

Abstract
The Streptococcus suis serotype 2 (S. suis 2) isolates 05ZYH33 and 98HAH33 have caused severe human infections in China. Using a strand-specific RNA-seq analysis, we compared the in vitro transcriptomes of these two Chinese isolates with that of a reference strain (P1/7). In the 89K genomic island that is specific to these Chinese isolates, a toxin–antitoxin system showed relatively high levels of transcription among the S. suis. The known virulence factors with high transcriptional activity in these two highly-pathogenic strains are mainly involved in adhesion, biofilm formation, hemolysis and the synthesis and transport of the outer membrane protein. Furthermore, our analysis of novel transcripts identified over 50 protein-coding genes with one of them encoding a toxin protein. We also predicted over 30 small RNAs (sRNAs) in each strain, and most of them are involved in riboswitches. We found that six sRNA candidates that are related to bacterial virulence, including cspA and rli38, are specific to Chinese isolates. These results provide insight into the factors responsible for the difference in virulence among the different S. suis 2 isolates.
Keywords: Streptococcus suis serotype 2, Transcriptome, Novel transcripts, sRNA, Virulence factor
Introduction
The Streptococcus suis serotype 2 (S. suis 2) are Gram-positive bacteria and represent the leading cause of porcine diseases [1]. They can also infect humans that have direct contact with infected swine, causing meningitis, hearing loss and septic shock [1–3]. In China, two large-scale outbreaks of severe human infections of S. suis 2 were reported in 1998 and 2005, respectively [4]. S. suis 2 is also one of the major pathogens associated with bacterial meningitis in other Asian areas, such as Hong Kong and Thailand [5–7]. Therefore, it poses a significant threat to public health [8].
The known markers of S. suis 2 include capsular polysaccharide (CPS), muramidase release protein (MRP), elongation factor (EF) and suilysin (SLY) [9]. The capsule is one of the key factors involved in bacterial pathogenicity. For instance, S. suis capsular polysaccharide inhibits phagocytosis through destabilization of lipid microdomains and prevents lactosylceramide-dependent recognition [9]. The capsule also protects the bacteria against phagocytosis. The Ssa protein of S. suis 2, a surface-anchored fibronectin-binding protein, also facilitates epithelial cell invasion [7]. In bacteria, two-component systems (TCSs) are crucial devices that enable their adaption to changing growth conditions, and hence affect bacterial virulence. More than 15 TCSs have been reported in S. suis 2 so far [4]. They are believed to facilitate bacterial adherence to mucosal epithelium cells, to participate in the process of capsular wall formation, and they have been shown to help bacteria survive and proliferate in in vivo mouse models [10–15]. Moreover, other regions of the S. suis 2 genome may contribute to its virulence as well [16].
In 2007, Chen et al. sequenced the whole genomes of the Chinese isolates of two S. suis 2 strains 05ZYH33 and 98HAH33, which were involved in the two aforementioned outbreaks, respectively [4]. Comparison of the genomes of these two Chinese strains with that of the reference genome, P1/7, suggested that an 89K genomic island (GI) is responsible for the altered pathogenicity of the Chinese isolates. Further analyses revealed that this 89K GI contains important virulence factors, including zeta toxin, TCS and ATP-binding cassette (ABC) transporters. Functional experiments revealed a dramatic decrease in the virulence of 05ZYH33 following the disruption of the TCS in the 89K GI [15]. Moreover, this 89K GI can be transmitted across different strains, and thus might represent a mechanism that enables S. suis 2 to adapt quickly to the local environment [17].
Although gene prediction methods have been widely applied in bacterial genome annotation, the use of next-generation sequencing (NGS) technology often reveals various novel transcripts, especially those belonging to the diverse small RNA (sRNA) families [18]. The regulatory role of these sRNAs in gene expression and in bacterial virulence is only now being slowly elucidated [19–22]. Here, we provide the first RNA-seq datasets from two Chinese isolates of S. suis 2.
Results
Transcriptome analysis pipeline and data statistics
We performed a strand-specific sequencing of the in vitro transcriptomes of three S. suis 2 strains, including the two highly-virulent isolates 05ZYH33 and 98HAH33, and the reference strain P1/7. About 27–40 million uniquely-mapped reads were obtained for each transcriptome, while achieving a good sequencing depth for further analysis (Table 1 and Figure S1). The regions with a single-nucleotide resolution sequencing coverage greater than 3 × accounted for over 93%, 92% and 95% of the whole genome of the 05ZYH33, 98HAH33 and P1/7 strains, respectively (data not shown). These percentages were similar to those previously reported for Bacillus anthracis (94%), Burkholderia mallei (95%) and Sulfolobus solfataricus (89.5%) [23–25], demonstrating the reliability of our datasets.
Table 1.
Transcriptome sequencing and mapping statistics for the three S. suis 2 strains
| Strain | No. of raw reads | No. of reads after rRNA removal | No. of mapped reads | No. of uniquely-mapped reads |
No. of TARs |
|
|---|---|---|---|---|---|---|
| + strand | − strand | |||||
| 05ZYH33 | 50,554,946 | 50,553,160 (99.999%) | 47,527,320 (94.01%) | 39,020,066 (82.1%) | 1285 | 1216 |
| 98HAH33 | 33,429,474 | 33,406,466 (99.93%) | 31,581,564 (94.54%) | 27,182,870 (86.07%) | 1332 | 1211 |
| P1/7 | 47,162,906 | 47,139,616 (99.95%) | 44,877,096 (95.2%) | 40,313,416 (92.43%) | 1222 | 1216 |
Note: Different coverage thresholds were used for identification of TARs (>70 bp in length) in the three strains, which are 9 × for 05ZYH33, 7 × for 98HAH33 and 16 × for P1/7, respectively. All the percentages in the parenthesis beside the absolute read No. were obtained by dividing the target read number by the one listed in the preceding column on its left. For example, percentage of “No. of reads after rRNA removal” = “No. of reads after rRNA removal” (50,553,160)/“No. of raw reads” (50,554,946) = 99.99%. TAR, transcriptionally-active region.
