

Abstract
In this work we report synthetic adhesins (SAs) enabling the rational design of the adhesion properties of E. coli. SAs have a modular structure comprising a stable β-domain for outer membrane anchoring and surface-exposed immunoglobulin domains with high affinity and specificity that can be selected from large repertoires. SAs are constitutively and stably expressed in an E. coli strain lacking a conserved set of natural adhesins, directing a robust, fast, and specific adhesion of bacteria to target antigenic surfaces and cells. We demonstrate the functionality of SAs in vivo, showing that, compared to wild type E. coli, lower doses of engineered E. coli are sufficient to colonize solid tumors expressing an antigen recognized by the SA. In addition, lower levels of engineered bacteria were found in non-target tissues. Therefore, SAs provide stable and specific adhesion capabilities to E. coli against target surfaces of interest for diverse applications using live bacteria.
Keywords: adhesins, cell surface antigens, E. coli, synthetic biology, tumor targeting
One of the aims of synthetic biology is the rational design of microorganisms for biotechnological applications.1 An important aspect for the design of these tailored microorganisms is the molecular composition of their cell surface, which determines their interaction with the environment. Incorporation of affinity ligands on the surface of the engineered cell could help to program its adhesion properties toward a target surface or cell of interest. Natural bacteria encode in their genomes diverse types of adhesive proteins, termed adhesins, which play a fundamental role in multiple processes (e.g., biofilm formation, host-colonization, cell invasion) by recognizing molecular structures found in abiotic and biotic surfaces of other bacteria, plant, and animal cells.2,3 However, the variety and redundancy of natural adhesins, together with the widespread distribution of their receptors among different cell types and surfaces, preclude their use as functional units conferring predictable adhesion capabilities.
E. coli is a common host for biotechnological applications due to its amenability for genetic manipulation and heterologous protein expression.4,5 Although some E. coli isolates have acquired pathogenicity traits, most strains of this Gram-negative bacterium are nonpathogenic commensals of the gastrointestinal (GI) tract of vertebrates.6 Interestingly, E. coli has shown potential for in vivo biomedical applications against infections agents and cancer. Specific E. coli strains have been used in humans as probiotics competing with pathogenic bacteria causing infections in the mucosal surfaces of the GI and urinary tracts.7,8 In addition, commensal and probiotic E. coli strains administered systemically have been shown to colonize solid tumors grafted subcutaneously in mice.9,10 Hence, engineering the adhesion properties of these nonpathogenic E. coli strains may improve their colonization efficiency and specificity for target mucosal surfaces and tumors in vivo.
In this work we report synthetic adhesins (SAs) that allow the rational design of robust and specific adhesion capacities to E. coli. SAs have a modular structural organization resembling that of natural E. coli adhesins, which are commonly composed of an anchor β-barrel module that is embedded in the bacterial outer membrane (OM) and an adhesion module bearing an immunoglobulin (Ig)-like fold that is exposed to the extracellular milieu.2,11 The designed SAs combine a highly stable β-barrel domain derived from intimin, an adhesin found in enterohemorrhagic and enteropathogenic E. coli strains (EHEC and EPEC, respectively),12,13 with Ig domains of high affinity, solubility, and stability based on the variable domain of heavy chain-only antibodies (HCAbs), known as VHHs (VH of HCAbs) or nanobodies.14 VHHs of defined specificity can be selected against antigens of interest from large VHH repertories cloned and expressed in E. coli and its bacteriophages.14,15
We show that SAs integrated in the chromosome of E. coli can be constitutively expressed, being nontoxic and stably maintained throughout multiple bacterial generations in vitro and in vivo in the absence of inducer and selection pressure. Expression of SAs in an engineered E. coli chassis devoid of a set of natural adhesins directed the specific adhesion of bacteria to abiotic surfaces and mammalian cells expressing on their surface the target molecule recognized by the SA. Importantly, in vivo experiments showed that low doses of the engineered bacteria colonize solid tumors expressing the target molecule with higher efficiency than the wild type E. coli strain, having also lower retention in normal organs.
Results and Discussion
Structure of Synthetic Adhesins and Targeted Adhesion of E. coli to Antigenic Surfaces
Although Ig-fragments have been displayed on the surface of E. coli with various expression systems,11 such as lipoprotein fusions16 and β-domains of autotransporters,17 adhesion of E. coli to target surfaces has not been observed with those systems. Lipoprotein fusions severely disturb the integrity of the OM causing significant leakiness and toxicity,18 whereas autotransporters are prone to proteolysis as part of their secretion mechanism.19 Recently, we reported that libraries of VHH can be displayed on the surface of E. coli cells fused to an N-terminal fragment of intimin from EHEC (residues 1–659; named Neae), allowing the isolation of high affinity clones binding an antigen of interest.15 The Neae polypeptide (ca. 69 kDa) comprises the native intimin signal peptide (SP), LysM domain for peptidoglycan binding, a 12-stranded β-barrel domain for OM insertion, and a surface-exposed Ig-like domain (named D0) lacking adhesion capacity (Figure 1a).12,13 VHH domains (ca. 13 kDa) are fused to the C-end of Neae generating protein fusions termed NVHH with a total mass of ca. 84 kDa and tagged with E- and myc-tag epitopes (Figure 1a).
Figure 1.
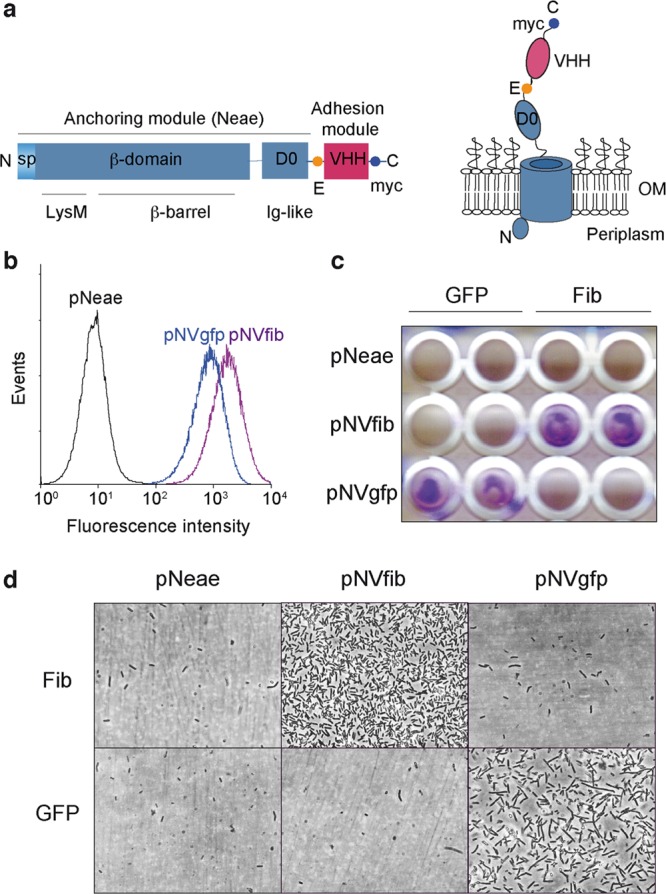
Synthetic adhesins and targeting of E. coli cells to antigens immobilized on a plastic surface. (a) Scheme of the primary structure of SAs (left) showing the N-terminal domain of intimin (Neae) as anchoring module, comprising the signal peptide (SP), LysM, β-barrel, and D0 Ig-like domains, fused to a variable Ig domain from heavy-chain-only antibodies (VHH) as adhesion module. The E-tag and myc-tag epitopes flanking the VHH domain are also indicated. Model of a SA fusion protein (right) in the bacterial outer membrane (OM) with the VHH exposed to the extracellular milieu. (b) Flow cytometry analysis of IPTG-induced E. coli EcM1 bearing pNeae (control), pNVgfp, or pNVfib plasmids. Histograms show the fluorescence intensity of bacteria stained with anti-myc mAb and secondary anti-mouse IgG-Alexa 488. (c) Induced E. coli EcM1 expressing the Neae polypeptide control (pNeae) or SAs against GFP (pNVgfp) or human fibrinogen (pNVfib) were incubated with plastic surfaces coated with GFP or human fibrinogen (Fib), as indicated. Bacterial adhesion was assessed by crystal violet staining. (d) Adhesion of E. coli bacteria to target antigen-coated plastic surface (as in panel c) observed under the light microscope.
