

Abstract
Liver diseases are one of the leading causes of mortality in the world. The hepatic illnesses, which include inherited metabolic disorders, hemophilias and viral hepatitides, are complex and currently difficult to treat. The maturation of gene therapy has heralded new avenues for developing effective intervention for these diseases. DNA modification using gene therapy is now possible and available technology may be exploited to achieve long term therapeutic benefit. The ability to edit DNA sequences specifically is of paramount importance to advance gene therapy for application to liver diseases. Recent development of technologies that allow for this has resulted in rapid advancement of gene therapy to treat several chronic illnesses. Improvements in application of derivatives of zinc finger proteins (ZFPs), transcription activator-like effectors (TALEs), homing endonucleases (HEs) and clustered regularly interspaced palindromic repeats (CRISPR) and CRISPR associated (Cas) systems have been particularly important. These sequence-specific technologies may be used to modify genes permanently and also to alter gene transcription for therapeutic purposes. This review describes progress in development of ZFPs, TALEs, HEs and CRISPR/Cas for application to treating liver diseases.
Keywords: Gene therapy, Zinc fingers, Transcription activator-like effectors, Clustered regularly interspaced short palindromic repeats, Homing endonucleases, Liver diseases
Core tip: The treatment of liver diseases is varied and often complicated. Gene editing is being developed to treat a variety of chronic disorders and has exciting potential for curing hepatic diseases. Engineering of derivatives of zinc finger proteins, transcription activator-like effectors, homing endonucleases and clustered regularly interspaced palindromic repeats potentially enables sequence-specific gene editing. These DNA binding proteins may be used to alter genes permanently or to influence the epigenetic status of liver cells for therapeutic benefit.
LIVER DISEASES AND THE APPLICATION OF GENE THERAPY
The liver plays an important role in metabolism and the etiology of numerous inherited and acquired disorders and can be traced to this organ. Liver diseases are varied and suitable options for therapy are often limited[1]. Treatment outcomes are dependent not only on patient history and disease type, but also disease progression and prior treatment[2]. Gene therapy, which offers the potential for improving currently available therapy for liver diseases, is defined as the introduction of nucleic acids into cells to treat a disease. Gene therapy may cause: (1) substitution or correction of genetic material; (2) changes in gene expression through augmentation; (3) post-transcriptional regulation; (4) cell death as a result of expression of toxic genes; and (5) transcriptional repression or activation (Figure 1)[3]. Applications of the approach may be expanded through use in concert with cell-based therapies. Collectively these versatile technologies have the potential to improve efficacy and expand the repertoire of strategies for management of liver diseases. Gene therapy is a rapidly developing field of research that has application to treatment and prophylaxis of many liver diseases, such as is caused by persistent hepatitis B virus (HBV) infection. Selected examples of gene therapy for various liver diseases are shown in Table 1 and Table 2.
Figure 1.
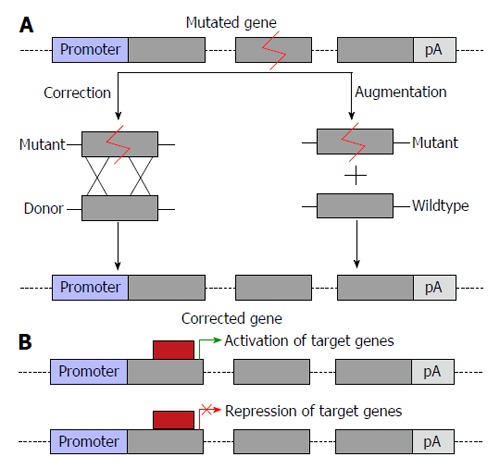
Potential outcomes of gene therapy. A: Correction of mutations or augmentation of gene function by introduction of a second copy of the gene of interest into a safe harbor locus; B: Changes to transcriptional regulation of a gene of interest, allowing for correction of gene function either by transcriptional activation or suppression. The grey boxes represent exons and linking lines represent introns.
Table 1.
Selected examples of liver-specific gene therapies for complex genetic and hereditary monogenic disorders
| Disease | Therapy | Rationale | Stage of development | Ref. |
| Gene therapy for complex genetic diseases | ||||
| Hepatocellular carcinoma | Recombinant human adenovirus type 5 administration followed by TACE | Adenovirus is highly infectious and when it is used in conjunction with TACE it improves tissue penetration and thus tumor shrinkage | Clinical (phase I and II) | [18] |
| Gene therapy for hereditary monogenic diseases | ||||
| Crigler-Najjar syndrome type I | AAV neonatal mouse hUGT1A1 gene transfer | AAV has low immunogenicity and is highly infectious in hepatocytes. Thus, in this study expression of bilirubin UDP glucuronosyl-transferase was augmented a large number of hepatocytes following transduction with hUGT1A1 | Preclinical | [19] |
| Familial hyper- cholesterolemia | Hepatocytes corrected with retroviruses expressing the low-density lipoprotein receptor | The transplantation of hepatocytes allows the slow repopulation of the liver with a desirable phenotype. In this study this method was used to introduce hepatocytes expressing the low-density lipoprotein receptor | Clinical (phase I) | [20] |
| Hemophilia A | Recombinant factor VIII fused to Fc domain of IgG1 (rFVIIIFc) | Coagulation factor replacement therapy requires the regular replacement of factor VIII (FVIII) with recombinant FVIII products or plasma-derived concentrates. The use of a long-lasting recombinant FVIII protein would reduce the need for frequent injections. The fusion of the human Fc domain of IgG1 to FVIII extends the half-life of FVIII and may reduce the injection frequency by 50% when compared with current treatments | Clinical (phase III) | [21] |
TACE: Trans-catheter arterial chemoembolization; AAV: Adeno-associated virus; UDP: Uridine 5’-diphospho.
Table 2.
Selected examples of liver-specific gene therapies for viral hepatitides
| Disease | Therapy | Rationale | Disadvantages | Stage of development | Ref. |
| HBV | Expression of anti-HBV primary microRNA (pri-miR)-122- and pri-miR-31-based mimics | Using artificial HBV-targeting pri-miRNAs it was possible to silence viral genes selectively, thus reducing their expression and causing inhibition of viral replication | Expression of pri-miRs must be maintained over prolonged periods | Preclinical | [22] |
| HBV | Administration of increasing doses of HB-110 DNA vaccine with fixed doses of adefovir dipivoxil | HB-110 is a DNA vaccine used in chronic hepatitis B infections. This vaccine induces antigen production and stimulates the immune response; helping to clear infected cells from the liver. Adefovir dipivoxil is a chemical therapeutic which blocks reverse transcriptase preventing viral replication. When used in conjunction, adefovir dipivoxil prevents viral replication and thus infection of healthy hepatocytes while the stimulated immune system clears infected hepatocytes, thus reducing the symptoms of chronic HBV | Possibility of escape mutants and viral rebound after therapeutic withdrawal | Clinical (phase I) | [23] |
| HBV | Administration of a single dose ARC-520 with entecavir | ARC-520 is an RNAi therapeutic that will be used in combination with the inhibitor of HBV DNA polymerase, entecavir, in patients with chronic HBV infection. The decline of HBsAg along with other measures will be evaluated to determine the efficacy of treatment in response to a single dose of ARC-520 | The efficacy of RNAi-therapeutics must be maintained over prolonged periods | Clinical (phase IIa) | [24] |
| HCV | Administration of Miravirsen to target miR-122 with LNA–modified oligonucleotide | miR-122 expression is specific to the liver and plays an important role in the regulation of HCV replication. Disrupting miRNAs can be difficult but one of the tools used to silence their activity are LNA-modified oligonucleotides. Their bridge modification significantly increases their hybridization properties, making disassociation more difficult. In this study they produced a LNA-oligonucleotide which is complementary to miR-122, inhibiting its activity; this decreases HCV replication and reduces infection | LNA-oligonucleotide expression needs to be maintained for long periods | Clinical (phase II) | [25] |
HBV: Hepatitis B virus; HCV: Hepatitis C virus; RNAi: RNA interference; HBsAg: Hepatitis B surface antigen; LNA: Locked nucleic acid.
