

Abstract
Background
Due to the indolent nature of prostate cancer, new prognostic measures are needed to identify patients with life threatening disease. SAM pointed domain-containing Ets transcription factor (SPDEF) has been associated with good prognosis and demonstrates an intimate relationship with the androgen receptor (AR), however its role in prostate cancer progression remains unclear.
Methods
A tissue microarray constructed from cores of 713 consecutive radical prostatectomy specimens were immunohistochemically stained for SPDEF and correlated with progression free and metastatic free survival. In vitro studies assessed growth rate, migration, and sensitivity to bicalutamide to explore mechanisms behind the tissue microarray observations.
Results
Patients with high SPDEF demonstrate longer metastases free survival after receiving the standard of care (HR = 9.80, P = 0.006). SPDEF expression corresponded with bicalutamide growth inhibition and apoptosis induction in all cell lines studied. In addition, a feed-forward loop of AR-SPEF expression regulation is observed.
Conclusions
SPDEF may be clinically useful to identify patients who will have extended benefits from androgen deprivation therapy. In vitro observations suggest SPDEF mediates initial sensitivity to androgen deprivation therapy through both AR regulation and downstream events.
Keywords: prostate, SPDEF, androgen receptor, metastasis
Introduction
Prostate cancer (CaP) is the most common malignancy in the US today, and is often treated successfully by radical prostatectomy, but ∼15% of patients will subsequently recur [1]. Androgen deprivation therapy (ADT) is the most common intervention for both recurrent and metastatic CaP, and has shown to extend life, relieve pain, and suppress PSA levels in 80–90% of patients [2,3]. About 90% of patients will relapse within 2–3 years, at which point expected survival is 16–18 months [4]. However, 10% of patients will survive for 10+ years after ADT, which suggests a curative response [5].
Huggins and Hodges observed in 1942 that prostate luminal epithelial cells depend on AR for survival and proliferation [6]. Upon removal of androgens, apoptosis is induced in normal and malignant prostate cells alike, this observation led to the use of ADT for the management of advanced CaP [6]. Studies have shown proliferation and apoptosis indices in both malignant and benign tissues return to baseline levels by 7–10 days after castration, suggesting that ADT effects are transient and castrate resistance may be driven by cells in the tumor that avoid apoptosis and therefore are not AR dependent [7].
Both AR-dependent luminal and non-AR-dependent basal epithelial cells are capable of being tumor-initiating cells [8]. Both luminal and basal tumor-initiating cells have shown the ability to give rise to well-differentiated carcinoma, which indicate that histological differentiation may not always reflect the molecular phenotype of the tumor [9–11]. Gleason score, a histological rubric of CaP prognosis, is an independent predictor of death from CaP, but has a relatively low positive predictive value [12]. Potentially, CaP tumors arising from the intermediate or basal cell population may be resistant to ADT, but may look similar in gross histological appearance to a tumor arising from a luminal epithelial cell. Desensitization to ADT and acquisition of aggressive tumor characteristics could be explained by a lack of luminal epithelial differentiation. However, many lines of evidence indicate castration recurrent prostate cancer (CRPC) is in some ways AR-dependent and hijacks AR networks to keep signaling active despite castrate conditions. These mechanisms include, but are not limited to, AR hypersensitivity, amplification, and constitutive activation, and intra-tumoral androgen production [1,2,13–16]. Therefore, it is apparent that a clearer understanding of AR signaling in sensitivity to ADT and the formation of CRPC is needed to establish biomarkers relevant to disease progression.
A protein intimately involved with AR gene regulation and may itself be an AR target gene is SAM pointed domain-containing Ets transcription factor (SPDEF). SPDEF was found to bind AR and NKX 3.1 to regulate prostate specific antigen [17]. SPDEF has shown to be necessary and sufficient to mediate terminal epithelial differentiation in airway and gastrointestinal epithelia, and evidence supports a similar role in CaP [18–20]. In cancer, some studies have shown SPDEF expression is associated with good prognosis and inhibits many oncogenic events, while others have shown that SPDEF can transform premalignant cells and is up-regulated in hyperplastic and neoplastic specimens [21–27]. Furthermore, SPDEF has shown to regulate apoptosis through suppression of survivin [28,29]. Given its readily apparent, but poorly understood role in CaP more study is warranted to discern the exact role of SPDEF in CaP progression.
The goal of the present study is to determine the effect SPDEF plays in the development of metastatic CRPC. Therefore, SPDEF expression was analyzed using immunohistochemistry (IHC) in a tissue micro-array (TMA) constructed from formalin fixed paraffin embedded (FFPE) radical prostatectomy (RP) specimens, for associations with clinical outcomes to determine the significance of SPDEF expression in early stage CaP and the formation of CRPC. To explore the mechanism behind observations made in the TMA specimens, SPDEF expression was modulated through overexpression and shRNA silencing and the effects on cell growth, migration, and the sensitivity of cells to bicalutamide induced growth inhibition and apoptosis induction were assessed. In addition, in silico observations on published ChIP-seq data sets indicate both transcription factors, AR and SPDEF, contain binding sites for one another either within the gene or in close proximity, and thus may regulate each other's transcription therefore SPDEF was also studied as a novel regulator of AR [30,31]. Studies were performed in models of multiple disease states, which included androgen sensitive LNCaP, castrate recurrent LNCaP C4-2, and AR-negative PC-3-M.