To investigate whether NVHH fusions could modify the adhesion properties of E. coli, we first examined the specific adhesion of bacteria to abiotic surfaces coated with the antigen recognized by the VHH. To this end, NVHH fusions binding GFP (NVgfp) and human fibrinogen (NVfib) were expressed in E. coli K-12 strain EcM1 (MG1655ΔfimA-H; Supplementary Table 1). This strain has a deletion of the fimA-H operon encoding type 1 fimbriae, a natural adhesin found in most E. coli strains and involved in recognition of mannosylated glycoproteins found on epithelial cell surfaces.20 Cultures of EcM1 bacteria bearing plasmids encoding NVgfp, NVfib, and Neae (Supplementary Table 1), were induced with IPTG, and the display of the NVHH protein fusions on the surface of E. coli was confirmed by flow cytometry with anti-myc monoclonal antibody (mAb) (Figure 1b). Induced bacteria were incubated on GFP- and fibrinogen (Fib)-coated plastic surfaces, washed with PBS, and stained with crystal violet to reveal the presence of bound bacteria. This experiment demonstrated the specific binding to those surfaces coated with the antigen recognized by the corresponding NVHH fusion (Figure 1c). Specific adhesion of E. coli bacteria was confirmed by light microscopy (Figure 1d). These results indicated that NVHH fusions could act as SAs driving the adhesion of E. coli bacteria to a target surface.
Constitutive, Stable, and Nontoxic Expression of SAs from the Chromosome of E. coli.
To further investigate E. coli targeting mediated by SAs, we developed constitutive gene expression cassettes that can be integrated as a single copy in the chromosome of E. coli. This strategy would allow constant expression of the SA in the absence of exogenous inducers and antibiotics for plasmid maintenance. For site-specific integration in the chromosome of E. coli, we adapted a marker-less gene deletion strategy that enables sequential manipulation of multiple gene loci leaving no antibiotic resistance genes or any other vector sequences in the chromosome.21 This technology is based on the insertion of a suicide plasmid bearing homology regions (HRs) of the targeted gene and the subsequent generation of double-strand breaks by expression of I-SceI restriction endonuclease in vivo, which are repaired, resulting in the specific modification of the targeted gene (Supplementary Figure S1). For this we constructed a suicide vector with I-SceI sites, named pGE (Supplementary Table 1), and generated derivatives for integration of SAs having VHHs binding GFP (pGEflu-SAgfp) or binding TirM (pGEflu-SAtir).15 TirM is the extracellular domain (residues 252–360) of the translocated intimin receptor of EHEC.22 These constructs incorporated a constitutive promoter, PN25,23 driving the expression of the SA, and HRs flanking the flu gene of E. coli K-12 strain MG1655. The flu gene encodes Antigen 43, an adhesin conserved in most E. coli strains that is involved in biofilm formation and bacterial self-aggregation.24 Using these vectors the flu gene of E. coli EcM1 was replaced by the gene cassettes expressing SAgfp or SAtir (Figure 2a), generating strains EcM1SAgfp and EcM1SAtir (Supplementary Table 1). We also generated with suicide plasmid pGEflu the control strain EcM1Δflu, an isogenic EcM1 strain not expressing SAs and having a deletion of flu identical to that generated by the insertion of SAs. Finally, these strains were tagged with a bioluminescent reporter, the luxCDABE operon from Photorhabdus luminescens(25) under the control of a constitutive promoter (P2), which was inserted in the matBCDEF operon with suicide vector pGEmat-lux (Supplementary Table 1). This operon encodes the meningitis-associated and temperature-regulated Mat fimbriae, also termed E. coli common pilus, which are produced by most E. coli pathogroups, being involved in early stage biofilm development and host cell recognition.26 The final bioluminescent strains were named EcM1luxΔflu, EcM1luxSAgfp, and EcM1luxSAtir (Supplementary Table 1).
Figure 2.

Constitutive, stable, and nontoxic expression of synthetic adhesins from the chromosome of E. coli. (a) Scheme of a SA gene fusion integrated in the flu gene of E. coli chromosome under the control of the constitutive promoter PN25. (b) Flow cytometry analysis of E. coli EcM1luxΔflu, EcM1luxSAgfp, or EcM1luxSAtir bacteria. Histograms show the fluorescence intensity of bacteria stained with anti-myc mAb and secondary anti-mouse IgG-Alexa 488. (c) Growth curves of bacterial cultures of the same strains shown in panel b. (d) Western blot analysis of the expression of SAs in EcM1luxSAgfp and EcM1luxSAtir strains grown in LB cultures for the indicated time. SAs were immunodetected with anti-myc tag mAb. Cytoplasmic GroEL chaperone was used as loading control (lower panel) and was detected with anti-GroEL mAb.
Flow cytometry analysis of EcM1luxSAgfp and EcM1luxSAtir with anti-myc mAb (Figure 2b) indicated that both SAs were highly and homogeneously expressed on the surface of bacteria, SAgfp being expressed at higher levels than SAtir. EcM1luxSAgfp and EcM1luxSAtir strains showed identical growth curves as EcM1luxΔflu in LB media at 37 °C with agitation (Figure 2c), and nearly identical colony forming units (CFU) per unit of optical density at 600 nm (OD600) were determined by plating (∼1.0 × 109 CFU/OD600), demonstrating that bacterial growth and viability was not affected by the constitutive expression of the SAs. In addition, EcM1luxSAgfp and EcM1luxSAtir strains were grown at 37 °C for 5 days with a daily dilution (1:2000) in fresh LB medium lacking antibiotics. Whole-cell protein extracts were prepared from each culture harvested every day and analyzed by Western blot with anti-myc mAb (to detect the SA) and anti-GroEL mAb, as loading control, demonstrating that the expression of the SAs was stably maintained throughout the length of the experiment (ca. 55 bacterial generations) without the need of any inducer or antibiotic pressure (Figure 2d). Similarly, the lux operon was stably expressed in both strains along the experiment (Supplementary Figure S2).
