(P)-279 phosphopeptide resins with blank resins, each from independent experiments. The strain used was SCY249. D, summary of candidate Thr(P)-90- and Thr(P)-279-associated proteins identified by quantitative MS.
A common feature of Xrs2, Rad53, Dun1, Dma1, and Dma2 is that they contain an FHA domain, which is known to specifically interact with phosphothreonine ligands (36, 37). To examine the involvement of the FHA domain in binding to Sae2, each FHA domain of Dma1, Dma2, Xrs2, Rad53, and Dun1 was expressed in bacteria, and cell extract was used to incubate with Thr(P)-90- or Thr(P)-279-containing peptide of Sae2. We found that the Rad53 FHA1 domain is strongly enriched by Thr(P)-90- and Thr(P)-279-containing phosphopeptides but not when these phosphopeptides were dephosphorylated by λ-phosphatase (Fig. 5A, top panel), indicating that these bindings require phosphorylated Thr-90 and Thr-279 of Sae2. The R70A mutation removes a conserved arginine residue in the FHA1 domain that is directly involved in binding to phosphorylated ligand (36). This mutation largely eliminates binding between Rad53 FHA1 domain and Thr(P)-90- and Thr(P)-279- containing phosphopeptides of Sae2 (Fig. 5A, bottom panels), further supporting a phosphorylation-mediated interaction between Sae2 and Rad53 FHA1 domain. In contrast, the Rad53 FHA2 domain does not show detectable binding to either Thr(P)-90 or Thr(P)-279 phosphopeptides (Fig. 5B). Thus, binding between Rad53 and Sae2 is specific to the Rad53 FHA1 domain, and it involves both phosphorylated Thr-90 and Thr-279 of Sae2.
FIGURE 5.

Phosphothreonine peptides of Sae2 interact directly with FHA domains or Rad53, Dun1, Xrs2, Dma1, and Dma2. A, top, Sae2 phosphopeptides containing either Thr(P)-90 or Thr(P)-279, treated with or without λ-phosphatase, and mock beads incubated with overexpressed Rad53-FHA1 from bacterial lysate. Bottom, Thr(P)-90 or Thr(P)-279 phosphopeptide and mock beads incubated with either overexpressed Rad53-FHA1 or its FHA mutant (Rad53-R70A-FHA1) from bacterial lysate. B, Thr(P)-90 or Thr(P)-279 phosphopeptide and mock beads incubated with either overexpressed Rad53-FHA2 or its FHA mutant (Rad53-R605A-FHA2) from bacterial lysate. C, same as in A, except with Dun1 FHA domain and its FHA mutant (Dun1-R60A-FHA). D, Xrs2-FHA domain overexpressed in bacterial lysate was incubated with Thr(P)-90 or Thr(P)-279, treated with or without λ-phosphatase and mock beads (top). Thr(P)-90 phosphopeptide and mock beads were incubated with either overexpressed Xrs2-FHA or its FHA mutant (Xrs2-R32G,S47A-FHA) from bacterial lysate (bottom). E, Sae2 phosphopeptides containing either Thr(P)-90 or Thr(P)-279 and mock beads incubated with either overexpressed Dma1-FHA or Dma2-FHA from bacterial lysate.
We next examined the binding between Sae2 phosphopeptides and the Dun1 FHA domain. Dun1 FHA domain was enriched by both Thr(P)-90- and Thr(P)-279-containing phosphopeptides of Sae2, and this binding were eliminated by a prior treatment of λ-phosphatase to dephosphorylate these Sae2 phosphopeptides (Fig. 5C, top panel). Once again, the R60A mutation of the conserved arginine in Dun1 FHA domain involved in ligand binding eliminated binding between Dun1 FHA domain and either Thr(P)-90 or Thr(P)-279 phosphopeptide (Fig. 5C, bottom panels), suggesting that this binding between Dun1 and Sae2 is phosphorylation-specific. Consistent with the above MS finding that Xrs2 was identified using Thr(P)-90- but not Thr(P)-279-containing phosphopeptide (Fig. 4), we found that Xrs2 FHA domain was enriched by Thr(P)-90- and not Thr(P)-279-containing phosphopeptides (Fig. 5D). This binding was eliminated by both λ-phosphatase treatment and the R32G and S47A mutations in Xrs2 FHA domain, indicating a phosphorylation-specific interaction. Similarly, both Dma1 and Dma2 FHA domains were enriched by Thr(P)-90 and not Thr(P)-279 phosphopeptides, in agreement with the above MS findings (Fig. 5E). Collectively, these findings showed that Sae2 interacts directly with FHA domains of Rad53, Dun1, Xrs2, Dma1, and Dma2 via its phosphorylated Thr-90 and Thr-279, in agreement with the above MS findings.