Although the application of gene therapy is far reaching, not all liver diseases are currently amenable to this type of treatment. Reversal of certain hepatic disorders, such as alcohol induced cirrhosis, is not feasible at present. With other diseases, such as tuberculosis of the liver, suitable technology is not yet available to effect treatment. For these reasons gene therapy is focused on developing treatment for hereditary monogenic diseases, certain viral infections, some acquired and complex genetic cellular gene defects. Complex genetic diseases are caused by multiple gene mutations which accumulate over time. An example of such a disease is hepatocellular carcinoma (HCC)[4]. Hereditary monogenic diseases are the result of a single inherited gene defect[5,6] and include disorders such as Crigler-Najjar syndrome type 1[5] and tyrosinemia type 1[6]. These diseases are caused by mutations in the genes encoding uridine 5’-diphospho-glucouronosyl transferase[7] and fumarylacetoacetate hydrolase (Fah) respectively[6], and result in hepatic accumulation of toxic metabolites. Other hereditary monogenic diseases such as hemophilia A and B, caused by gene mutations in coagulation factors VIII[8] and IX[9] respectively, result in excessive bleeding and associated extra hepatic manifestations[10]. Viral infections of the liver include hepatitis B[11] and hepatitis C[12]. These viral hepatitides are major risk factors for chronic liver diseases, with carriers exhibiting increased susceptibility to cirrhosis and hepatocellular carcinoma[11,12]. Gene therapy can be applied to treating these diseases by altering gene expression through gene editing, augmentation of gene expression, transcriptional repressive or activational mechanisms (Figure 1)[3].
Availability of DNA editing tools will expand the application of gene therapy to the treatment of liver diseases. Currently available technologies have limitations. For example, application of RNA interference-based silencing to treatment of HBV infection may not be sufficiently durable and specific to achieve a safe curative effect. Genome modification technologies provide the means for overcoming some of these shortcomings. Currently gene editing tools are based on engineered derivatives of zinc finger proteins (ZFPs)[13], transcription activator-like effectors (TALEs)[14], homing endonucleases (HEs)[15] and clustered regularly interspaced palindromic repeat (CRISPR) and CRISPR associated (Cas) protein arrays[16,17]. This review will explore the application of these sequence-specific DNA modification technologies to treatment of liver diseases.
SEQUENCE-SPECIFIC DNA EDITING TECHNOLOGIES: A TOOL FOR GENE THERAPY
The ability to sequence entire genomes has facilitated identification of genetic defects that underlie diseases, and this has assisted with prevention and cure of illness. Sequence-specific DNA editing technologies thus present a novel, versatile tool for modifying the genome and treating disease. Early applications of this approach were hampered by several factors. These included low efficiency, the need for complicated screening to identify suitable gene editors and the potential for adverse effects caused by lack of specificity[16,26]. During the past decade, characterization of derivatives of sequence-specific proteins and RNA-guided nucleases[27], which include ZFPs[28], TALEs[29], HEs[30,31] CRISPR/Cas[17] has allowed investigators to develop tools to edit almost any gene.
ZFPs
ZFP arrays rely on the sequence-specific interaction of the His2-Cys2 protein motif present in individual fingers with specific sequences in the target DNA[32]. Zinc finger (ZF) domains constitute one of the most common DNA-binding motifs in eukaryotes and prokaryotes. Moreover they are the second most commonly encoded family of proteins in the human genome. Each finger consists of 30 amino acids with 3 amino acids from an α-helical region interacting with 3 consecutive base pairs (bp) in the major groove of the target DNA[13]. Naturally DNA-binding domains comprise tandem repeats of up to three fingers, and thus allow recognition of up to 9 bp of the target sequence. The ability to link more than 3 adjacent fingers together has been useful for application of this technology and was made possible through the discovery of a structurally conserved linker region of sequence amino acid TGEKP[33]. This conserved linker region provides additional stability to the protein-DNA complexes[33]. It is possible to build modular arrays that recognize 9-18 bp, which would provide sufficient specificity for targeting within the human genome[34,35]. Through use of libraries consisting of combinations of ZFs that recognize almost all 64 possible nucleotide triplets, it was possible to select ZFPs with the intended target binding specificity[13,36]. A drawback of the approach was that combinations of the fingers did not always bind to targets with intended affinity[13,36]. This phenomenon is the result of context dependency of the individual fingers[13,36]. That is, binding affinity of individual fingers is influenced by neighboring modules, and strength of interaction of the fingers with their intended cognates depends on properties of surrounding sequences of the ZFP[13,36]. The development of the Oligomerized Pool ENgineering protocols, which provides a publically available tool for the development of ZFPs, improved the ability of researchers to select proteins with good target-binding properties[37]. Subsequently, an approach was developed that takes into account the effect of neighboring individual ZFs when generating an array. This method, context dependent assembly (CoDA), selects for active arrays by using combinations derived from known existing functional ZFPs[38]. In CoDA the N and C-terminal fingers from different arrays, which have a common middle finger, can be joined together to make new active ZFPs. The method is rapid and does not require a selection process, thus reducing the screening required for identification of functional ZFPs[35,38]. As a result of the influence of context dependency, and the experimentally observed affinity of ZFPs for G-rich sequences, not all DNA sequences are suitable targets for binding by ZFPs. It has been estimated that one pair of efficient ZFPs may be generated for approximately every 100 bp of the human genome[16]. The context dependency of the ZFs also has implications for off-target activity. Tools that aid in target selection and prediction of off-target activity have been developed to assist with generating functional ZFPs[28,39]. Despite the difficulties associated with use of ZFPs they are currently the most widely applied of the genome editing technologies. This is illustrated by their recent application in preclinical and clinical settings in which ZF nucleases (ZFNs) were used to disrupt the human immunodeficiency virus (HIV) co-receptor CC-chemokine receptor 5 gene in CD4+ T cells to produce cells that are resistant to HIV infection[40-42].
TALEs
TALEs are a unique family of DNA-binding proteins that have been isolated from the phytopathogenic bacteria of Xanthomonas species and Ralstonia solanacearum[43]. The characterization of native TALEs led to discovery of the DNA-binding region, which is a highly repetitive protein sequence[43-45]. Each repeat comprises 33-35 amino acids[43] that collectively constitute a protein of approximately 122 kDa[46]. The repeats of the TALE each recognize and bind to a single nucleotide. The nucleotide specificity is conferred by two consecutive variable residues at positions 12 and 13 of the repeated unit, and are termed the repeat variable diresidues (RVDs) (Figure 2). Each RVD interacts specifically with either A, T, G or C nucleotides[47], and a simple code determines nucleotide binding. There are 20 known RVDs and certain RVDs are found more commonly than others in native TALEs[47]. Individual RVD recognition of its cognate nucleotide is independent of both the preceding and succeeding RVDs[43,47]. This lack of context dependence is a distinct advantage over ZFPs, and allows for convenient construction of TALE subunits that recognize specific DNA sequences. A requirement of efficient TALE binding is the presence of a T nucleotide at the base 0 position of the cognate (T0)[48]. Substitution of T0 with another nucleotide reduces the binding of the TALE[49] and places a minor restriction on the number of target sites within the genome that can be recognized by custom TALEs. TALE recognition sequences are up to 18 nucleotides in length and alterations at a single nucleotide can have a significant impact on binding efficiency. However, the average binding contribution of individual RVDs is minimal. Nevertheless, choice of RVD may have an impact on the electrostatic interactions between the TALE and DNA, which in turn may influence the stability of the DNA-TALE complex. The length of the target sequence is approximately equal to the number of RVD repeats and single repeats each bind to 1 nucleotide of DNA[48]. Mismatches at the 5’ end of the recognized sequence are more detrimental to TALE binding than they are at the 3’ end of a target[47]. This suggests that the generation of synthetic designer TALEs should be biased to favor selection of unique targets within 5’ regions[47]. Apart from RVD choice and target sequence selection, other factors influencing TALE binding efficacy include modifications to the DNA target and chromatin structure variations[47]. Valton et al[50] showed that TALE arrays cannot bind 5-methylcytosine (5mC) residues, which may pose barrier to application in mammalian systems where DNA methylation commonly occurs as a transcriptional regulatory mechanism[51]. The RVDs NG and N* both have some affinity for 5mC, and these RVDs may thus be incorporated into engineered TALEs to target DNA that is methylated[50]. Development of web-based tools, which take all of these parameters into account, has simplified TALE array design and facilitated their more wide spread application[52]. TALEs are large proteins, and with addition of the nuclease or transcriptional regulation domains, DNA encoding the engineered DNA binding proteins may be as long as 4 kb[43,53,54]. This makes them significantly larger than ZFPs and considerably larger than HEs. Although the TALE binding arrays have been shown to be robust and easily programmable, the size of the TALEs makes their delivery challenging for therapeutic application.
