

Abstract
Objective
The lymphatic vasculature is a well-established conduit for metastasis, but the mechanisms by which tumor cells interact with lymphatic endothelial cells (LECs) to facilitate escape remain poorly understood. Elevated levels of the lymphangiogenic peptide adrenomedullin (AM) are found in many tumors and we previously characterized that its expression is necessary for lymphatic vessel growth within both tumors and sentinel lymph nodes and for distant metastasis.
Approach and Results
This study used a tumor cell-LEC co-culture system to identify a series of AM-induced events that facilitated transendothelial migration (TEM) of the tumor cells through a lymphatic monolayer. High levels of AM expression enhanced adhesion of tumor cells to LECs and further analysis revealed that AM promoted gap junction coupling between LECs as evidenced by spread of Lucifer yellow dye. AM also enhanced heterocellular gap junction coupling as demonstrated by Calcein dye transfer from tumor cells into LECs. This connexin-mediated gap junction intercellular communication (GJIC) was necessary for tumor cells to undergo TEM since pharmacological blockade of this heterocellular communication prevented the ability of tumor cells to transmigrate through the lymphatic monolayer. Additionally, treatment of LECs with AM caused nuclear translocation of β-catenin, a component of endothelial cell junctions, causing an increase in transcription of the downstream target gene C-MYC. Importantly, blockade of GJIC prevented β-catenin nuclear translocation.
Conclusions
Our findings indicate that maintenance of cell-cell communication is necessary to facilitate a cascade of events that lead to tumor cell migration through the lymphatic endothelium.
Keywords: lymphatics, endothelial cell, metastasis, gap junction, connexins
Introduction
Lymphatic vessels serve as a conduit by which tumor cells can escape to colonize distant organs. Seminal studies implicating the lymphangiogenic growth factors VEGF-C and VEGF-D in promoting tumor metastasis through enhanced lymphangiogenesis have provided insight into how lymphatics promote cancer progression 1-3. For example, VEGF-C was shown to activate the lymphatic endothelium leading to vessel destabilization and thereby enabling tumor cell entry into the lymphatic vessel 4. In addition to VEGF family members, other processes such as chemoattraction 5, lymph flow 5, 6, enhanced adhesion 7, and lymph node lymphangiogenesis 8, 9 are also implicated in promoting entry of tumor cells into the lymphatic vasculature and facilitating metastasis. However, the precise mechanisms of how this occurs at the critical interface between the tumor cells and lymphatic endothelial cells (LECs) remains to be elucidated.
One mechanism by which neighboring cells interact to communicate and exchange information is through the formation of gap junction channels. Oligomerized connexin hexamers termed connexons from juxtaposed cells form channels that allow for cytoplasmic exchange of small signaling molecules such as metabolites, ions, and secondary messengers. Establishment of gap junctional intercellular communication (GJIC) through these channels is critical to maintain homeostasis during development and in adulthood 10. Of particular relevance, proper development of the lymphatic vasculature, specifically the lymphatic valves, is dependent on precise regulation and expression of certain members of the connexin protein family such as connexins (Cx) 37, and Cx43 11. In fact, missense mutations in the gap junction gene GJC2 (encoding Cx47) have been identified in families with dominantly inherited lymphedema 12. This finding is significant because it links impaired lymphatic activity with a mutation that alters gap junction function. These defects emphasize the critical role that connexins play in lymphatic function and disease 13.
Connexins appear to play diverse roles in cancer. Some studies suggest that expression of connexins confers a tumor suppressor function 14-16. Along these lines, mice heterozygous for Cx43 (Cx43+/−) had an increased susceptibility to urethane-induced lung tumors 17. More recent evidence, however, proposes that connexins are dynamically regulated depending on the stage of tumorigenesis, and therefore elevated levels may be important in promoting angiogenesis 18 and invasion 19-24. These data suggest that increased connexin expression in later stages of tumorigenesis enables tumor cells to penetrate the vessels and thus promote colonization of distant tissues. Moreover, connexin proteins also have channel-independent functions 25 such as serving as adhesion sites which can mediate the invasion of glioma cells through the parenchyma 26.