FHA Domains of Rad53, Dun1, and Xrs2 Exhibit Phosphorylation-specific Interaction with Sae2
To further characterize the interactions between Sae2 and Rad53, Dun1, and Xrs2, which are known to have important functions in the DNA damage response, we performed pull-down experiments by incubating FHA domains of Rad53, Dun1, and Xrs2 with cell extracts derived from Sae2-3HA strain and its variants. We found that Rad53 FHA1, Dun1, and Xrs2 FHA domains specifically enriched Sae2, and this enrichment was eliminated when Sae2-3HA cell extract was pretreated by λ-phosphatase (Fig. 6A), indicating that phosphorylation of Sae2 is required for its binding to these FHA domains. Next, we examined mutations of various phosphorylation sites of SAE2 and their effects on binding to various FHA domains. We found that the sae2-T90A mutation partially reduced the amount of Sae2 bound to Rad53 FHA1 domain (Fig. 6B, lane 6), whereas sae2-T279A mutation caused a further enrichment of Sae2 by Rad53 FHA1 domain (Fig. 6B, lane 7), which was completely eliminated by sae2-2AQ mutation and R70A mutation in Rad53 FHA1 domain (Fig. 6B, lanes 8 and 9). Thus, both Thr(P)-90 and Thr(P)-279 of Sae2 are involved in binding to Rad53 FHA1 domain, in agreement with the above findings (Figs. 4 and 5A). The sae2-T279A mutation might enhance this interaction by increasing the amount of Sae2 that is phosphorylated at Thr-90 (Fig. 1B). Like the Rad53 FHA1 domain, the Dun1 FHA domain also specifically enriched Sae2 (Fig. 6C), which was partially reduced by sae2-T90A and sae2-T279A mutations. The binding between Dun1 FHA domain and Sae2 is completely eliminated by the sae2-2AQ mutation and the R60A mutation of Dun1 (Fig. 6C). Thus, binding between Sae2 and Dun1 involves both phosphorylated Thr-90 and Thr-279 of Sae2 and the FHA domain of Dun1. Consistent with the binding between Xrs2 FHA domain and Sae2 Thr(P)-90-containing phosphopeptide (Fig. 5D), Xrs2 FHA domain specifically enriched Sae2 (Fig. 6D), which was largely eliminated by T90A, but not T279A mutation of Sae2. Again, mutations of the conserved residues Arg-32 and Ser-47 in the Xrs2 FHA domain, which are expected to interact with phosphothreonine ligands, abolished the interaction between the Xrs2 FHA domain and Sae2 (Fig. 6D), indicating the binding specificity.
FIGURE 6.

FHA domains of Rad53, Dun1, and Xrs2 specifically enriched phosphorylated Sae2 induced by phleomycin treatment in a Thr-90- and Thr-279-dependent manner. A, pull-down of phleomycin-treated Sae2-3HA with recombinant Rad53-FHA1, Dun1-FHA, and Xrs2-FHA domains from lysates incubated with or without λ-phosphatase. B, pull-down assay using recombinant GST-FHA1 of Rad53 (amino acids 2–279) to detect the specific enrichment of Sae2-3HA and various sae2 mutant proteins, revealing the involvement of both Thr-90 and Thr-279 of Sae2 and that the binding is eliminated by the R70A mutation in the Rad53 FHA1 domain. C, pull-down assay using GST-FHA (amino acids 1–150) of Dun1 to enrich Sae2 showed the involvement of both Thr-90 and Thr-279 of Sae2 and that this binding is essentially eliminated by the R60A mutation in the Dun1 FHA domain. D, pull-down assay using Xrs2 FHA-BRCT-TAF (amino acids 1–331) showed the involvement of Thr-90 of Sae2 and that this binding was diminished by the R32G and S47A mutations in the FHA domain of Xrs2. Strains used were JLY030, JLY032, JLY048, JLY050, and JLY053.