Figure 2.
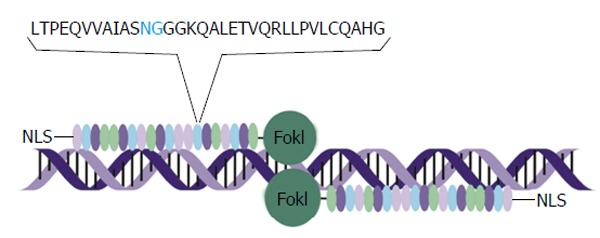
Schematic representation of transcription activator-like effector nuclease interaction with DNA. A pair of transcription activator-like effector (TALE) nucleases interacts with its target through a tandem arrangement of DNA-binding modules, represented by oval structures in the schematic. Each DNA-binding module comprises 34 amino acids, with a repeat variable diresidue (RVD), in blue, at amino acids 12 and 13. The RVD specifies the DNA base to be targeted. There is a nuclear localisation signal on the N-terminal and a FokI effector bound to the C-terminal. As well as FokI nuclease domains, TALEs may also be bound to transcriptional regulators such as Krüppel-associated box or VP16/64. NLS: Nuclear localisation signal.
CRISPR/Cas
CRISPR/Cas systems are naturally RNA-mediated adaptive defense mechanisms that are found in some prokaryotic organisms. Using short CRISPR RNAs (crRNAs) and Cas proteins, the prokaryote is able to identify and eliminate invading DNA elements[55]. CRISPR/Cas complexes identify targeted genomic loci using a 20 nucleotide RNA guide (gRNA), which is complementary to its target DNA sequence. CRISPR/Cas systems are classified into three types (I-III) on the basis of the structure and sequence of their Cas proteins[56]. All three have three essential components: the CRISPR array, the upstream leader sequence and the cas genes[57]. The most commonly used CRISPR/Cas system for gene editing applications is type II CRISPR/Cas[58]. The CRISPR array is composed of identical repeats that are 23-47 bp in length[59]. The CRISPR/Cas leader region acts as a promoter for the transcription of the CRISPR array[60]. The cas genes encode the Cas proteins. This protein contains RuvC-like and HNH-like catalytic domains, which cleave the targeted DNA. The HNH-like domain cleaves the strand that is complementary to the gRNA, while the RuvC region cuts the other non-complementary DNA strand of the target[61]. Recognition of the target sequence by the Cas proteins is facilitated by presence of the proto-spacer-associated motif (PAM). This sequence is usually 2-4 nucleotides long and flanks the target site. It is absent from the endogenous loci and thus prevents CRISPR/Cas auto cleavage and adds specificity to targeting[60].
The type II CRISPR/Cas systems use the Cas9 protein, which recognizes a PAM sequence of 5’NGG3’. In type II systems a small non-coding RNA, known as the trans-activating crRNA (tracrRNA), which is partially complementary to the CRISPR repeats, forms an RNA duplex with crRNA. This RNA hybrid is recognized and processed to form mature gRNA and, subsequently, associates with Cas9. The complex, including the tracrRNA, recognizes invading DNA and inactivates it by cleavage[60,62] (Figure 3). When using an engineered CRISPR/Cas system cognates for new gRNAs are identified within the targeted gene. Binding sites for these artificial gRNAs are preceded by the PAM, which is required by the associated Cas protein. Optimization of the CRISPR/Cas guide architecture, to enhance specificity and broaden their application, is a highly active field of research[63]. As with other gene editing technologies, the CRISPR/Cas system faces several developmental challenges, including potential off-target activity which may result in adverse mutation.
Figure 3.
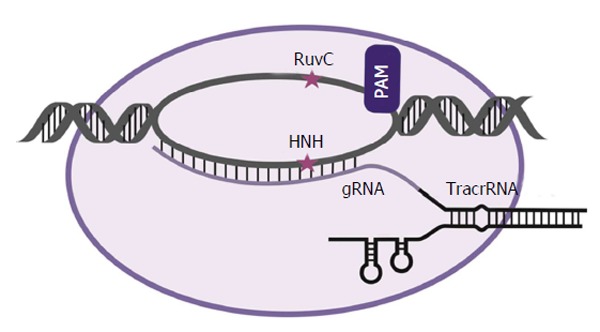
Schematic representation of the clustered regularly interspaced palindromic repeats/clustered regularly interspaced palindromic repeats associated 9 complex and its interaction with DNA. With the CRISPR/Cas system, a 20 nucleotide guide RNA is processed and bound to the Cas9 protein. When adjacent to a proto-spacer–associated motif element, the guide recognises and binds a complementary sequence in the DNA to direct target cleavage by the Cas9 protein. A catalytically inactive Cas9 protein may also be coupled to other effectors such as Krüppel-associated box or VP16/64 domains. CRISPR/Cas: Clustered regularly interspaced palindromic repeats/CRISPR associated. CRISPR: Clustered regularly interspaced palindromic repeats; Cas: CRISPR associated; tracrRNA: Trans-activating CRISPR RNA; gRNA: RNA guide.
HEs
HEs are named as a result of their native endonuclease and homing activities. Homing is the transfer of introns or inteins into alleles that themselves do not have introns or inteins[64]. These endonucleases are encoded by open reading frames (ORFs) within the mobile sequence. They avoid disrupting the genetic function of the host by exclusively moving the mobile sequences into inteins and introns. HEs have an ability to recognize DNA target sites of 14-44 bp, which correspond to the intron/intein insertion site. They create a double-strand break (DSB) or single-stranded nick to promote insertion of sequences encoding the HEs within introns or inteins into the target allele[15]. Group I introns, large self-splicing ribozymes, encode the most well characterized HEs. In the case of group I introns, the HEs are translated prior to transposition of the mobile element[15]. The resulting endonuclease is specific for the “intronless” target site. Host-mediated homology-directed repair (HDR) results in the unidirectional transfer of the HE-encoding intron into the cleaved allele with disruption of the homing site. Although less well studied, the process for HEs comprising inteins is likely to be similar to that of HEs encoded within group I introns[65]. Six families of group I HEs have been identified, namely: LAGLIDADG; GIY-YIG; HNH; His-Cys box; EDxHD and PD-(D/E) xK[15]. These families are classified according to the presence of conserved amino acid sequence motifs within their active sites. The largest family of the HEs is the LAGLIDADG HEs (LHEases). LHEases generate staggered DSBs in the DNA at the target site to produce four nucleotide 3’ overhangs[15] which induce DNA repair.
HEs are attractive gene editing tools because of their high sequence specificity and were applied as gene modifiers in murine cells 20 years ago[66]. Since then, studies have been performed targeting human genes[67] and certain monogenic diseases[30]. Theoretically, their high sequence specificity should reduce off-target effects, which is supported by their limited toxicity when expressed in cells over prolonged periods[31]. A further advantage to HEs is that they are significantly smaller than ZFPs and TALEs, which simplifies their delivery. However, engineering HEs is significantly more difficult than it is for ZFPs, or TALEs[16]. This is because enzymatic and target recognition sequences are located within the same domains of the HEs, and altering target specificity without compromising enzymatic function is complicated to achieve[15]. Despite these limitations, studies continue to advance the development of HEs[68,69]. It is possible that improvements in use of HEs may lead to their use for gene editing purposes and for liver-specific applications. However, with advances in other gene editing technologies use of HEs are currently not in favor.