Materials and Methods
Patients
All patients studied had a diagnosis of clinically localized prostatic adenocarcinoma and underwent RP between 1993 and 2005 at Roswell Park Cancer Institute (RPCI, Buffalo, NY). Under an IRB approved protocol, tissue specimens were incorporated into TMAs as described below and clinical information was abstracted from medical records; clinical information included clinical stage and Gleason score, surgery date, pathological stage and Gleason score, margin status, all PSA values and any CaP-related treatments and dates, distant metastatic disease and diagnostic date, date of death and whether death was a result of CaP. PSA values were used to determine recurrence using both the American Urological Association (AUA) definition, which defines a recurrence as an initial PSA value ≥ 0.2 ng/ml followed by a subsequent confirmatory PSA value ≥ 0.2 ng/ml, and the National Comprehensive Cancer Center Network (NCCN) definition, a detectable PSA that rises on two or more subsequent determinations.
IHC Staining of SPDEF
Three, 0.6 mm tissue cores of FFPE prostate tissue per patient obtained at RP were arrayed, using the Beecher Manual Tissue Microarrayer Model MTA-I (Beecher Instruments, Silver Spring, MD). Specimens for controls consisted of cores from 10 different organs including lung, breast, kidney, liver, spleen, ovary, testes, colon, tonsil, and brain and represented slightly more than 20% of all cores in each TMA block. TMA blocks were sectioned and IHC stained by the Pathology Resource Network within the Department Pathology at RPCI. In brief, TMA blocks were cut in 5 mm sections, placed on charged slides, and dried at 60°C for l hr. Slides were cooled to room temperature, deparaffinized in three changes of xylene, and rehydrated using graded alcohols. Endogenous peroxidase was quenched twice with aqueous 3% H2O2 and washed with PBS/T. For antigen retrieval, the TMAs were immersed in Target Retrieval Solution, pH 9 (Dako) and heated in a vegetable steamer, followed by a 20 min cool down and a PBS/T wash. Slides were then loaded on a DAKO Autostainer and blocked with serum-free protein block (Dako). The protein block was blown off and the primary rabbit Ab for SPDEF (made in lab and previously referenced [29]) was applied at a concentration of 5.6 mg/ml for l hr [29]. An isotype-matched control (5.6 mg/ml rabbit IgG) was used on a duplicate slide in place of the primary Ab as a negative control. Slides were rinsed with PBS/T and anti-rabbit Envision + reagent (Dako) was applied for 30 min. PBS/T was used as a wash and the chromagen DAB+ (Dako) was applied for 10 min. After IHC staining, slides were counterstained with Hematoxylin, dehydrated, cleared and cover-slipped. Adenocarcinoma was identified in each core and given a score (0–5) based on the intensity of SPDEF staining. The scoring system for SPDEF was as follows; 0 = undetectable staining, l = very faint, 2 = faint, 3 = moderate, 4 = intense, 5 = very intense. Scores were averaged and patients were then stratified into tertiles based on SPDEF expression. AR was previously stained by the RPCI Pathology Resource Network, is available for institutional analysis, and was scored similarly to SPDEF.
Cell Culture
Human CaP cell lines, LNCaP, LNCaP C4-2, and PC-3-M were cultured in RMPI 1640 medium (Cellgro) with 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Cellgro) at 37°C and 5% CO2. Androgen starvation was performed by culturing the LNCaP cells in phenol-red free RMPI 1640 with 10% charcoal stripped serum (Atlanta Biologicals). Bicalutamide (Casodex, Sigma) was diluted in complete media to reach indicated concentrations before being applied to cells and incubated for indicated timepoints.
Lentiviral shRNA and cDNA Overexpression Preparation and Delivery
pGIPz Lentivius
Bacterial stocks of pGIPz lentiviral particles encoding shRNA towards AR (oligo ID #s V2LHS_239220; V2LHS_239574; V2LHS_149850; and V2LHS_149847, Open Biosystems) or SPDEF (oligo ID#s V2LHS_48718; V2LHS_48716; V2LHS_48714, Open Biosystems) were obtained from the shRNA core facility at RPCI. LNCaP and LNCaP C4-2 cells were allowed to reach 80% confluence before incubation with virus containing supernatant overnight. Media is changed the second day and cells are allowing to recover and express the vector for 48 hr before selection. Stable cells were selected with puromycin (2 ug/ml) for 1 week. Stable cell lines were verified by western blot using antibodies for AR (Santa Cruz, Ab# N-20), SPDEF (homemade), and HSP70 (Santa Cruz, AB# K-20) as a loading control. HSP70 was chosen so that all three proteins could be probed on the same blot, some literature suggests HSP70 may be androgen regulated, however it was not observed in this study (data not shown) [32].
pcDNA3.1-SPDEF overexpression
SPDEF cDNA was encoded in pcDNA3.1 and transfected in human CaP cell lines LNCaP, LNCaP C4-2, and PC-3-M with LipoD293 (SignaGen). Media was changed after overnight incubation with transfection media and cells were allowed to recover and expression vector for 48 hr. Stably expressing cells were selected for with neomycin (200 ug/ml) for 1 week. Stable cell lines were verified by Western blot using antibodies for SPDEF (homemade) and HSP70 (Santa Cruz, AB# K-20).
Cell Growth Estimation
Stable lines were plated equally after selection and both plating amounts and cell growth at time points of 24, 48, and 72 hr was determined by trypan blue exclusion using Vi-Cell XR Cell Viability Analyzer (Beckman Coulter). To assess cell growth inhibition by bicalutamide, stable lines were plated equally and allowed to attach overnight before treatment with 50 mM bicalutamide for 72 hr. 3-[4,5-dimethylthiazol-2-yl]-2,5-dipheny ltetrazoliumbromide (MTT) was used for an estimation of cell population after bicalutamide treatment and absorbance was measured using a Synergy 2 optical plate reader (Bio-tek). No significant difference was observed between either of the empty vector controls; therefore for clarity one column was omitted. The shRNA knockdown data is representative of three different shRNA constructs. Apoptosis was confirmed assessing caspase-7 cleavage (Cell signaling, AB#9491) by Western blot.
