

Abstract
Patterns of amino-acid polymorphism in human mitochondrial genes have been interpreted as evidence for divergent selection among populations that inhabit climatically distinct environments. If similar patterns are mirrored in other broadly distributed mammalian species, then adaptive modifications of mitochondrial protein function may be detected in comparisons among locally adapted populations of a single wide-ranging species, or among closely related species that have adapted to different environments. Here, we test for evidence of positive selection on cytochrome b variation within and among species of the ecologically diverse rodent genus Peromyscus. We used likelihood-based comparisons of synonymous and nonsynonymous substitution rates to test for evidence of divergent selection between high- and low-altitude haplogroups of the deer mouse, Peromyscus maniculatus. We also tested for evidence of divergent selection among different species of Peromyscus that inhabit different thermal environments. In contrast to the purported evidence for positive selection on mitochondrial proteins in humans and other nonhuman mammals, results of our tests suggest that the evolution of cytochrome b in Peromyscus is chiefly governed by purifying selection.
Keywords: adaptation, cytochrome b, deer mouse, McDonald–Kreitman test, positive selection, Peromyscus
Introduction
The mitochondrial cytochrome b gene encodes an integral membrane protein component of the cytochrome bc1 complex, which catalyzes the redox transfer of electrons from ubiquinone to cytochrome c in the mitochondrial electron transport chain. As the efficiency of the electron transport chain governs key aspects of aerobic energy metabolism (reviewed by Rolfe and Brown, 1997), several investigators have suggested that functional modifications of redox proteins, such as cytochrome b, may be involved in physiological adaptation to different thermal environments (Mishmar et al., 2003; Ruiz-Pesini et al., 2004; Fontanillas et al., 2005). For example, Mishmar et al. (2003) and Ruiz-Pesini et al. (2004) argued that patterns of amino-acid polymorphism in human mitochondrial genes reflect a history of local adaptation to different climatic regimes. Global surveys of nucleotide variation in human mitochondrial genes show that ratios of nonsynonymous to synonymous substitution rates are significantly higher in European/ temperate and Asian (Siberian)/Arctic haplogroups than in the African/tropical haplogroups (Mishmar et al., 2003; Ruiz-Pesini et al., 2004; but see also Sun et al., 2007).
This pattern is consistent with positive selection or a relaxation of purifying selection in the high-latitude populations (Elson et al., 2004; Kivisild et al., 2006). A second pattern that has emerged from phylogenetic analysis of human mtDNA variation is that physico-chemically radical amino-acid substitutions are generally more common in terminal branches of the haplotype genealogies than in the older, internal branches (Moilanen and Majamaa, 2003). This pattern further suggests a history of purifying selection against mildly deleterious amino-acid variants and is consistent with a commonly observed excess of low-frequency amino-acid polymorphisms in mitochondrial genes of humans and other animals (Nachman et al., 1996; Hasegawa et al., 1998; Nachman, 1998; Rand and Kann, 1996, 1998; Fry, 1999). However, in human mtDNA haplogroups that are characteristic of temperate and Arctic regions, a significantly disproportionate number of physicochemically radical amino-acid substitutions were concentrated in the internal branches of the haplotype genealogies, a pattern consistent with a history of lineage-specific positive selection (Ruiz-Pesini et al., 2004). In cytochrome b haplogroups characteristic of north-temperate Eurasia, Ruiz-Pesini et al. (2004) identified physicochemically radical substitutions in highly conserved coenzyme Q (CoQ)-binding sites. As the CoQ-binding sites of cytochrome b play a critical role in the complex III Q cycle, these substitutions may influence the allocation of energy derived from oxidative phosphorylation to metabolic heat production.
If patterns of amino-acid polymorphism in human mitochondrial genes reflect a history of divergent selection among populations that inhabit climatically distinct environments, as suggested by Mishmar et al. (2003) and Ruiz-Pesini et al. (2004), then it stands to reason that parallel patterns of adaptive variation may be observed in other broadly distributed mammalian species that inhabit a similar diversity of thermal environments. Modifications of mitochondrial protein function may be evident in comparisons between locally adapted populations of a wide-ranging species, or between closely related species that have adapted to different environments.
Here, we test for evidence of positive selection on cytochrome b variation within and among species of the ecologically diverse rodent genus, Peromyscus. Mice in the genus Peromyscus inhabit an extremely diverse range of habitat types, ranging from alpine and subarctic environments to subtropical cloud forests in Central America (Hall, 1981; Carleton, 1989). An appreciable fraction of this environmental heterogeneity is also contained within the range of the most widely distributed species in the genus, Peromyscus maniculatus (Hall, 1981). As might be expected, this ecological variation within and among Peromyscus species is mirrored by extensive variation in basal metabolic rates and thermal tolerances (MacMillen and Garland, 1989).
If the highly conserved functions of the electron transport chain have undergone adaptive modifications in human populations that have colonized cold, high-latitude environments within the last 10 000–50 000 years, then similar modifications of redox proteins, such as cytochrome b, may be evident in Peromyscus species or subspecies that have inhabited high altitude or high latitude environments for much longer periods of evolutionary time. In addition to differences in the likely timescale of environmental adaptation, species like P. maniculatus are characterized by much larger effective population sizes than humans, so weak selection on amino-acid variation is more likely to dominate drift.
To test for evidence of divergent selection on cytochrome b variation in deer mice, we compared ratios of synonymous and nonsynonymous substitution among the branches of species- and genus-level cytochrome b phylogenies. To test for evidence of divergent selection within P. maniculatus, we also made use of neutrality tests that compare ratios of polymorphism to divergence at both synonymous and nonsynonymous sites. Finally, we used an alignment of cytochrome b from 632 mammalian species together with structural models of the bc1 complex to ascertain the functional importance of amino-acid substitutions occurring within P. maniculatus and between Peromyscus species.