Building upon our previous study which identified adrenomedullin (AM) as a factor which promotes tumor lymphangiogenesis and distant metastasis 27, we investigated the role of GJIC in this process. By focusing on the tumor cell – endothelial cell interactions, we identified a series of AM-induced events that promote the transendothelial migration of tumor cells including functional GJIC and subsequent β-catenin nuclear translocation. To our knowledge, this is the first study to detail how tumor cells and LECs physically interact to facilitate tumor spread through the lymphatics. This study reinforces the often overlooked role that the lymphatic endothelium plays in actively promoting the metastatic process.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
AM promotes the adhesion of tumor cells to the lymphatic endothelium and enhances their transendothelial migration
To test whether AM is involved in mediating adhesion of tumor cells to the lymphatic vasculature, we utilized AM-dosed LLC murine tumor cells that either express a 2-fold increase in Adm expression (AM OExp), a 92% reduction in Adm expression (AM RNAi) or maintain basal levels (EV; empty vector control) 27. Importantly, the LLC tumor cells have negligible expression of the AM receptor Calcrl 27, therefore the tumor cells are insensitive to manipulation of their AM levels since the AM-induced signal cannot be transduced through CLR. Rather, any resultant biological effects due to low or elevated AM levels can be attributed to AM-mediated signaling in other cell types in the microenvironment, such as LECs which express abundant CLR. The genetically AM-dosed cells were labeled with Cell Tracker Green (CTG) dye and added to an LEC monolayer. After 15 minutes of co-culture, fluorescence intensity was measured and we detected an AM dose dependent increase in the adhesion of tumor cells to the lymphatic monolayer (Figure 1A,B). We confirmed equal CTG fluorescence to ensure that altering Adm dosage does not affect CTG dye labeling (Figure 1C). Next, we utilized a pharmacologic approach to confirm that AM was mediating this adhesion. We treated the LEC monolayer with 1nM murine AM (mAM) peptide and the AM receptor antagonist AM22-52 and then added CTG-labeled LLC cells. Again, there was increased adhesion of tumor cells to LECs in the presence of AM and this adhesion was dramatically reduced in the presence of the AM inhibitor (Figure 1D). To corroborate these results, we analyzed the CTG-labeled human tumor cell line MCF-7 (Figure 1E) and similarly found that stimulation of LECs with 10nM human AM (hAM) peptide promoted the adhesion of the MCF-7 cells to the LECs (Figure 1F).
Figure 1.

Adrenomedullin promotes the adhesion and transendothelial migration (TEM) of tumor cells to LECs. A. AM-dosed LLC cells were labeled with Cell Tracker Green (CTG) dye and incubated with a monolayer of LECs. After 15 minutes, non-adhered cells were aspirated and fluorescence of adhered cells was measured. B. Representative images of CTG-labeled tumor cells (black arrows) adhered to an LEC monolayer. Magnification: 10X. Scale bars: 150µm. Phase contrast images are an optical zoom of the area surrounded by a white box. C. Graph depicts equal CTG labeling of the AM-dosed LLC cells. D. LECs were treated with vehicle or 1nM murine AM with or without a 30 minute pretreatment with the AM inhibitor AM22-52 (1µM) followed by addition of CTG-labeled LLC tumor cells for a 15 minute incubation. A,D. Fluorescence readings indicated an AM dose dependent increase in tumor cell adhesion to the LEC monolayer. Reduced AM levels through either RNAi depletion of Adm in tumor cells or pre-treatment with the AM inhibitor AM22-52 prevented the increased adhesion. E. A standard curve of CTG-labeled LLC and MCF-7 cells. F. CTG-labeled MCF-7 cells also exhibit an AM-induced increase in adhesion to LECs after 2 hour incubation. G. CTG-labeled AM-dosed LLC cells were seeded atop a confluent LEC monolayer and after a 6 hour co-culture, filters were removed from the transwell inserts. Image threshold analysis of 4 fields/ filter revealed that AM promoted TEM. H. Using a pharmacological approach, a confluent LEC monolayer was treated with vehicle or 1nM murine AM +/− AM22-52 followed by addition of CTG- labeled unmodified LLC cells similarly showed that AM promoted TEM. Analysis of 2 additional tumor cell lines, CTG-labeled MCF-7 (I) and SK-MEL-2 (J) cells showed similar trends of AM enhanced TEM within 24 and 3 hours, respectively. G,H,I,J. Magnification: 10X. Scale bar: 100µm. The data shown are means ± SE and are representative of 3 independent experiments that yielded similar results.