Flow Cytometry
Apoptotic, live, and dead cell populations were quantified by flow cytometry using Hoescht dye (HD) and propidium iodide (PI) staining from the Chromatin Condensation/Dead Cell Apoptosis Kit with Hoechst 33342 and PI for Flow Cytometry (Invitrogen). A LSRII (Becton Dickinson) flow cytometer was used in the analysis.
Migration Assay
Migration was assessed using the Cultrex Cell Migration Assay (Trevigen Inc.) with manufacturer protocols. In brief, cells were serum starved overnight before 25,000 cells/well were plated in the top of a 96-well transwell chamber plate and allowed to migrate into FBS for 24 hr. The bottom chamber was then washed and labeled with Calcien AD (Trevigen Inc.), provided in kit, and fluorescence was measured using a Synergy 2 optical plate reader (Bio-tek).
Results
TMA Patient Summary
A TMA was assembled of 0.6 mm cores from RP specimens according to materials and methods. Of the 713 patients initially studied, 125 patients received neo-adjuvant hormone therapy prior to RP and were analyzed separately, and 5 patients did not have follow-up. Additionally, SPDEF scores could not be obtained for 54 patients due to missing cores or cores with insufficient adenocarcinoma. 536 patients were represented in the main analysis. Of the 125 patients who received neo-adjuvant ADT prior to RP and were excluded from the main analysis, 54 had experienced a biochemical recurrence by the AUA definition, 51 by NCCN definition, and 12 experienced distant metastases.
Clinico-pathological parameters, SPDEF stratification, and resulting patient population percentages for each are described in Supplemental Table I. Clinico-pathological parameters in this patient population were typical of patients treated at RPCI. A median of 105 months of patient follow-up time was available. Evidence of recurrent disease was observed in 148 patients: 117 patients recurred by AUA guidelines and 132 recurred by NCCN guidelines. 106/148 patients that recurred received at least one treatment post-radical prostatectomy RP. Of those, 82% received radiation, and 15% received hormonal therapy and 3% either received combination radiation/hormonal therapy or chemotherapy. Forty-two patients received a second treatment, of which 95% received hormone therapy and 5% received radiation. Eighteen patients experienced confirmed distant metastasis.
Loss of SPDEF Expression Associates with the Incidence of Metastatic Disease
Tumor cores from RP specimens on the TMA containing adenocarcinoma were given scores based on SPDEF intensity, which were then tabulated and averaged across triplicate TMAs. An average staining intensity of 1.71 was observed. SPDEF staining was seen in the nuclei of epithelial cells, with some cores also having positive cytoplasmic staining (Fig. 1A and B). Univariate and multivariate analyses using Cox proportional hazard modeling showed that neither age nor body mass index (BMI) associated with recurrence (Supplemental Tables II and III). Standard clinico-pathologic features (e.g., Gleason sum, extra-capsulary disease, positive margins, and peak pre-op PSA) independently correlated with biochemical recurrence by either definition, and in both univariate and multivariate analysis (Supplemental Tables II and III). Loss of SPDEF expression did not correlate with biochemical recurrence by either definition (Fig. 1C).
Fig. 1.
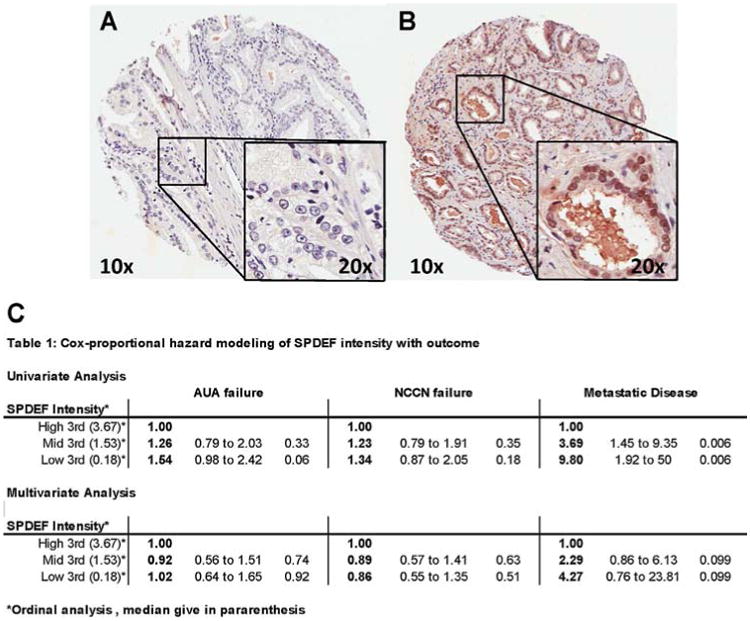
Example SPDEF IHC staining of TMA tumor core. TMA was constructed and IHC stained by RCPI PRN. A: Analysis consisted of 0.6 mm tumor and normal tissue cores from 713 patients arrayed in paraffin blocks. A: Example of SPDEF intensity = 0, (B) example of SPDEF intensity = 5. C: Cox proportional hazard modeling of SPDEF staning intensity and outcome rates. SPDEF does not associate with biochemical failure, however significantly associates with metastatic disease incidence in the univaritate analysis; the multivariate shows a trend, although it is not significant.
Among standard clinico-pathologic variables, only high Gleason sum (hazard ratio (HR) = 60.08; P < 0.0001) and the presence of extra-capsulary disease (HR = 12.48; P < 0.001) associated with increased incidence of metastatic disease. These associations were observed in both univariate and multivariate analysis (Supplemental Tables IV and V). Loss of SPDEF expression (HR = 3.69, SPDEF mid; to HR = 9.80, SPDEF low; P = 0.006) significantly associated with a coordinated increased incidence of metastatic disease, however while a trend was seen (SPDEF low, HR 4.27) in multivariate analysis, it was not statistically significant. Kaplan–Meier survival analysis confirmed the Cox proportional hazard results; patients whose tumors were SPDEF high demonstrated significantly longer metastatic disease-free survival (P = 0.0097) (Fig. 2).