Analysis of differential gene expression in homologous genes
To compare the gene expression levels among the three isolates, we first identified their homologous genes (Materials and methods and Figure 1). Overall, 1672 homologous genes were identified in the three stains, and the differentially expressed genes (DEGs) were determined using three software packages (Materials and methods). In total, 318 genes were differentially expressed between 05ZYH33 and P1/7 strains, and 231 DEGs were identified between 98HAH33 and P1/7 strains. There were 120 DEGs common to these two comparisons (Table S1). Among them, expression of about 50% genes was upregulated 2–4 folds in the two Chinese isolates, and these genes shared similar expression patterns in the two Chinese strains when compared with that in P1/7 (Figure 2). Interestingly, most upregulated genes encode surface-anchored proteins, whereas the downregulated genes were enriched in the β-glucoside-specific phosphotransferase system (PTS) (Table S1). The most downregulated gene in the Chinese strains was SSU05_1491/SSU98_1502 (SSU1310, the homologous gene in P1/7), which encodes a transcriptional anti-terminator (Table S1).
Figure 1.
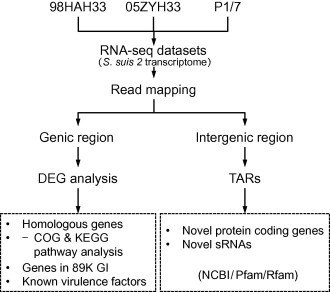
The pipeline for S. suis 2 transcriptome analysis
Sequencing reads were first mapped to reference genome. Sequencing reads in genic region are used to calculate the expression levels of each gene, and then DEGs are identified among three isolates. Sequencing reads found in intergenic regions are used to define novel TARs, which are annotated based on homology analysis. DEG, differentially-expressed gene; TAR, transcriptionally-active region; GI, genomic island.
Figure 2.
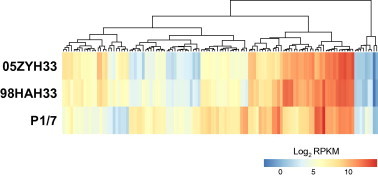
DEGs in the Chinese isolates shared a similar expression pattern
In total 120 DEGs are shared by both Chinese isolates when compared with P1/7. The hierarchical heatmap (R package: pheatmap) illustrates transcriptional levels (log2 ratios of RPKM, scale is shown at the bottom) for all the DEGs in three strains. The expression levels of DEGs in two Chinese isolates are more similar compared to P1/7.
The two Chinese isolates shared 246 additional homologous genes (not shown). Among the 10 most highly-expressed genes comparing with the reference isolate, eight were seen in both isolates. For example, SSU05_0582/SSU98_0586 encodes the IS5 family transposase, whereas SSU05_0179/SSSSU98_0181 are exoprotein A genes from the RTX family and the encoded protein was reported to cause hemolysis [26].
Clusters of orthologous group analysis
For all the genes annotated in previous studies, further scrutiny of the gene expression data revealed that 2092, 2094 and 1890 genes were identified with reads per kilo bases per million reads (RPKM) >1, which accounted for 92.8%, 92.9% and 94% of all genes in 05ZYH33, 98HAH33 and P1/7 strains, respectively. Among them, 1737, 1712 and 1559 genes were annotated with known clusters of orthologous groups (COG) functions in 05ZYH33, 98HAH33 and P1/7, respectively. A similar gene distribution in COGs was observed for all three strains (Figure S2), which indicates the similar biological processes can be observed in all three isolates.
To investigate the biological processes and show different activity in two Chinese isolates comparing with P1/7 isolate, we further analyzed the classification of the 120 aforementioned DEGs and found that 96 genes were classified into four major functional categories with 16 subgroups (Figure 3). Among the homologous genes that were upregulated in the Chinese isolates, 15 were significantly enriched in the functional group of translation, ribosomal structure and biogenesis (P < 0.000005, Fisher’s exact test). Meanwhile, among the homologous genes that were downregulated in the Chinese isolates, 22 significantly-enriched genes (P < 0.00000005, Fisher’s exact test) fall into the COG subgroup of carbohydrate transport and metabolism.
Figure 3.
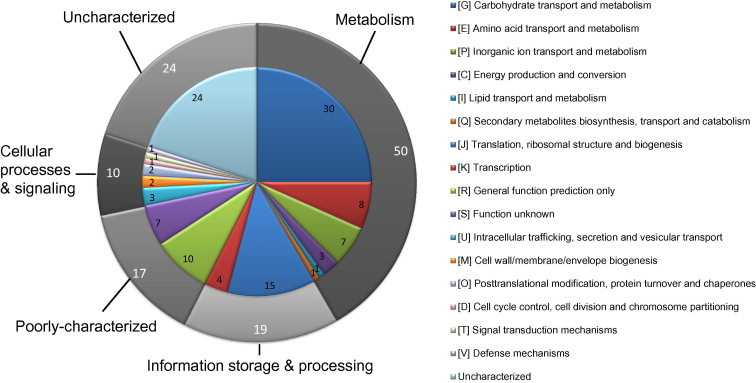
COG analysis of homologous DEGs
The pie chart showed the COG analysis for the classification of homologous DEGs in the 05ZYH33 and 98HAH33 isolates, compared to the P1/7 reference strain. The general categories are indicated on the outer circle and the subgroups are indicated in the inner circle with the number of genes belonging to each group listed.
KEGG pathway analysis
KEGG pathway mapping revealed that the biological processes that were abundant in the DEGs mainly include PTS, glycolysis/gluconeogenesis and ribosome (Table 2 and Figures S3−S5) (P < 0.01).
Table 2.