We then wanted to determine whether the AM-induced increase in tumor cell-LEC adhesion consequently results in enhanced transendothelial migration (TEM). Using a standard transwell assay, CTG-labeled AM-dosed tumor cells were added to transwell inserts which contained a confluent monolayer of LECs. Confluency was verified by H&E stained filters (data not shown). After 6 hours of co-culture, the filters from the inserts were removed and the fluorescent threshold area was determined. There was a significant increase in fluorescence as tumor derived AM dose increased, indicative of transmigrated CTG-labeled tumor cells (Figure 1G). To confirm that AM was responsible for the increased TEM, we repeated this experiment using a pharmacological approach. First we pre-treated the LEC monolayer with the AM inhibitor AM22-52 followed by addition of 10nM human or 1nM murine AM peptide. After the LEC monolayer was treated with AM for 5 minutes, CTG-labeled murine LLC tumor cells or human MCF-7 and SK-MEL-2 tumor cells were added to the transwell inserts. Again, we observed an AM-induced increase in TEM in all 3 tumor lines and the AM inhibitor reduced the transmigration to levels comparable to vehicle treatment (Figure 1H-J). Interestingly, the time course of TEM of the 2 human cells lines ranges from 3 to 24 hours for the SK-MEL-2 and MCF-7 cells, respectively. These data concur with the characterization that SK-MEL-2 cells are a highly metastatic cell line whereas the MCF-7 cells are reported to be weakly metastatic. In both instances, however, treatment with AM promotes TEM. Importantly, comparable to the LLC tumor cells, the MCF-7 and SK-MEL-2 cell lines also had negligible expression of the AM receptor Calcrl (Ct values ~32, data not shown). Therefore, AM is stimulating the LECs, rather than the tumor cells, to promote TEM.
Lymphatic endothelial cells form functional gap junctions upon AM treatment.
To assess whether gap junction formation is enhanced by AM treatment we performed a scrape loading dye transfer assay using the gap junction permeable dye Lucifer Yellow (LY). Treatment of LECs with 10nM hAM for 30 minutes resulted in a dramatic spread (3-fold increase) of LY between the cells indicative of gap junction coupling (Figure 2). Importantly, this dye transfer was abrogated by pretreatment with the AM inhibitor AM22-52 confirming that this was an AM-mediated effect. Furthermore, to verify that the spread of LY dye was through gap junctions rather than nonspecific dye transmission, we treated cells with the gap junction inhibitor carbenoxolone (CBX) 28 and subsequently detected no transfer of dye to adjacent cells (Figure 2). Taken together, these results suggest that AM treatment enhances gap junction mediated intercellular communication in LECs.
Figure 2.
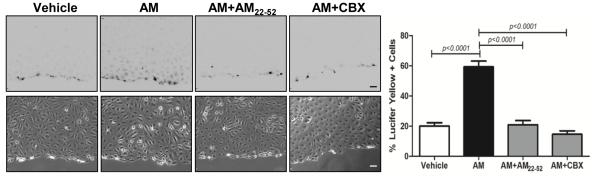
Adrenomedullin promotes the formation of gap junctions in LECs. Transfer of Lucifer yellow dye into neighboring LECs was determined after scrape-loading. Cells were pretreated with the AM inhibitor AM22-52 (1µM) or the gap junction inhibitor CBX (100µM) for 30 minutes, followed by treatment with 10nM human AM for 10 minutes. The percent of dye coupled cells was quantified by dividing the number of Lucifer yellow positive cells by the total number of cells in the field. Data represent means ± SE (n=3). Magnification: 10X. Scale bar: 100µm.
Connexins assemble to form gap junctions upon AM treatment.
We assessed the expression of connexins 37, 43, and 47 (Cx37, Cx43, Cx47) given their importance in lymphatic vascular development 11. Using real-time qPCR, we found that 10nM hAM treatment of LECs caused an increase in GJA1, a decrease in GJA4, and no change in expression of GJC2 (Figure 3A-C). These dynamic changes in mRNA transcript were verified by western blot analysis of AM-treated LECs (Figure 3D,E). Using immunofluorescence analysis, we found that vehicle treated LECs displayed punctate Cx43 staining throughout the cytoplasm with only minimal localization to the cell membranes (Figure 3F). By contrast, LECs treated for 15 minutes with 10nM hAM had Cx43 localized to regions of cell-cell contacts (Figure 3F). Analysis of Cx37 and Cx47 revealed no changes in localization upon hAM treatment (Figure 3G,H). The striking AM-induced membrane localization of Cx43 was prevented by treatment with the AM inhibitor AM22-52 (Figure 3F). Interestingly, LECs pretreated with the GJ inhibitor CBX before addition of hAM still had localization of Cx43 to regions of cell-cell contact (Figure 3F). This suggests that gap junction coupling itself is not required for AM-induced Cx43 membrane localization. Collectively, these data show that stimulation of LECs with AM causes an upregulation of Cx43 expression and changes its sub-cellular localization to areas of cell-cell contact.