Materials and methods
Taxon sampling
For the analysis of species-level variation (within the P. maniculatus species complex), we used cytochrome b sequences from a total of 311 specimens collected from 34 localities across North America (Table S1, Supplementary material Online). We sequenced cytochrome b from a total of 143 specimens and we obtained the remaining 168 sequences from GenBank (NCBI). Four sequences from P. keeni and two sequences from P. melanotis were also included in the alignment, as previous studies suggested that these taxa may be part of a ‘species complex’ that includes several genetically distinct lineages that are at present regarded as subspecies of P. maniculatus (Carleton, 1989; Hogan et al., 1993; Dragoo et al., 2006).
For analyses of cytochrome b variation among different species of Peromyscus, we used a total of 38 sequences representing 26 nominal species with diverse altitudinal and latitudinal ranges (Table S2, Supplementary material online). This set of species was selected with the goal of maximizing phylogenetic coverage, while maintaining a relatively even distribution of branch lengths in the reconstructed tree. In the cases of P. betae, P. boylii, P. maniculatus and P. truei, we included specimens that were representative of multiple, genetically distinct subspecies because physiological traits that may be influenced by efficiency of the electron transport chain are known to vary at the intraspecific level (Cook and Hannon, 1954; Chappell and Snyder, 1984; MacMillen and Garland, 1989; Storz, 2007).
As explained below (see Results), the species-level and genus-level phylogeny reconstructions both showed that P. melanotis was basal to a monophyletic group that comprised the entire P. maniculatus species complex. We therefore sequenced cytochrome b from 13 additional P. melanotis specimens (Table S1, Supplementary material online) for the purpose of making outgroup comparisons with P. maniculatus in McDonald-Kreitman tests (McDonald and Kreitman, 1991).
PCR and sequencing
Genomic DNA was isolated from tissue samples with the DNeasy Tissue Kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's specifications. We amplified the cytochrome b gene using the primers Cytochrome-b 15334L (5′-CTTCATTTTTGGTTTACAA GAC-3′) and L14724 (5′-TGATATGAAAAACCATCG TTG-3′) with a 5 min denaturing period (95 °C) followed by 30 cycles of denaturing (94 °C×30 s), annealing (55°Cx30s) and extension (72°C × 60s). PCRs were performed using 2.5 μl of 10×Buffer II (Applied Biosystems, Foster City, CA, USA), 2.5 μl of 25 mm MgCl2, 2.5 μl of 10 mm dNTPs (2.5 μm each, dATP, dGTP, dCTP, dTTP), approximately 100 ng template DNA (0.5-2 μl DNeasy elution), 1U of Ampli-Taq Gold polymerase (Applied Biosystems) and ddH2O to make a total volume of 25 μl. PCR products were purified using ExoSAP-IT (USB Corporation, Cleveland, OH, USA), cycle-sequenced with Big-Dye chemistry and run on an ABI 3730 sequencer (Applied Biosystems). The resulting chromatograms were visually inspected and aligned using Sequencher (Genecodes Corporation, Ann Arbor, MI, USA), and amino-acid translations of DNA sequence were conducted with MacClade (Maddison and Maddison, 1989).
Phylogenetic reconstruction
We used Bayesian and maximum likelihood methods to reconstruct phylogenetic relationships among nonredun-dant cytochrome b haplotypes of P. maniculatus, and among species of Peromyscus. Sequences for Reithrodontomys raviventer and P. leucopus obtained from Genbank were used as outgroups for genus-level (Peromyscus) and species-level (P. maniculatus) data sets, respectively (for accession numbers, refer to Tables S1 and S2, Supplementary material online). Owing to incomplete sequences from some representatives of the P. maniculatus species complex, the species-level analysis was restricted to codons 19–374 of the cytochrome b gene. The genus-level data set included only Peromyscus species for which complete sequences were available, and analyses were therefore performed on the complete 381 codon sequence of the gene.
We used Modeltest (Posada and Crandall, 1998) to select substitution models for our likelihood analyses. For both genus-level and species-level alignments, the best-fitting model was the parameter-rich GTR+G+I. We implemented this same model in our Bayesian analyses to allow for direct comparisons of tree topologies, and because overparameterization is unlikely to introduce type-one or type-two errors into Bayesian phylogenetic reconstructions (Lemmon and Moriarty, 2004).
Bayesian phylogeny reconstructions were conducted using MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003). We fit the GTR+G+I model to each codon position and conducted two simultaneous independent runs of 10 000 000 generations with four simultaneous chains, sampling every 1000th tree. Burn-in values were varied according to the time required for likelihood values to converge. To obtain 50% consensus trees, we discarded the first 1500 sampled trees in the species-level analysis and the first 500 trees in the genus-level analysis.
Maximum likelihood reconstructions were conducted using TreeFinder (Jobb et al., 2004). We pruned the species-level data set to 40 sequences by selecting representative haplotypes from each well-supported clade in the Bayesian phylogeny. We used 1000 pseudoreplicates to compute bootstrap support values.
Summary statistics of DNA polymorphism and divergence were calculated using the program DnaSP (Rozas et al., 2003), and levels of nucleotide divergence between P. maniculatus clades were estimated using MEGA 3.1 (Kumar et al., 2004).
Codon-based analysis of selection pressure
To test for natural selection on cytochrome b coding sequence, we used a maximum likelihood approach to examine variation in the ratio of nonsynonymous and synonymous substitutions, dN/dS (=ω) (Goldman and Yang, 1994; Nielsen and Yang, 1998; Yang and Nielsen, 2000; Bielawski and Yang, 2004). Positive selection is implicated when ω>1, whereas purifying selection is implicated when ω<1. Analyses of the species-level and genus-level cytochrome b alignments were conducted with the codeml program in the PAML 3.15 package (Yang, 1997; http://abacus.gene.ucl.ac.uk/software/ paml.html). All analyses were run three times with different starting values for the ω ratio (0.5, 1 and 2). Maximum likelihood scores for nested models were compared using likelihood ratio tests (Felsenstein, 1981).