Due to inherent differences in the experiments in Figs. 5 and 6, it is difficult to compare the relative binding efficiency between them. It is further possible that phosphopeptides of Sae2 may not behave the same as phosphorylated Sae2 protein, which is phosphorylated at Thr-90 and Thr-279 at unknown levels. Moreover, the binding affinity between full-length Rad53, Dun1, and Xrs2 (together with Mre11 and Rad50) with Sae2 remains to be investigated. Nevertheless, these findings here show that phosphorylation of Thr-90 and Thr-279 of Sae2 specifically mediates its interactions with FHA domains of Rad53, Dun1, and Xrs2 via the known phosphorylation-dependent FHA recognition mechanism. These findings also provided indirect evidence for phosphorylation of Thr-90 and Thr-279 of Sae2.
Genetic Relationships of RAD53 and DUN1 with SGS1 and EXO1
Considering the lethality of the sae2-2AQ sgs1Δ exo1Δ triple mutant and that Rad53 and Dun1 interact with Sae2 via Thr-90 and Thr-279 (Figs. 4–6), we reasoned that the rad53 and dun1 mutants might share genetic interaction profiles similar to that of the sae2-2AQ mutant. To test this, we examined the effect of mutating RAD53 and DUN1 in the sgs1Δ and exo1Δ mutants. We found that the dun1Δ rad53-R70A,N107A sgs1Δ triple mutant was dead (Fig. 7A), whereas the dun1Δ exo1Δ sgs1Δ and rad53-R70A,N107A exo1Δ sgs1Δ triple mutants were viable (Fig. 7A). Thus, the loss of both the Dun1 and Rad53 FHA1 domain causes lethality in cells lacking SGS1. We note here that the sae2-2AQ mutant is lethal in cells lacking both Sgs1 and Exo1 (Fig. 3A). The stronger genetic interaction between rad53 dun1Δ and sgs1Δ could be attributed to the fact that Rad53 and Dun1 FHA domains are known to have additional ligands besides Sae2.
FIGURE 7.

Genetic interactions between the dun1Δ and rad53-R70A,N107A mutations with the sgs1Δ and exo1Δ mutations. A, tetrad dissection of diploids containing the following heterozygous mutations: dun1Δ, rad53 (rad53-R70A,N107A), and sgs1Δ; exo1Δ, sgs1Δ and dun1Δ; exo1Δ, sgs1Δ, and rad53. Strains used were JLY171, JLY496, and JLY497. B and C, camptothecin sensitivity of sae2-2AQ, dun1Δ, rad53 (rad53-R70A,N107A), and their combined mutations at various concentrations. The ratio of the slope of the curve between treated (SlopeC) and untreated (SlopeC = 0) is plotted for each concentration (see “Experimental Procedures”). The average result of three independent experiments is shown with S.D. represented by error bars. Strains used were HZY1077, HZY1244, JLY221, JLY223, JLY635, JLY637, JLY639, and JLY641. D, rates of accumulating GCRs comparing the sae2-2AQ, dun1Δ, and rad53 (rad53-R70A) mutants. Strains used were HZY2672, HZY2709, HZY2908, HZY2909, HZY2912, and HZY2913.