Figure 3.

Connexin expression and localization in response to adrenomedullin. A. Gene expression analysis revealed that treatment of LECs with 10nM human AM (4 hrs) caused an increase in GJA1 (A), a decrease in GJA4 (B), and no change in expression of GJC2 (C). D, E. Western blot analysis confirms these trends in expression following AM treatment (24 hrs) . F. LECs were pretreated with inhibitors (1µM AM22-52 or 100µM carbenoxolone) followed by a 15 minute treatment with 10nM hAM and then stained for Cx43. Immunofluorescence analysis revealed that AM and AM+CBX treatments caused a distinct linearization of Cx43 at cell-cell contacts as compared to a more punctuate pattern in vehicle and AM inhibitor treatments. G, H. AM treatment of LECs failed to cause a change in localization of Cx37 and Cx47. Magnification: 40X. Scale bar: 50µm.
AM-overexpressing tumors have increased expression of Cx43 in lymphatic vessels
Studies have reported that levels of Cx43 mRNA or protein are dynamically regulated and for example, are elevated in estrogen stimulated endometrium 29 and metastatic tissue 22-24, 30. To determine if AM can similarly modulate Cx43 expression in vivo, we subcutaneously injected mice with 1x106 AM-dosed LLC cells and harvested tumors after 14 days 27. Lymphatic vessels were identified by LYVE1 staining, and interestingly, in the presence of high tumor derived AM (AM OExp), integrated density analysis revealed that the lymphatic vessels had increased expression of Cx43 (Figure 4). Furthermore, tumors expressing low levels of AM had only minimal Cx43 staining in the lymphatic vessels (Figure 4). These data demonstrate that in vivo tumor lymphatic vasculature responded to high levels of tumor-derived AM by upregulating Cx43 expression.
Figure 4.

Mice were injected subcutaneously with AM-dosed LLC tumor cells (1x106) and after 14 days tumors were collected. Tumors over-expressing AM (AM OExp) had increased expression of Cx43 within lymphatic vessels (LYVE1). Graph depicts integrated density analysis of Cx43 and reveals dose dependent expression of Cx43 with increased tumor AM expression. *One way ANOVA analysis: p=0.012. Magnification: 40X. Scale bars: 50µm.
AM promotes heterocellular GJIC and blockade of this communication prevents TEM.
To address whether AM has the ability to promote heterocellular gap junction formation we performed a dye transfer assay. LLC tumor (donor) cells were loaded with the gap junction permeable dye Calcein while the LECs (acceptor) were loaded with the gap junction impermeable dye DiI. After a 3 hour co-culture, we observed almost 80% of hAM treated LECs (acceptor) were Calcein positive (Figure 5). Treatment with the gap junction inhibitor CBX completely blocked any transfer of Calcein from the tumor cells to LECs and confirmed that functional gap junction channels were responsible for the observed dye transfer. Next, we wanted to determine the functional consequences of this AM-induced heterotypic cellular communication and we hypothesized that GJIC may be necessary for the AM-induced TEM observed in Figure 2. Consistent with these results, we found that AM promoted the TEM of CTG- labeled tumor cells and intriguingly, CBX prevented the ability of tumor cells to migrate through an LEC monolayer (Figure 5B). These findings demonstrate that AM induced formation of functional gap junctions between tumor cells and LECs is necessary for tumor cell migration through an LEC monolayer.
Figure 5.

Heterocellular dye transfer and TEM are enhanced by adrenomedullin and require functional GJIC. A. LLC tumor cells (donor) loaded with 5µM calcein were co-cultured with mLECs (acceptor) that were labeled with 5µM DiI. mLECs were treated with vehicle (TC water), 1nM mAM or mAM + CBX (100um) for 30 minutes prior to addition of calcein labeled LLC cells. After 3 hour co-culture, cells were fixed and analyzed for the number of tumor cells that transferred calcein to adjacent mLECs. Representative images are shown. Magnification: 40X. Scale bar: 50µm. B. Dye coupled cells were scored as positive if a calcein+ LLC tumor cell was in the same field and adjacent to a DiI+ and calcein+ mLEC. Dye coupling was expressed as a percent of the total number of tumor cells counted (~30-40 cells/treatment). Graph depicts pooled data from 3 independent experiments. C. A confluent mLEC monolayer was treated with the gap junction inhibitor CBX (100µM) and murine AM (1nM) or vehicle for 30 minutes. CTG-labeled LLC cells were added to the monolayer and after a 6 hour co-culture, TEM was determined by fluorescent microscopy. The data shown are representative of 3 independent experiments that yielded similar results. Magnification: 10X. Scale bar: 50µm.