For the species-level P. maniculatus data set, we first tested for heterogeneity in ω ratios among branches of the inferred phylogeny by comparing log-likelihood scores for a model that allowed a single ω ratio (model M0) vs a model that allowed separate ω ratios for each branch in the phylogeny (model M1). We then tested for evidence of positive selection (as indicated by ω>1) in specific branches of the P. maniculatus phylogeny. We estimated ω for distinct haplogroups that were characteristic of the Great Plains (the low-altitude haplogroup) and montane environments of the Southern Rocky Mountains (the high-altitude haplogroup). We included 1–3 representative sequences from each branch of the 50% consensus Bayesian phylogeny in these analyses, and compared log-likelihood scores for three evolutionary models. The first model allowed two ω ratios, one for the pooled sample of high- and low-altitude haplogroups, and a second ω ratio for the remaining ‘background’ branches of the intraspecific gene genealogy. The second model allowed ω ratios to vary among haplogroups by assuming three separate ω ratios: one for the high-altitude haplogroup, one for the low-altitude haplogroup and a third one for the remaining background branches. The third model allowed five separate ω ratios for the following partitions of the P. maniculatus gene genealogy: (i) the stem of the high-altitude haplogroup; (ii) the crown of the high-altitude haplogroup; (iii) the stem of the low-altitude hap-logroup; (iv) the crown of the low-altitude haplogroup; and (v) the remaining background branches of the phylogeny. Here and elsewhere, the stem of a given clade refers to the ancestral branch leading to the most recent common ancestor of the set of sequences under consideration, whereas the crown group includes the most recent common ancestor and all descendent sequences. By allowing separate ω ratios for the stem and the crown of a given clade, it may be possible to detect an early acceleration of the nonsynonymous substitution rate that was eventually superseded by purifying selection (Goodman, 1982).
Using the genus-level data set, we designed two intermediate nested models to investigate variation in the ω ratio during the diversification of the P. maniculatus species complex. The first model allowed one ω ratio for the P. maniculatus clade and a second ω ratio for the remaining background branches of the phylogeny. The second model allowed three ω ratios: one for the stem of the P. maniculatus clade, a second for the crown and a third for the remaining background branches of the phylogeny. One limitation of the intermediate nested models is that each assumes a single ω ratio for all amino-acid sites in the cytochrome b alignment, and may therefore fail to detect positive selection on a small subset of sites. We therefore fitted a branch-site model (Yang and Nielsen, 2002; Zhang et al., 2005) to the Peromyscus data set that allowed us to test for rate heterogeneity across sites within the P. maniculatus clade. Model A allowed four classes of sites: site class 0 included codons that were subject to stringent functional constraints with 0<ω0<1 estimated from the data, site class 1 included codons that were unconstrained with ω1 =1 fixed and site classes 2a and 2b included codons that were conserved or neutral on the background branches but were allowed to undergo a shift to positive directional selection in the foreground lineage, with ω2 estimated as a free parameter. We used model A to construct a likelihood ratio test in which the null hypothesis was a simplified version of model A with ω2 =1 fixed, following Zhang et al. (2005). This null model allowed a shift from purifying selection on the background branches to relaxed constraint on the foreground branches. Consequently, the likelihood ratio test involving the comparison of model A vs the null model allowed us to test whether an accelerated rate of nonsynonymous substitution in the foreground lineage could be attributed to positive selection or a relaxation of functional constraint.
Results and discussion
Cytochrome b variation in the P. maniculatus species complex
The phylogeographic structure of cytochrome b variation within P. maniculatus is largely concordant with results obtained in earlier studies of mtDNA variation in this species complex (Avise et al., 1983; Lansman et al., 1983; Dragoo et al., 2006). Our phylogeny reconstruction of the 311 cytochrome b sequences from the P. maniculatus species complex revealed six distinct clades (Figure 1), which we designate as clades 1–6 (following Dragoo et al., 2006). Whereas Dragoo et al. (2006) found P. melanotis to be nested within the P. maniculatus species complex, we found P. melanotis to be sister to a monophyletic group containing all P. maniculatus haplo-types and the nominal species P. keeni. Monophyly of the maniculatus + keeni clade was supported by 78.3% of maximum likelihood bootstrap replicates and by a Bayesian posterior probability of 0.96.
Figure 1.
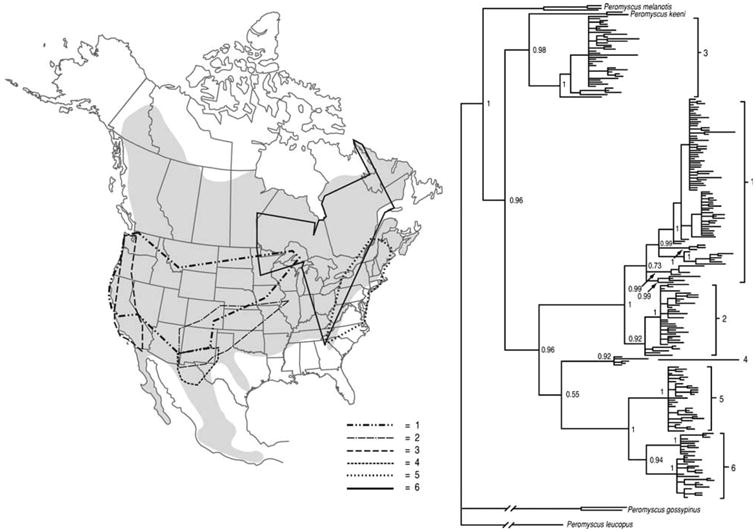
Bayesian phylogeny of cytochrome b sequences from 311 P. maniculatus specimens on the basis of a GTR+I+G model of nucleotide substitution. Bayesian posterior probabilities are given above each branch. A similar topology was recovered in a maximum likelihood phylogeny of 40 randomly selected sequences; all labeled nodes were supported by >50% bootstrap values in 1000 pseudo replicates. The inset map of North America shows the geographic distribution of P. maniculatus (shaded region) and the six major cytochrome b clades.