Fig. 2.
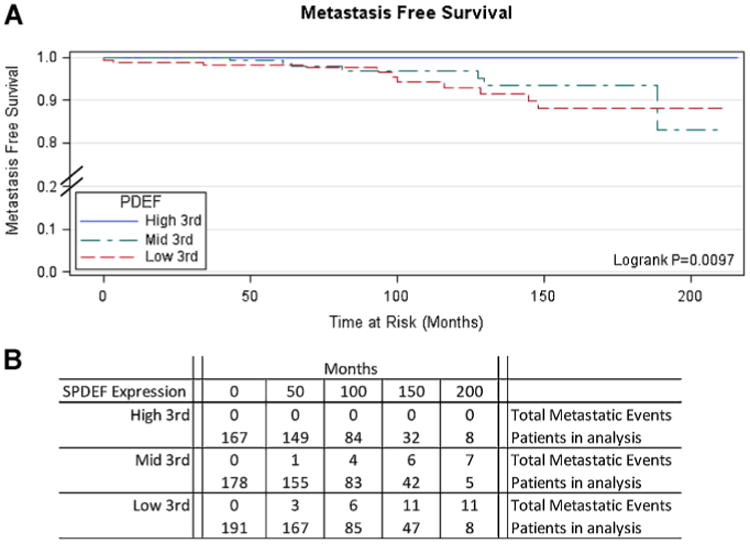
KaplanˆMeier survival analysis demonstrate significantly longer metastasis free survival in patients with high SPDEF. TMA was assembled from consecutive RP specimens of CaP patients at RPCI treated with the standard of care (n = 713). Cores IHC stained in triplicate with a-SPDEF and given a score on a1ˆ5 scale based on intensity. Metastasis disease defined as distant metastasis to lymph or organ sites. A median of 105 months follow-up was available for analysis. Analysis performed with SAS. A: Kaplan survival analysis. B: Numerical representation of patients and events analyzed at each time point.
These analyses indicate that although all clinico-pathological parameters associate with a greater incidence of biochemical recurrence, many parameters do not associate with metastatic disease. Consistent with clinical observations, Gleason score and the presence of extra-capsullary disease were most strongly associated with metastatic disease. Patients with high SPDEF expression demonstrated a lower incidence and greater metastatic disease-free survival time. Therefore, high SPDEF expression could have potential as a clinical biomarker of patients at a lower risk of developing metastatic disease. Analysis of patients who received neo-adjuvant ADT further supports SPDEF's role as a clinical biomarker. While not significant, patients with high SPDEF tended to have longer metastatic disease-free survival (Supplemental Fig. 1).
In order to clarify the observations made in the patients represented on the TMA, the relationship between SPDEF and AR expression was explored in the patients that received ADT. AR and SPDEF expression were seen to significantly correlate (Supplemental Fig. 2; Spearman correlation coefficients, P < 0.0001). This observation was further supported by publicly available ChIP-seq data suggesting that AR and SPDEF bind to genomic regions proximal to each other and thus may play a role in regulating one another's expression [30,31]. Interestingly, while SPDEF significantly associated with incidence of metastatic disease in patients who received ADT, AR expression did not (Supplemental Tables VI and VII), suggesting SPDEF may play a role downstream of AR. Therefore the relationship between AR and SPDEF expression and SPDEF's role in the sensitivity to bicalutamide was explored in vitro.
SPDEF and AR Act in a Feed-Forward Mechanism
Therefore to test whether AR regulates SPDEF expression, androgen sensitive LNCaP cells were treated with 10 nM DHT + 10% FBS, 10% FBS (complete media; CM), 10% CSS-FBS (androgen starved; AS), or treated with 20 mM bicalutamide (non-steroidal AR inhibitor) for 24 hr. 10 nM DHT modestly induced SPDEF expression compared to CM, and either androgen starvation or inhibition of AR resulted in downregulation of SPDEF protein (Fig. 3A) and mRNA (Fig. 3B). A similar downregulation of both SPDEF protein and mRNA was seen after shRNA AR knockdown (Fig. 3C and D).
Fig. 3.
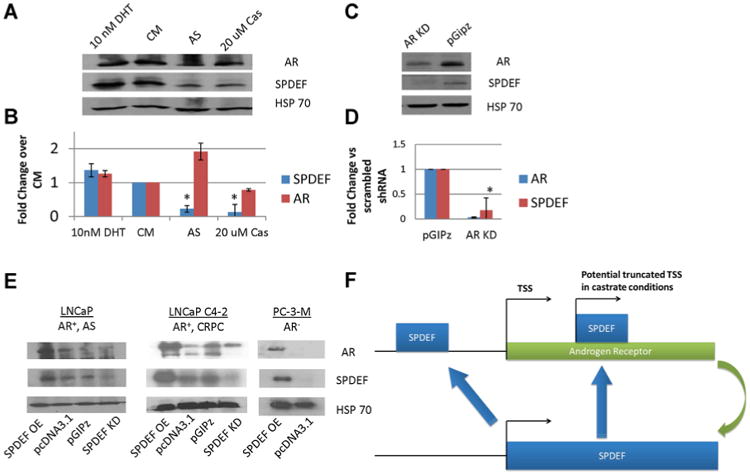
ARˆSPDEF Feed-forward Loop. Cells were grown in RPMI 1640 + 10% with either 10 nM DHT or 20 mM bicalutamide or 10% CSS FBS and SPDEF and AR expression is indicated by (A) protein by western blot, or (B) mRNA by RT-qPCR. Cells infected with pGIPz lentivirus particles encoding shRNA towards AR results in a similar down-regulation of SPDEF expression as indicated by (C) protein by western blot and (D) mRNA by qPCR. AS = Androgen Starvation, *P < 0.05, OE= overexpression, EV=empty vectors, KD = knockdown. E: Reciprocal regulation of AR by SPDEF Cells were either transfected with pcDNA3.1-SPDEF (SPDEF OE) or infected with pGIPz-shRNA SPDEF (SPDEF KD). Western blot was used to detect protein expression for AR, SPDEF, and HSP70. SPDEF OE can be seen to increase AR expression in all three-cell lines tested, however SPDEF KD only downregulated the short form of AR expressed in LNCaPC4-2 cells. Error bars indicate standard deviation and *P < 0.05 by students t-test. F: Hypothetical mechanism for SPDEFˆAR regulation. AR activation induces SPDEF expression. SPDEF potentially binds elements in AR promoter to induce full-length receptor in normal conditions In castrate conditions SPDEF may bind at elements within the AR gene to activate the truncated form of the receptor.