Adhesion of Engineered E. coli to Antigens Expressed on the Surface of Mammalian Cells
We investigated whether EcM1luxSAgfp and EcM1luxSAtir bacteria adhered specifically to in vitro cultured mammalian cells expressing on their surface the cognate antigens recognized by the SAs. As an experimental model, we stably transfected HeLa cells with plasmids pDisplay-GFP-tm and pDisplay-TirM-tm (Supplementary Table 1), which expressed GFP and TirM antigens, respectively, on the extracellular side of the plasma membrane by means of an N-terminal signal peptide and a C-terminal transmembrane (tm) anchor domain from the platelet derived growth factor receptor (PDGFR) (Supplementary Figure S3a). In addition, the protein fusion containing TirM incorporates, before the tm domain, the monomeric green fluorescent protein mWasabi.27,28 The primary sequence of mWasabi differs from that of GFP and is not recognized by the VHH cloned in SAgfp.28,29 Thus, expression of GFP-tm and TirM-tm fusion proteins on the surface of HeLa cells can be monitored by their green fluorescence emission. Upon stable transfection, fluorescence-positive HeLa cells were enriched by fluorescence-activated cell sorting (FACS) and expanded in vitro. The expanded cell populations, named HeLa-GFP-tm and HeLa-TirM-tm, contained cells with different expression levels of these antigens on their surface, including some cells with undetectable levels of the antigens, especially in HeLa-TirM-tm (Supplementary Figure S3b).
We performed in vitro adhesion assays of HeLa-GFP-tm and HeLa-TirM-tm cultures infected with EcM1luxSAgfp or EcM1luxSAtir bacteria (MOI 300:1). After 1 h infection, unbound bacteria were removed with PBS, and the samples were stained with anti-E. coli polyclonal antibodies and DAPI (to stain DNA). Inspection of the samples by light and fluorescence microscopy (Figure 3) revealed very high numbers of EcM1luxSAgfp and EcM1luxSAtir bacteria (red fluorescence) specifically adhered to HeLa cells expressing GFP or TirM, respectively (green fluorescence). Target HeLa cells were largely covered by bound bacteria expressing the corresponding SA, as can be also visualized by three-dimensional reconstruction of confocal images (Supplementary Video 1). In contrast, EcM1luxSAgfp and EcM1luxSAtir bacteria were not bound to HeLa cells expressing the nonspecific antigen (i.e., TirM for EcM1luxSAgfp; GFP for EcM1luxSAtir) (Figure 3a and b). Importantly, bacteria adhered to cells with different expression levels of the target antigen on their surface, but not to antigen-negative cells that can be found in both HeLa-GFP-tm and HeLa-TirM-tm cultures (labeled with asterisks in Figure 3c). Similar results were observed with lower MOI and infection times (see below). We also tested whether the bioluminescent reporter could provide a sensitive readout of the bacterial adhesion process. To this end, HeLa, HeLa-GFP-tm, and HeLa-TirM-tm cells were grown in a multiwell tissue culture plate and infected as above with EcM1luxSAgfp, EcM1luxSAtir, or EcM1luxΔflu. After 1 h infection, the plate was washed with PBS and monitored for light emission. Bioluminescence signals were observed only in those wells in which bacteria expressing SAs infected HeLa cells expressing the cognate target antigen (Supplementary Figure S4).
Figure 3.

Adhesion of E. coli expressing synthetic adhesins to target mammalian cells. (a) Light and fluorescence microscopy images of HeLa-GFP-tm cells grown in culture and infected with EcM1luxSAgfp or EcM1luxSAtir bacteria (MOI 300:1), as indicated. Expression of GFP-tm fusion on the surface of these cells is detected by the green fluorescence emission. (b) Light and fluorescence microscopy images of HeLa-TirM-tm cells grown in culture and infected with EcM1luxSAgfp or EcM1luxSAtir bacteria (MOI 300:1), as indicated. Expression of TirM-tm fusion on the surface of these cells is detected by the green fluorescence emission of its mWasabi domain. Bacteria were stained with anti-E. coli polyclonal serum and anti-IgG-rabbit-Alexa-594 (red). (c) Confocal microscopy images of HeLa-GFP-tm cells infected with EcM1luxSAgfp (top panels) and HeLa-TirM-tm cells infected with EcM1luxSAtir (bottom panels). Infection and staining was carried out as in panels a and b. In addition, cell nuclei and bacterial chromosomes were stained with DAPI (blue). The selected images contain antigen-negative cells (labeled with asterisks) to show the absence of bound bacteria to them.
To gain further insight into the adhesion process mediated by the SAs, we performed time-lapse live microscopy recording the adhesion of EcM1luxSAgfp bacteria to HeLa-GFP-tm cells, selecting a field in the microscope that allow comparison of the behavior of bacteria encountering antigen-positive and antigen-negative cells. For this experiment infection of HeLa-GFP-tm culture was performed with a lower MOI (100:1) to reduce bacteria in the medium that could hinder the visualization of cells. The result from this experiment (Supplementary Video 2) showed that antigen-positive HeLa-GFP-tm cells (green) were covered with bacteria within 5 min, whereas almost no bacteria were attached to antigen-negative cells. Interestingly, a significant number of bacteria attached in just 2 min to antigen-positive cells. In fact, adhesion of an individual bacterium occurs in a very short time period (<30 s) after an apparent random collision of a swimming bacterium with the surface of a mammalian cell. After this initial contact with the target cell, bacteria can be observed moving and tumbling in close proximity to the surface of the mammalian cell, eventually establishing a permanent adhesion phenotype and stopping its active motility. In contrast, bacteria that contact with an antigen-negative cell move and tumble close to the cell surface but then swim in a different direction away from the cell (see Supplementary Video 2). This behavior suggests that a productive adhesion of the SAs to the target surface is somehow transduced by the bacterium to stop flagella. Interestingly, an arrest of flagellum rotation has been described in the adhesion of Caulobacter crescentus to surfaces during its life cycle.30 In E. coli, intracellular levels of c-di-GMP second messenger have been proposed to regulate changes in flagellar rotation during the transition between motile and surface-attached multicellular communities.31 Taken together, our experiments demonstrated that the engineered E. coli strains with constitutive expression of SAs were able to adhere fast, in high numbers, and in a highly specific manner to mammalian cells expressing on their surface the target antigen recognized by the SA. In addition, the mechanisms underlying the behavior of engineered E. coli with SAs to a target surface might be related to those found in natural adhesion processes that trigger arrest of flagellar rotation.