With the exception of clade 3, which is restricted to the southwestern United States, inferred relationships of all major cytochrome b clades were well supported in Bayesian and maximum likelihood trees. Net nucleotide divergences between clades ranged from 1.7 to 4.4% (Table 1). At the interface between the Great Plains and the Southern Rocky Mountains, we observed a clear phylogeographic break between a high-altitude hap-logroup (contained within clade 1) and a low-altitude haplogroup (contained within clade 2; Figure 2).
Table 1. Pairwise levels of net nucleotide divergence between Peromyscus maniculatus clades depicted in Figure 1, obtained using the Kimura 2-parameter substitution model.
| DNA haplogroup | Clade 1 | Clade 2 | Clade 3 | Clade 4 | Clade 5 | |
|---|---|---|---|---|---|---|
| Clade 1 | Central and western USA | |||||
| Clade 2 | Central USA | 0.017 (0.003) | ||||
| Clade 3 | Western USA | 0.034 (0.006) | 0.035 (0.006) | |||
| Clade 4 | Southwestern USA | 0.044 (0.006) | 0.043 (0.006) | 0.029 (0.005) | ||
| Clade 5 | Northeastern USA and eastern Canada | 0.043 (0.007) | 0.040 (0.007) | 0.040 (0.006) | 0.037 (0.005) | |
| Clade 6 | Northcentral USA and central Canada | 0.038 (0.006) | 0.034 (0.006) | 0.041 (0.006) | 0.032 (0.006) | 0.015 (0.004) |
Values in parentheses represent standard errors computed from 10 000 bootstrap replicates.
Figure 2.
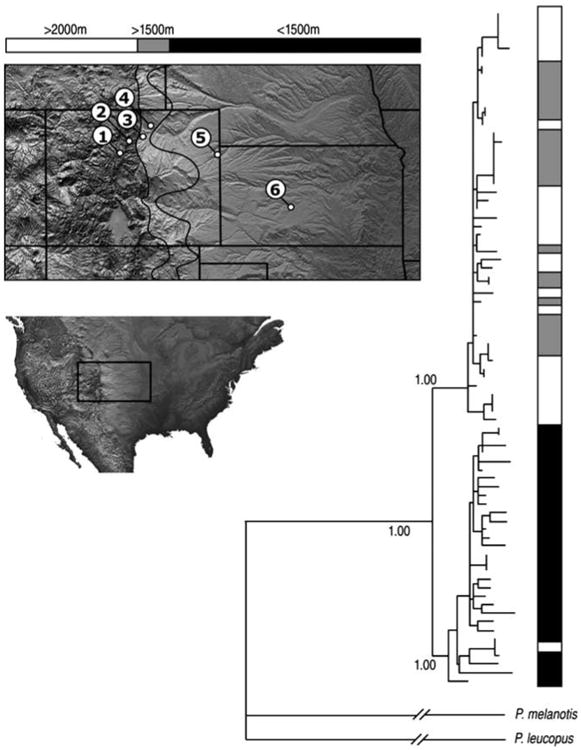
Bayesian phylogeny of cytochrome b sequences from deer mice that were collected along an altitudinal gradient. The gradient spanned the interface between the southern Rocky Mountains and the Great Plains. The inset map shows the location of six collecting localities in Colorado and Kansas, USA: (1) Summit of Mt. Evans, Clear Creek Co., Colorado (4347 m); (2) Niwot Ridge, Boulder Co., CO (2900 m); (3) Mesa Reservoir, Boulder Co., Colorado (1672 m); (4) Nunn, Weld Co., Colorado (1578 m); (5) Bonny Reservoir, Yuma Co., Colorado (1158m); and (6) Fort Larned National Monument, Pawnee Co., Kansas (620 m). The tree was based on a GTR+I+G model of nucleotide substitution. Bayesian posterior probabilities are given above each of the main nodes. The haplogroup containing specimens from montane localities (1–4) is nested within clade ‘1’, and the haplogroup that primarily consists of specimens from plains localities (5–6) is nested within clade ‘2’ (see Figure 1).
Consistent with the findings of Dragoo et al. (2006), all haplogroups were characterized by an excess of low-frequency polymorphisms, as reflected by negative values for Tajima's D and Fu and Li's D* (Table 2). With the exception of clade 4, which was represented in our data set by only six individuals, the values for Tajima's D and Fu and Li's D* were significantly negative in each of the haplogroups from the Great Plains and western North America. This same excess of low-frequency polymorphisms was also observed at multiple, unlinked autosomal loci in a large sample of mice that are representative of clades 1 and 2 (Storz et al., 2007; Storz and Kelly, 2008). The consistently negative skews in the site-frequency distributions of nuclear and mitochondrial loci most likely reflect a recent history of population expansion.
Table 2. Levels and patterns of nucleotide variation in six cytochrome b clades of Peromyscus maniculatus depicted in Figure 1.