These results indicate that SPDEF is regulated by AR, but do not rule out that SPDEF could play a role in AR regulation. Therefore, western blot was used to analyze AR expression after SPDEF overexpression and knockdown in two models of AR+ cells, LNCaP and the castrate recurrent subline LNCaP C4-2, and after SPDEF overexpression in AR− PC-3-M cells (Fig. 3E). Overexpression of SPDEF induced AR expression in all models including the AR− PC-3-M cells, in contrast to SPDEF knockdown, which only seemed to preferentially down-regulate the constitutively active short form of AR LNCaP C4-2 cells. These results indicate SPDEF may be a novel regulator of AR and also suggest that SPDEF and AR act in a feed-forward mechanism (Fig. 3F).
Role of SPDEF in CaP Disease Progression
To determine SPDEF effects on disease progression the growth rate of cells after SPDEF overexpression and shRNA KD was studied. Results show SPDEF KD significantly inhibited growth rate in LNCaP and LNCaP C4-2 cells while overexpression of SPDEF increased the growth of LNCaP C4-2 cells (Fig. 4A and B). In contrast, overexpression of SPDEF inhibited growth of PC-3-M cells, which is consistent with the established role of ectopically expressing AR in this cell type [33] (Fig. 4C). Taken together these results suggest SPDEF plays a role in androgen sensitive growth of CaP cell lines.
Fig. 4.
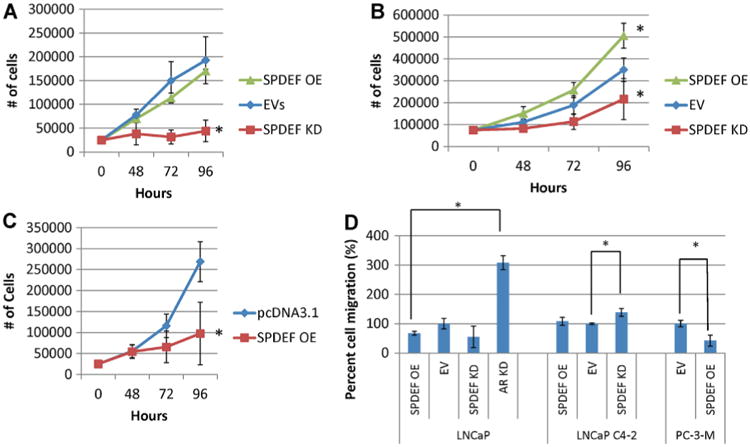
SPDEF effects on disease progression. After selection, cells were plated at 25,000 cells well and allowed to grow for 96 hr in RPMI1640 + 10% FBS and antibiotic. At indicated timepoints wells were harvested in triplicate. Cell number was assessed by trypan blue exclusion in (A) LNCaP, (B) LNCaP C4-2, and (C) PC-3-M. D: SPDEF inhibits migration in androgen independent CaP lines After infection/transfection and subsequent selection, 20,000 cells were plated in RPMI1640 without FBS in the top well of a 96-well modified Boyden-chamber assay plate and let migrate into FBS overnight. Cell number was estimated by Calcein-AD fluorescence. Error bars indicate standard deviation. *P < 0.05 OE= Overexpression, EV=empty vectors, KD = knockdown, AS= androgen sensitive.
A modified Boyden chamber assay was used to assess the migration of stable cells either ectopically expressing SPDEF cDNA, or shRNA against SPDEF or AR (Fig. 4D). Neither overexpression nor knockdown of SPDEF had a significant effect on the migration of androgen sensitive LNCaP cells. Only knockdown of SPDEF in LNCAP C4-2 cells significantly increased migration and ectopic expression of SPDEF in PC-3-M cells significantly inhibited migration compared to empty vector controls. These results may indicate that SPDEF effects on motility are context dependent and may depend on AR regulation. To explore this relationship AR was knocked down in LNCaP cells. LNCaP AR KD cells demonstrate significantly increased migration compared to EV controls, which is consistent with previous literature reports [34]. Given SPDEF's apparent intimate role in AR regulation and function SPDEF role in sensitivity to bicalutamide was studied.