AM-induced GJIC causes β-catenin nuclear translocation.
AM was recently identified as a Wnt/β-catenin target 31 and we examined whether this pathway was implicated in the TEM process by assessing β-catenin localization in LECs after treatment with hAM. In contrast to the jagged staining pattern observed at cell-cell contacts in control cells, 10nM hAM treatment caused a more linear arrangement of β-catenin (Figure 6A). Given that β-catenin is part of a complex at the endothelial junction, this finding is consistent with the linearization observed in VE-cadherin staining upon AM treatment 32. Furthermore, treatment with AM induced nuclear localization of β-catenin in 26% of hAM-treated LECs (Figure 6A,B). Importantly, the AM inhibitor AM22-52 prevented β-catenin translocation to the nucleus confirming that this translocation is triggered by AM signaling. The gap junction inhibitor CBX also failed to cause β-catenin accumulation at the nucleus, but interestingly, staining shows it was aligned at cell-cell contacts as seen in AM treated LECs (Figure 6A). This suggests that functional gap junctions are necessary for β-catenin nuclear translocation which then leads to an increase in gene transcription of the downstream target C-MYC (Figure 6C). Our model summarizes how AM activates the lymphatic endothelium to promote tumor cell dissemination (Figure 6D). In the presence of AM, tumor cells exhibit increased adhesion to the LECs and transmigration through the monolayer requires functional gap junction mediated intercellular communication and subsequent β-catenin translocation to the nucleus.
Figure 6.

β-catenin nuclear translocation is promoted by adrenomedullin and is dependent on GJIC. A. Treatment of LECs with 10nM human AM caused an alignment of β-catenin at the membrane along with its nuclear translocation. These changes were prevented by pretreatment with AM inhibitor AM22-52 (1µM). The gap junction inhibitor CBX (100µM) prevented β-catenin nuclear translocation indicating that GJIC is necessary for its translocation. Magnification: 40X. Scale bar: 50µm. B. Quantification showing the percentage of cells with β-catenin nuclear translocation relative to the total number of cells in the field. 80-120 cells were analyzed for each treatment from 3 independent experiments and expressed as means ± SE. C. RNA isolated from LECs treated with vehicle or 10nM human AM (4hrs) was analyzed by qPCR for expression of C-MYC. D. The model depicts how high tumor-derived AM promotes increased tumor cell adhesion to LECs, connexin-mediated GJIC, β-catenin nuclear translocation and subsequent gene transcription to facilitate transendothelial migration.
Discussion
It has been speculated that tumor cells utilize similar mechanisms as immune cells to home to and transverse through the lymphatic endothelium. For example, gradients of CCR7 and CCL21 allow immune cells to chemoattract towards lymphatic vessels 33 and integrins have been shown to participate in the transit of immune cells across lymphatic endothelium under inflammatory conditions 34. In support of these findings, several groups have shown similar chemokine gradients contribute to tumor cell migration and invasion towards lymphatics 35-37. Furthermore, Garmy-Susini and colleagues identified integrin α4β1 as a marker of tumor lymphatic endothelium and the downstream signaling cascade it elicits promotes the adhesion and invasion of LECs 38. We find that AM promotes the adhesion of tumor cells to the lymphatic endothelium since blockade of AM signaling reduces the ability of tumor cells to adhere to the endothelium and establish functional heterocellular gap junction channels. It remains unclear whether the adhesion is directly mediated by integrins or perhaps through the gap junction protein, connexins which are expressed by both the tumor cells and the vasculature.
In addition to increased peri-tumoral lymphatics, enhanced lymphatic flow has also been reported to contribute to lymphatic metastasis 5, 6, 9. In a B16 melanoma tumor model, induction of lymph node lymphangiogenesis has been associated with a dramatic increase in lymph flow to the tumor-draining lymph node (LN) 9. We have previously shown that high levels of tumor-derived AM induces LN lymphangiogenesis 27 and literature suggests that the resultant enhanced flow could serve to promote initial tumor cell entry into the lymphatic vessels and then tumor cell transit into and through the draining LN. Intriguingly, disrupted lymph flow due altered gap junction function has been linked to mutations in GJC2 (encodes for the connexin 47 protein) 12. Similarly, conditional knockout mice for the AM receptor (Calclr) exhibit dysfunctional lymphatic flow and permeability 39. These results warrant further investigation to determine whether and how gap junctions facilitate lymph flow and importantly, how compromised lymph flow affects a pathological process like tumor metastasis.