| Cytochrome b haplogroup | Class | S | H | HD | π | θ | Tajima's D | Fu and Li's D* | |
|---|---|---|---|---|---|---|---|---|---|
| Clade 1 | Central and western USA (N=123) | Synonymous | 121 | 88 | 0.975 | 0.024 | 0.074 | −2.153** | −4.210** |
| Nonsynonymous | 53 | 44 | 0.801 | 0.019 | 0.077 | −2.331** | −4.765** | ||
| Total | 133 | 91 | 0.977 | 0.008 | 0.024 | −2.177** | −4.495** | ||
| Clade 2 | Central USA (N = 47) | Synonymous | 64 | 35 | 0.984 | 0.018 | 0.046 | −2.156* | −3.006* |
| Nonsynonymous | 27 | 20 | 0.798 | 0.012 | 0.042 | −2.401** | −3.931** | ||
| Total | 74 | 35 | 0.984 | 0.006 | 0.016 | −2.246** | −3.305* | ||
| Clade 3 | Western USA (N = 35) | Synonymous | 84 | 37 | 0.986 | 0.027 | 0.065 | −2.100* | −3.597* |
| Nonsynonymous | 48 | 29 | 0.962 | 0.031 | 0.084 | −2.222** | −4.311** | ||
| Total | 103 | 40 | 0.995 | 0.010 | 0.024 | −2.129* | −3.856** | ||
| Clade 4 | Southwestern USA (N = 6) | Synonymous | 10 | 5 | 0.933 | 0.380 | 0.438 | −0.795 | −0.851 |
| Nonsynonymous | 2 | 2 | 0.333 | 0.333 | 0.438 | −1.132 | −1.155 | ||
| Total | 12 | 5 | 0.933 | 0.004 | 0.005 | −0.913 | −0.967 | ||
| Clade 5 | Northeast USA and eastern Canada (N = 61) | Synonymous | 51 | 31 | 0.953 | 0.015 | 0.034 | −1.855* | −1.409 |
| Nonsynonymous | 15 | 13 | 0.569 | 0.006 | 0.022 | −2.157* | −2.250 | ||
| Total | 53 | 32 | 0.954 | 0.005 | 0.011 | −1.886* | −1.461 | ||
| Clade 6 | Northcentral USA and central Canada (N = 39) | Synonymous | 61 | 29 | 0.979 | 0.192 | 0.226 | −0.528 | −0.694 |
| Nonsynonymous | 12 | 13 | 0.587 | 0.071 | 0.226 | −2.047* | −2.779* | ||
| Total | 73 | 32 | 0.982 | 0.012 | 0.016 | −0.999 | −1.220 | ||
| Total (clades 1–6) (N = 311) | Synonymous | 245 | 226 | 0.993 | 0.099 | 0.141 | −0.914 | −3.448** | |
| Nonsynonymous | 119 | 126 | 0.947 | 0.065 | 0.157 | −1.767* | −6.966** | ||
| Total | 286 | 235 | 0.994 | 0.031 | 0.049 | −1.115 | −4.762** |
Abbreviations: S, number of polymorphic sites; H, number of haplotypes; HD, haplotype diversity; π, nucleotide diversity; θ, proportion of segregating sites.
Tajima's D and Fu and Li's D* compare observed allele frequency distributions with expectations based on neutral theory.
P<0.05;
P<0.01.
Of the 311 cytochrome b sequences we analyzed from the P. maniculatus species complex (defined here as P. maniculatus + P. keeni to the exclusion of P. gossypinus, P. leucopus and P. melanotis), we recovered 236 unique haplotypes at the nucleotide level and 95 unique haplotypes at the amino-acid level. Twenty-two of the amino-acid haplotypes were recovered more than once in the sample. These nonunique haplotypes were distinguished by a total of 37 amino-acid substitutions at 22 sites.
We obtained conflicting results concerning the monophyly of P. maniculatus in species-level and genus-level phylogenies. Phylogenetic reconstructions of the species-level data set indicated that P. keeni and a subset of haplotypes recovered from P. maniculatus gambelli form a monophyletic clade that is sister to the remaining P. maniculatus haplotypes. Reconstructions of the genus-level data set supported monophyly of P. maniculatus with a Bayesian posterior probability of 0.72 and placed P. keeni as the most closely related congener (Figure 3). Overall, species relationships within our genus-level phylogeny were consistent with a recent, independent phylogeny of Peromyscus cytochrome b sequences (Bradley et al., 2007).
Figure 3.
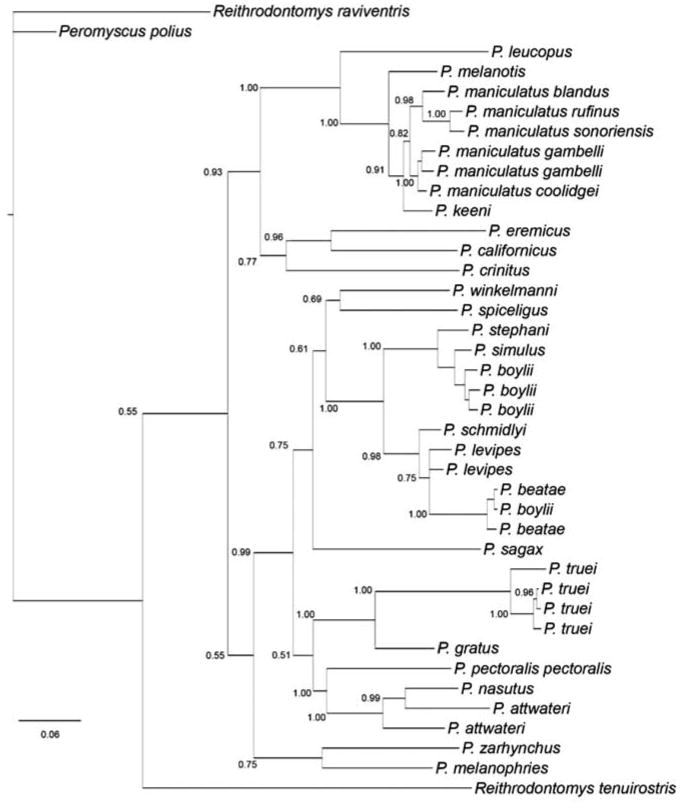
Bayesian phylogeny of cytochrome b sequences from 26 Peromyscus species. Bayesian posterior probabilities are given above each branch. A similar topology was recovered from a maximum likelihood reconstruction using the GTR+I+G model implemented in Treefinder (Jobb et al., 2004); all labeled nodes had bootstrap support values >50%.