SPDEF Mediates Bicalutamide Growth Inhibition in CaP
Stably selected cells either expressing SPDEF cDNA or SPDEF shRNA were treated with 50 mM bicalutamide for 72 hr. Knockdown of SPDEF decreased bicalutamide growth inhibition in both LNCaP and LNCaP C4-2 cells, while ectopic expression of SPDEF increased the sensitivity of LNCaP C4-2 and PC-3-M cells to bicalutamide (Fig. 5A). Flow cytometry was used to assess live, dead and apoptotic cell populations after 72 hr of 50 mM bicalutamide treatment. Overexpression of SPDEF in LNCaP C4-2 and PC-3-M cells increased the populations of both dead cells and cells in early apoptosis that were induced by bicalutamide treatment (Fig. 5B). Additionally, a slight trend was seen where the SPDEF knockdown decreased bicalutamide induced dead and apoptotic cell populations in both LNCaP and LNCaP C4-2 cells (Fig. 5B). Apoptosis was confirmed in LNCaP and LNCaP C4-2 cells by western blot for caspase-7 cleavage. Consistent with flow cytometry results, SPDEF expression level correlated with amount of caspase-7 induced, however the phenotype was more pronounced in LNCaP C4-2 cells compared to LNCaP cells (Fig. 6). While modest these results indicate that SPDEF plays a role in bicalutamide induced apoptosis and growth inhibition and support IHC results.
Fig. 5.
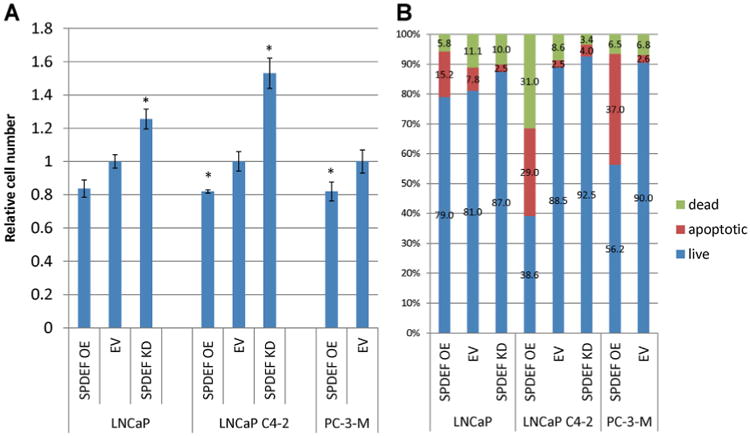
A: SPDEF expression corresponded with sensitivity to bicalutamide growth inhibition. After SPDEF transfection/infection and selection, cells were treated with 50 mM bicalutamide for 72 hr. Cell number was estimated by MTT. No significant difference was observed between EV controls, one EV bar was omitted. All numbers are reported compared to untreated control. B: SPDEF expression correlates with sensitivity to bicalutamide. Cells treated with 50 mM Casodex for 72 hr in triplicate, stained with HD and PI, and then analyzed by flow cytometry. Error bars indicate standard deviation.*P < 0.05, OE=overexpression, EV=empty vectors, KD = knockdown.
Fig. 6.
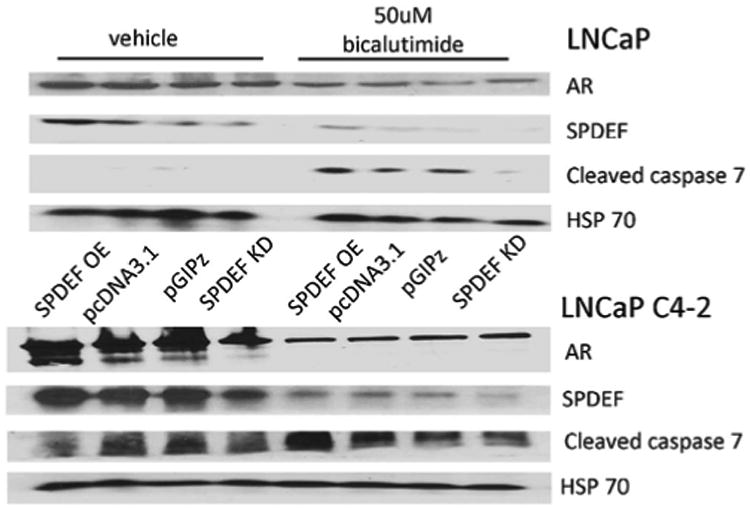
Bicalutamide induces caspase 7 cleavage in cells expression SPDEF. LNCaP and LNCaP C4-2 cells were transfected/infected with vectors containing SPDEF cDNA or shRNA or control vectors and subsequently treated with 50 mM bicalutamide. SPDEF, AR, and cleaved caspase 7 protein expression were detected by western blot. HSP 70 was used as a loading control. OE= overexpression, EV= empty vectors, KD = knockdown.
Discussion
Identification of patients at risk of developing metastatic disease is the largest obstacle faced by clinicians in management of CaP today. In both patients receiving ADT post-biochemical failure and patients receiving neo-adjuvant ADT prior to RP, high SPDEF expression associated with greater metastatic disease-free survival after therapy, which is consistent with previous findings that SPDEF expression was a positive prognostic factor in several malignancies [22,28]. However, due to the fact that the patients who developed metastatic disease in both cohorts received ADT at some point in their treatment, conclusions could not be made to whether observed associations were the result of natural disease progression or sensitivity to ADT without additional mechanistic evidence. Therefore, the second goal of this study was to determine the effect of SPDEF expression on disease progression and ADT response separately, to support or rule out what role SPDEF plays in the development of metastatic CRPC.
Modulation of SPDEF expression demonstrated mixed effects on disease progression, which could indicate that SPDEF functions are dependent on cell type and AR status. In LNCaP cells, which are derived from human luminal epithelial androgen sensitive CaP, overexpression of SPDEF resulted in increased AR expression, but KD of SPDEF had no effect on AR expression. This is in contrast to the effects of SPDEF on the growth rate of LNCaP cells after SPDEF modulation, where KD of SPDEF significantly inhibited growth, but overexpression of SPDEF largely had no effect. Taken together, these results suggest that a feed forward loop may exist between AR and SPDEF, and that SPDEF may be a downstream effector of androgen stimulated growth (Fig. 3F). In support of this, overexpression of SPDEF slightly increased sensitivity of cells to both bicalutamide growth inhibition and apoptosis; conversely KD SPDEF antagonized the effect but to a larger degree. No observable difference was seen in migration after SPDEF modulation. These results suggest SPDEF plays a role in androgen dependent proliferation and survival in androgen sensitive LNCaP cells.