DEGs in PTS and glycolysis/gluconeogenesis pathways
| Strain | Gene symbol | Gene ID | FC | Protein |
|---|---|---|---|---|
| PTS pathway | ||||
| 98HAH33 | LacF | SSU98_1051 | 3.05 | PTS cellobiose-specific component IIA |
| Pts/Crr | SSU98_0384 | 1.92 | PTS IIC component, glucose | |
| AgaW | SSU98_1233 | −1.37 | PTS, mannose | |
| UlaA | SSU98_0191 | −1.38 | Hypothetical protein | |
| ManY | SSU98_0439 | −1.43 | PTS, mannose | |
| LacE | SSU98_0210 | −1.49 | PTS cellobiose-specific component IIC | |
| CelB | SSU98_2074 | −1.62 | PTS cellobiose-specific component IIC | |
| CelB | SSU98_0712 | −1.66 | PTS cellobiose-specific component IIC | |
| AgaE | SSU98_1232 | −1.71 | PTS, mannose | |
| ManZ | SSU98_0440 | −1.80 | PTS, mannose | |
| BglF | SSU98_1501 | −6.74 | PTS IIC component, glucose | |
| 05ZYH33 | LacF | SSU05_1038 | 1.11 | PTS cellobiose-specific component IIA |
| Pts/Crr | SSU05_0398 | 1.56 | PTS IIC component, glucose/maltose/N-acetylglucosamine-specific | |
| AgaW | SSU05_1218 | −2.59 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IIC | |
| UlaA | SSU05_0189 | −1.77 | PTS system ascorbate-specific transporter subunit IIC | |
| ManY | SSU05_0451 | −2.19 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IIC | |
| LacE | SSU05_0212 | −3.43 | PTS cellobiose-specific component IIC | |
| CelB | SSU05_2071 | −1.67 | PTS cellobiose-specific component IIC | |
| CelB | SSU05_0713 | −3.26 | PTS cellobiose-specific component IIC | |
| AgaE | SSU05_1217 | −3.05 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IID | |
| ManZ | SSU05_0452 | −2.33 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IID | |
| BglF | SSU05_1490 | −5.40 | PTS IIC component, glucose/maltose/N-acetylglucosamine-specific | |
| Strain | EC ID | Gene ID | FC | Protein |
| Glycolysis/gluconeogenesis pathway | ||||
| 98HAH33 | 2.7.1.69 | SSU98_0384 | 1.92 | PTS IIC component, glucose |
| 3.2.1.86 | SSU98_2083 | −1.02 | Alpha-galactosidase | |
| 1.1.1.1 | SSU98_0274 | −1.63 | AdhA | |
| 1.1.1.1 | SSU98_0275 | −1.92 | Bifunctional acetaldehyde-CoA | |
| 3.2.1.86 | SSU98_1500 | −6.14 | Beta-glucosidase | |
| 05ZYH33 | 2.7.1.69 | SSU05_0398 | 1.56 | PTS IIC component, glucose/maltose/N-acetylglucosamine-specific |
| 3.2.1.86 | SSU05_2079 | −1.69 | Alpha-galactosidase/6-phospho-beta-glucosidase | |
| 1.1.1.1 | SSU05_0279 | −2.29 | Alcohol dehydrogenase | |
| 1.1.1.1 | SSU05_0280 | −3.01 | Bifunctional acetaldehyde-CoA/alcohol dehydrogenase | |
| 3.2.1.86 | SSU05_1489 | −5.05 | Beta-glucosidase/6-phospho-beta-glucosidase/beta-galactosidase | |
Note: FC stands for fold change, which was calculated by log2 expression of the respective Chinese isolate/expression of P1/7. PTS, phosphotransferase system. EC ID refers to the enzyme commission number in KEGG.
Although all three strains shared the species-specific components in the PTS pathway, fewer transcripts of the Man family genes were detected in 98HAH33 and 05ZYH33 (Figure S3). Conversely, most Gk family genes except BgIF (PTS system, β-glucosides-specific II component) had more transcripts detected in the two Chinese isolates. However, it is of note that BgIF is the most downregulated gene in PTS pathway, suggesting a reduced conversion from β-glucosides to phospho-β-glucosides.
In addition, the transcriptional level of the genes (SSU05_0398/SSU98_0384) responsible for transforming arbutin and salicin into arbutin-6p and salicin-6p, respectively, was increased, whereas the transcriptional activity of downstream 6-phospho-β-glucosidase (Enzyme Commission number: 3.2.1.86, encoded by SSU05_1489/SSU98_1500 and SSU05_2079/SSU98_2083) was extensively repressed in the two Chinese isolates (Figure S4). Therefore, their products arbutin-6p and salicin-6p may accumulate in the bacterial metabolites of the two Chinese isolates.
Differential expression levels of known virulence factors
It is believed that two Chinese isolates are more pathogenic than P1/7 isolate, and we next analyzed transcriptional level of all known virulence factors in three isolates. Our analysis indicated that these three isolates had 96 known virulence factors in common according to VFDB. However, only the gene encoding ORF46-putative ABC transporter membrane protein (SSU05_0111/SSU98_0114/SSU0114, VFG1497) was highly expressed in the Chinese isolates; whereas expression of three genes including SSU05_0148/SSU98_0152/SSU0147, SSU05_1381/SSU98_1396/SSU0466 and SSU05_1387/SSU98_1401/SSU1215, were downregulated (Table 3). We then analyzed all the DEGs encoding virulence factors shared by P1/7 and either 05ZYH33 or 98HAH33 rather than both of them. The KEGG analysis revealed that the ABC transporter pathway was enriched in the highly-expressed virulence factors in the 98HAH33 and 05ZYH33 (Table S2).
Table 3.
Differentially-expressed virulence factors
| Category | VFDB ID | Gene ID | RPKM | Gene symbol | Protein |
|---|---|---|---|---|---|
| All three isolates | |||||
| Upregulated | VFG1497 | SSU05_0111/SSU98_0114/SSU0114 | 152.6/292.4/79.3 | ORF46 | Putative ABC transporter membrane protein |
| Downregulated | VFG1855 | SSU05_0148/SSU98_0152/SSU0147 | 2680.4/2435.8/3243.0 | htpB | Hsp60, 60 K heat shock protein HtpB |
| VFG0869 | SSU05_1381/SSU98_1396/SSU0466 | 147.1/212.3/679.6 | aatC | AatC ATB binding protein of ABC transporter | |
| VFG0594 | SSU05_1387/SSU98_1401/SSU1215 | 135.3/359.7/912.5 | pipD | Pathogenicity island encoded protein: SPI3 | |
| 05ZYH33/98HAH33-specific | |||||
| VFG0317 | SSU05_1281/SSU98_1295 | 119.3/90.4 | rfaJ | Lipopolysaccharide 1,2-glucosyltransferase | |
| VFG2194 | SSU05_2090/SSU98_2093 | 64.5/81.1 | fsrA | Unknown | |
| VFG1462 | SSU05_0588/SSU98_0592 | 23.4/33.5 | l0015 | Putative IS protein | |
| VFG1513 | SSU05_0587/SSU98_0591 | 15.2/16.1 | s0025 | IS66-like transposase | |
| VFG0284 | SSU05_0973/SSU98_0987 | 0.8/1.9 | cag5 (virD4) | Cag pathogenicity island protein, DNA transfer protein | |
| VFG2173 | SSU05_0965/SSU98_0978 | 0.6/1.0 | asal | Aggregation substance Asa1 | |
| VFG1453 | SSU05_0979/SSU98_0993 | 0.2/0.7 | ORF2 | Putative DNA methylase | |
| Well-known factors | |||||
| Upregulated | – | SSU05_0753/SSU98_0756/SSU0706 | 4575.6/6524.3/2045.4 | mrp | Muramidase release protein |
| – | SSU05_0155/SSU98_0158/SSU0153 | 19380.7/20694.6/16530.14 | gapdh | Glyceraldehyde-3-phosphate dehydrogenase | |
| Downregulated | – | SSU05_0841/SSU98_0841/SSU0785 | 423.3/339.3/506.326 | lepA | Elongation factor |
| Mixed | – | SSU05_1403/SSU98_1416/SSU1231 | 137.9/677.642/440.004 | sly | Suilysin |
| – | SSU05_0564/SSU98_0567/SSU0515 | 1026.3/441.9/704.4 | cps2A | Capsular polysaccharide | |
Note: ID for the genes, which essentially encode the same virulence factor in the strains and their corresponding RPKM, were listed in the order of 05ZYH33, 98HAH33 and P1/7. For example, Gene ID SSU05_0111/SSU98_0114/SSU0114 represent SSU05_0111 in 05ZYH33, SSU98_0114 in 98HAH33 and SSU0114 in P1/7, which all encode ORF46, and their expression level in terms of RPKM is 152.6 in 05ZYH33, 292.4 in 98HAH33 and 79.3 in P1/7, respectively. Mixed means that the expression of genes was upregulated in one Chinese isolate but downregulated in the other isolate.