The identification of mutations in the connexin gene family which cause alterations in gap junction mediated functions begs the question of whether this gene family can be exploited as a possible therapeutic target. The ability of the lymphatic vasculature to form heterocellular gap junctions with tumor cells to facilitate transendothelial migration has potentially important implications due to the phenomenon known as the bystander effect. The concept that a signal or molecule is transferred among neighboring cells to elicit a biological response has been used for example, as a treatment strategy for eradication of brain tumors 40. The commonly used approach involves retroviral mediated transfer of the herpes simplex virus thymidine kinase that sensitizes the transduced tumor or endothelial cells to the anti-viral ganciclovir to cause cell death. In a 9L brain tumor model using this strategy, researchers discovered a decrease in tumor vasculature and tumor regression 41. Intriguingly, evidence suggests that gap junction mediated intercellular communication is one mechanism that contributes to bystander cytotoxicity 42, 43. In fact, some have proposed to target connexins for gene therapy since their expression, localization at the plasma membrane, and assembly into functional gap junctions would improve the efficacy of bystander effect 44. Our results clearly demonstrate that AM signaling promotes the organization of connexins into functional gap junctions to promote intercellular communication and subsequent TEM through a lymphatic monolayer. The development of therapies that rely upon functional coupling of cells and subsequent transfer of molecules among adjacent cells could provide a novel therapeutic strategy to disrupting or reducing the ability of tumor cells to escape through the lymphatics.
Significance
The lymphatic vascular system contributes to the distant spread of tumor cells but the molecular mechanism of how this occurs is unclear. We provide evidence that the lymphangiogenic peptide adrenomedullin, found to be elevated in many tumors, promotes the adhesion and subsequent connexin mediated gap junction intercellular communication between tumor cells and lymphatic endothelial cells. This coupling or ability to communicate promotes transendothelial migration and we identify β-catenin nuclear translocation as a mediator of this process. This study provides novel insight into how the lymphatic endothelium communicates with tumor cells through gap junction channels to facilitate escape through the lymphatic vasculature.
Acknowledgments
a) Acknowledgments: The authors thank Dan O. Kechele and Dr. C. Robert Bagnell and members of the UNC Microscopy Services Laboratory for assistance.
b) Sources of Funding: UNC-CH University Cancer Research Innovation Award and U.S. National Institutes of Health grants HD060860, DK099156 to K.M.C.
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- AM
adrenomedullin
- LEC
lymphatic endothelial cell
- TEM
transendothelial migration
- GJIC
gap junction intercellular communication
Footnotes
c) Disclosures: none
References
- 1.Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, Pepper MS. Vascular endothelial growth factor-c-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 2001;20:672–682. doi: 10.1093/emboj/20.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG. Vegf-d promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–191. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 3.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by vegf-c promotes breast cancer metastasis. Nat Med. 2001;7:192–198. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 4.He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, Harding T, Jooss K, Takahashi T, Alitalo K. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer research. 2005;65:4739–4746. doi: 10.1158/0008-5472.CAN-04-4576. [DOI] [PubMed] [Google Scholar]
- 5.Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine ccr7 signaling. Cancer Cell. 2007;11:526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, Fukumura D, Padera TP, Jain RK. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-c increases metastasis by increasing delivery of cancer cells to lymph nodes: Therapeutic implications. Cancer research. 2006;66:8065–8075. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- 7.Irjala H, Alanen K, Grenman R, Heikkila P, Joensuu H, Jalkanen S. Mannose receptor (mr) and common lymphatic endothelial and vascular endothelial receptor (clever)-1 direct the binding of cancer cells to the lymph vessel endothelium. Cancer research. 2003;63:4671–4676. [PubMed] [Google Scholar]