Colonization of Tumors in Vivo by Engineered E. coli with Synthetic Adhesins
To test the functionality of the SAs in vivo, we evaluated whether the engineered bacteria exhibited an improved colonization of solid tumors expressing the target antigen on the surface of tumor cells. It is well established from animal models and clinical trials that intravenous administration of facultative anaerobic bacteria such as Salmonella and E. coli strains results in the preferential bacterial colonization of solid tumors.32 This natural ability of anaerobic bacteria for tumor colonization has encouraged research aimed to engineer the expression of therapeutic proteins within the tumor (e.g., cytotoxins, pro-drug converting enzymes) as well as gene reporters for in vivo tumor diagnosis (e.g., bioluminescence, magnetic resonance imaging).32,33 Clinical trials using systemic administration of Salmonella for human cancer therapy revealed that the highest bacterial dose that was tolerated was still insufficient for the effective colonization of solid tumors found in those patients.34,35 Therefore, it would be of interest to engineer bacteria with improved tumor colonization capabilities (i.e., requiring lower doses of administered bacteria and showing an increased ratio of bacteria in tumors versus healthy organs).32
It has been previously shown that systemic administration of a single bacterial dose of 5 × 106 CFU of wild type E. coli K-12 (strain MG1655) to tumor-bearing mice elicited colonization of >90% of the tumors.9 Three to 5 days after its intravenous injection, E. coli K-12 bacteria were recovered in high numbers from the tumor mass (∼108 CFU/g), whereas organs such as liver or spleen had low bacterial numbers (<103 CFU/g). Following a similar approach, we established an in vivo tumor xenograft model based on HeLa-GFP-tm cells. Athymic Nude mice that received subcutaneously ∼106 HeLa-GFP-tm cells developed solid tumors of ∼200–400 mm3 in about 10–15 days. These animals were divided in two experimental groups (n = 6) and received intravenously in their lateral tail vein a single dose of 1 × 107 CFU of EcM1luxSAtir or EcM1luxSAgfp. Four days after the intravenous injection, live imaging of the animals showed bioluminescence signals in the tumors from both groups, and bacteria were found in high numbers (∼108–109 CFU/g) in all tumors from each group (6/6) (Supplementary Figure S5a), whereas livers and spleens did not contain bacteria (detection limit ∼5 × 101 CFU/g) or had low bacterial numbers (<103 CFU/g) (Supplementary Figure S5b). The level of expression of the SAs and bioluminescence signals were identical in the inoculated bacteria and in tumor-recovered bacteria, showing the stability of the expression of SAs in vivo (Supplementary Figure S6). Histological cross sections of colonized HeLa-GFP-tm tumors stained with anti-E. coli antibodies revealed that bacteria from both strains localized preferentially at inner regions of the tumors (Supplementary Figure S7), especially at the border with outer regions where tumor cells preferentially proliferate. A similar pattern of bacterial distribution was reported for tumors colonized with wild type E. coli MG1655.9
Hence, the above data indicated that the engineered E. coli strains stably expressed the SAs in vivo and maintained the capacity to colonize solid tumors of HeLa-GFP-tm cells regardless of the specificity of the SA when a high bacterial dose was administered (1 × 107 CFU). To determine whether SAs could favor tumor colonization with lower bacterial doses, first we showed that a bacterial dose of 1 × 105 CFU was suboptimal for the colonization of HeLa-GFP-tm tumors by wild type E. coli MG1655 as only one-third of the tumors (3/9) were colonized 4 days after its systemic administration (Figure 4a). This dose represents 2% of the dose reported for optimal tumor colonization with wild type E. coli MG1655.9 Nevertheless, those tumors that were colonized had bacterial titers (∼108 CFU/g) similar to those found with high bacterial doses, suggesting that once E. coli succeeded at an early stage of tumor colonization, bacteria can proliferate, reaching a final titer that is independent of the inoculated dose. In contrast, livers and spleens of these animals had undetectable levels of bacteria (Figure 4b).
Figure 4.

In vivo colonization of tumors with low doses of E. coli expressing synthetic adhesins. (a) Bacterial colonization of HeLa-GFP-tm or HeLa tumors, as indicated on top, by the following E. coli strains: wild type K-12 (MG1655), EcM1luxSAtir (SAtir), and EcM1luxSAgfp (SAgfp). Each bacterial strain was intravenously administered (1 × 105 CFU/mouse) to tumor-bearing mice (experimental groups n = 9) and 4-days postadministration the number of CFU in each tumor was determined. Each circle in the graph represents the CFU determined per gram of tumor (Log10 CFU/g) for each animal in the different experimental groups. The ratio of colonized tumors in each group is shown at the bottom along with the statistical analyses between groups connected with lines. Two-tailed P values of Fisher’s exact test are indicated with one asterisk (*) when P < 0.05 or two asterisks (**) when P = 0.015. On the right, bioluminescence live imaging of HeLa-GFP-tm tumor-bearing mouse infected with 1 × 105 CFU of EcM1luxSAtir (left image) or EcM1luxSAgfp (right image). Images are overlays of photographic white-light and bioluminescence signals from a representative tumor-bearing mouse infected with the strains, as indicated on top. The intensities of the bioluminescence signals are represented in pseudocolor according to the scale bar. (b) Graphs showing bacterial titers in livers (left) and spleens (right) from those animals with a HeLa-GFP-tm tumor colonized in panel a by wild-type K-12 (MG1655), EcM1luxSAtir (SAtir), or EcM1luxSAgfp (SAgfp) strains. Each circle in the graph represents the CFU determined per gram of tissue (log10 CFU/g).
Therefore, a suboptimal dose of 1 × 105 CFU was chosen to compare the tumor colonization capacity of the engineered E. coli strains. As above, EcM1luxSAtir or EcM1luxSAgfp bacteria were administered intravenously to two groups of Nude mice (n = 9) bearing subcutaneous HeLa-GFP-tm tumors (≥200 mm3). Four days after inoculation, most animals (8/9) of the group inoculated with EcM1luxSAgfp had their tumors colonized with high bacterial titers (≥108 CFU/gr), whereas less than one-third of the animals (2/9) in the group inoculated with EcM1luxSAtir had colonized tumors (Figure 4a). This differential colonization was also detected by bioluminescence in vivo imaging of the animals (Figure 4a). As a control for specificity, a dose of 1 × 105 CFU of EcM1luxSAgfp strain was also administered systemically to a group (n = 9) of Nude mice bearing tumors derived from untransfected HeLa cells. Four days after inoculation, HeLa tumors were colonized with low efficiency (2/9) by EcM1luxSAgfp strain, identical to that found with EcM1luxSAtir strain in HeLa-GFP-tm tumors (Figure 4a). In addition, livers and spleens of animals with colonized tumors had undetectable levels of bacteria (Figure 4b). Taken together, these results demonstrated that engineered E. coli with SAs were able to colonize solid tumors expressing a target cell surface antigen with high efficiency and specificity using low bacterial doses that are suboptimal for wild type E. coli or engineered bacteria with a SA of a different specificity.
Several factors have been proposed to be involved in the colonization of solid tumors by E. coli and other facultative and anaerobic bacteria (e.g., Salmonella, Listeria, Bifidobacterium). The initial bacterial entry seems to be due to the highly permeable and abnormal vasculature of the tumor.36 Infecting bacteria also induce pro-inflammatory signals (e.g., tumor necrosis factor α; TNFα) that increase blood flooding into the tumor mass assisting bacterial entry.37 At this stage, bacterial adhesion to tumor cells mediated by SAs may facilitate successful tumor colonization. At later stages, bacterial replication is favored in the intratumoral microenvironment, rich in certain nutrients (e.g., amino acids, ribose) and having a low oxygen concentration that impairs clearance of bacteria by the immune system.32 In the colonized tumors, we found similar bacterial titers and intratumoral distribution irrespective of the specificity of the SA or antigen expressed by the tumor, indicating that SAs appear to have less influence in the replication and intratumoral distribution of bacteria at a later stage of the colonization.
Lower Retention of Engineered E. coli in Liver and Spleen
Next, we investigated whether the engineered bacteria with SAs and lacking natural adhesins, in addition to the improved colonization of target tumors shown previously, could have a lower retention in normal organs compared to wild type E. coli. For this purpose, we analyzed bacterial titers in livers and spleens of two groups (n = 9) of Nude mice without implanted tumors in which a high dose (1 × 107 CFU) of wild type E. coli MG1655 or engineered EcM1luxSAgfp was inoculated. Three mice from each group were euthanized at 4, 24, and 48 h after inoculation, and the bacterial titers in liver and spleen were determined (Figure 5). This experiment revealed that, at all the time points analyzed, the titers of the engineered E. coli strain in these organs were lower than those in the case of the wild type E. coli strain, especially in liver where the titers of engineered bacteria were ∼10–30 times lower than the wild type strain (Figure 5). In the spleen, the engineered bacteria had titers ∼2–4-fold lower than the wild type strain. Therefore, these data indicate that the engineered bacteria exhibit a lower retention in healthy organs than the wild type E. coli strain. This phenotype might be due to the deletion of natural adhesins in the engineered strain (i.e., type 1 fimbriae, Antigen 43, and MAT fimbriae) that have the potential to bind receptors widely expressed in the surface of multiple host cells and tissues.11,24 Hence, expression of SAs into an E. coli chassis lacking of a set of natural adhesins generates bacteria with selective adhesion to target tissues and lower adhesion to non-target organs.