USE OF SEQUENCE-SPECIFIC GENE EDITING TECHNOLOGIES FOR TREATMENT OF LIVER DISEASES
Although gene editing as a mode of treating liver diseases is still limited, application of the technologies to management of other diseases has useful implications for hepatology. Gene editing initially focused on creation of DNA-modifying nucleases derived from ZFPs[28,70], TALEs[29,71], CRISPR/Cas[17,58] and HEs[15]. The native endonuclease activity of CRISPR/Cas and HEs means that no additional effectors are required for their application as nucleases. However TALEs and ZFPs require that nuclease domains be added to the DNA-binding regions, and FokI is most commonly used. FokI is a type IIS restriction enzyme that recognizes its target site as a monomer and nicks a single-strand of DNA[72], but requires dimerization for double-stranded DNA-cleaving activity[73]. The implication of this is that a ZFN and TALE nuclease (TALEN) pairs are required to effect complete cleavage of the duplex. To engineer a site-specific ZFN or TALEN, independent subunits need to be designed to nick each strand of the DNA target. To achieve intended cleavage of the DNA duplex, the subunits need to be delivered to a target cell simultaneously[16] (Figures 2 and 4). This is not necessarily a limitation as the addition of a second nuclease improves sequence-specificity, which is especially desirable in a clinical setting.
Figure 4.
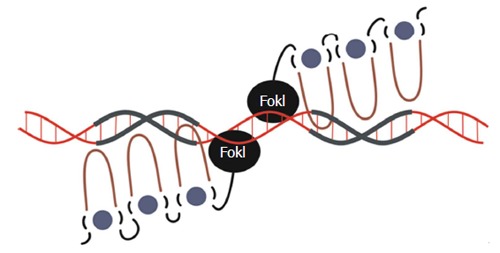
Schematic representation of zinc finger nuclease interactions with DNA. Each zinc finger interacts with 3-4 bp in the major groove of the DNA double helix. The Hys2Cys2 side chains (depicted as the loop) bind to the DNA, which is facilitated by the interaction between the zinc finger protein (ZFP) and a Zinc ion (depicted as green circle) which stabilizes the protein structure. As well as FokI nuclease domains, ZFPs may also be bound to other effectors such as Krüppel-associated box or VP16/64.
DNA-modifying nucleases enable genome modification by introducing DSBs in the DNA and stimulation of the mutagenic non-homologous end joining (NHEJ) and high fidelity HDR pathways[26] (Figure 5). Repeated activation of NHEJ typically results in generation of small insertions or deletions (indels) at a targeted site that may be used to disable specific genes (Figure 5)[26]. Introduction of a gene modifying nuclease along with donor DNA (plasmid, single-stranded or double-stranded oligonucleotides), which has homology to sites flanking the target, induces HDR[74,75]. By activating HDR it is possible to make precise alterations to the genome: correction of single point mutations, facilitation of integration of single and multiple transgenes into specific loci can be achieved[76,77] (Figure 5). As a result permanent correction, silencing or augmentation of cellular genes may be achieved. When two duplex DNA-cleaving nucleases are simultaneously introduced into a cell, inversions, translocations, large insertions and deletions may be induced within the intervening sequences (Figure 5).
Figure 5.
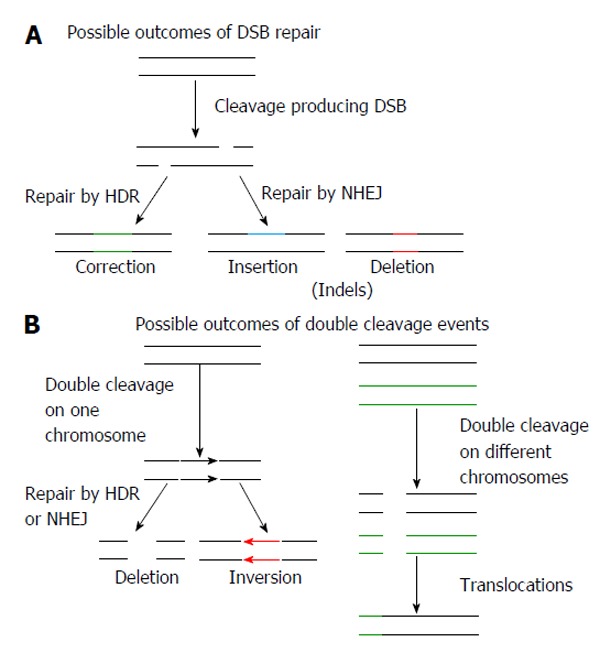
Potential effects of using genome editing endonucleases. A: Nuclease induced double strand breaks (DSB) may be repaired by homology directed repair (HDR), resulting in corrections that may be to a single base pair or thousands of base pairs. Repair by non-homologous end joining (NHEJ) may result in non-specific insertions and deletions (indels); B: The introduction of two nucleases targeting the same chromosome may result in large deletions or inversions of the DNA. Chromosomal translocations may occur when two DSBs are introduced on different chromosomes.
Early investigations focused on the nuclease activity of gene modifiers. However, their potential for off-target mutagenic effects, accumulation of cytotoxic DSBs and reliance on host repair machinery, led investigators to explore therapeutic utility of transcriptional activators and repressors[16] to modulate genes selectively without changing the sequence of the DNA[71]. A commonly used transcriptional repressor is the Krüppel-associated box (KRAB) domain, which is a 75 amino acid motif originally found on the N-terminus of ZFPs[78,79]. The Herpes simplex virus type l-derived 65 kDa VP16[80] domain, or its synthetic tetrameric derivative VP64[81], are commonly used to effect transcriptional activation.
TALEs, ZFPs and CRISPR/Cas constructs have all been used to create artificial transcription factors[82,83]. ZFPs or TALEs may be fused to an activator or repressor domain to create ZF-transcription factors or TALE-transcription factors. These transcription factors have the advantage of functioning as monomeric proteins, although multiple transcription factors might improve their efficacy. Modification of CRISPR/Cas arrays to act as artificial transcription factors is achieved by modifying the Cas9 endonuclease to render it catalytically inactive, and form the so-called dead Cas9 (dCas9). dCas9 may still be recruited by gRNAs to target specific DNA sites[84], but when modified with an effector domain such as KRAB or VP64, it is able to alter the epigenetic state of its target[85].
In addition to deciding on which type of gene modifier to apply for therapy, there are disease-specific considerations that must be evaluated before a new treatment may be developed. These include an assessment of the proportion of hepatocytes that needs to be treated, duration of expression of a gene modifier, the potential for off-target activity and the type of gene alteration that is required to attain a therapeutic effect. Thus, treatment of a viral hepatitis would be vastly different from that required for a hereditary or complex genetic disorder.
APPLICATION OF GENE EDITING TECHNOLOGIES TO THE TREATMENT OF VIRAL HEPATITIS
Zimmerman et al[86] investigated the ability of ZFPs to target the episomal covalently closed circular DNA (cccDNA) reservoir of duck HBV (DHBV). This replication intermediate is a template for the transcription of viral genes and the production of pre-genomic RNA[86]. The persistence of cccDNA is responsible for chronic HBV infection and currently there are no available therapies that directly target this replication intermediate. The anti-DHBV ZFPs were designed to target the enhancer 1 region of the cccDNA, as this element controls transcription from both the core and surface promoters. After introduction of the ZFPs into cells in culture, a decrease in total viral RNA and pre-genomic RNA with ZFPa (61.2% and 57.2% respectively) and ZFPb (45.3% and 73.5% respectively) was observed. Furthermore, a reduction in DHBV core and surface protein expression and production of virus progeny was also reported. Importantly, the introduction of the ZFPs did not result in any significant toxicity[86]. In 2010 Cradick et al[87] evaluated the ability of ZFNs to target and disrupt HBV DNA. The ZFNs used were designed by searching the HBV genome for recognition sites for pairs of three-finger ZFNs separated by six nucleotides. One pair of ZFNs was selected from 18. This ZFN pair cleaved within the HBV core ORF and reduced the pre-genomic RNA by 29%. About 26% of targeted DNA was linearized in the presence of the ZFNs and approximately 10% showed aberrant re-ligation. The re-ligated products all showed indels and subsequent disruption of viral replication markers[87]. Although this study demonstrated the ability of ZFNs to cleave HBV DNA targets, there was no evidence to suggest that the ZFN pair was able to disable the HBV cccDNA.