The three main structural domains of the cytochrome b protein were characterized by pronounced differences in levels of amino-acid variation (Table 3). Within P. maniculatus, the matrix domain was the most variable and the intermembrane domain was the least variable. This suggests that the intermembrane domain is characterized by more stringent functional constraints, which is consistent with the findings of previous studies (Degliesposti et al., 1993; McClellan et al., 2005).
Table 3. Amino-acid changes in the three functional domains of Peromyscus maniculatus cytochrome b.
| Domain | Proportion of polymorphic sites | Proportion of physicochemically radical substitutions | Proportion of replacement sites with multiple substitutions in the P. maniculatus phylogeny |
|---|---|---|---|
| Matrix | 0.07 (3/41) | 1.00 (10/10) | 0.90 (37/41) |
| Transmembrane | 0.09 (18/202) | 0.17 (4/24) | 0.29 (59/202) |
| Intermembrane | 0.03 (3/106) | 0.33 (1/3) | 0.00 (0/106) |
| χ2 (df=2) | 3.387 (P>0.05) | 35.777 (P<0.001)* | 27.157 (P<0.001)* |
Significant after Bonferronni correction (α = 0.017; P<0.05/3).
Polymorphism and divergence
McDonald–Kreitman tests (McDonald and Kreitman, 1991) comparing polymorphism within P. maniculatus with divergence from P. melanotis revealed an excess of replacement polymorphism within the low-altitude haplogroup, within the high-altitude haplogroup, within keeni-like and maniculatus-like clades and within the pooled sample of all P. maniculatus cytochrome b sequences (Table 4). The excess of replacement polymorphism was statistically significant in the low-altitude haplogroup and in clade 3. Repeating McDonald–Kreit-man tests with cytochrome b sequence from a more distantly related outgroup, P. leucopus (Figures 1 and 3), yielded similar outcomes for each clade (results not shown). This excess of low-frequency amino-acid polymorphism (reflected by neutrality indices>1) is a pattern that is commonly observed in mitochondrial genes of humans and other animals (Nachman et al., 1996; Hasegawa et al., 1998; Nachman, 1998; Rand and Kann, 1996, 1998; Fry, 1999) and is typically interpreted as evidence for purifying selection against mildly deleterious mutations.
Table 4.
McDonald-Kreitman (1991) tests and neutrality indices (Rand and Kann, 1996) on the basis of cytochrome b polymorphism in Peromyscus maniculatus relative to divergence from P. melanotis.
| Haplogroup | Site class | Polymorphic sites | Divergence to P. melanotis | Neutrality index | P-value |
|---|---|---|---|---|---|
| All P. maniculatus | Nonsynonymous | 47 | 1 | 3.57 | 0.31 |
| Synonymous | 194 | 13 | |||
| Clades 1–6, excluding clade 3 | Nonsynonymous | 49 | 1 | 3.14 | 0.32 |
| Synonymous | 157 | 16 | |||
| Clade 3 | Nonsynonymous | 38 | 1 | 13.78 | <0.01 |
| Synonymous | 80 | 29 | |||
| High-altitude haplogroup (subset of clade 1) | Nonsynonymous | 11 | 4 | 3.13 | 0.09 |
| Synonymous | 36 | 41 | |||
| Low-altitude haplogroup (subset of clade 2) | Nonsynonymous | 14 | 2 | 5.22 | 0.03 |
| Synonymous | 55 | 41 |
Phylogenetic relationships and geographic distributions of P. maniculatus haplogroups are depicted in Figure 1.
P-values are derived from Fisher's exact test.
Molecular evolution of cytochrome b within P. maniculatus The free-ratio model of codon substitution provided a slightly better fit to the P. maniculatus data than the one-ratio model, although the difference in log-likelihood scores was not statistically significant (2Δℓ = 107.98, df = 87, P<0.063). As this result suggests the possibility of heterogeneity in the ω ratio among the different P. maniculatus haplogroups, we proceeded with additional tests to determine if the ω ratio differed between high-and low-altitude haplogroups. A model that allowed two ω ratios, one for the pooled sample of high- and low-altitude haplogroups and one for the remaining back-ground branches, provided a better fit to the data than the one-ratio model (2Δ ℓ = 4.09, df=1, P= 0.04). In this two-ratio model, estimates of ω for each group were <1, consistent with a history of purifying selection within both groups. More complex models that allowed for additional sources of rate variation within the high- and low-altitude haplogroups did not significantly improve likelihood scores (Table 5). Thus, there is no evidence for different rates of nonsynonymous substitution in high-and low-altitude haplogroups.
Table 5. Maximum-likelihood analysis of variation in the dN/dS ratio (=ω) across branches of species- and genus-level cytochrome b phylogenies.
| Model | No. of parameters | Parameter estimates | Likelihood |
|---|---|---|---|
| Lineage model: Peromyscus maniculatus haplogroups | |||
| M0 | 1 | ωb=0.041 | −3793.93 |
| M1 (free-ratio) | 88 | — | −3739.94 |
| (Background) + (high and low) | 2 | ωb = 0.034 | −3791.88 |
| ωhl =0.063 | |||
| (Background) + (high) + (low) | 3 | ωb = 0.034 | −3791.80 |
| ωh = 0.070; ωl =0.0058 | |||
| (Background) + (high stem) + (high crown) + (low stem) + (low crown) | 5 | ωb = 0.034 | −3790.94 |
| ωhs =0.119; ωhc =0.064 | |||
| ωls = 1×10-4; ωlc = 0.062 | |||
| Lineage model: Peromyscus species | |||
| M0 | 1 | ω=0.028 | −9289.81 |
| M1 (free-ratio) | 76 | — | −9220.34 |
| (Background) + (P. maniculatus total group) | 2 | ωb = 0.028 ωPm = 0.031 | −9289.76 |
| (Background) + (P. maniculatus stem) + (P. maniculatus crown) | 3 | ωb = 0.028 | −9286.72 |
| ωs = 2/0 | |||
| ωc = 0.025 |
‘High’ and ‘low’ refer to the high- and low-altitude haplogroups of P. maniculatus (see text for details). Subscripts denote the following: b=background branches, h=high-altitude haplogroup, l=low-altitude haplogroup, s=stem, c=crown, and Pm=P. maniculatus clade.