To further explore the genetic relationships between sae2-2AQ and the rad53 and dun1Δ mutations, we examined the effect of rad53 and dun1Δ mutations on CPT sensitivity of the sae2-2AQ mutant. We found that the sae2-2AQ dun1Δ double mutant has essentially the same sensitivity to CPT as the sae2-2AQ mutant (Fig. 7B), whereas the rad53-R70A,N107A sae2-2AQ double mutant is modestly more sensitive to CPT than either the rad53-R70A,N107A or sae2-2AQ single mutants (Fig. 7C). The sae2-2AQ rad53 dun1Δ triple mutant is more sensitive to CPT than either the sae2-2AQ or rad53 dun1Δ double mutant. Once again, we note here that this elevated CPT sensitivity of the sae2-2AQ rad53 dun1Δ triple mutant could be complicated by the fact that Rad53 and Dun1 FHA domains are known to have additional ligands.
Given that phosphorylation of Thr-90 and Thr-279 of Sae2 plays a role in preventing GCRs (Fig. 3C and Table 2), we compared the rate of accumulating GCRs in the sae2-2AQ mutant with those in the rad53 and dun1Δ mutants. As reported previously, deletion of DUN1 causes an increase of GCRs (∼8-fold) compared with a wild-type strain (38). Mutation of rad53-R70A resulted in a GCR rate similar to that of sae2-2AQ mutant, whereas the rate of accumulating GCRs in dun1Δ rad53-R70A double mutant is comparable with that in the dun1Δ, rad53, and sae2-2AQ single mutants (Fig. 7D and Table 2). Thus, the rates of accumulating GCRs in these mutants are modest and comparable with each other.
Genetic Relationships of Xrs2 FHA Domain with SGS1 and EXO1
Sae2 is known to function together with the Mre11-Rad50-Xrs2 complex. It is interesting to detect a specific interaction between phosphorylated Thr-90 and Xrs2 FHA domain here (Figs. 4–6). To explore this further, we examined the effect of R32G and S47A mutations in the Xrs2 FHA domain, which specifically disrupts its binding to phosphorylated Thr-90 of Sae2. We found that this xrs2 mutation does not cause an appreciable increase in sensitivity to a lower concentration (10 μm) of CPT treatment, although a higher sensitivity was observed at 20 μm CPT (Fig. 8A). This xrs2 mutation does not appreciably alter CPT sensitivity of sgs1Δ, unlike sae2-2AQ (Fig. 8A), although we note here that Xrs2 FHA domain only interacts with phosphorylated Thr-90 of Sae2. On the other hand, this xrs2 FHA domain mutation does cause an elevated sensitivity to CPT in cells lacking Exo1 (Fig. 8B), unlike sae2-2AQ (Fig. 3D). Taken together, the findings presented here suggest that phosphorylation of Sae2 and its recruitment of Rad53, Dun1, and Xrs2 might have multiple functions in DNA repair-related processes.
FIGURE 8.
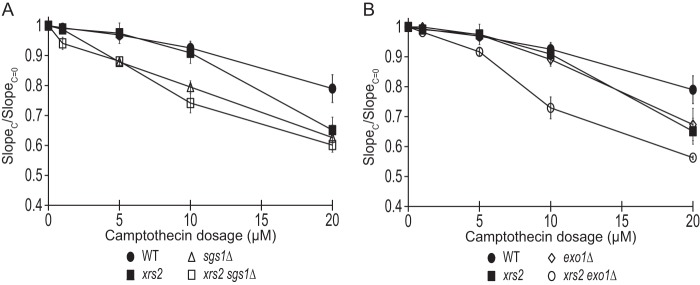
Genetic interaction of the xrs2-fha mutation with sgs1Δ and exo1Δ. A and B, camptothecin sensitivity of xrs2 (xrs2-R32G,S47A), exo1Δ, sgs1Δ, and their combined mutations at various concentrations. The ratio of the slope of the curve between treated (SlopeC) and untreated (SlopeC = 0) is plotted for each concentration (see “Experimental Procedures”). Strains used were HZY1077, JLY391, JLY235, JLY335, JLY230, and JLY395.