Zhao et al[88] evaluated the use of a ZF-artificial repressor (ATF) to down-regulate an integrated hepatitis B X gene (HBx) in the Hep3B hepatocellular carcinoma cell line. During normal HBV infection, HBx gene integration is associated with the progression of chronic HBV to HCC and is responsible for the dysregulation of expression of a number of cellular genes. The ZFP-based artificial repressor recognized 18 bp in the enhancer 1 region of the HBx gene and used the KRAB domain to effect knockdown. Investigators saw a marked decrease in luciferase production from an HBx reporter construct. When the ATF was stably expressed in the Hep3B cell line there was significant cell cycle arrest. In a cell lines without the integrated HBx gene no cell cycle arrest was observed[88]. This study presents a novel approach to inhibiting HCC progression caused by HBV and illustrates the potential of applying gene editing technologies to the functional repression of disease associated genes.
In 2013, Bloom et al[89] were the first group to demonstrate the potential of anti-HBV TALENs for treatment of chronic HBV infection. The investigators successfully designed four pairs of TALENs. Two of these pairs target the surface (S) and core (C) ORFs of HBV. The S and C targeting TALENs efficiently caused indels in these ORFs including those found within the cccDNA of HepG2.2.15 cells which stably replicates HBV[90]. In this assay, the cccDNA was isolated using a hirt extraction coupled to use of an ATPase dependent nuclease, which selectively degrades nicked or linear DNA. Following this isolation, deep sequencing was used to evaluate targeted disruption of the TALEN cleavage sites. The S TALEN disrupted 31% of the cccDNA, while the C TALEN disrupted 12% of the HBV cccDNA. Hydrodynamic injection (HDI) was used to introduce HBV DNA into mice to simulate HBV replication in vivo. A significant reduction in markers of viral replication following the HDI-mediated introduction of the TALENs was observed in this model. The S TALEN effected knockdown of HBV surface antigen (HBsAg) by more than 90% on both days 3 and 5 after HDI. Circulating viral particle equivalents decreased by approximately 70% with the introduction of either the S or C TALEN. Immunohistochemical detection of the HBV core antigen in the liver of treated mice was only decreased with the addition of the C TALEN. These results not only showed that the TALENs had an effect in a murine model of HBV infection but that the effect of each TALEN was limited to its specific target. Furthermore, intrahepatic mRNA concentrations remained constant after TALEN treatment, demonstrating that inhibition of viral replication markers was the result of inactivating the targeted genes rather than transcriptional repression. It was noted that the other two pairs of TALENs, targeting the polymerase ORF (P1 and P2) did not result in target cleavage despite reducing HBsAg expression. These observations suggested that the P1 and P2 TALENs may function through a different mechanism of action, such as by targeted transcriptional repression. Measurements of aspartate aminotransferase and alanine aminotransferase activity in the murine liver indicated that there were minimal toxic effects associated with TALEN treatment[89]. However, as cccDNA is not produced by mice, the evaluation of TALEN mediated cccDNA disruption in vivo could not be carried out in this study.
A subsequent study by Chen et al[91] supported the findings made by Bloom et al[89]. Chen et al[91] designed a unique set of TALENs against conserved regions within HBV genomic DNA. Application of their TALEN pairs resulted in the reduction of HBV core, surface and e antigen expression; viral knockdown and a reduction in the cccDNA. cccDNA was produced at low levels in Huh7 cells using a transfection method. cccDNA levels were evaluated by quantitative polymerase chain reaction using HBV cccDNA specific primers, which were designed to amplify DNA across the gap that is present in relaxed circular DNA (rcDNA). This allowed for discrimination between the two HBV replication intermediates. cccDNA was reduced by approximately 10%-20% with TALENs-L1/R1 and twofold in TALENs-L2/R2. Furthermore, a synergistic effect on the inhibition of HBV transcription was observed when TALENs and interferon-á were co-administered[91]. The studies by Bloom et al[89] and Chen et al[91] provide evidence for TALEN applications to treating chronic diseases that have a DNA reservoir within the human liver. While the findings of these studies are promising the need to test TALEN efficacy in a model that simulates the human condition more closely remains important. The prolonged nuclease activity required, which would ensure eradication of all of the cccDNA, and thus successful elimination of HBV infection may also result in unwanted side effects. Therefore, the development complementary technologies to combat HBV such as TALE transcriptional regulators which may be used in their place or in conjunction with their TALEN counterparts should also be considered.
Most recently, Lin et al[92] evaluated the potential of a CRISPR/Cas approach to eradicating HBV replication intermediates (rcDNA and cccDNA) both in vitro and in vivo. The investigators designed gRNAs to the HBV A genotype and of the eight gRNAs, four had good anti-HBV activity. Improved efficacy was observed when the gRNAs and Cas9 were co-expressed from the same plasmid, with the most active gRNA reducing intracellular HBsAg levels by 96%. This gRNA targeted the surface region and was designated as the S1 guide. When Lin et al[92] evaluated effects of gRNAs in combination, anti-HBV efficacy was augmented and large deletions in the viral DNA could be introduced between the two gRNA recognition sites. The ability to delete larger sections of DNA has implications for the eradication of integrated HBV genomes, and has been successful when using a set of CRISPR/Cas gRNAs to eliminate HIV-1 provirus infection[92]. To evaluate the ability of the CRISPR/Cas gRNAs to eliminate HBV-cccDNA, the investigators used a DHBV replication model with new DHBV-specific gRNAs. With this avian model of HBV replication, more cccDNA is formed in transfected cells. Results indicated that cccDNA and rcDNA production was significantly diminished. This study is the first to demonstrate the application of CRISPR/Cas constructs against HBV. This early evidence suggests that as with TALENs and ZFNs, CRISPR/Cas may be a useful tool against viral hepatitis[93].
APPLICATION OF GENE EDITING TECHNOLOGIES IN VIVO AND EX VIVO
In 2011, Li et al[94] investigated the potential of ZFNs for gene editing of liver progenitor cells and the correction of gene mutations responsible for hemophilia B in the murine model. In this study ZFNs targeting intron 1 of the human F9 gene were shown to induce DSB and HDR at the intended sites in both the human erythroleukemia K-562 cells and the Hep3B human hepatocyte line[94]. An in vivo study was performed in which the ZFNs and a gene targeting vector with a donor sequence were delivered to the liver using an adeno-associated virus (AAV), serotype 8 vector. The ZFNs and gene targeting vector mediated correction and resultant prolonged clotting times of the mice. When liver regeneration was induced in these animals, the effects of the treatment were maintained[94].
The ability to repair genetic defects has been furthered by improvements in technologies related to use of induced pluripotent stem cells (iPSCs). There is potential for the application of patient-derived iPSCs for the correction of underlying genetic errors while autologous transplant reduces problems of immune-mediated graft rejection[95,96]. Coupling this methodology to gene editing may be used to correct gene defects and induce disease-resistant phenotypes. After modification ex vivo, differentiation into hepatocytes and autologous transplant, it may be feasible to generate livers that no longer manifest a disease phenotype.
Rio et al[97] were the first to demonstrate the potential of DNA modification technologies to correct DNA repair deficiency syndromes. In this study, a pair of ZFNs targeting the AAV 1 locus were introduced into iPSCs using recombinant AAVs. These ZFNs, introduced by an integrase defective lentiviral vector, induced cleavage of the target and improved integration of a new Fanconi anemia, complementation group A cassette by HDR. The ZFNs were central to the correction of fanconi anaemia (FA) phenotype. Corrected human iPSCs were re-programmed, re-differentiated and introduced into the bone marrow of FA sufferers. These re-differentiated cells were able to produce disease-free liver cells[97].
In an earlier study, Yusa et al[98], used the piggyBac transposon and ZFN-based genome modification in human iPSCs to achieve the biallelic correction of the Z mutation (342Glu to Lys) associated with á1-antitrypsin deficiency. ZFN pairs were designed to target the site of a single point mutation in the á1-antitrypsin gene. Following expression of the ZFN pair and introduction of donor constructs, 11% of the screened iPSC colonies exhibited biallelic excision and repair. Subsequent sequencing analyses showed that the Z mutation had been corrected on both alleles in these colonies. When the human iPSCs were transplanted and differentiated they produced normal liver cells, with no á1-antitrypsin deficiency[98]. In 2013, Choi et al[99] compared the efficacy of the ZFN pair used in the study by Yusa et al[98] to the efficacy of TALEN pairs in patient-derived iPSCs. The TALEN pair was designed to target regions adjacent to the Z mutation of the á1-antitrypsin gene. Following introduction of the TALEN pair and donor sequences, all 66 iPSC clones showed the desired corrective integration. Twenty five to thirty three percent of these clones lacked the endogenous allele, suggesting the simultaneous targeting of both alleles, which was confirmed by subsequent sequence analyses. When comparing the efficacy of the TALENs to the ZFNs, Choi et al[99] demonstrated that their TALEN pair achieved comparable or higher gene targeting efficiencies (100% single allele cleavage efficiency with 25%-33% biallelic targeting) than were observed with ZFNs pair (54% single allele cleavage efficiency with 4% biallelic targeting). These studies highlight the potential for gene editing technologies in the correction of inborn genetic disorders while illustrating the point that gene editor choice has an impact on therapeutic outcome.