In LNCaP C4-2 cells, which are a castration recurrent subline of LNCaP cells, SPDEF overexpression results in increased expression of both the full length and short form AR, although SPDEF KD only decreased the short form of AR expression. This is consistent with the apparent feed-forward loop observed in LNCaP cells, with the additional step of SPDEF driving expression of the short form of the receptor. Consistent with these observations, both overexpression and shRNA KD of SPDEF showed a concordant change in growth rate, and both bicalutamide sensitivity and apoptotic induction. SPDEF KD was also observed to increase migration in LNCaP C4-2 cells. All effects were more pronounced in the LNCaP C4-2 cells compared to LNCaP cells, which may be due to regulation of the short form of AR.
The CaP cell line PC-3-M expresses a very low level of SPDEF and is considered to be AR negative. Over-expression of SPDEF was seen to induce AR expression, which in turn decreased the growth rate of PC-3-M cells, consistent with results observed after ectopic AR expression in PC-3 cells [33]. Overexpression of SPDEF also resulted in sensitization to bicalutamide growth inhibition and apoptosis induction and decreased migration rate.
Taken together these data support the potential of a feed-forward loop between SPDEF and AR in all cell types studied. However, while a correlation was observed in the TMA between tumor AR and SPDEF expression, AR expression itself did not associate with metastatic disease incidence post-ADT, further suggesting SPDEF plays a role in sensitivity to ADT. SPDEF KD made LNCaP and LNCaP C4-2 cells resistant to bicalutamide; AR was not lost in these cells. Therefore these results implicate SPDEF in a downstream role in AR dependence, rather than through AR expression regulation itself.
In the current study, all cells ectopically expressing SPDEF were more sensitive to both growth inhibition and apoptosis induction by bicalutamide compared to controls as well as SPDEF KD cells. Sensitivity to ADT induced apoptosis is consistent with a role for SPDEF in the terminal differentiation of prostate luminal epithelial cells which is also consistent with functions demonstrated in the colon, stomach, and airway epithelium [19,20,35]. Furthermore, overexpression of SPDEF induced AR expression in PC-3-M cells considered AR− and sensitized these cells to apoptosis; suggesting that ectopic expression of SPDEF caused PC-3-M cells to become more luminal like. Definitive studies would need to assess cytokeratin expression patterns to confirm the presence of luminal markers.
These results support the idea that SPDEF may be involved in AR dependence and suggest a feed-forward mechanism of AR and SPDEF regulation, where AR is required for SPDEF expression, but not vice versa. Paradoxically, SPDEF KD was observed to ablate the constitutively active short form of the AR in LNCaP C4-2 cells, which may indicate the SPDEF-AR feed-forward loop is hijacked in castrate conditions. Publicly available ChIP-seq data shows extensive SPDEF binding across the AR gene, which could provide a mechanism of SPDEF mediated transcription of the short form of AR [31]. Additional experiments could determine if SPDEF activates secondary transcriptional start sites in the AR gene in castrate conditions, thus activating the expression of the short form of AR.
Supplementary Material
Statement of novelty and impact.
SAM pointed domain-containing Ets transcription factor (SPDEF) is an androgen receptor target gene that is intimately involved in normal epithelial cell differentiation but however has shown both oncogenic and tumor suppressor roles in cancer. We demonstrate utility of tumor SPDEF expression as a predictive biomarker for patients that will experience extended benefit from androgen deprivation therapy. Mechanistic in vitro studies indicate that the benefits are likely due to SPDEF mediating sensitivity to apoptosis induced by androgen withdrawal and inhibition of migration. In addition, we show that SPDEF's function is dependent on AR status, likely due to the fact that SPDEF is shown to be a novel regulator of both full length and truncated androgen receptor expression.
Acknowledgments
This work was sponsored by NIH R01 Grant (CA133241) to Dr. Fengzhi Li, and by shared resources supported by NCI Cancer Center Core Support Grant to Roswell Park Cancer Institute (CA016056). We would like to thank the RPCI shRNA Core Facility for providing the SPDEF shRNA, the RPCI Pathology Research Network for performing the immunohistochemistry, and the RPCI Clinical Data Network for data mining the clinical data.
Grant sponsor: NIH R01 Grant; Grant number: CA133241; Grant sponsor: NCI Cancer Center Core Support Grant; Grant number: CA016056.
Footnotes
Conflict of interest: None.
Supporting Information: Additional supporting information may be found in the online version of this article.