DISCUSSION
Sae2 and its ortholog CtIP have a key function in the initial processing of damaged DNA, which influences subsequent DNA repair pathway choices. A conserved feature of Sae2 and CtIP is their post-translational modifications; in particular, phosphorylation of Sae2/CtIP has been shown to be critical for its functions in diverse organisms (9, 13, 15–18, 20, 39–41). However, the molecular mechanism concerning phosphorylation regulation of Sae2 remains insufficiently understood. Here we showed that two conserved residues, threonine 90 and threonine 279, of Sae2 redundantly regulate its functions in the DNA damage response and suppression of gross chromosomal rearrangements. We further show that phosphorylation of these residues of Sae2 mediates specific interactions with multiple FHA domain-containing proteins, including Rad53, Dun1, and Xrs2, which are known to function in the DNA damage response. The initial analyses presented here reveal that these biochemical interactions are highly specific, and they probably contribute to the function of Sae2 phosphorylation in not only DNA damage checkpoint but also DNA repair.
Conserved Threonines of Sae2 Have a Redundant Role in the DNA Damage Checkpoint and DNA Repair
Among those Mec1 and Tel1 phosphorylation sites of Sae2, Thr-279 is conserved in vertebrate CtIP and phosphorylated by ATR and ATM, orthologs of Mec1 and Tel1, respectively. Thr-90 of Sae2 is conserved among most fungal species but not mammalian CtIP or S. pombe Ctp1. Despite this sequence divergence, Thr-90 of Sae2 has a redundant role with Thr-279 in regulating its functions in the DNA damage response. Mutations of both Thr-90 and Thr-279 of Sae2 cause a persistent and elevated Rad53 phosphorylation, similar to deletion of SAE2 (9, 13). The synthetic lethal interaction between sae2-2AQ mutation and sgs1Δ exo1Δ mutation is striking (Fig. 3A), and it suggests that phosphorylation of Sae2 may have an important function in DNA end processing, considering the role of Sgs1 and Exo1 in DNA end processing. Further study is needed to address this more specifically.
Interestingly, despite the finding that sae2-S249A mutation has a significant effect on Sae2 electrophoretic mobility shift (Fig. 1B), it does not cause any detectable phenotype in the assays performed in this study. This does not exclude the possibility that Ser-249 phosphorylation of Sae2 might have another function yet to be identified. However, our findings here suggest that evolutionary conservation of the phosphorylated threonine residues of Sae2, rather than stoichiometry of phosphorylation, may provide a better prediction of potential functions of Sae2 phosphorylation.
Potential Functions of Sae2-associated FHA Domain-containing Proteins
Although it is evident that phosphorylation of Sae2 and its ortholog CtIP have important roles in regulating their functions in DNA damage checkpoint and DNA repair, the precise function of their phosphorylation has been insufficiently understood. Here we show that threonine-specific phosphorylation of Sae2 by Mec1 and Tel1 can recruit several FHA domain-containing proteins, including Rad53, Dun1, Xrs2, Dma1, and Dma2. The biochemical data presented here demonstrated the specificity of these phosphorylation-mediated interactions, consistent with the known ligand-binding property of FHA domains. It should be noted that these interactions are expected to have low micromolar affinity (36), thus requiring the use of recombinant FHA domain or abundant phosphopeptides of Sae2 to facilitate the detection of these interactions. For this reason, we cannot rule out the possibility that some of the binding interactions could occur as a biochemical artifact. On the other hand, similar FHA-mediated interactions have been shown to be important for the DNA damage checkpoint (30, 36, 41, 42). Therefore, it is reasonable to suggest that some of these interactions may have important roles in Sae2 functions. Among them, Rad53, Dun1, and Xrs2 are particularly interesting because they have known roles in the DNA damage response. Unlike genetic analysis of various sae2 phosphorylation-defective mutants, analysis of rad53, dun1Δ, and xrs2 mutants could be complicated by the fact that FHA domains in these proteins are known to have other ligands, which are critical for their diverse functions in the DNA damage response. For example, both FHA domains of Rad53 are known to interact with Rad9 and Mrc1, which are important for its activation (29, 30). Additionally, many other ligands of the Rad53 FHA1 domain have been identified (22). Similarly, the FHA domain of Dun1 is known to be critical for its interaction with Rad53 and trans-activation by Rad53 (42, 43). The Xrs2 FHA domain has also been shown to interact with Lif1 to promote nonhomologous end joining (44). These other functions of Rad53, Dun1, and Xrs2 may complicate the genetic analysis here. Despite these caveats, several observations here suggest that Rad53 and Dun1 may be involved in the function of Sae2 phosphorylation in DNA repair. For example, the rad53 dun1Δ mutation is synthetic lethal with sgs1Δ, although sae2-2AQ is lethal in cells lacking both Sgs1 and Exo1. Obviously, further studies are needed to dissect the functions of the biochemical interactions uncovered here, which point to a complex role of Sae2 phosphorylation in several processes.