Furthermore, seven other virulence factors were found to be specific to the two virulence strains (Table 3). For instance, The RPKM value of SSU05_1281/SSU98_1295 ranked the top among those of the seven factors. SSU05_1281/SSU98_1295 encodes lipopolysaccharide 1,2-glucosyltransferase (RfaJ), which adds a glucose (II) group on the galactose (I) group of lipopolysaccharides (LPS). The gene expression of well-known factors such as cps, mrp, lepA (EF4) and sly are also listed in Table 3.
Transcriptional activity of the 89K GI in 98HAH33 and 05ZYH33
89K GI is specific to two pathogenic Chinese isolates. In this region, 98HAH33 and 05ZYH33 strains have 78 homologous genes. Among them, 59 genes had ⩾ 3 sequencing reads recovered in the RNA-seq datasets. The distribution of the RPKM values for all of these genes in the two strains showed that highest number of genes had RPKM values < 1, indicating a low transcriptional activity during in vitro culture (Figure 4). Three known virulence factors specific to the Chinese isolates, including VFG0284, VFG2173 and VFG1453 (Table 3), were located in this GI as well.
Figure 4.
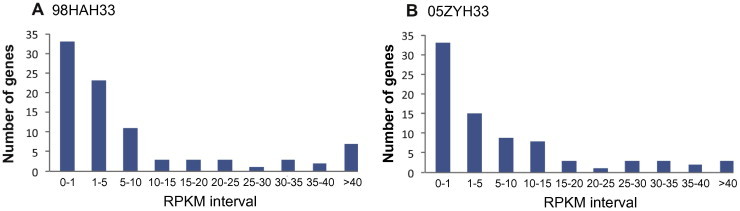
Transcriptional activity of genes located in 89K GI of the Chinese isolates
The histogram demonstrates the distribution of genes at different transcription level (RPKM values) for 98HAH33 (A) and 05ZYH33 (B). The group “RPKM <1” contains the highest number of genes, indicating the low transcriptional activity of 89K GI region during in vitro incubation of these isolates.
Six genes with high transcriptional activity were seen in both strains (Table 4). According to the STRING database (Search Tool for the Retrieval of Interacting Genes/Proteins, http://www.string-db.org/), a signal recognition particle GTPase encoded by SSU05_0936/SSU98_0943 and a transcriptional regulator encoded by SSU05_0937/SSU98_0944 can form a toxin–antitoxin (TA) system. This TA system showed a high homology with the PezAT chromosomal TA system of the human pathogen Streptococcus pneumoniae [27].
Table 4.
Top 10 most highly-expressed genes specific to the Chinese isolates
| Strain | Rank | Gene ID | RPKM | COG ID | Protein |
|---|---|---|---|---|---|
| 05ZYH33 | 1 | SSU05_0582 | 2067.935 | COG3039L | IS5 family transposase |
| 2 | SSU05_1895 | 1461.981 | – | Hypothetical protein | |
| 3 | SSU05_1896 | 1428.723 | COG3871R | Hypothetical protein | |
| 4 | SSU05_0177 | 1284.939 | – | Extracellular protein | |
| 5 | SSU05_0178 | 1190.736 | – | Epf-like protein | |
| 6 | SSU05_0179 | 1149.505 | – | RTX family exoprotein A gene | |
| 7 | SSU05_0373 | 1073.18 | COG1335Q | Nicotinamidase-like amidase | |
| 8 | SSU05_0372 | 782.191 | COG1670J | Histone acetyltransferase HPA2-like acetyltransferase | |
| 9 | SSU05_0553 | 664.709 | – | Hypothetical protein | |
| 10 | SSU05_1990 | 660.201 | COG3481R | HD-superfamily hydrolase | |
| 98HAH33 | 1 | SSU98_0181 | 2124.663 | – | RTX family exoprotein A gene |
| 2 | SSU98_0586 | 1986.35 | COG3039L | IS5 family transposase | |
| 3 | SSU98_0179 | 1799.871 | – | Extracellular protein | |
| 4 | SSU98_0180 | 1684.485 | – | Epf-like protein | |
| 5 | SSU98_1014 | 1076.934 | – | Hypothetical protein | |
| 6 | SSU98_0364 | 1049.139 | COG1335Q | Nicotinamidase-like amidase | |
| 7 | SSU98_1026 | 986.816 | COG0448G | ADP-glucose pyrophosphorylase | |
| 8 | SSU98_1897 | 972.083 | – | Hypothetical protein | |
| 9 | SSU98_0363 | 876.425 | COG1670J | Histone acetyltransferase HPA2-like acetyltransferase | |
| 10 | SSU98_1898 | 796.214 | COG3871R | Hypothetical protein | |
Note: The toxin–antitoxin system components with high transcription level in both virulent isolates are highlighted in bold. RPKM, reads per kilo bases per million reads; COG, clusters of orthologous groups.
Novel transcripts identified in the transcriptomes of three isolates
Gene annotation of these three isolates is mainly based on prediction method, and we used transcriptome data to find novel genes in three genomes. In all, over 50 novel transcriptionally-active regions (TARs) were identified in intergenic regions of the two Chinese isolates (see Material and Method). Most of them were homologous to genes that only encode hypothetical proteins (Table S3). Only 9 and 8 TARs had homologous genes with known functions for the 05ZYH33 and 98HAH33, respectively (Table 5). In particular, one TAR predicted to encode a toxin protein was identified in both isolates.
Table 5.