References
- 1.Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223–234. doi: 10.1007/978-0-387-69080-3_21. [DOI] [PubMed] [Google Scholar]
- 2.Azzouni F, Mohler J. Biology of castration-recurrent prostate cancer. Urol Clin North Am. 2012;39:435–52. doi: 10.1016/j.ucl.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–1671. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 5.Tangen CM, Faulkner JR, Crawford ED, Thompson IM, Hirano D, Eisenberger M, Hussain M. Ten-year survival in patients with metastatic prostate cancer. Clin Prostate Cancer. 2003;2:41–45. doi: 10.3816/cgc.2003.n.011. [DOI] [PubMed] [Google Scholar]
- 6.Huggins C. Effect of orchiectomy and irradiation on cancer of the prostate. Ann Surg. 1942;115:1192–1200. doi: 10.1097/00000658-194206000-00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohlson N, Wikstrom P, Stattin P, Bergh A. Cell proliferation and apoptosis in prostate tumors and adjacent non-malignant prostate tissue in patients at different time-points after castration treatment. Prostate. 2005;62:307–315. doi: 10.1002/pros.20139. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 11.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, Patay Z, Scoggins M, Ogg R, Pei Y, Yang ZJ, Brun S, Lee Y, Zindy F, Lindsey JC, Taketo MM, Boop FA, Sanford RA, Gajjar A, Clifford SC, Roussel MF, McKinnon PJ, Gutmann DH, Ellison DW, Wechsler-Reya R, Gilbertson RJ. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andren O, Fall K, Franzen L, Andersson SO, Johansson JE, Rubin MA. How well does the Gleason score predict prostate cancer death? A 20-year followup of a population based cohort in Sweden. J Urol. 2006;175:1337–1340. doi: 10.1016/S0022-5347(05)00734-2. [DOI] [PubMed] [Google Scholar]
- 13.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–319. [PubMed] [Google Scholar]
- 14.Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: Locking up the molecular escape routes. Clin Cancer Res. 2009;15:3251–3255. doi: 10.1158/1078-0432.CCR-08-1171. [DOI] [PubMed] [Google Scholar]
- 15.Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, Wilson EM. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486–1496. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Titus MA, Zeithaml B, Kantor B, Li X, Haack K, Moore DT, Wilson EM, Mohler JL, Kafri T. Dominant-negative androgen receptor inhibition of intracrine androgen-dependent growth of castration-recurrent prostate cancer. PLoS ONE. 2012;7:e30192. doi: 10.1371/journal.pone.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Nandi AK, Li X, Bieberich CJ. NKX-3.1 interacts with prostate-derived Ets factor and regulates the activity of the PSA promoter. Cancer Res. 2002;62:338–340. [PubMed] [Google Scholar]
- 18.Steffan JJ, Koul S, Meacham RB, Koul HK. The transcription factor SPDEF suppresses prostate tumor metastasis. J Biol Chem. 2012;287:29968–29978. doi: 10.1074/jbc.M112.379396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noah TK, Kazanjian A, Whitsett J, Shroyer NF. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp Cell Res. 2010;316:452–465. doi: 10.1016/j.yexcr.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, Gregorieff A, Clevers H, Whitsett JA. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest. 2009;119:2914–2924. doi: 10.1172/JCI39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sood AK, Kim H, Geradts J. PDEF in prostate cancer. Prostate. 2012;72:592–596. doi: 10.1002/pros.21461. [DOI] [PubMed] [Google Scholar]
- 22.Steffan JJ, Koul HK. Prostate derived ETS factor (PDEF): A putative tumor metastasis suppressor. Cancer Lett. 2011;310:109–117. doi: 10.1016/j.canlet.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Turner DP, Findlay VJ, Moussa O, Semenchenko VI, Watson PM, LaRue AC, Desouki MM, Fraig M, Watson DK. Mechanisms and functional consequences of PDEF protein expression loss during prostate cancer progression. Prostate. 2011;71:1723–1735. doi: 10.1002/pros.21389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson TR, Koul S, Kumar B, Khandrika L, Venezia S, Maroni PD, Meacham RB, Koul HK. Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer: Regulation of MMP 9 by PDEF. Mol Cancer. 2010;9:148. doi: 10.1186/1476-4598-9-148. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Schaefer JS, Sabherwal Y, Shi HY, Sriraman V, Richards J, Minella A, Turner DP, Watson DK, Zhang M. Transcriptional regulation of p21/CIPl cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J Biol Chem. 2010;285:11258–11269. doi: 10.1074/jbc.M109.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sood AK. PDEF and PDEF-induced proteins as candidate tumor antigens for T cell and antibody-mediated immunotherapy of breast cancer. Immunol Res. 2010;46:206–215. doi: 10.1007/s12026-009-8129-2. [DOI] [PubMed] [Google Scholar]
- 27.Gu X, Zerbini LF, Otu HH, Bhasin M, Yang Q, Joseph MG, Grail F, Onatunde T, Correa RG, Libermann TA. Reduced PDEF expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res. 2007;67:4219–4226. doi: 10.1158/0008-5472.CAN-06-3689. [DOI] [PubMed] [Google Scholar]
- 28.Ghadersohi A, Sharma S, Zhang S, Azrak RG, Wilding GE, Manjili MH, Li F. Prostate-derived Ets transcription factor (PDEF) is a potential prognostic marker in patients with prostate cancer. Prostate. 2011;71:1178–1188. doi: 10.1002/pros.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghadersohi A, Pan D, Fayazi Z, Hicks DG, Winston JS, Li F. Prostate-derived Ets transcription factor (PDEF) downregulates survivin expression and inhibits breast cancer cell growth in vitro and xenograft tumor formation in vivo. Breast Cancer Res Treat. 2007;102:19–30. doi: 10.1007/s10549-006-9314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolrna A, Varjosalo M, Gehrke AR, Yan J, Talukder S, Turunen M, Taipale M, Stunnenberg HG, Ukkonen E, Hughes TR, Bulyk ML, Taipale J. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu S, Tan Z, Wortman M, Dong Z. Regulation of heat shock protein 70-1 expression by androgen receptor and its signaling in human prostate cancer cells. Int J Oncol. 2010;36:459–467. [PMC free article] [PubMed] [Google Scholar]
- 33.Heisler LE, Evangelou A, Lew AM, Trachtenberg J, Elsholtz HP, Brown TJ. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol Cell Endocrinol. 1997;126:59–73. doi: 10.1016/s0303-7207(96)03970-6. [DOI] [PubMed] [Google Scholar]
- 34.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci USA. 2008;105:12182–12187. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park KS, Korfhagen TR, Bruno MD, Kitzmiller JA, Wan H, Wert SE, Khurana Hershey GK, Chen G, Whitsett JA. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest. 2007;117:978–988. doi: 10.1172/JCI29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.