We speculate that phosphorylated Sae2 may recruit Rad53, Dun1, and Xrs2 to the sites of DNA damage for different functions. Rad53 and Dun1 may phosphorylate proteins at the sites of DNA damage in a Sae2-dependent manner. For example, Dun1 was implicated in suppressing DSB-induced chromosomal translocations in the same study that demonstrated a role for Sae2 and its DNA damage-induced phosphorylation (14). Clearly, identification of the relevant substrates of Rad53 and Dun1 that are specifically mediated by Sae2 would be essential to understand the function of the interactions reported here.
Acknowledgments
We thank Dr. Claudio Albuquerque for help with the MS analysis of phosphopeptide-binding proteins and Dr. Veronica Baldo for discussion and for sharing yeast genetic techniques during the course of this study. We thank Dr. Marco Foiani for the anti-Rad53 antibody used in this study. We thank Dr. Lorraine Symington for helpful discussions and critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM080469 (to H. Z. and R. S.). This work was also supported by Ludwig Cancer Research (to H. Z. and R. S.).
- DSB
- double-stranded break
- GCR
- gross chromosomal rearrangement
- CPT
- camptothecin
- CDK
- cyclin-dependent kinase
- FHA
- Forkhead-associated
- 3HA
- 3× HA
- TAF
- His6-FLAG-TEV-Protein A
- MMS
- methyl methanesulfonate
- SILAC
- stable isotope labeling via amino acid in culture
- HU
- hydroxyurea.
REFERENCES
- 1. Harrison J. C., Haber J. E. (2006) Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40, 209–235 [DOI] [PubMed] [Google Scholar]
- 2. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 3. Mimitou E. P., Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakada D., Matsumoto K., Sugimoto K. (2003) ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 17, 1957–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 7. Brush G. S., Morrow D. M., Hieter P., Kelly T. J. (1996) The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc. Natl. Acad. Sci. U.S.A. 93, 15075–15080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gatei M., Young D., Cerosaletti K. M., Desai-Mehta A., Spring K., Kozlov S., Lavin M. F., Gatti R. A., Concannon P., Khanna K. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 25, 115–119 [DOI] [PubMed] [Google Scholar]
- 9. Baroni E., Viscardi V., Cartagena-Lirola H., Lucchini G., Longhese M. P. (2004) The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell Biol. 24, 4151–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Craven R. J., Greenwell P. W., Dominska M., Petes T. D. (2002) Regulation of genome stability by TEL1 and MEC1, yeast homologs of the mammalian ATM and ATR genes. Genetics 161, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pennaneach V., Kolodner R. D. (2004) Recombination and the Tel1 and Mec1 checkpoints differentially effect genome rearrangements driven by telomere dysfunction in yeast. Nat. Genet. 36, 612–617 [DOI] [PubMed] [Google Scholar]
- 12. Myung K., Datta A., Kolodner R. D. (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104, 397–408 [DOI] [PubMed] [Google Scholar]
- 13. Clerici M., Mantiero D., Lucchini G., Longhese M. P. (2006) The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 7, 212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee K., Zhang Y., Lee S. E. (2008) Saccharomyces cerevisiae ATM orthologue suppresses break-induced chromosome translocations. Nature 454, 543–546 [DOI] [PubMed] [Google Scholar]
- 15. Li S., Ting N. S., Zheng L., Chen P. L., Ziv Y., Shiloh Y., Lee E. Y., Lee W. H. (2000) Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 406, 210–215 [DOI] [PubMed] [Google Scholar]
- 16. Peterson S. E., Li Y., Wu-Baer F., Chait B. T., Baer R., Yan H., Gottesman M. E., Gautier J. (2013) Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 49, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., Shi L. Z., Wong C. C., Han X., Hwang P. Y., Truong L. N., Zhu Q., Shao Z., Chen D. J., Berns M. W., Yates J. R., 3rd, Chen L., Wu X. (2013) The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 9, e1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huertas P., Cortés-Ledesma F., Sartori A. A., Aguilera A., Jackson S. P. (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu Q., Chow J., Bernstein K. A., Makharashvili N., Arora S., Lee C. F., Person