In a study by Yin et al[100] the CRISPR/Cas system was used to correct a Fah U1 mutation in the hepatocytes of a murine model for the hereditary monogenic disease, tyrosinemia. The correction of the G to A splicing mutation, which restores correct processing of mRNA transcripts, in the endogenous Fah locus was achieved by HDR. A single guide RNA targeting the Fah gene, was introduced in conjunction with the Cas9 protein and a 199 nucleotide single-stranded DNA donor. The DNA donor contained the correcting wild-type sequence which was flanked on both sides by sequences that were homologous to the DNA of the mutant gene. The correction of the Fah gene resulted in expression of the wild-type Fah protein in murine hepatocytes. Subsequent proliferation of the Fah-positive hepatocytes and replacement of diseased cells in mice improved their survival[100].
CURRENT LIMITATIONS AND FUTURE PROSPECTS OF SEQUENCE-SPECIFIC DNA MODIFYING TECHNOLOGIES FOR THE TREATMENT OF LIVER DISEASES
The treatment of liver diseases is often complex, which has been a motivating factor for new and better technologies. While gene editing does not provide a cure-all therapeutic for liver diseases, the approach has potential for application to monogenic disorders and some viral hepatic infections. However, three significant challenges need to be addressed before clinical application of gene editing for hepatic diseases is realized: ensuring (1) specific targeting; (2) safe and efficient delivery to target cells; and (3) limited immunogenicity of the gene modifier and its delivery agent are all important.
Off-target activity by gene modifiers may result in cytotoxicity and the repair of DSBs generated from this activity may result in undesired deletions, inversions, and translocations[28]. Furthermore, the off-target activity from gene modifiers is potentially carcinogenic. When fused to activator, repressor and/or nuclease domains, gene editors may cause inactivation of tumor suppressor genes or activation of oncogenes. To alleviate these concerns, a number of studies evaluating the off-target effects of gene editing technologies have been carried out[101,102]. These investigations have sought to improve the design of the DNA recognition motifs. Pattanayak et al[103] developed new design rules to alleviate off-target activity of ZFNs. Other groups have investigated improvement of the architecture, length and composition of the CRISPR/Cas chimeric gRNAs[63,102,104]. New tools which evaluate potential off-target activity in silico have also been described[105], and more sensitive methods of identifying and characterizing off-target activity in vivo have been reported[102]. When using exome and deep sequencing, TALENs have been shown to have significantly fewer off-target effects than ZFNs[29,106]. This was corroborated in 2014 when a study by Suzuki et al[107] found that TALENs did not increase the overall mutation load of a targeted genome. In 2014 Smith et al[108] demonstrated that both CRISPR/Cas and TALENs had almost no off-target mutagenic effects in iPSCs. These studies suggest that off-target activity of TALENs and CRISPR/Cas may be more limited than originally anticipated. However, guidelines for acceptable levels of off-target activity need to be developed. Currently, limited off-target activity in introns and non-coding sections of DNA are considered acceptable[29,107,108], but with advancing understanding of the functional roles of non-coding DNA this may not always be true. Development of tools of bioinformatics which aid target choice has simplified the design of recognition sequences with low homology to exons and important regulatory elements[52,105]. It should also be noted that these studies focus on developing technologies with off-target activity below the detection of deep sequencing technologies. Deep sequencing technologies are currently the most sensitive tool available for evaluating off target effects[29,102]. Current limitations in technologies for DNA sequencing mean that this requirement is presently sufficient but will need to be re-examined as more sensitive technologies are developed. Potential off-target activity of therapeutic artificial transcription factors still needs to be evaluated. The growing body of evidence and tools for design and analysis suggests that while off-target effects will need to be evaluated for any clinical application, this need not be a universally limiting factor.
The second obstacle to efficient application of gene editing technologies is their specific delivery to intended target cells. This specific delivery may be further complicated by the need for multiple administrations of a targeted therapy due to their lower efficacy rates (around 50%) in a clinical setting. Viral vectors, which have been widely used in other gene therapy applications, may be employed as an efficient delivery mechanism for gene modifiers. Studies have shown potential for the use of lentivirus-, adenovirus- and baculovirus-based delivery methods, and AAV vectors have also been used to deliver gene editing tools successfully[109,110]. These viral vectors have some limitations with respect to the type of gene editing technology that may be delivered, the specific circumstance under which the vectors may be used and whether delivery may be carried out in vivo or ex vivo[109,110]. Challenges, such as immunogenicity and potential mutation or integration that may be associated with the use of viral vectors, have led to the exploration of other avenues for delivery. Liu et al[111] explored the possibility of conjugating TALENs to cell penetrating peptides such as the Tat protein to facilitate the systemic delivery of TALENs in vitro. Gaj et al[112] have assessed the applicability of a Salmonella-derived delivery system. Both of these studies were moderately successful. mRNA delivery offers another potential solution, which has the advantage of improving the regulation of the duration of transgene expression. mRNA needs only to be delivered to the cytoplasm, does not need to be transcribed, and therefore enables realization of the therapeutic effect more quickly. Moreover, since mRNA is unable to integrate into the host genome concerns about exogenous DNA integration are alleviated[113]. mRNA may be delivered using non-viral vectors which are less immunogenic than viral vectors and recombinant proteins[114]. They are also amenable to large scale preparation that is required for clinical use. There are some TALEN and ZFN studies which have applied this approach, although with moderate success to date[115,116]. The vectors used for delivery of sequences encoding gene editors may be engineered to target specific tissues. Using different serotypes, receptors or lipids it is possible to ensure specific delivery of the transgene payload. This flexibility also allows for modification of the delivery vehicle to avoid immune detection, should repeat administrations be necessary in a clinical setting. The choice of delivery system will eventually depend on a combination of the type of gene editing technology being used, its specific application, requirements for dose regulation, duration of expression and minimizing of off target effects.
The potential of an immune reaction to gene editors may be a concern for application of the technology. This immunostimulation may result from either the gene modifier itself[117] or from the mechanism of delivery[118]. In either case it is important to assess immune activation as it may diminish efficacy following repeat administrations and cause toxicity. Immune stimulation has significantly hampered gene therapies in the past[119] and will be important for clinical application of gene editing.
CONCLUSION
This review has discussed ZFPs, TALEs, CRISPR/Cas and HEs highlighting their advantages and disadvantages for applications to the treatment of liver diseases. Gene editing has a promising future as a clinical therapeutic and as these techniques continue to be refined, off-target activity will become more limited and delivery more feasible. Several studies have already demonstrated that these technologies may be used in a tissue- and patient-specific manner to disable, augment and correct gene function. Other studies have established their potential to expand the use of iPSCs in the treatment of disease. Ultimately gene modifiers and cell-based therapies will increase the number of disorders that may be permanently corrected and alter the way in which inborn liver diseases and viral hepatic infections are treated. These technologies present a novel, versatile tool for the treatment and cure of many hepatic illnesses.
ACKNOWLEDGMENTS
The authors wish to acknowledge Dr. Abdullah Ely for his assistance in the preparation of the manuscript.
Footnotes
P- Reviewer: Ampuero J, Mikolasevic I, Rippe RA S- Editor: Gong XM L- Editor: A E- Editor: Liu SQ
Supported by The South African National Research Foundation (NRF, GUNs 81768, 81692, 68339, 85981 and 77954); Poliomyelitis Research Foundation; Claude Leon Foundation (SAN); The University of the Witwatersrand Research Council (BM) and Medical Research Council.