Molecular evolution of cytochrome b within the genus Peromyscus
The free-ratio model provided a better fit to the genus-level data than the one-ratio model (2Δ ℓ = 138.94; df= 75, P<0.001). As this result indicated rate heterogeneity among the branches of the genus-level Peromyscus phylogeny, we tested for evidence of an accelerated rate of nonsynonymous substitution within the clade representing the most broadly distributed Peromyscus species, P. maniculatus. A model that allowed three separate ω ratios (one for the stem of the P. maniculatus clade, one for the crown and one for the background branches) provided a significantly better fit to the data than a more simple model that allowed two separate ratios (one for the entire P. maniculatus clade (stem + crown) and one for the background branches; 2Δ ℓ=6.07; df = 1; P<0.014) (Table 5). This heterogeneity in ω ratios is attributable to the fact that the stem of the P. maniculatus clade was characterized by two nonsynonymous substitutions and zero synonymous substitutions. Ancestral sequence reconstruction (using the codeml program) revealed that the two substitutions occurred at residue positions 110 and 345. The substitution at residue position 110 involved replacement of a polar threonine with a hydrophobic methionine in the bc-loop of the matrix domain, and the substitution at position 345 involved replacement of an uncharged tyrosine with a positively charged histidine in the H helix of the transmembrane domain. Neither of these two substitutions is predicted to have important effects on cyto-chrome b function. Residue positions 110 and 345 are among the most variable sites in cytochrome b of mammals (Figure S1, Supplementary material online), which suggests that these two sites are not subject to particularly stringent functional constraints. Sites that were identified in earlier studies as potential targets of positive selection in human cytochrome b (Mishmar et al., 2003; Ruiz-Pesini et al., 2004) were invariant both within P. maniculatus and among Peromyscus species.
Branch-site models revealed no evidence for positive selection in the P. maniculatus lineage. The model allowing ω>1 for a proportion of sites in the stem of the P. maniculatus clade did not identify any sites under positive selection, and had the same log-likelihood score (lnL= –9198.96) as a simplified model in which the entire cytochrome b gene was constrained to ω = 1 in foreground and background lineages.
Conclusion
We detected no evidence of positive selection on cytochrome b variation among phylogeographically distinct haplogroups of P. maniculatus or among different species of Peromyscus. Results of our McDonald-Kreit-man tests revealed evidence for purifying selection against mildly deleterious amino-acid mutations in P. maniculatus, and results of our phylogenetic analysis of codon-substitution patterns revealed that the evolution of cytochrome b in Peromyscus is chiefly governed by purifying selection. Our results stand in contrast to reports of positive selection on cytochrome b in recent human history (Mishmar et al., 2003; Ruiz-Pesini et al., 2004) and during the diversification of anthropoid primates (Andrews et al., 1998; Grossman et al., 2004). If it is true that human cytochrome b has undergone adaptive modifications in response to metabolic challenges associated with the colonization of temperate and sub-Arctic environments, our results demonstrate that such patterns are not universal among broadly distributed, temperate zone mammals. The evolutionary pattern described for humans is clearly not mirrored by species, such as deer mice, that inhabit an even more diverse range of thermal environments.
Supplementary Material
Acknowledgments
We thank S Sabatino and C Dingle for help in the lab, F Hoffmann for help with data analysis and H Moriyama for help with protein structure modeling. F Hoffman, T Miller, H Moriyama, R Nichols, G Orti, A Runk, T Zera and two anonymous reviewers provided helpful comments on the manuscript. This work was funded by grants to JFS from the National Science Foundation (DEB-0614342), the National Institutes of Health (R01 HL087216) and the Nebraska Research council and grants to EJG from the American Society of Mammalogists and the University of Nebraska (LSIGRP, School of Biological Sciences).
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
References
- Andrews TD, Jermiin LS, Easteal S. Accelerated evolution of cytochrome b in simian primates: adaptive evolution in concert with other mitochondrial proteins? J Mol Evol. 1998;47:249–257. doi: 10.1007/pl00006382. [DOI] [PubMed] [Google Scholar]
- Avise JC, Shapira JF, Daniel SW, Aquadro CF, Lansman RA. Mitochondrial DNA differentiation during the specia-tion process in Peromyscus. Mol Biol Evol. 1983;1:38–56. doi: 10.1093/oxfordjournals.molbev.a040301. [DOI] [PubMed] [Google Scholar]
- Bielawski JP, Yang Z. A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J Mol Evol. 2004;59:121–132. doi: 10.1007/s00239-004-2597-8. [DOI] [PubMed] [Google Scholar]
- Bradley RD, Durish ND, Rogers DS, Miller JR, Engstrom MD, Kilpatrick CW. Towards a molecular phylogeny for Peromyscus: evidence from mitochondrial cytochrome-b sequences. J Mammal. 2007;88:1146–1159. doi: 10.1644/06-MAMM-A-342R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton MD. Systematics and evolution. In: Kirkland GL, Layne JN, editors. Advances in the Study of Peromyscus (Rodentia) Texas Tech University Press; Lubbock, Texas: 1989. pp. 7–141. [Google Scholar]
- Chappell MA, Snyder LR. Biochemical and physiological correlates of deer mouse alpha-chain hemoglobin polymorphisms. Proc Natl Acad Sci USA. 1984;81:5484–5488. doi: 10.1073/pnas.81.17.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SF, Hannon JP. Metabolic differences between three strains of Peromyscus maniculatus. J Mammal. 1954;35:553–560. [Google Scholar]