- 8.Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. Vegf-a induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201:1089–1099. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrell MI, Iritani BM, Ruddell A. Tumor-induced sentinel lymph node lymphangiogenesis and increased lymph flow precede melanoma metastasis. Am J Pathol. 2007;170:774–786. doi: 10.2353/ajpath.2007.060761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herve JC, Derangeon M. Gap-junction-mediated cell-to-cell communication. Cell Tissue Res. 2013;352:21–31. doi: 10.1007/s00441-012-1485-6. [DOI] [PubMed] [Google Scholar]
- 11.Kanady JD, Dellinger MT, Munger SJ, Witte MH, Simon AM. Connexin37 and connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Developmental biology. 2011;354:253–266. doi: 10.1016/j.ydbio.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, Meriney SD, Feingold E, Finegold DN. Gjc2 missense mutations cause human lymphedema. Am J Hum Genet. 2010;86:943–948. doi: 10.1016/j.ajhg.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meens MJ, Sabine A, Petrova TV, Kwak BR. Connexins in lymphatic vessel physiology and disease. FEBS letters. 2014;588:1271–1277. doi: 10.1016/j.febslet.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Rose B, Mehta PP, Loewenstein WR. Gap-junction protein gene suppresses tumorigenicity. Carcinogenesis. 1993;14:1073–1075. doi: 10.1093/carcin/14.5.1073. [DOI] [PubMed] [Google Scholar]
- 15.Mesnil M. Connexins and cancer. Biol Cell. 2002;94:493–500. doi: 10.1016/s0248-4900(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 16.Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nat Rev Cancer. 2010;10:435–441. doi: 10.1038/nrc2841. [DOI] [PubMed] [Google Scholar]
- 17.Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Mori CM, da Silva TC, Oloris SC, Garate AP, Massironi SM, Yamasaki H, Dagli ML. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis. 2004;25:1973–1982. doi: 10.1093/carcin/bgh193. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, DeMattia JA, Song H, Couldwell WT. Communication between malignant glioma cells and vascular endothelial cells through gap junctions. J Neurosurg. 2003;98:846–853. doi: 10.3171/jns.2003.98.4.0846. [DOI] [PubMed] [Google Scholar]
- 19.Ito A, Katoh F, Kataoka TR, Okada M, Tsubota N, Asada H, Yoshikawa K, Maeda S, Kitamura Y, Yamasaki H, Nojima H. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. The Journal of clinical investigation. 2000;105:1189–1197. doi: 10.1172/JCI8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, Nwagwu C, Le DM, Yong VW, Song H, Couldwell WT. Increased invasive capacity of connexin43-overexpressing malignant glioma cells. J Neurosurg. 2003;99:1039–1046. doi: 10.3171/jns.2003.99.6.1039. [DOI] [PubMed] [Google Scholar]
- 21.Pollmann MA, Shao Q, Laird DW, Sandig M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005;7:R522–534. doi: 10.1186/bcr1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanczuga-Koda L, Sulkowski S, Lenczewski A, Koda M, Wincewicz A, Baltaziak M, Sulkowska M. Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J Clin Pathol. 2006;59:429–433. doi: 10.1136/jcp.2005.029272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoletov K, Strnadel J, Zardouzian E, Momiyama M, Park FD, Kelber JA, Pizzo DP, Hoffman R, VandenBerg SR, Klemke RL. Role of connexins in metastatic breast cancer and melanoma brain colonization. Journal of cell science. 2013;126:904–913. doi: 10.1242/jcs.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang B, Peng ZH, Yu PW, Yu G, Qian F, Zeng DZ, Zhao YL, Shi Y, Hao YX, Luo HX. Aberrant expression of cx43 is associated with the peritoneal metastasis of gastric cancer and cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PloS one. 2013;8:e74527. doi: 10.1371/journal.pone.0074527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou JZ, Jiang JX. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions - an update. FEBS letters. 2014;588:1186–1192. doi: 10.1016/j.febslet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JH, Takano T, Cotrina ML, Arcuino G, Kang J, Liu S, Gao Q, Jiang L, Li F, Lichtenberg-Frate H, Haubrich S, Willecke K, Goldman SA, Nedergaard M. Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J Neurosci. 2002;22:4302–4311. doi: 10.1523/JNEUROSCI.22-11-04302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karpinich NO, Kechele DO, Espenschied ST, Willcockson HH, Fedoriw Y, Caron KM. Adrenomedullin gene dosage correlates with tumor and lymph node lymphangiogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:590–600. doi: 10.1096/fj.12-214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldberg GS, Moreno AP, Bechberger JF, Hearn SS, Shivers RR, MacPhee DJ, Zhang YC, Naus CC. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp Cell Res. 1996;222:48–53. doi: 10.1006/excr.1996.0006. [DOI] [PubMed] [Google Scholar]