Conflict-of-interest: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 13, 2014
First decision: September 28, 2014
Article in press: January 20, 2015
References
- 1.Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–526. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs F, Gordts SC, Muthuramu I, De Geest B. The liver as a target organ for gene therapy: state of the art, challenges, and future perspectives. Pharmaceuticals (Basel) 2012;5:1372–1392. doi: 10.3390/ph5121372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blum HE. Molecular therapy and prevention of liver diseases. Adv Med Sci. 2007;52:29–36. [PubMed] [Google Scholar]
- 4.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crigler JF, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169–180. [PubMed] [Google Scholar]
- 6.Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci USA. 1977;74:4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 8.Graham JB, Miller CH, Reisner HM, Elston RC, Olive JA. The phenotypic range of hemophilia A carriers. Am J Hum Genet. 1976;28:482–488. [PMC free article] [PubMed] [Google Scholar]
- 9.Yang MY, Ragni MV. Clinical manifestations and management of labor and delivery in women with factor IX deficiency. Haemophilia. 2004;10:483–490. doi: 10.1111/j.1365-2516.2004.00946.x. [DOI] [PubMed] [Google Scholar]
- 10.Wahlberg T. Carriers and noncarriers of haemophilia A. Evaluation of bleeding symptoms registered by a self-administered questionnaire with binary (no/yes) questions. Thromb Res. 1982;25:415–422. doi: 10.1016/0049-3848(82)90131-1. [DOI] [PubMed] [Google Scholar]
- 11.McMahon BJ. Chronic hepatitis B virus infection. Med Clin North Am. 2014;98:39–54. doi: 10.1016/j.mcna.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364:2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- 13.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 14.Maeder ML, Linder SJ, Reyon D, Angstman JF, Fu Y, Sander JD, Joung JK. Robust, synergistic regulation of human gene expression using TALE activators. Nat Methods. 2013;10:243–245. doi: 10.1038/nmeth.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoddard BL. Homing endonucleases from mobile group I introns: discovery to genome engineering. Mob DNA. 2014;5:7. doi: 10.1186/1759-8753-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bikard D, Marraffini LA. Control of gene expression by CRISPR-Cas systems. F1000Prime Rep. 2013;5:47. doi: 10.12703/P5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong J, Li W, Dong A, Mao S, Shen L, Li S, Gong X, Wu P. Gene therapy for unresectable hepatocellular carcinoma using recombinant human adenovirus type 5. Med Oncol. 2014;31:95. doi: 10.1007/s12032-014-0095-4. [DOI] [PubMed] [Google Scholar]
- 19.Bortolussi G, Zentillin L, Vaníkova J, Bockor L, Bellarosa C, Mancarella A, Vianello E, Tiribelli C, Giacca M, Vitek L, et al. Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum Gene Ther. 2014;25:844–855. doi: 10.1089/hum.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossman M, Raper SE, Kozarsky K, Stein EA, Engelhardt JF, Muller D, Lupien PJ, Wilson JM. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat Genet. 1994;6:335–341. doi: 10.1038/ng0494-335. [DOI] [PubMed] [Google Scholar]
- 21.Mahlangu J, Powell JS, Ragni MV, Chowdary P, Josephson NC, Pabinger I, Hanabusa H, Gupta N, Kulkarni R, Fogarty P, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317–325. doi: 10.1182/blood-2013-10-529974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ely A, Naidoo T, Mufamadi S, Crowther C, Arbuthnot P. Expressed anti-HBV primary microRNA shuttles inhibit viral replication efficiently in vitro and in vivo. Mol Ther. 2008;16:1105–1112. doi: 10.1038/mt.2008.82. [DOI] [PubMed] [Google Scholar]
- 23.Yoon SK, Seo YB, Im SJ, Bae SH, Song MJ, You CR, Jang JW, Yang SH, Suh YS, Song JS, et al. Safety and immunogenicity of therapeutic DNA vaccine with antiviral drug in chronic HBV patients and its immunogenicity in mice. Liver Int. 2015;35:805–815. doi: 10.1111/liv.12530. [DOI] [PubMed] [Google Scholar]
- 24.Arrowhead Research. Arrowhead begins dosing in phase 2a trial of rnai therapeutic arc-520 in chronic hepatitis b patients. [Updated 2014 March 24]. Available from: http: //www.arrowheadresearch.com/press-releases/arrowhead-begins-dosing-phase-2a-trial-rnai-therapeutic-arc-520-chronic-hepatitis-b.
- 25.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29:196–201. doi: 10.1042/0300-5127:0290196. [DOI] [PubMed] [Google Scholar]
- 27.Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annu Rev Genomics Hum Genet. 2013;14:135–158. doi: 10.1146/annurev-genom-091212-153435. [DOI] [PubMed] [Google Scholar]
- 28.Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22:539–548. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol. 2013;31:251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- 30.Arnould S, Chames P, Perez C, Lacroix E, Duclert A, Epinat JC, Stricher F, Petit AS, Patin A, Guillier S, et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol. 2006;355:443–458. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 31.Aubert M, Boyle NM, Stone D, Stensland L, Huang ML, Magaret AS, Galetto R, Rawlings DJ, Scharenberg AM, Jerome KR. In vitro Inactivation of Latent HSV by Targeted Mutagenesis Using an HSV-specific Homing Endonuclease. Mol Ther Nucleic Acids. 2014;3:e146. doi: 10.1038/mtna.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 33.Laity JH, Dyson HJ, Wright PE. DNA-induced alpha-helix capping in conserved linker sequences is a determinant of binding affinity in Cys(2)-His(2) zinc fingers. J Mol Biol. 2000;295:719–727. doi: 10.1006/jmbi.1999.3406. [DOI] [PubMed] [Google Scholar]
- 34.Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF, Segal DJ, Weitzman MD, Cathomen T. Custom zinc-finger nucleases for use in human cells. Mol Ther. 2005;12:610–617. doi: 10.1016/j.ymthe.2005.06.094. [DOI] [PubMed] [Google Scholar]
- 35.Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, et al. Highly active zinc-finger nucleases by extended modular assembly. Genome Res. 2013;23:530–538. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez CL, Foley JE, Wright DA, Müller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4:1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HJ, Kim E, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, Zheng Z, Cotte J, Carpenito C, Wood T, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. 2013;24:245–258. doi: 10.1089/hum.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boch J, Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- 44.White FF, Yang B. Host and pathogen factors controlling the rice-Xanthomonas oryzae interaction. Plant Physiol. 2009;150:1677–1686. doi: 10.1104/pp.109.139360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai J, Choi SH, Ponciano G, Leung H, Leach JE. Xanthomonas oryzae pv. oryzae avirulence genes contribute differently and specifically to pathogen aggressiveness. Mol Plant Microbe Interact. 2000;13:1322–1329. doi: 10.1094/MPMI.2000.13.12.1322. [DOI] [PubMed] [Google Scholar]
- 46.Scholze H, Boch J. TAL effectors are remote controls for gene activation. Curr Opin Microbiol. 2011;14:47–53. doi: 10.1016/j.mib.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Meckler JF, Bhakta MS, Kim MS, Ovadia R, Habrian CH, Zykovich A, Yu A, Lockwood SH, Morbitzer R, Elsäesser J, et al. Quantitative analysis of TALE-DNA interactions suggests polarity effects. Nucleic Acids Res. 2013;41:4118–4128. doi: 10.1093/nar/gkt085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 49.Römer P, Recht S, Lahaye T. A single plant resistance gene promoter engineered to recognize multiple TAL effectors from disparate pathogens. Proc Natl Acad Sci USA. 2009;106:20526–20531. doi: 10.1073/pnas.0908812106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valton J, Dupuy A, Daboussi F, Thomas S, Maréchal A, Macmaster R, Melliand K, Juillerat A, Duchateau P. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J Biol Chem. 2012;287:38427–38432. doi: 10.1074/jbc.C112.408864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun N, Liang J, Abil Z, Zhao H. Optimized TAL effector nucleases (TALENs) for use in treatment of sickle cell disease. Mol Biosyst. 2012;8:1255–1263. doi: 10.1039/c2mb05461b. [DOI] [PubMed] [Google Scholar]