Figure 5.

Low retention of the engineered E. coli strain in healthy organs. Bacterial titers (log10 CFU/g) recovered from livers (left) and spleens (right) of tumor-less Nude mice at 4, 24, and 48 h after intravenous administration of 1 × 107 CFU of wild type E. coli K-12 (MG1655) or EcM1luxSAgfp, as indicated. Each bar represents the average CFU/g with standard deviation determined from three mice from each group at the indicated time postinoculation.
Conclusions
We have engineered a stable and specific adhesion of E. coli bacteria with SAs based on the β-domain from intimin12 and the Ig-domain from VHHs.14 Constitutive expression of SAs from the chromosome did not affect growth and viability of the engineered E. coli strain, being stably maintained through multiple generations without selection pressure. The modular architecture of the SAs makes possible the simple modification of their specificity by an exchange of the VHH sequence, which can be selected from repertoires employing phage and cell display technologies.14,15 This could allow the generation of SAs against virtually any antigen of interest, enabling the design of E. coli targeting any chosen surface or cell. In addition, intimin β-domain and VHHs are resistant to proteases and denaturant agents (e.g., SDS, urea, temperature).13,14 Altogether, these properties make SAs extremely robust, inducing the stable attachment of a great number of E. coli bacteria to the targeted surface or cell. Furthermore, we have demonstrated the functionality of SAs in vivo showing that the engineered E. coli strain colonizes more efficiently solid tumors expressing a target antigen. The higher efficiency introduced by the SAs allowed a significant reduction (ca. 2 orders of magnitude) of the bacterial dose needed for optimal tumor colonization in vivo, an important issue in order to increase the biosafety of bacterial therapies.32 Tumors cells often deregulate the expression and/or post-translational modification of cell surface proteins (e.g., growth factor receptors, mucins), which has been used as targets for developing therapeutic antibodies against tumor cells.38,39 Hence, SAs against these validated targets could improve bacterial colonization of specific tumors.
Engineered E. coli expressing SAs may be of interest for other biomedical and industrial applications. For instance, SAs could help in the localized delivery of antigens to specific immune cells for more effective vaccines based on bacterial vectors.40 SAs could also be targeted to antigens expressed by viral and bacterial pathogens, which could facilitate the therapeutic intervention of engineered bacteria against pathogens.41,42 In addition, targeting of other bacteria could generate defined bacterial consortia for industrial bioprocesses,43 whereas adhesion to abiotic surfaces could improve the development of biosensors.44 Given the conservation of the cellular machineries used for folding and insertion of β-barrel OM proteins,45,46 it is likely that the reported SAs of E. coli could be functional in other Gram-negative bacterial species, including alternative nonpathogenic bacteria used in synthetic biology such as Pseudomonas putida.47 In conclusion, SAs could be applied in multiple synthetic biology projects, and future work should expand their use in E. coli and other bacteria for specialized applications.
Methods
Bacterial Strains and Growth Conditions
The E. coli strains used in the experiments described in this work are listed in Supplementary Table S1. Bacteria were grown in Luria–Bertani (LB) liquid medium and agar plates (1.5% w/v), at 37 °C, unless otherwise indicated. When needed for plasmid or strain selection, antibiotics were added to the media at the following concentrations: chloramphenicol (Cm) at 30 μg/mL, ampicillin (Ap) at 100 μg/mL, and kanamycin (Km) at 50 μg/mL. E. coli DH10B-T1R strain was used as host for cloning and propagation of plasmids with a pBR origin of replication (e.g., pAK-Not, pNV-derivatives). For cloning and propagation of suicide pGE-plasmid derivatives, containing the conditional pi-dependent R6K origin of replication,48 the E. coli strains BW25141 or CC118-λpir were used. For inducible expression of the SAs from plasmid vectors with plac promoter (i.e., pNVfib, pNVgfp), EcM1 bacteria bearing the corresponding plasmid were grown in LB-Cm at 30 °C, and 0.05 mM isopropyl-thio-β-d-galactoside (IPTG) was added at an optical density 600 nm (OD600) of 0.5. The cultures were further grown for 2 h at 30 °C with agitation (160 rpm). Bacteria with constitutive expression of the synthetic adhesins and the lux operon from the chromosome were always grown statically at 37 °C in LB, except for analysis of their growth curve in which cultures were grown at 37 °C with agitation (160 rpm). Bioluminescence of bacterial colonies grown on LB plates were monitored in a Chemi-doc XRS+ (Bio-Rad).
Plasmids, DNA Constructs, and Oligonucleotides
The plasmids constructed and used in this study are summarized in Supplementary Table S1. Oligonucleotides were synthesized by Sigma Genosys and are listed in Supplementary Table S2. PCRs were performed with Taq DNA polymerase for standard amplifications in screenings and with proof-reading DNA polymerases, Vent DNA polymerase (New England Biolabs) and High-expand fidelity (Roche) for cloning purposes. All plasmid constructs were fully sequenced (Secugen). Details of plasmid constructions are described as Supporting Information.
E. coli Genome Modification and Strain Construction
Site-specific deletions and insertions in the chromosome of E. coli were done with the marker-less strategy of genome edition based on expression of I-SceI endonuclease.4,21 Briefly, the E. coli strain to be modified was initially transformed with plasmid pACBSR (CmR),49 expressing I-SceI and λ Red proteins under the control of PBAD promoter (inducible with l-arabinose), and subsequently electroporated with the corresponding pGE-based suicide vector (KmR). Cointegrants were selected on LB-Cm-Km plates incubated at 37 °C. Individual colonies were isolated and grown for 6 h in LB-Cm liquid medium containing l-arabinose 0.4% (w/v) with agitation (160 rpm). After this period, a ∼1 μL sample of these cultures was streaked on LB-Cm plates using an inoculating loop and incubated overnight. Individual colonies were replicated in LB-Cm and LB-Cm-Km plates to screen for Km-sensitive colonies that have performed resolution of the cointegrant vector after I-SceI induction. Individual Km-sensitive colonies were screened by PCR with specific oligonucleotides to identify those with the desired modification in their chromosome (i.e., deletion, insertion, substitution). Plasmid pACBSR was cured from the final strain by growth in liquid LB and streaking on LB plates. Individual colonies were replicated in LB and LB-Cm plates to screen for Cm-sensitive colonies. Construction details of individual E. coli strains are described as Supporting Information.
In Vitro Cell Culture and Plasmid Transfection
The human epithelial tumor cell line HeLa (ATCC, CCL-2) was grown as a monolayer in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine (complete DMEM), at 37 °C with 5% CO2. For transfection, HeLa cells were seeded in 24-well tissue culture plates (BD Falcon) (∼105 cells/well), grown for 20 h at 37 °C with 5% CO2, and transfected with 0.6 μg/well of plasmids pDisplay-GFP-tm or pDisplay-TirM-tm following the calcium phosphate method.50 After 22 h incubation at 37 °C with 5% CO2, the medium was removed, and wells were washed three times with phosphate-buffered saline (PBS) and filled with complete DMEM medium containing G418 (0.5 mg/mL, Invitrogen, Life Technologies). After 24 h incubation, cells were passed to a T75 flask (BD Falcon) with the same medium and further grown. G418-resistant HeLa cells expressing GFP or mWasabi fusion proteins were selected from these cultures by fluorescence activated cell sorting (FACS) using an Epics-Altra Cell Sorter (Beckman Coulter). HeLa-GFP-tm and HeLa-TirM-tm positive cells populations were expanded on complete DMEM medium.