M. D., Rothstein R., Paull T. T. (2014) Phosphorylation-regulated transitions in an oligomeric state control the activity of the Sae2 DNA repair enzyme. Mol. Cell Biol. 34, 778–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Williams R. S., Dodson G. E., Limbo O., Yamada Y., Williams J. S., Guenther G., Classen S., Glover J. N., Iwasaki H., Russell P., Tainer J. A. (2009) Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 22. Smolka M. B., Chen S. H., Maddox P. S., Enserink J. M., Albuquerque C. P., Wei X. X., Desai A., Kolodner R. D., Zhou H. (2006) An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J. Cell Biol. 175, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vance J. R., Wilson T. E. (2002) Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl. Acad. Sci. U.S.A. 99, 13669–13674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deng C., Brown J. A., You D., Brown J. M. (2005) Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics 170, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Putnam C. D., Kolodner R. D. (2010) Determination of gross chromosomal rearrangement rates. Cold Spring Harb. Protoc. 10.1101/pdb.prot5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen S. H., Albuquerque C. P., Liang J., Suhandynata R. T., Zhou H. (2010) A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 285, 12803–12812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 28. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vialard J. E., Gilbert C. S., Green C. M., Lowndes N. F. (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 17, 5679–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun Z., Hsiao J., Fay D. S., Stern D. F. (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281, 272–274 [DOI] [PubMed] [Google Scholar]
- 31. Alcasabas A. A., Osborn A. J., Bachant J., Hu F., Werler P. J., Bousset K., Furuya K., Diffley J. F., Carr A. M., Elledge S. J. (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3, 958–965 [DOI] [PubMed] [Google Scholar]
- 32. Tanaka K., Russell P. (2001) Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 3, 966–972 [DOI] [PubMed] [Google Scholar]
- 33. Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., Boone C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 34. Mimitou E. P., Symington L. S. (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Putnam C. D., Pallis K., Hayes T. K., Kolodner R. D. (2014) DNA repair pathway selection caused by defects in TEL1, SAE2, and de novo telomere addition generates specific chromosomal rearrangement signatures. PLoS Genet. 10, e1004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Durocher D., Henckel J., Fersht A. R., Jackson S. P. (1999) The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell 4, 387–394 [DOI] [PubMed] [Google Scholar]
- 37. Durocher D., Taylor I. A., Sarbassova D., Haire L. F., Westcott S. L., Jackson S. P., Smerdon S. J., Yaffe M. B. (2000) The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell 6, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 38. Putnam C. D., Hayes T. K., Kolodner R. D. (2009) Specific pathways prevent duplication-mediated genome rearrangements. Nature 460, 984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu X., Chen J. (2004) DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 24, 9478–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cartagena-Lirola H., Guerini I., Viscardi V., Lucchini G., Longhese M. P. (2006) Budding yeast Sae2 is an in vivo target of the Mec1 and Tel1 checkpoint kinases during meiosis. Cell Cycle 5, 1549–1559 [DOI] [PubMed] [Google Scholar]
- 41. Dodson G. E., Limbo O., Nieto D., Russell P. (2010) Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle 9, 1516–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen S. H., Smolka M. B., Zhou H. (2007) Mechanism of Dun1 activation by Rad53 phosphorylation in Saccharomyces cerevisiae. J. Biol. Chem. 282, 986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bashkirov V. I., Bashkirova E. V., Haghnazari E., Heyer W. D. (2003) Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol. Cell Biol. 23, 1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palmbos P. L., Wu D., Daley J. M., Wilson T. E. (2008) Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics 180, 1809–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]