- 52.Heigwer F, Kerr G, Walther N, Glaeser K, Pelz O, Breinig M, Boutros M. E-TALEN: a web tool to design TALENs for genome engineering. Nucleic Acids Res. 2013;41:e190. doi: 10.1093/nar/gkt789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morbitzer R, Elsaesser J, Hausner J, Lahaye T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011;39:5790–5799. doi: 10.1093/nar/gkr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mussolino C, Morbitzer R, Lütge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westra ER, Nilges B, van Erp PB, van der Oost J, Dame RT, Brouns SJ. Cascade-mediated binding and bending of negatively supercoiled DNA. RNA Biol. 2012;9:1134–1138. doi: 10.4161/rna.21410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mulepati S, Bailey S. Structural and biochemical analysis of nuclease domain of clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 3 (Cas3) J Biol Chem. 2011;286:31896–31903. doi: 10.1074/jbc.M111.270017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res. 2012;40:9887–9896. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dujon B. Sequence of the intron and flanking exons of the mitochondrial 21S rRNA gene of yeast strains having different alleles at the omega and rib-1 loci. Cell. 1980;20:185–197. doi: 10.1016/0092-8674(80)90246-9. [DOI] [PubMed] [Google Scholar]
- 65.Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757–3774. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA. 1994;91:6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva GH, Belfort M, Wende W, Pingoud A. From monomeric to homodimeric endonucleases and back: engineering novel specificity of LAGLIDADG enzymes. J Mol Biol. 2006;361:744–754. doi: 10.1016/j.jmb.2006.06.063. [DOI] [PubMed] [Google Scholar]
- 68.Grosse S, Huot N, Mahiet C, Arnould S, Barradeau S, Clerre DL, Chion-Sotinel I, Jacqmarcq C, Chapellier B, Ergani A, et al. Meganuclease-mediated Inhibition of HSV1 Infection in Cultured Cells. Mol Ther. 2011;19:694–702. doi: 10.1038/mt.2010.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ulge UY, Baker DA, Monnat RJ. Comprehensive computational design of mCreI homing endonuclease cleavage specificity for genome engineering. Nucleic Acids Res. 2011;39:4330–4339. doi: 10.1093/nar/gkr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Segal DJ, Beerli RR, Blancafort P, Dreier B, Effertz K, Huber A, Koksch B, Lund CV, Magnenat L, Valente D, et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry. 2003;42:2137–2148. doi: 10.1021/bi026806o. [DOI] [PubMed] [Google Scholar]
- 71.Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skowron P, Kaczorowski T, Tucholski J, Podhajska AJ. Atypical DNA-binding properties of class-IIS restriction endonucleases: evidence for recognition of the cognate sequence by a FokI monomer. Gene. 1993;125:1–10. doi: 10.1016/0378-1119(93)90738-o. [DOI] [PubMed] [Google Scholar]
- 73.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, Hu Y, Luo Z, Huang P, Wu Q, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods. 2013;10:329–331. doi: 10.1038/nmeth.2374. [DOI] [PubMed] [Google Scholar]
- 75.Certo MT, Gwiazda KS, Kuhar R, Sather B, Curinga G, Mandt T, Brault M, Lambert AR, Baxter SK, Jacoby K, et al. Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat Methods. 2012;9:973–975. doi: 10.1038/nmeth.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Orlando SJ, Santiago Y, DeKelver RC, Freyvert Y, Boydston EA, Moehle EA, Choi VM, Gopalan SM, Lou JF, Li J, et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010;38:e152. doi: 10.1093/nar/gkq512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, Collingwood TN, Frodin M, Davis GD. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bellefroid EJ, Marine JC, Matera AG, Bourguignon C, Desai T, Healy KC, Bray-Ward P, Martial JA, Ihle JN, Ward DC. Emergence of the ZNF91 Krüppel-associated box-containing zinc finger gene family in the last common ancestor of anthropoidea. Proc Natl Acad Sci USA. 1995;92:10757–10761. doi: 10.1073/pnas.92.23.10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen HJ, Rauscher FJ. Krüppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci USA. 1994;91:4509–4513. doi: 10.1073/pnas.91.10.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 81.Beerli RR, Segal DJ, Dreier B, Barbas CF. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beerli RR, Dreier B, Barbas CF. Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci USA. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beerli RR, Barbas CF. Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol. 2002;20:135–141. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- 84.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zimmerman KA, Fischer KP, Joyce MA, Tyrrell DL. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J Virol. 2008;82:8013–8021. doi: 10.1128/JVI.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947–954. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao X, Zhao Z, Guo J, Huang P, Zhu X, Zhou X, Yang Z, Zhao L, Xu L, Xu J, et al. Creation of a six-fingered artificial transcription factor that represses the hepatitis B virus HBx gene integrated into a human hepatocellular carcinoma cell line. J Biomol Screen. 2013;18:378–387. doi: 10.1177/1087057112463066. [DOI] [PubMed] [Google Scholar]
- 89.Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21:1889–1897. doi: 10.1038/mt.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sells MA, Zelent AZ, Shvartsman M, Acs G. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J Virol. 1988;62:2836–2844. doi: 10.1128/jvi.62.8.2836-2844.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen J, Zhang W, Lin J, Wang F, Wu M, Chen C, Zheng Y, Peng X, Li J, Yuan Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2014;22:303–311. doi: 10.1038/mt.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, Lin YY, Wang HY, Wang CC, Shen YC, et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tolar J, McGrath JA, Xia L, Riddle MJ, Lees CJ, Eide C, Keene DR, Liu L, Osborn MJ, Lund TC, et al. Patient-specific naturally gene-reverted induced pluripotent stem cells in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2014;134:1246–1254. doi: 10.1038/jid.2013.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Armijo E, Soto C, Davis BR. HIV/AIDS: modified stem cells in the spotlight. Cell Mol Life Sci. 2014;71:2641–2649. doi: 10.1007/s00018-014-1572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rio P, Baños R, Lombardo A, Quintana-Bustamante O, Alvarez L, Garate Z, Genovese P, Almarza E, Valeri A, Díez B, et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med. 2014;6:835–848. doi: 10.15252/emmm.201303374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordóñez A, Hannan NR, Rouhani FJ, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, Liu JO, Deng C, Ye Z, Jang YY. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology. 2013;57:2458–2468. doi: 10.1002/hep.26237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sander JD, Ramirez CL, Linder SJ, Pattanayak V, Shoresh N, Ku M, Foden JA, Reyon D, Bernstein BE, Liu DR, et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013;41:e181. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Suzuki K, Yu C, Qu J, Li M, Yao X, Yuan T, Goebl A, Tang S, Ren R, Aizawa E, et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell. 2014;15:31–36. doi: 10.1016/j.stem.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, Recchia A, Cathomen T, Gonçalves MA. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu H, Lau CH, Goh SL, Liang Q, Chen C, Du S, Phang RZ, Tay FC, Tan WK, Li Z, et al. Baculoviral transduction facilitates TALEN-mediated targeted transgene integration and Cre/LoxP cassette exchange in human-induced pluripotent stem cells. Nucleic Acids Res. 2013;41:e180. doi: 10.1093/nar/gkt721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu J, Gaj T, Patterson JT, Sirk SJ, Barbas CF. Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS One. 2014;9:e85755. doi: 10.1371/journal.pone.0085755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods. 2012;9:805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Van Tendeloo VF, Ponsaerts P, Berneman ZN. mRNA-based gene transfer as a tool for gene and cell therapy. Curr Opin Mol Ther. 2007;9:423–431. [PubMed] [Google Scholar]
- 114.Bettinger T, Carlisle RC, Read ML, Ogris M, Seymour LW. Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 2001;29:3882–3891. doi: 10.1093/nar/29.18.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim JS, Lee HW. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 2013;31:23–24. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- 116.Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao Q, Temsamani J, Iadarola PL, Jiang Z, Agrawal S. Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem Pharmacol. 1996;51:173–182. doi: 10.1016/0006-2952(95)02177-9. [DOI] [PubMed] [Google Scholar]
- 118.Bessis N, GarciaCozar FJ, Boissier MC. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 2004;11 Suppl 1:S10–S17. doi: 10.1038/sj.gt.3302364. [DOI] [PubMed] [Google Scholar]
- 119.Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286:2244–2245. doi: 10.1126/science.286.5448.2244. [DOI] [PubMed] [Google Scholar]