Flow Cytometry Analysis
Bacteria (equivalent to a final OD600 of 1.0) were harvested from cultures by centrifugation (4000 × g, 3 min), washed with PBS, and resuspended in 1 mL of PBS containing 10% (v/v) goat serum (Sigma). An aliquot sample of 200 μL was incubated for 1 h on ice with anti-myc mAb clone 9B11 (1:200; Cell Signaling Technology). Next, bacteria were washed in PBS, resuspended in 500 μL of PBS containing 10% (v/v) goat serum, and stained for 40 min in the dark with Alexa 488-conjugated anti-mouse IgGs (1:500; Molecular Probes, Life Technologies). Bacteria were finally washed and resuspended in a final volume of 1 mL of PBS and analyzed in a flow cytometer (Gallios, Beckman Coulter).
Protein Extract Preparation, SDS-PAGE, and Western Blots
Whole-cell protein extracts were prepared in urea-SDS sample buffer as described previously.13 SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting conditions to polyvinylidene difluoride membrane (PVDF, Immobilon-P; Millipore) have been reported.13,15 For immunodetection, the PVDF membranes were incubated with anti-myc mAb clone 9B11 (1:2000; Cell Signaling Technology) to detect myc-tagged SAs and anti-mouse IgG-peroxidase (POD) conjugate (1:5000; Sigma) as secondary antibody. GroEL was detected with anti-GroEL mAb-POD conjugate (1:5000; Sigma). Membranes were developed by chemiluminescence using a mixture in 100 mM Tris-HCl (pH 8.0) containing 1.25 mM luminol (Sigma), 0.22 mM cumaric acid (Sigma), and 0.0075% (v/v) H2O2 (Sigma) and exposed to an X-ray film (Curix, Agfa).
Adhesion Assays to Antigens Immobilized on a Plastic Surface
Purified GFP (Upstate, Millipore) or human fibrinogen (Enzyme Research Laboratories) as indicated was diluted in PBS to 10 μg/mL, and 100 μL was adsorbed onto ELISA plates (Maxisorb, Nunc) overnight at 4 °C. Antigen-coated plates were washed with PBS and blocked for 1 h at room temperature with PBS containing 3% (w/v) skimmed milk. Next, a bacterial suspension (100 μL) of the indicated strain was added at an OD600 of 3.0 in PBS and incubated for an additional 1 h. The plates were then washed with PBS three times, and the presence of bound bacteria was visualized by light microscopy and macroscopically detected by staining with crystal violet solution (Sigma).
Adhesion Assays to in Vitro Cultured HeLa Cells and Immunofluorescence Microscopy
Bacteria of the indicated E. coli strain were harvested by centrifugation (4000 × g, 3 min) from static liquid LB cultures grown overnight at 37 °C. An OD600 of 1.0 from these cultures grown statically was determined to contain ∼3 × 108 CFU/mL by plating in LB agar. Bacteria were washed in PBS and resuspended at 3 × 107 CFU/mL in PBS or DMEM. For an infection at MOI 300:1, a 1 mL sample of this bacterial suspension was added to a single well of a 24-well tissue culture plate having the indicated HeLa cell line (∼105 cells/well). HeLa cells were grown on sterile coverslips (13 mm diameter, VWR international) placed at the bottom of the well. After 1 h infection at 37 °C, the wells were aspirated and washed five times with 1 mL of PBS at room temperature. The coverslips were fixed for 20 min at room temperature with 0.5 mL of a paraformaldehyde 4% (w/v) solution in PBS and washed three times with 1 mL of PBS. Coverslips were blocked and stained for 1 h at room temperature in a wet chamber with 50 μL of PBS/10% goat serum solution having a rabbit polyclonal serum anti-E. coli O and K antigenic serotypes (1:1000; Biodesign). The coverslips were washed by immersion 15 times in a large volume of PBS (100 mL), placed again the wet chamber, and incubated for 40 min at room temperature with 50 μL of PBS/10% goat serum solution having a goat anti-rabbit IgG-Alexa 594 conjugated secondary antibody (1:500; Molecular Probes, Life Technologies) and DAPI (1:500; Sigma). Next, the coverslips were washed with PBS as above, the excess of liquid was removed by touching a kimwipe with the edge of the coverslip, and the coverslips were mounted with 2 μL of Prolong (Invitrogen) on glass slides. The samples were examined by epifluorescence microscopy (Zeiss Axio imager microscope) or by confocal microscopy (Leica TCS SP5 multispectral confocal system). For time-lapse live cell video microscospy, HeLa-GFP-tm cells, grown on 24-well tissue culture plates, were infected with 1 × 107 CFU of EcM1luxSAgfp in a final volume ∼0.3 mL/well (MOI 100:1) and monitored using an Olympus IX71 microscope equipped with an Olympus cell∧R motorized TIRF system.
Infection of Tumor-Bearing Mice and Recovery of Bacteria from Tissues
All animals experiments were done in accordance with protocols approved by the CNB Ethics Committee for Animal Experimentation (ref 11034) and by the Hospital Universitario Puerta de Hierro Animal Care and Use Committee, in compliance with Spanish and European Union legislation. Five-week-old athymic female Hsd:Athymic Nude-Foxn1nu were obtained from Harlan (Harlan Ibérica). Tumor-bearing mice were generated by subcutaneous injection of ∼1 × 106 HeLa-GFP-tm or HeLa cells, as indicated, in 100 μL of PBS containing 20% (v/v) Matrigel (BD Biosciences), into the abdominal right flank of 6-week-old Nude mice. Tumor volumes were estimated with calipers according to the formula: width2 × length × 0.52. When tumors reached a volume between 200 and 400 mm3, the mice were randomly divided into experimental groups. Bacteria were grown and harvested as described above for the in vitro adhesion assays and resuspended in sterile PBS at 1 × 108 CFU/mL. For each independent experiment the number of CFU in this bacterial suspension was determined, showing an experimental error below 10%. This bacterial stock was directly used for systemic inoculation of mice receiving a dose of 1 × 107 CFU by injection of 100 μL into the lateral tail vein using a 0.5 mL syringe with a 29G needle (Becton Dickinson). Alternatively, for mice receiving a dose of 1 × 105 CFU, the original bacterial stock was diluted in PBS to 1 × 106 CFU/mL, and 100 μL was injected per mouse as above. These bacterial doses did not cause any apparent disease symptoms in the animals, which showed normal phenotype throughout the entire experiment (i.e., mobility, weight, feeding behavior). For determination of bacterial CFU per gram (CFU/g) of tumors, livers, and spleens, animals were euthanized, and tumors and organs were excised, placed individually into (preweighed) sterile tubes containing 5 mL of PBS, and weighed. Samples were then transferred to sterile sampling bags (VWR), and Triton X-100 (Sigma) was added to a final concentration of 0.2% (v/v). Samples were homogenized by soft mechanical squeezing. Next, a 100-μL sample of the homogenates was serially diluted in LB, plated in LB agar, and incubated overnight at 37 °C to determine CFU. Bacterial titers were expressed as CFU/g of tissue. For statistical analysis a minimal bacterial titer of 50 CFU/g, corresponding to the detection limit of the assay, was considered in those cases in which no bacteria were detected by plating.