- Degliesposti M, Devries S, Crimi M, Ghelli A, Patarnello T, Meyer A. Mitochondrial cytochrome-b—evolution and structure of the protein. Biochim Biophys Acta. 1993;1143:243–271. doi: 10.1016/0005-2728(93)90197-n. [DOI] [PubMed] [Google Scholar]
- Dragoo JW, Lackey JA, Moore KE, Lessa EP, Cook JA, Yates TL. Phylogeography of the deer mouse (Peromyscus maniculatus) provides a predictive framework for research on hantaviruses. J Gen Virol. 2006;87:1997–2003. doi: 10.1099/vir.0.81576-0. [DOI] [PubMed] [Google Scholar]
- Elson JL, Turnbull DM, Howell N. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection. Am J Hum Genet. 2004;74:229–238. doi: 10.1086/381505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Fontanillas P, Depraz A, Giorgi MS, Perrin N. Non-shivering thermogenesis capacity associated to mitochondrial DNA haplotypes and gender in the greater white-toothed shrew, Crocidura russula. Mol Ecol. 2005;14:661–670. doi: 10.1111/j.1365-294X.2004.02414.x. [DOI] [PubMed] [Google Scholar]
- Fry AJ. Mildly deleterious mutations in avian mitochondrial DNA: evidence from neutrality tests. Evolution. 1999;53:1617–1620. doi: 10.1111/j.1558-5646.1999.tb05426.x. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;5:725–726. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Goodman M. Positive selection causes purifying selection. Nature. 1982;295:630. doi: 10.1038/295630a0. [DOI] [PubMed] [Google Scholar]
- Grossman LI, Willdman DE, Schmidt TR, Goodman M. Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet. 2004;20:579–584. doi: 10.1016/j.tig.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Hall ER. The Mammals of North America. I & II. John Wiley & Sons; New York: 1981. [Google Scholar]
- Hasegawa M, Cao Y, Yang Z. Preponderance of slightly deleterious polymorphism in mitochondrial DNA: nonsynonymous/synonymous rate ratio is much higher within species than between species. Mol Biol Evol. 1998;15:1499–1505. doi: 10.1093/oxfordjournals.molbev.a025877. [DOI] [PubMed] [Google Scholar]
- Hogan KM, Hedin MC, Koh HS, Davis SK, Greenbaum AF. Systematic and taxonomic implications of karyo-typic, electrophoretic, and mitochondrial-DNA variation in Peromyscus from the Pacific Northwest. J Mammal. 1993;74:819–831. [Google Scholar]
- Grossman LI, Wildman DE, Schmidt TR, Goodman M. Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet. 2004;20:579–584. doi: 10.1016/j.tig.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Jobb G, von Haeseler A, Strimmer K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004;4:18. doi: 10.1186/1471-2148-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kivisild T, Shen P, Wall DP, Do B, Sung R, Davis K, et al. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lansman RA, Avise JC, Aquadro CF. Extensive genetic variation in mitochondrial DNA's among geographic populations of the deer mouse, Peromyscus maniculatus. Evolution. 1983;37:1–16. doi: 10.1111/j.1558-5646.1983.tb05509.x. [DOI] [PubMed] [Google Scholar]
- Lemmon AR, Moriarty EC. The importance of proper model assumption in Bayesian phylogenetics. Syst Biol. 2004;53:265–277. doi: 10.1080/10635150490423520. [DOI] [PubMed] [Google Scholar]
- MacMillen RE, Garland TJ. Adaptive physiology. In: Kirkland GL, Layne JN, editors. Advances in the Study of Peromyscus (Rodentia) Texas Tech University Press; Lubbock, Texas: 1989. pp. 143–168. [Google Scholar]
- Maddison WP, Maddison DR. Interactive analysis of phylogeny and character evolution using the computer program MacClade. Folia Primatol (Basel) 1989;53:190–202. doi: 10.1159/000156416. [DOI] [PubMed] [Google Scholar]
- McClellan DA, Palfreyman EJ, Smith MJ, Moss JL, Christensen RG, Sailsbery JK. Physicochemical evolution and molecular adaptation of the cetacean and artiodactyl cyto-chrome b proteins. Mol Biol Evol. 2005;22:437–455. doi: 10.1093/molbev/msi028. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, et al. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci USA. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moilanen JS, Majamaa K. Phylogenetic network and physicochemical properties of nonsynonymous mutations in the protein-coding genes of human mitochondrial DNA. Mol Biol Evol. 2003;20:1195–1210. doi: 10.1093/molbev/msg121. [DOI] [PubMed] [Google Scholar]
- Nachman MW. Deleterious mutations in animal mito-chondrial DNA. Genetica. 1998;102–103:61–69. [PubMed] [Google Scholar]
- Nachman MW, Brown WM, Stoneking M, Aquadro CF. Nonneutral mitochondrial DNA variation in humans and chimpanzees. Genetics. 1996;142:953–963. doi: 10.1093/genetics/142.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Droso-phila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Mutation and selection at silent and replacement sites in the evolution of animal mitochondrial DNA. Genetica. 1998;102–103:393–407. [PubMed] [Google Scholar]
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- Storz JF. Hemoglobin function and physiological adaptation to hypoxia in high-altitude mammals. J Mammal. 2007;88:24–31. [Google Scholar]
- Storz JF, Kelly JK. Effects of spatially varying selection on nucleotide diversity and linkage disequilibrium: Insights from deer mouse globin genes. Genetics. 2008;180:367–379. doi: 10.1534/genetics.108.088732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF, Sabatino SJ, Hoffmann FG, Gering EJ, Moriyama H, Ferrand N, et al. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 2007;3:e45, 0448–0459. doi: 10.1371/journal.pgen.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Kong Q, Zhang Y. The role of climate in human mitochondrial DNA evolution: a reappraisal. Genomics. 2007;89:338–342. doi: 10.1016/j.ygeno.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. CABIOS. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.