- 29.Tanmahasamut P, Sidell N. Up-regulation of gap junctional intercellular communication and connexin43 expression by retinoic acid in human endometrial stromal cells. J Clin Endocrinol Metab. 2005;90:4151–4156. doi: 10.1210/jc.2004-0663. [DOI] [PubMed] [Google Scholar]
- 30.Elzarrad MK, Haroon A, Willecke K, Dobrowolski R, Gillespie MN, Al-Mehdi AB. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008;6:20. doi: 10.1186/1741-7015-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodar C, Assar R, Colombres M, Aravena A, Pavez L, Gonzalez M, Martinez S, Inestrosa NC, Maass A. Genome-wide identification of new wnt/beta-catenin target genes in the human genome using cart method. BMC Genomics. 2010;11:348. doi: 10.1186/1471-2164-11-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunworth WP, Fritz-Six KL, Caron KM. Adrenomedullin stabilizes the lymphatic endothelial barrier in vitro and in vivo. Peptides. 2008;29:2243–2249. doi: 10.1016/j.peptides.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis. 2014;17:335–345. doi: 10.1007/s10456-013-9407-0. [DOI] [PubMed] [Google Scholar]
- 34.Teijeira A, Garasa S, Pelaez R, Azpilikueta A, Ochoa C, Marre D, Rodrigues M, Alfaro C, Auba C, Valitutti S, Melero I, Rouzaut A. Lymphatic endothelium forms integrin-engaging 3d structures during dc transit across inflamed lymphatic vessels. J Invest Dermatol. 2013;133:2276–2285. doi: 10.1038/jid.2013.152. [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi H, Fujimoto A, Tanaka M, Yamano T, Hsueh E, Hoon DS. Ccl21 chemokine regulates chemokine receptor ccr7 bearing malignant melanoma cells. Clin Cancer Res. 2004;10:2351–2358. doi: 10.1158/1078-0432.ccr-03-0195. [DOI] [PubMed] [Google Scholar]
- 36.Shields JD, Emmett MS, Dunn DB, Joory KD, Sage LM, Rigby H, Mortimer PS, Orlando A, Levick JR, Bates DO. Chemokine-mediated migration of melanoma cells towards lymphatics--a mechanism contributing to metastasis. Oncogene. 2007;26:2997–3005. doi: 10.1038/sj.onc.1210114. [DOI] [PubMed] [Google Scholar]
- 37.Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA. Vascular endothelial growth factor-c and c-c chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer research. 2009;69:349–357. doi: 10.1158/0008-5472.CAN-08-1875. [DOI] [PubMed] [Google Scholar]
- 38.Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, Feral C, Papayannopoulou T, Lowy A, Blair SL, Cheresh D, Ginsberg M, Varner JA. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer research. 2010;70:3042–3051. doi: 10.1158/0008-5472.CAN-09-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoopes SL, Willcockson HH, Caron KM. Characteristics of multi-organ lymphangiectasia resulting from temporal deletion of calcitonin receptor-like receptor in adult mice. PloS one. 2012;7:e45261. doi: 10.1371/journal.pone.0045261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 41.Ram Z, Walbridge S, Shawker T, Culver KW, Blaese RM, Oldfield EH. The effect of thymidine kinase transduction and ganciclovir therapy on tumor vasculature and growth of 9l gliomas in rats. J Neurosurg. 1994;81:256–260. doi: 10.3171/jns.1994.81.2.0256. [DOI] [PubMed] [Google Scholar]
- 42.Fick J, Barker FG, 2nd, Dazin P, Westphale EM, Beyer EC, Israel MA. The extent of heterocellular communication mediated by gap junctions is predictive of bystander tumor cytotoxicity in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11071–11075. doi: 10.1073/pnas.92.24.11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spray DC, Hanstein R, Lopez-Quintero SV, Stout RF, Jr., Suadicani SO, Thi MM. Gap junctions and bystander effects: Good samaritans and executioners. Wiley Interdiscip Rev Membr Transp Signal. 2013;2:1–15. doi: 10.1002/wmts.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sirnes S, Lind GE, Bruun J, Fykerud TA, Mesnil M, Lothe RA, Rivedal E, Kolberg M, Leithe E. Connexins in colorectal cancer pathogenesis. International Journal of Cancer. 2014:1–11. doi: 10.1002/ijc.28911. [DOI] [PubMed] [Google Scholar]