Novel TARs with homologous genes having known functions
| Strain | TAR ID | Strand | Start position | End position | Length | Homologous gene | Genome ID | Protein |
|---|---|---|---|---|---|---|---|---|
| 05ZYH33 | 05zyh33_new1 | + | 24,422 | 24,911 | 490 | GI: 386587281 | NC_017622.1 | Rod shape-determining protein MreD |
| 05zyh33_new6 | + | 84,830 | 84,947 | 118 | GI: 403060703 | NC_018526.1 | 50S ribosomal protein L36 | |
| 05zyh33_new7 | + | 208,869 | 209,049 | 181 | GI: 403060703 | NC_018526.1 | Copper-transporting ATPase | |
| 05zyh33_new8 | − | 464,948 | 465,239 | 292 | GI: 386587281 | NC_017622.1 | Transposase | |
| 05zyh33_new16 | + | 566,368 | 566,536 | 169 | GI: 403060703 | NC_018526.1 | N-acylneuraminate cytidylyltransferase | |
| 05zyh33_new22 | − | 933,091 | 933,265 | 175 | GI: 386585202 | NC_017621.1 | Lantibiotic protein | |
| 05zyh33_new23 | − | 999,593 | 999,674 | 82 | GI: 403060703 | NC_018526.1 | Helicase subunit of the DNA excision repair complex | |
| 05zyh33_new34 | − | 1,578,674 | 1,578,830 | 157 | GI: 403060703 | NC_018526.1 | Serum opacity factor | |
| 05zyh33_new48 | − | 1,915,147 | 1,915,405 | 259 | GI: 403060703 | NC_018526.1 | Toxin–antitoxin system, toxin protein | |
| 98HAH33 | 98hah33_new5 | + | 84,826 | 84,943 | 118 | GI: 403060703 | NC_018526.1 | 50S ribosomal protein L36 |
| 98hah33_new7 | + | 208,714 | 208,894 | 181 | GI: 403060703 | NC_018526.1 | Copper-transporting ATPase | |
| 98hah33_new17 | + | 566,231 | 566,399 | 169 | GI: 403060703 | NC_018526.1 | N-acylneuraminate cytidylyltransferase | |
| 98hah33_new24 | − | 932,796 | 932,970 | 175 | GI: 386585202 | NC_017621.1 | Lantibiotic protein | |
| 98hah33_new25 | − | 999,285 | 999,366 | 82 | GI: 403060703 | NC_018526.1 | Helicase subunit of the DNA excision repair complex | |
| 98hah33_new27 | − | 1,116,314 | 1,116,593 | 280 | GI: 403060703 | NC_018526.1 | Membrane associated protein | |
| 98hah33_new45 | − | 1,892,560 | 1,892,725 | 166 | GI: 403060703 | NC_018526.1 | 50S ribosomal protein L33 | |
| 98hah33_new48 | − | 1,914,521 | 1,914,779 | 259 | GI: 403060703 | NC_018526.1 | Toxin–antitoxin system, toxin protein | |
Note: GI number is listed for each homologous gene. TAR, transcriptionally-active region.
We also predicted 56 sRNAs in three isolates. Among them, 12 sRNAs related to bacterial virulence are listed in Table 6. It is of note that rliD and RatA were detected in all three isolates. In addition, the two Chinese isolates also had the sRNAs cspA and rli38 in common and the latter was located at the 89K GI. For the rest sRNAs, over 27% (12/44) of the novel sRNAs were involved in riboswitches (Table S4).
Table 6.
Novel sRNAs potentially regulating bacterial virulence
| Strain | TAR ID | Strand | TAR | Mapped with homologous genes | sRNA | Description | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | Length | Start | End | ||||
| 05ZYH33 | 05seq336 | − | 389,484 | 392,352 | 389,489 | 389,850 | 362 | 1 | 428 | cspA: cspA thermoregulator |
| 05seq972 | + | 1,825,119 | 1,830,751 | 1,830,583 | 1,830,751 | 169 | 1 | 169 | rliD: Listeria sRNA rliD | |
| 05seq1101 | + | 1,993,916 | 1,994,224 | 1,993,916 | 1,993,992 | 77 | 10 | 91 | RatA: RNA anti-toxin A | |
| 05seq710 | + | 961,698 | 961,817 | 961,698 | 961,817 | 120 | 15 | 134 | rli38: Listeria sRNA rli38 | |
| 98HAH33 | 98seq331 | − | 391,826 | 392,204 | 391,827 | 392,185 | 359 | 1 | 428 | cspA: cspA thermoregulator |
| 98seq1086 | − | 2,057,227 | 2,057,829 | 2,057,231 | 2,057,296 | 66 | 1 | 71 | STnc370: Enterobacterial sRNA STnc370 | |
| 98seq1021 | + | 1,824,549 | 1,830,052 | 1,829,956 | 1,830,052 | 97 | 1 | 97 | rliD: Listeria sRNA rliD | |
| 98seq1137 | + | 1,993,284 | 1,993,592 | 1,993,284 | 1,993,360 | 77 | 10 | 91 | RatA: RNA anti-toxin A | |
| 98seq696 | + | 961,400 | 961,519 | 961,400 | 961,519 | 120 | 15 | 134 | rli38: Listeria sRNA rli38 | |
| P1/7 | 17seq501 | − | 635,186 | 635,918 | 635,187 | 635,263 | 77 | 1 | 77 | isrK: isrK Hfq binding RNA |
| 17seq1095 | − | 1,905,111 | 1,905,186 | 1,905,111 | 1,905,186 | 76 | 1 | 91 | RatA: RNA anti-toxin A | |
| 17seq916 | + | 1,736,325 | 1,741,929 | 1,741,790 | 1,741,929 | 140 | 1 | 140 | rliD: Listeria sRNA rliD | |
Note: TAR, transcriptionally-active region. Mapped starts and ends indicate the region of TARs that is aligned with homologous genes. Starts and ends of sRNA indicate the corresponding part of sRNA. TARs that are shared in all three strains are highlighted in bold in description.
Discussion
Known virulence factors contributing to the pathogenicity of S. suis 2
Although in vitro transcriptomes do not resemble in vivo ones, this analysis nonetheless illustrated the overall expression patterns for all the known virulence factors of S. suis 2 during in vitro incubation. The genes encoding the five well-known factors, mrp, gapdh, sly, lepA (EF4) and cps2A, all had relatively high transcription levels comparing to other virulence factors (Table 3). In particular, although it was previously reported that inactivation of mrp is related to an increase in bacterial virulence [28], the two pathogenic strains had even higher transcription levels of mrp.