Bioluminescence Imaging
Bacterial light emission from LB plates or from tissue culture plates was captured using a ChemiDoc XRS system (Bio-Rad). For live imaging of bioluminescent bacteria in tumor-bearing mice, animals were anesthetized with 4% Isoflurane (Forene, Abbott) and maintained under 1% Isoflurane in a thermostated chamber with a high-resolution charge-coupled-device (CCD) cooled digital camera ORCA-2BT (Hamamatsu Photonics). Imaging analysis was done with the Hokawo software (Hamamatsu Photonics).
Statistics
Statistical analyses comparing the ratio of colonized tumors between experimental groups were conducted with Fisher’s exact test to determine two-tailed P values using Prism 5.0 (GraphPad software Inc.). Data were considered significantly different when P < 0.05.
Histology
For ex vivo histological analyses, resected tumors were fixed with 4% (w/v) paraformladehyde in PBS for 16 h at 4 °C and embedded in paraffin. Serial 6 μm sections were cut at the tumor center with a microtome (Leica RM2155) and placed onto glass slides. The samples were deparaffinized with xylene and ethanol solutions, rehydrated in PBS, and incubated with 1 M citrate buffer (pH 6.0) and PBS-Tween (PBS, 0.1% Tween 20) to unmask antigens. Tissue sections were blocked with 10% goat serum in PBS-Tween and incubated with anti-E. coli polyclonal serum (1:100) 1 h at room temperature in the same buffer. Next, samples were washed with PBS-Tween and incubated with goat anti-rabbit IgG-Alexa 594 conjugated secondary antibody (1:500) in PBS-Tween with 10% goat serum for 1 h at room temperature. Samples were mounted in Vectashield media (Vector Laboratories), and images were obtained with a fluorescence microscope (Leica DMI6000B fluorescence system with Orca R2 digital camera from Hamamatsu).
Acknowledgments
We thank Prof. Víctor de Lorenzo for valuable discussion and support. We especially thank the excellent technical assistance of Sylvia Gutiérrez Erlandsson (Confocal Microscopy facility of the CNB) and Francisco Javier Martin Torre (Animal House facility of the CNB). Work in the laboratory of LAF is supported by research grants from the Spanish “Ministerio de Economía y Competitividad” (MINECO) (BIO2011-26689), “Comunidad Autónoma de Madrid” (S2010-BMD-2312), “La Caixa” Foundation, and the European Research Council (ERC-2012-ADG_20120314). C.P.-L. was supported by a Ph.D. FPI contract BES-2009-024051 from MINECO. Work in the laboratory of L.A.-V. is supported by research grants from MINECO (BIO2011-22738) and “Comunidad Autónoma de Madrid” (S2010-BMD-2312). R.F.-P. was supported by a Ph.D. FPI contract BES-2009-027649 from MINECO.
Supporting Information Available
Contains further details about plasmid and bacterial strain constructions, supplemental tables, references and figures. Video 1: Sequential Z stack images of HeLa-GFP-tm cells infected with EcM1luxSAgfp bacteria (MOI 300:1) obtained by confocal laser scanning microscopy. After 1 h of infection, cells and bacteria were fixed with paraformaldehyde. Mammalian cells expressing GFP (green) and bacteria (red) were labeled with anti-E. coli rabbit polyclonal antibodies and anti-rabbit-Alexa-594 antibodies. The bacterial and cellular genomes (blue) were stained with DAPI. Video 2. Infection of HeLa-GFP-tm cells with EcM1luxSAgfp bacteria (MOI 100:1) assessed by time-lapse live-cell video microscopy using an Olympus IX71 microscope equipped with an Olympus motorized CellR∧TIRF system. Addition of bacteria to the culture started at 00:00:27 s, and images were acquired for 5 min. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contribute equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Khalil A. S.; Collins J. J. (2010) Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korea C. G.; Ghigo J. M.; Beloin C. (2011) The sweet connection: Solving the riddle of multiple sugar-binding fimbrial adhesins in Escherichia coli: Multiple E. coli fimbriae form a versatile arsenal of sugar-binding lectins potentially involved in surface-colonisation and tissue tropism. Bioessays 33, 300–311. [DOI] [PubMed] [Google Scholar]
- Kline K. A.; Falker S.; Dahlberg S.; Normark S.; Henriques-Normark B. (2009) Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5, 580–592. [DOI] [PubMed] [Google Scholar]
- Posfai G.; Plunkett G. 3rd; Feher T.; Frisch D.; Keil G. M.; Umenhoffer K.; Kolisnychenko V.; Stahl B.; Sharma S. S.; de Arruda M.; Burland V.; Harcum S. W.; Blattner F. R. (2006) Emergent properties of reduced-genome Escherichia coli. Science 312, 1044–1046. [DOI] [PubMed] [Google Scholar]
- Huang C. J.; Lin H.; Yang X. (2012) Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 39, 383–399. [DOI] [PubMed] [Google Scholar]
- Tenaillon O.; Skurnik D.; Picard B.; Denamur E. (2010) The population genetics of commensal Escherichia coli. Nat. Rev. Micrbiol. 8, 207–217. [DOI] [PubMed] [Google Scholar]
- Salvador E.; Wagenlehner F.; Kohler C. D.; Mellmann A.; Hacker J.; Svanborg C.; Dobrindt U. (2012) Comparison of asymptomatic bacteriuria Escherichia coli isolates from healthy individuals versus those from hospital patients shows that long-term bladder colonization selects for attenuated virulence phenotypes. Infect. Immun. 80, 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenborn U.; Schulze J. (2009) The non-pathogenic Escherichia coli strain Nissle 1917–features of a versatile probiotic. Microb. Ecol. Health Dis. 21, 122–158. [Google Scholar]
- Weibel S.; Stritzker J.; Eck M.; Goebel W.; Szalay A. A. (2008) Colonization of experimental murine breast tumours by Escherichia coli K-12 significantly alters the tumour microenvironment. Cell. Microbiol. 10, 1235–1248. [DOI] [PubMed] [Google Scholar]
- Stritzker J.; Weibel S.; Hill P. J.; Oelschlaeger T. A.; Goebel W.; Szalay A. A. (2007) Tumor-specific colonization, tissue distribution, and gene induction by probiotic Escherichia coli Nissle 1917 in live mice. Int. J. Med. Microbiol. 297, 151–162. [DOI] [PubMed] [Google Scholar]
- Bodelón G.; Palomino C.; Fernández L. A. (2013) Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev. 37, 204–250. [DOI] [PubMed] [Google Scholar]
- Fairman J. W.; Dautin N.; Wojtowicz D.; Liu W.; Noinaj N.; Barnard T. J.; Udho E.; Przytycka T. M.; Cherezov V.; Buchanan S. K. (2012) Crystal structures of the outer membrane domain of intimin and invasin from enterohemorrhagic E. coli and enteropathogenic Y. pseudotuberculosis. Structure 20, 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelón G.; Marín E.; Fernández L. A. (2009) Role of periplasmic chaperones and BamA (YaeT/Omp85) in folding and secretion of Intimin from enteropathogenic Escherichia coli strains. J. Bacteriol. 191, 5169–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797. [DOI] [PubMed] [Google Scholar]