Glucose metabolism in bacteria not only reflects their priority for certain carbon sources, but also affects their virulence [29]. PTS functions as a sugar phosphorylating system and regulates diverse metabolic processes that affect the expression of many genes, especially those related to bacterial virulence [30]. In Listeria monocytogenes, the depletion of the mpt operon, which encodes the EII(t)(Man) PTS permease, resulted in increased expression of five genes in the prfA virulence gene cluster [31]. In this study, we observed the downregulation of four genes of the Man family in the PTS pathway, suggesting the involvement of the PTS pathway in changing the virulence of both Chinese isolates.
Downregulation of transcriptional anti-terminators may influence the virulence of the Chinese isolates
In the PTS pathway, the most significantly downregulated gene was SSU05_1490/SSU98_1501 (BgIF). In a recent study performed in Escherichia coli, BgIF has been shown to negatively regulate transcriptional anti-terminator proteins of the BgIG family [32]. Both Chinese isolates just have one gene that belongs to the BgIG family, SSU05_1491/SSU98_1502, with fewer transcripts identified compared to P1/7, rather than increased as predicted. Therefore, there may be other novel proteins in the BgIG family or distinctive regulatory mechanisms in S. suis 2, which have yet to be discovered. Nevertheless, previous studies showed that decreased activity of the transcriptional anti-terminator proteins might change the bacterial virulence [33]. For instance, the reduction in the expression of the transcriptional anti-terminator RfaH may increase the Ag43 expression level, and thereby stimulate biofilm formation [33]. The repressed transcription of SSU05_1491/SSU98_1502 indicates that some mechanism is responsible for the change in virulence in the two Chinese isolates.
Conclusion
Here, we provided the first publicly-available RNA-seq data for three S. suis 2 isolates, and we highlighted the biological processes in the two highly pathogenic isolates, providing clues for mechanistic research. Furthermore, our detailed lists of novel protein-coding genes and sRNAs provide candidates for further functional investigations, especially in the regulation of bacterial virulence.
Materials and methods
Bacterial culture
The three S. suis 2 strains 98HAH33, 05ZYH33 and P1/7 were obtained from Institute of Microbiology, Chinese Academy of Sciences. Bacteria were grown in Todd-Hewitt Broth (THB) (Difco Laboratories, Detroit, MI, USA) supplemented with 2% yeast extract (THY) at 37 °C. The growth rates and bacterial concentrations for all strains were determined by measuring the optical density (OD) at 600 nm each 60 min (Figure S6). All isolates reached the log/exponential phase after incubation for 5 h.
Transcriptome sequencing
Total RNA was isolated from bacteria at log/exponential phase using the SV Total RNA Isolation system (Promega), according to the manufacturer’s instructions. Ribosomal RNAs were removed using the Ribo-Zero™ rRNA Removal Kit for Gram-Positive Bacteria (EPICENTRE Biotechnologies, Madison, WI). Strand-specific libraries were constructed and sequenced on the Illumina GAII platform by the Genomics and Bioinformatics Platform at the Beijing Institute of Genomics, Chinese Academy of Sciences. The raw sequencing datasets were uploaded to the NCBI SRA database (accession No: SRP043973).
Read mapping
The sequencing read quality was evaluated using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and data saturation analyses were carried out using the RSeQC software [34]. Qualified reads (quality score > 20) were first filtered for ribosomal RNAs using the BWA software (http://bio-bwa.sourceforge.net/), and the rest were subsequently mapped to reference genomes of 98HAH33,05ZYH33 and P1/7. The uniquely-mapped reads (MAPQ > 20) were used for further analysis. In addition, the sequencing coverage for single nucleotide of the genome as calculated using the genomeCoverageBed in BEDTool [35].
Gene expression analysis for homologous genes
Identification of homologous genes
Protein sequences were analyzed to identify homology across the three isolates by using the Inparanoid software [36] and a pair-wise analysis was also performed to identify the homology between any two strains by using the MultiParanoid software [37].
Differential gene expression
Based on the gene annotations in the database, the RPKM values were calculated for each gene using uniquely-mapped reads [38]. Raw read counts of genes were calculated by using HTSeq [39], BEDTools [35] and RSeQC [34]. Furthermore, DEGseq [40] was used to identify DEGs with the MARS model, with the criteria of fold change > 2 and P < 0.05.
Pathway analysis
Candidate genes were first mapped onto functional groups in COG; Gene Oncology and KEGG pathway analyses were performed online using the DAVID software [41] (Fisher’s exact test, P < 0.05).
Virulence factor analysis
Virulence factors were extracted from the virulence factor database (VFDB) [42]. A VFs.faa file was used for local blasting with BLASTP (v2.2.24, using default parameter with E value set as 1E−5), and the top hits were used as candidates for further analysis.
Gene annotation of the S. suis 2 genome using RNA-seq data
Identification of novel TARs in intergenic regions
The sequencing depth for each nucleotide was first calculated, and a nucleotide was defined to be transcriptionally-active when its sequencing depth was higher than the lower deciles of the sequencing depth in the transcriptome. Consecutive transcriptionally-active nucleotides form TARs. In this study, TARs > 70 bp in intergenic regions were further annotated [43].
Annotation of novel genes
The DNA sequences for all novel TARs were blasted against NCBI Bacterial Genomes (all.ffn.tar.gz) for homology identification using BLAT. The functions of the novel TARs in three S. suis 2 isolates were annotated based on the function of their known homologous genes in other bacteria.
Identification of sRNAs
TARs without identified homologous genes were further compared with sRNAs in both sRNAMAP [44] using BLAT(V35) [45] and Rfam (11.0) using INFERNAL(1.1rc2) [46] to identify the potential sRNAs.
Authors’ contributions
CZ conceived the project. DZ, ND and SM designed the experiments and performed the data analysis. SM and GL prepared all the samples. QH, GL and WC participated in sequencing experiments and data analysis. DZ, ND and CZ wrote the manuscript. All the authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgements
We thank Joel Haywood (Institute of Microbiology, Chinese Academy of Sciences, China) for proofreading and copyediting. This research was supported by the CAS Key Laboratory of Pathogenic Microbiology and Immunology of China (Grant No. 2009CASPMI-007) to DZ and the National Natural Science Foundation of China (Grant No. 81201700) to DZ.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Supplementary material
Growth curves for all three strains. Growth curves for the P1/7, 98HAH33 and 05ZYH33 are depicted in blue, red and green respectively. P1/7 seems grow a little faster than two Chinese isolates after incubation for 6 h.