- Salema V.; Marín E.; Martínez-Arteaga R.; Ruano-Gallego D.; Fraile S.; Margolles Y.; Teira X.; Gutierrez C.; Bodelón G.; Fernández L. Á. (2013) Selection of single domain antibodies from immune libraries displayed on the surface of E. coli cells with two β-domains of opposite topologies. PLoS One 8, e75126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco J. A.; Campbell R.; Iverson B. L.; Georgiou G. (1993) Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. U.S.A. 90, 10444–10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín E.; Bodelón G.; Fernández L. A. (2010) Comparative analysis of the biochemical and functional properties of C-terminal domains of autotransporters. J. Bacteriol. 192, 5588–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou G.; Stephens D. L.; Stathopoulos C.; Poetschke H. L.; Mendenhall J.; Earhart C. F. (1996) Display of beta-lactamase on the Escherichia coli surface: outer membrane phenotypes conferred by Lpp’-OmpA’-beta-lactamase fusions. Protein Eng. 9, 239–247. [DOI] [PubMed] [Google Scholar]
- Leyton D. L.; Rossiter A. E.; Henderson I. R. (2012) From self sufficiency to dependence: mechanisms and factors important for autotransporter biogenesis. Nat. Rev. Microbiol 10, 213–225. [DOI] [PubMed] [Google Scholar]
- Connell I.; Agace W.; Klemm P.; Schembri M.; Marild S.; Svanborg C. (1996) Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc. Natl. Acad. Sci. U.S.A. 93, 9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posfai G.; Kolisnychenko V.; Bereczki Z.; Blattner F. R. (1999) Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 27, 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel G.; Phillips A. D.; Trabulsi L. R.; Knutton S.; Dougan G.; Matthews S. (2001) Intimin and the host cell—is it bound to end in Tir(s)?. Trends Microbiol. 9, 214–218. [DOI] [PubMed] [Google Scholar]
- Deuschle U.; Kammerer W.; Gentz R.; Bujard H. (1986) Promoters of Escherichia coli: a hierarchy of in vivo strength indicates alternate structures. EMBO J. 5, 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude M. W.; Henderson I. R. (2008) Regulation and function of Ag43 (flu). Annu. Rev. Microbiol. 62, 153–169. [DOI] [PubMed] [Google Scholar]
- Winson M. K.; Swift S.; Hill P. J.; Sims C. M.; Griesmayr G.; Bycroft B. W.; Williams P.; Stewart G. S. (1998) Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol. Lett. 163, 193–202. [DOI] [PubMed] [Google Scholar]
- Rendon M. A.; Saldana Z.; Erdem A. L.; Monteiro-Neto V.; Vazquez A.; Kaper J. B.; Puente J. L.; Giron J. A. (2007) Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc. Natl. Acad. Sci. U.S.A. 104, 10637–10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov D. M.; Matz M. V.; Lukyanov S.; Lukyanov K. A. (2010) Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 90, 1103–1163. [DOI] [PubMed] [Google Scholar]
- Ai H. W.; Olenych S. G.; Wong P.; Davidson M. W.; Campbell R. E. (2008) Hue-shifted monomeric variants of Clavularia cyan fluorescent protein: identification of the molecular determinants of color and applications in fluorescence imaging. BMC Biol. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U.; Zolghadr K.; Muyldermans S.; Schepers A.; Cardoso M. C.; Leonhardt H. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteomics 7, 282–289. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick C. L.; Viollier P. H. (2012) Reflections on a sticky situation: how surface contact pulls the trigger for bacterial adhesion. Mol. Microbiol. 83, 7–9. [DOI] [PubMed] [Google Scholar]
- Fang X.; Gomelsky M. (2010) A post-translational, c-di-GMP-dependent mechanism regulating flagellar motility. Mol. Microbiol. 76, 1295–1305. [DOI] [PubMed] [Google Scholar]
- Forbes N. S. (2010) Engineering the perfect (bacterial) cancer therapy. Nat. Rev. Cancer 10, 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh J. H.; Kittleson J. T.; Arkin A. P.; Anderson J. C. (2013) Modular design of a synthetic payload delivery device. ACS Synth. Biol. 2, 418–424. [DOI] [PubMed] [Google Scholar]
- Toso J. F.; Gill V. J.; Hwu P.; Marincola F. M.; Restifo N. P.; Schwartzentruber D. J.; Sherry R. M.; Topalian S. L.; Yang J. C.; Stock F.; Freezer L. J.; Morton K. E.; Seipp C.; Haworth L.; Mavroukakis S.; White D.; MacDonald S.; Mao J.; Sznol M.; Rosenberg S. A. (2002) Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J. Clin. Oncol. 20, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimann D. M.; Rosenberg S. A. (2003) Continuous intravenous administration of live genetically modified Salmonella typhimurium in patients with metastatic melanoma. J. Immunother. 26, 179–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes N. S.; Munn L. L.; Fukumura D.; Jain R. K. (2003) Sparse initial entrapment of systemically injected Salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 63, 5188–5193. [PubMed] [Google Scholar]
- Leschner S.; Westphal K.; Dietrich N.; Viegas N.; Jablonska J.; Lyszkiewicz M.; Lienenklaus S.; Falk W.; Gekara N.; Loessner H.; Weiss S. (2009) Tumor invasion of Salmonella enterica serovar Typhimurium is accompanied by strong hemorrhage promoted by TNF-alpha. PLoS One 4, e6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwkowski M. X.; Mellman I. (2013) Antibody therapeutics in cancer. Science 341, 1192–1198. [DOI] [PubMed] [Google Scholar]
- Oliveira S.; Heukers R.; Sornkom J.; Kok R. J.; van Bergen En Henegouwen P. M. (2013) Targeting tumors with nanobodies for cancer imaging and therapy. J. Controlled Release 172, 607–617. [DOI] [PubMed] [Google Scholar]
- Bolhassani A.; Zahedifard F. (2012) Therapeutic live vaccines as a potential anticancer strategy. Int. J. Cancer 131, 1733–1743. [DOI] [PubMed] [Google Scholar]
- Saeidi N.; Wong C. K.; Lo T. M.; Nguyen H. X.; Ling H.; Leong S. S.; Poh C. L.; Chang M. W. (2011) Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol. Syst. Biol. 7, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I. Y.; Tan M. H.; Koh E.; Ho C. L.; Poh C. L.; Chang M. W. (2013) Reprogramming microbes to be pathogen-seeking killers. ACS Synth. Biol. 3, 228–237. [DOI] [PubMed] [Google Scholar]
- Minty J. J.; Singer M. E.; Scholz S. A.; Bae C.-H.; Ahn J.-H.; Foster C. E.; Liao J. C.; Lin X. N. (2013) Design and characterization of synthetic fungal-bacterial consortia for direct production of isobutanol from cellulosic biomass. Proc. Natl. Acad. Sci. U.S.A. 110, 14592–14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M.; Tsai S. L.; Chen W. (2013) Microbial biosensors: engineered microorganisms as the sensing machinery. Sensors 13, 5777–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N.; Kuszak A. J.; Gumbart J. C.; Lukacik P.; Chang H.; Easley N. C.; Lithgow T.; Buchanan S. K. (2013) Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan C. L.; Silhavy T. J.; Kahne D. E. (2011) β-Barrel membrane protein assembly by the Bam complex. Annu. Rev. Biochem. 80, 189–210. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia E.; de Lorenzo V. (2011) Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 13, 2702–2716. [DOI] [PubMed] [Google Scholar]
- Stalker D. M.; Kolter R.; Helinski D. R. (1982) Plasmid R6K DNA replication. I. Complete nucleotide sequence of an autonomously replicating segment. J. Mol. Biol. 161, 33–43. [DOI] [PubMed] [Google Scholar]
- Herring C. D.; Glasner J. D.; Blattner F. R. (2003) Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311, 153–163. [DOI] [PubMed] [Google Scholar]
- Jordan M.; Schallhorn A.; Wurm F. M. (1996) Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 24, 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.