Sequencing saturation analysis. The saturation analysis was performed using RseQC package. Genes were sorted according to the RPKM values from low to high, and classified into 4 groups based on quartiles (Q1−Q4). The X-axis represents the percentage of reads that were randomly extracted from total reads for calculation of expression level, and the Y-axis represents the error between the calculated express level and the actual expression level. The curve becomes flat when ⩾70% reads were extracted, indicating that the number of reads is more than enough for RPKM calculation for all genes, and the expression levels of genes are accurate.
All three strains shared similar distribution of expressed genes in each COG. All the expressed genes in three isolates are classified according to COG category. For each category, three strains have similar number of expressed genes.
DEGs in the PTS pathway. Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Different transcription levels between the two Chinese isolates and the reference P1/7 are depicted in different colors as follows: upregulated genes with fold change >2 (pink), upregulated genes with fold change >8 (red), downregulated genes with fold change >2 (yellow), downregulated genes with fold change >8 (purple) and no expression changes observed (green).
DEGs in glycolysis/gluconeogenesis pathway. Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Different transcription levels between the two Chinese isolates and the reference P1/7 are depicted in different colors as follows: upregulated genes with fold change >2 (pink), upregulated genes with fold change >8 (red), downregulated genes with fold change >2 (yellow), downregulated genes with fold change >8 (purple) and no expression changes observed (green).
Highly-expressed genes enriched in KEGG category (Ribosome). Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Genes that showed upregulated expression with fold change >2 between the two Chinese isolates and the reference P1/7 are boxed in red, whereas genes without altered expression between the two Chinese isolates and the reference P1/7 are boxed in green. The corresponding gene names in other S. suis isolates are indicated in purple background above.
Differentially-expressed homologous genes shared by all three strains.
DEGs in ABC pathway.
Novel TARs predicted to encode hypothetical proteins.
Novel sRNAs mainly related to riboswitches.
References
- 1.Wertheim H.F., Nghia H.D., Taylor W., Schultsz C. Streptococcus suis: an emerging human pathogen. Clin Infect Dis. 2009;48:617–625. doi: 10.1086/596763. [DOI] [PubMed] [Google Scholar]
- 2.Palmieri C., Varaldo P.E., Facinelli B. Streptococcus suis, an emerging drug-resistant animal and human pathogen. Front Microbiol. 2011;2:235. doi: 10.3389/fmicb.2011.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan J.H., Yeh B.I., Seet C.S. Deafness due to haemorrhagic labyrinthitis and a review of relapses in Streptococcus suis meningitis. Singapore Med J. 2010;51:e30–e33. [PubMed] [Google Scholar]
- 4.Chen C., Tang J., Dong W., Wang C., Feng Y., Wang J. A glimpse of streptococcal toxic shock syndrome from comparative genomics of S. suis 2 Chinese isolates. PLoS One. 2007;2:e315. doi: 10.1371/journal.pone.0000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma E., Chung P.H., So T., Wong L., Choi K.M., Cheung D.T. Streptococcus suis infection in Hong Kong: an emerging infectious disease? Epidemiol Infect. 2008;136:1691–1697. doi: 10.1017/S0950268808000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerdsin A., Dejsirilert S., Puangpatra P., Sripakdee S., Chumla K., Boonkerd N. Genotypic profile of Streptococcus suis serotype 2 and clinical features of infection in humans, Thailand. Emerg Infect Dis. 2011;17:835–842. doi: 10.3201/eid1705.100754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W., Wan Y., Tao Z., Chen H., Zhou R. A novel fibronectin-binding protein of Streptococcus suis serotype 2 contributes to epithelial cell invasion and in vivo dissemination. Vet Microbiol. 2013;162:186–194. doi: 10.1016/j.vetmic.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Feng Y., Zhang H., Ma Y., Gao G.F. Uncovering newly emerging variants of Streptococcus suis, an important zoonotic agent. Trends Microbiol. 2010;18:124–131. doi: 10.1016/j.tim.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Charland N., Harel J., Kobisch M., Lacasse S., Gottschalk M. Streptococcus suis serotype 2 mutants deficient in capsular expression. Microbiology. 1998;144:325–332. doi: 10.1099/00221287-144-2-325. [DOI] [PubMed] [Google Scholar]
- 10.Xu J., Fu S., Liu M., Xu Q., Bei W., Chen H. The two-component system NisK/NisR contributes to the virulence of Streptococcus suis serotype 2. Microbiol Res. 2014;169:541–546. doi: 10.1016/j.micres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Shen X., Zhong Q., Zhao Y., Yin S., Chen T., Hu F. Proteome analysis of the two-component SalK/SalR system in Epidemic Streptococcus suis serotype 2. Curr Microbiol. 2013;67:118–122. doi: 10.1007/s00284-013-0343-4. [DOI] [PubMed] [Google Scholar]
- 12.Wang H., Shen X., Zhao Y., Wang M., Zhong Q., Chen T. Identification and proteome analysis of the two-component VirR/VirS system in epidemic Streptococcus suis serotype 2. FEMS Microbiol Lett. 2012;333:160–168. doi: 10.1111/j.1574-6968.2012.02611.x. [DOI] [PubMed] [Google Scholar]
- 13.Han H., Liu C., Wang Q., Xuan C., Zheng B., Tang J. The two-component system Ihk/Irr contributes to the virulence of Streptococcus suis serotype 2 strain 05ZYH33 through alteration of the bacterial cell metabolism. Microbiology. 2012;158:1852–1866. doi: 10.1099/mic.0.057448-0. [DOI] [PubMed] [Google Scholar]
- 14.Li J., Tan C., Zhou Y., Fu S., Hu L., Hu J. The two-component regulatory system CiaRH contributes to the virulence of Streptococcus suis 2. Vet Microbiol. 2011;148:99–104. doi: 10.1016/j.vetmic.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Li M., Wang C., Feng Y., Pan X., Cheng G., Wang J. SalK/SalR, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS One. 2008;3:e2080. doi: 10.1371/journal.pone.0002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H., Fan H.J., Lu C.P. Identification and distribution of putative virulent genes in strains of Streptococcus suis serotype 2. Vet Microbiol. 2009;133:309–316. doi: 10.1016/j.vetmic.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Li M., Shen X., Yan J., Han H., Zheng B., Liu D. GI-type T4SS-mediated horizontal transfer of the 89K pathogenicity island in epidemic Streptococcus suis serotype 2. Mol Microbiol. 2011;79:1670–1683. doi: 10.1111/j.1365-2958.2011.07553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croucher N.J., Thomson N.R. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol. 2010;13:619–624. doi: 10.1016/j.mib.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romilly C., Lays C., Tomasini A., Caldelari I., Benito Y., Hammann P. A non-coding RNA promotes bacterial persistence and decreases virulence by regulating a regulator in Staphylococcus aureus. PLoS Pathog. 2014;10:e1003979. doi: 10.1371/journal.ppat.1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wootton L. Bacterial pathogenesis: sRNA clears the way for G4. Nat Rev Microbiol. 2013;11:146. doi: 10.1038/nrmicro2985. [DOI] [PubMed] [Google Scholar]
- 21.Lamichhane G., Arnvig K.B., McDonough K.A. Definition and annotation of (myco) bacterial non-coding RNA. Tuberculosis. 2013;93:26–29. doi: 10.1016/j.tube.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Mellin J.R., Cossart P. The non-coding RNA world of the bacterial pathogen Listeria monocytogenes. RNA Biol. 2012;9:372–378. doi: 10.4161/rna.19235. [DOI] [PubMed] [Google Scholar]
- 23.Wurtzel O., Sapra R., Chen F., Zhu Y., Simmons B.A., Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Passalacqua K.D., Varadarajan A., Ondov B.D., Okou D.T., Zwick M.E., Bergman N.H. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sittka A., Lucchini S., Papenfort K., Sharma C.M., Rolle K., Binnewies T.T. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lally E.T., Hill R.B., Kieba I.R., Korostoff J. The interaction between RTX toxins and target cells. Trends Microbiol. 1999;7:356–361. doi: 10.1016/s0966-842x(99)01530-9. [DOI] [PubMed] [Google Scholar]
- 27.Khoo S.K., Loll B., Chan W.T., Shoeman R.L., Ngoo L., Yeo C.C. Molecular and structural characterization of the PezAT chromosomal toxin–antitoxin system of the human pathogen Streptococcus pneumoniae. J Biol Chem. 2007;282:19606–19618. doi: 10.1074/jbc.M701703200. [DOI] [PubMed] [Google Scholar]
- 28.Boyle M.D., Raeder R., Flosdorff A., Podbielski A. Role of emm and mrp genes in the virulence of group A streptococcal isolate 64/14 in a mouse model of skin infection. J Infect Dis. 1998;177:991–997. doi: 10.1086/515241. [DOI] [PubMed] [Google Scholar]
- 29.Gorke B., Stulke J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol. 2008;6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 30.Deutscher J., Francke C., Postma P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev. 2006;70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vu-Khac H., Miller K.W. Regulation of mannose phosphotransferase system permease and virulence gene expression in Listeria monocytogenes by the EII(t)Man transporter. Appl Environ Microbiol. 2009;75:6671–6678. doi: 10.1128/AEM.01104-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothe F.M., Bahr T., Stulke J., Rak B., Gorke B. Activation of Escherichia coli antiterminator BglG requires its phosphorylation. Proc Natl Acad Sci U S A. 2012;109:15906–15911. doi: 10.1073/pnas.1210443109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beloin C., Michaelis K., Lindner K., Landini P., Hacker J., Ghigo J.M. The transcriptional antiterminator RfaH represses biofilm formation in Escherichia coli. J Bacteriol. 2006;188:1316–1331. doi: 10.1128/JB.188.4.1316-1331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 35.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien K.P., Remm M., Sonnhammer E.L. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–D480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexeyenko A., Tamas I., Liu G., Sonnhammer E.L. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics. 2006;22:e9–e15. doi: 10.1093/bioinformatics/btl213. [DOI] [PubMed] [Google Scholar]
- 38.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 39.Anders S., Pyl P.T., Huber W. HTSeq — a python framework to work with high-throughput sequencing data. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L., Feng Z., Wang X., Wang X., Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 41.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Chen L., Xiong Z., Sun L., Yang J., Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40:D641–D645. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy J.S., Kumar R., Watt J.M., Lawrence M.L., Burgess S.C., Nanduri B. Transcriptome profile of a bovine respiratory disease pathogen: Mannheimia haemolytica PHL213. BMC Bioinformatics. 2012;13:S4. doi: 10.1186/1471-2105-13-S15-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang H.Y., Chang H.Y., Chou C.H., Tseng C.P., Ho S.Y., Yang C.D. sRNAMap: genomic maps for small non-coding RNAs, their regulators and their targets in microbial genomes. Nucleic Acids Res. 2009;37:D150–D154. doi: 10.1093/nar/gkn852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent W.J. BLAT — the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burge S.W., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E.P. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth curves for all three strains. Growth curves for the P1/7, 98HAH33 and 05ZYH33 are depicted in blue, red and green respectively. P1/7 seems grow a little faster than two Chinese isolates after incubation for 6 h.
Sequencing saturation analysis. The saturation analysis was performed using RseQC package. Genes were sorted according to the RPKM values from low to high, and classified into 4 groups based on quartiles (Q1−Q4). The X-axis represents the percentage of reads that were randomly extracted from total reads for calculation of expression level, and the Y-axis represents the error between the calculated express level and the actual expression level. The curve becomes flat when ⩾70% reads were extracted, indicating that the number of reads is more than enough for RPKM calculation for all genes, and the expression levels of genes are accurate.
All three strains shared similar distribution of expressed genes in each COG. All the expressed genes in three isolates are classified according to COG category. For each category, three strains have similar number of expressed genes.
DEGs in the PTS pathway. Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Different transcription levels between the two Chinese isolates and the reference P1/7 are depicted in different colors as follows: upregulated genes with fold change >2 (pink), upregulated genes with fold change >8 (red), downregulated genes with fold change >2 (yellow), downregulated genes with fold change >8 (purple) and no expression changes observed (green).
DEGs in glycolysis/gluconeogenesis pathway. Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Different transcription levels between the two Chinese isolates and the reference P1/7 are depicted in different colors as follows: upregulated genes with fold change >2 (pink), upregulated genes with fold change >8 (red), downregulated genes with fold change >2 (yellow), downregulated genes with fold change >8 (purple) and no expression changes observed (green).
Highly-expressed genes enriched in KEGG category (Ribosome). Genes identified in the genomes of all three strains were highlighted with boxes in color, whereas genes do not exist in these strains are not highlighted. Genes that showed upregulated expression with fold change >2 between the two Chinese isolates and the reference P1/7 are boxed in red, whereas genes without altered expression between the two Chinese isolates and the reference P1/7 are boxed in green. The corresponding gene names in other S. suis isolates are indicated in purple background above.
Differentially-expressed homologous genes shared by all three strains.
DEGs in ABC pathway.
Novel TARs predicted to encode hypothetical proteins.
Novel sRNAs mainly related to riboswitches.